

Abstract
Background and Aims
β‐Caryophyllene (BCP) is a plant‐derived FDA approved food additive with anti‐inflammatory properties. Some of its beneficial effects in vivo are reported to involve activation of cannabinoid CB2 receptors that are predominantly expressed in immune cells. Here, we evaluated the translational potential of BCP using a well‐established model of chronic and binge alcohol‐induced liver injury.
Methods
In this study, we investigated the effects of BCP on liver injury induced by chronic plus binge alcohol feeding in mice in vivo by using biochemical assays, real‐time PCR and histology analyses. Serum and hepatic BCP levels were also determined by GC/MS.
Results
Chronic treatment with BCP alleviated the chronic and binge alcohol‐induced liver injury and inflammation by attenuating the pro‐inflammatory phenotypic `M1` switch of Kupffer cells and by decreasing the expression of vascular adhesion molecules intercellular adhesion molecule 1, E‐Selectin and P‐Selectin, as well as the neutrophil infiltration. It also beneficially influenced hepatic metabolic dysregulation (steatosis, protein hyperacetylation and PPAR‐α signalling). These protective effects of BCP against alcohol‐induced liver injury were attenuated in CB2 receptor knockout mice, indicating that the beneficial effects of this natural product in liver injury involve activation of these receptors. Following acute or chronic administration, BCP was detectable both in the serum and liver tissue homogenates but not in the brain.
Conclusions
Given the safety of BCP in humans, this food additive has a high translational potential in treating or preventing hepatic injury associated with oxidative stress, inflammation and steatosis.
Linked Articles
This article is part of a themed section on Inventing New Therapies Without Reinventing the Wheel: The Power of Drug Repurposing. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.2/issuetoc
Abbreviations
- 4‐HNE
4‐hydroxynonenal
- ALT
alanine aminotransferase
- Arg1
arginase 1
- BCP
β‐caryophyllene
- Clec7A
C‐type lectin domain family 7 member A
- F4/80
EGF‐like module‐containing mucin‐like hormone receptor‐like 1
- FAAH
fatty acid amide hydrolase
- Iba‐1
ionized calcium‐binding adapter molecule 1
- ICAM‐1
intercellular adhesion molecule 1
- LY6G
lymphocyte antigen 6 complex locus G6D
- NIAAA
National Institute on Alcohol Abuse and Alcoholism
- TBS‐T
Tris‐buffered saline with Tween 20
Tables of Links
| TARGETS |
|---|
| GPCRs a |
| CB1 receptors |
| CB2 receptors |
| Nuclear hormone receptors b |
| PPAR‐α |
| Enzymes c |
| 3‐Hydroxy‐3‐methylglutaryl‐CoA synthase 2 |
| Arginase I |
| COX‐2 |
| FAAH, fatty acid amide hydrolase |
| SIRT1, sirtuin‐1 |
| LIGANDS |
|---|
| 2‐Arachidonoylglycerol |
| Anandamide |
| Arachidonic acid |
| CCL2 |
| CCL4 |
| CXCL2 |
| ICAM1, intercellular adhesion molecule 1 |
| IL‐1β |
| IL‐6 |
| IL‐10 |
| N‐ Oleoylethanolamide |
| TNF‐α |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a,b,c).
Introduction
β‐Caryophyllene (BCP) is a bicyclic sesquiterpene found in larger amounts in numerous essential oils of food plants from cloves, basil and black pepper. Moreover, BCP is found in copaiba (Copaifera spp.) and marijuana/hemp (Cannabis spp.), which have been used in traditional medicine for centuries due to their anti‐inflammatory and analgesic effects (Gertsch et al., 2010). Due to its favourable taste and scent and apparent lack of toxicity, BCP is approved by the FDA as a food additive for flavouring. BCP, which is devoid of psychoactive effects, has been demonstrated to activate cannabinoid CB2 receptors, which are primarily expressed in immune and immune‐derived cells. This makes BCP a promising food‐derived agent that may be exploited therapeutically to treat various inflammatory diseases (Gertsch et al., 2008). BCP has been reported to exert protective effects in experimental animal models of inflammatory pain (Gertsch et al., 2008), kidney injury (Horvath et al., 2012b), ischaemic stroke (Choi et al., 2013), Parkinson's disease (Ojha et al., 2016), toxic hepatitis (D‐galactosamine‐ and endotoxin‐induced) (Cho et al., 2015), experimental liver fibrosis (Mahmoud et al., 2014) and colitis (Bento et al., 2011). BCP has been also proposed recently, to have anti‐addictive potential (Al Mansouri et al., 2014). BCP has been reported to act on targets other than CB2 receptors, including sirtuin 1 (SIRT‐1; Zheng et al., 2013), PPAR‐α (Wu et al., 2014), fatty acid amide hydrolase (FAAH) or COX‐2 (Chicca et al., 2014).
Endocannabinoids and cannabinoid receptor signalling play a central role in the development of liver diseases by influencing pivotal inflammatory and metabolic pathways (Teixeira‐Clerc et al., 2010; Tam et al., 2011; Silvestri and Di Marzo, 2013). Activation of hepatic CB1 receptors by endocannabinoids or synthetic ligands promotes alcoholic‐ (Jeong et al., 2008) and non‐alcoholic steatohepatitis (Osei‐Hyiaman et al., 2005; Tam et al., 2012), liver injury (Horvath et al., 2012a; Cao et al., 2013) and fibrosis (Teixeira‐Clerc et al., 2006). In contrast, CB2 receptor activation has tissue protective, anti‐inflammatory and antifibrotic effects in preclinical models of liver injury, inflammation and fibrosis (Batkai et al., 2007; Teixeira‐Clerc et al., 2010; Louvet et al., 2011; Horvath et al., 2012a; Cao et al., 2013). However, despite the promise of selective CB2 receptor agonists in liver disease based on preclinical studies, no such agonists are available suitable for human testing in liver disease to date. Unlike the potent synthetic CB2 receptor agonists currently used in animal models, the phytochemical BCP could be more readily tested in humans as it is a FDA approved food additive, thus having immediate translational potential.
Inflammation plays a crucial role in the development and progression of alcoholic liver disease. In this study, we investigated if BCP treatment exerts beneficial effects against liver injury and inflammation induced by chronic plus binge ethanol feeding in mice, and whether these effects were mediated via CB2 receptors.
Methods
Animals and chemicals
All animal care and experimental protocols conformed to the National Institutes of Health (NIH) guidelines and were approved by the Institutional Animal Care and use Committee of the National Institute on Alcohol Abuse and Alcoholism (Bethesda, MD). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015). Ten‐week‐old male C57BL/6J mice were obtained from the Jackson Laboratory (Bar Harbor, ME). Male CB2−/− mice on C57BL/6J background and their wild‐type controls (CB2+/+) were used in the study (termed CB2−/− and CB2+/+ mice).
(E)‐BCP was obtained as previously described (Gertsch et al., 2008). Analytical measurements by GC–MS showed that it was 95% pure with β‐caryophyllene oxide and α‐humulene as the major impurities.
Alcoholic steatohepatitis model
Male C57BL/6J mice, weighing more than 20 g were used for ad libitum ethanol feeding, as described as the chronic plus binge alcohol feeding‐induced steatohepatitis model used and developed at the National Institute on Alcohol Abuse and Alcoholism (NIAAA) (Bertola et al., 2013a). Lieber‐DeCarli ‘82 Shake and Pour control liquid diet (Bio‐Serv, product no. F1259SP) and Lieber‐DeCarli ‘82 Shake and Pour ethanol liquid diet (Bio‐Serv, product no. F1258SP) were used for diet preparation. Mice were fed liquid control diet (Bio‐Serv, Frenchtown, NJ) for 5 days, and from day 5, mice were switched either to a liquid diet containing 5% ethanol for 10 days, or were pair‐fed a control diet for 10 days. BCP (10 mg·kg−1 dose dissolved in DMSO‐Tween‐saline in a ratio of 1:1:18) or vehicle were administered i.p. every day. At day 11, mice in the ethanol groups were gavaged with a single dose of ethanol (5 g·kg−1 body weight, 30% ethanol), whereas the mice in the control groups were gavaged with isocaloric maltodextrin solution. All mice were killed 9 h after gavage.
Determination of BCP pharmacokinetics in vivo
We studied the pharmacokinetic properties of BCP, in two set of experiments. To test the effect of chronic BCP treatment and the potential influence of ethanol feeding, in a separate set of experiments, BCP (10 mg·kg−1·day−1 dose dissolved in DMSO‐Tween‐saline in a ratio of 1:1:18) or vehicle were administered i.p. to male C57BL/6J mice for 10 days in accordance with our Lieber‐DeCarli ethanol feeding protocol, described above. Liver, brain and serum samples were collected at 30, 60, 120, and 360 min after the administration of the last dose of BCP.
In a separate set of experiment BCP (10 mg/kg single dose dissolved in DMSO‐Tween‐Saline in a ratio of 1:1:18) or vehicle were administered i.p. or orally to male C57BL/6J mice and serum samples were collected at 30, 60, 120 and 360 min after drug administration.
BCP was quantified in liver, kidney, brain and serum. Snap‐frozen tissues were weighed and transferred into a 2 mL tube containing three chrome‐steel beads and 0.1 M formic acid and homogenized using a mini bead beater (except for the serum). An aliquot of the homogenized tissues (100 μL) was rapidly transferred into plastic tube containing 90 μL of ethyl acetate and 10 μL of α‐humulene (used as internal standard), strongly vortexed for 30 s and sonicated in ice‐cold bath for 5 min. Then, samples were centrifuged at 10 000 × g for 10 min at 4°C and kept for 1 h at −20°C to facilitate the recovery of the upper organic phase. Samples were analysed by gas chromatography (GC)/electron ionization (EI)‐mass spectrometry using an Agilent 6890 N GC equipped with a 30 m HP‐5MS column and a 5975 C EI‐MS with triple‐axis detector. As carrier gas, helium was used at a constant flow rate of 1.0 mL·min−1 with splitless injection. Separation of BCP and its internal standard was achieved with the following oven programme: initial temp. of 100°C followed by an increase to 120°C at 15°C min−1, kept for 0.5 min before increasing to 180°C at 7°C min−1. Oven temperature was finally increased to 310°C at 20°C min−1 for a total time of 18.9 min. The following specific ions were used for selected ion monitoring: m/z 93 for α‐humulene and m/z 91 for BCP.
Determination of liver injury
After venous blood collection, serum was prepared (centrifugation for 10 min at 2500x g) immediately followed by the determination of the serum levels of alanine aminotransferase (ALT) using a clinical chemistry analyser – Idexx VetTest 8008 (Idexx Laboratories, Westbrook, ME, USA).
Reverse transcription and real‐time PCR
Hepatic tissues were homogenized in Trizol (Invitrogen, Carlsbad, CA, USA), and total RNA was isolated with Direct‐zol™ RNA MiniPrep Kit (Zymo Research, Irvine, CA, USA). All RNA samples were DNAse digested, and RNA concentration was measured with NanoDrop (Thermo Scientific, Waltham, MA USA). Approximately 2 μg RNA was reverse‐transcribed (High‐Capacity cDNA Reverse Transcription Kit, Applied Biosystems, Foster City, CA, USA), and the target genes were amplified using the standard SyberGreen based real‐time PCR kit (SYBR® Select Master Mix, Applied Biosystems, Foster City, CA). Primers sequences are provided in Supplementary Table S1.
Histology and immunohistochemistry
After routine formalin‐fixed, paraffin‐embedded tissue sample specimen processing, 5 μm thick liver sections were prepared and stained with haematoxylin and eosin for histological evaluation of liver injury.
For immunohistochemistry, deparaffinized sections underwent antigen retrieval (pH = 6 citrate buffer, at 95°C for 10 min or Proteinase K (20 μg·mL−1 in Tris‐EDTA buffer pH = 8) digestion for F4/80 staining, at 37°C for 15 min followed by 10 min additional digestion at room temperature). After blocking endogenous peroxidase activity (3% H2O2 solution in PBS), the sections were blocked in appropriate sera (2.5% goat, or horse serum in PBS and 2% milk powder or bovine serum albumin). Primary antibodies (4‐hydroxynonenal (4‐HNE) (Japan Institute for the Control of Aging, Nikken SEIL Co., Fukuroi, Shizuoka, Japan), Iba‐1 (Wako Pure Chemical Industries, Chuo‐Ku, Osaka, Japan), F4/80 (eBioscience, San Diego, CA, USA) and Ly6‐G (Abcam, Cambridge, MA, USA)) were incubated with the sections overnight in diluted blocking solution at 4°C. After primary antibody incubations, the sections were washed three times in PBS and incubated for an hour either with biotinylated secondary antibody (Vectastain ABC kit, Vector Laboratories, Burlingame, CA, USA) or with an anti‐rat IgG/anti‐rabbit IgG conjugated with a peroxidase polymer (ImmPress reagents, Vector Laboratories, Burlingame, CA, USA). Secondary antibodies were washed three times for 10 min, and the specific signal was developed with diaminobenzidine (ImmPACT DAB EqV Peroxidase (HRP) Substrate, Vector Laboratories, Burlingame, CA, USA). The specific staining was visualized and images were acquired using BX‐41 microscope (Olympus, Tokyo, Japan).
For confocal imaging, the sections were incubated with a goat anti‐rabbit IgG secondary antibody, conjugated to Alexa Fluor® 594 or Alexa Fluor® 488 (Thermo Scientific, Waltham, MA USA). Nuclei were stained with the far‐red emitting DRAQ5 stain (Cell Signalling Technology, Danvers, MA, USA) and visualized under a Zeiss LSM710 confocal microscope (Jena, Germany).
Liver samples embedded in optimal cutting temperature compound were cut (10 μm) and stained with Oil Red O (Sigma‐Aldrich, St. Louis, MO, USA) dissolved in isopropanol to evaluate hepatic lipid accumulation.
Determination of hepatic triglyceride content
Triglyceride content was measured from frozen liver tissues by the Triglyceride Quantification Colorimetric Kit (Biovision, San Francisco, CA, USA) according to the manufacturer's instructions.
Determination of hepatic endocannabinoid content
Endocannabinoids were measured from frozen liver tissues by stable isotope dilution liquid chromatography/tandem mass spectrometry (LC–MS/MS) as described previously (Mukhopadhyay et al., 2011).
Western blots
Frozen liver samples were homogenized in RIPA lysis buffer (150 mM NaCl, 50 mM Tris, 1%NP‐40). Protein concentrations were determined by the bicinchoninic acid method, using bovine serum albumin as standard (Pierce, Rockford, USA). 20 μg of protein was loaded from each sample onto 4–20% polyacrylamide gel. After separation by electrophoresis, proteins were transferred (Trans‐Blot Turbo Blotting System, BioRad, Hercules, CA, USA) onto the PVDF membrane. Successful transfer was controlled by using Ponceau dye. The membrane was blocked with 5% non‐fat dry milk in 0.05% Tris‐buffered saline with Tween 20 (TBS‐T) for 1 h at room temperature. After the blocking step, the membrane was incubated with a primary antibody (dissolved in 1% non‐fat dry milk–TBS‐T, 1:1000 dilution) against either SIRT‐1 (Cell Signalling Technology, Danvers, MA, USA), PPAR‐α (Abcam, Cambridge, MA, USA) or acetyl‐lysine (Cell Signalling Technology, Danvers, MA, USA) for 2 h at room temperature, followed by washing with 0.05% TBS‐T (3 × 10 min). After washing, the membrane was incubated with a secondary antibody (horseradish peroxidase‐conjugated affinity purified goat anti‐rabbit, 1/5000 dilution) in 1% non‐fat dry milk in TBS‐T for 1 h at room temperature. Then the membrane was washed again three times for 10 min. For detection of the bands, the membrane was incubated with enhanced chemiluminescence reagent (SuperSignal West Pico Substrate, Thermo Scientific ‐ Pierce, Rockford, IL, USA) for 5 min and the signal was recorded in a gel documentation system (Versadoc 4000MP, Bio‐Rad Imaging System, Hercules, USA). Band densities were evaluated by using Quantity One software (Bio‐Rad Imaging System, Hercules, USA). Loading control was done by determining the actin content of each sample. Briefly, after stripping the membrane, it was probed with a primary antibody directly conjugated with HRP that recognizes actin (1/10000 dilution – Abcam, Cambridge, MA, USA) for 1 h at room temperature, followed by washing with TBS‐T. Actin band visualization and evaluation of band densities were done as described above. There was no significant difference in actin between the groups.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All the values are presented as mean ± SEM. Statistical analysis of the data was performed by one or two‐way ANOVA followed by Tukey's post hoc test for multiple comparisons, as appropriate. The analysis was conducted using GraphPad‐Prism4 software. P < 0.05 was considered statistically significant.
Results
Treatment with BCP protects against alcoholic steatohepatitis
We aimed to study the potential hepatoprotective action of BCP in setting of alcoholic liver disease. We employed the chronic plus binge ethanol feeding model, developed in the National Institute on Alcohol Abuse and Alcoholism (Bertola et al., 2013a), resembling major pathological features of early alcoholic liver disease, involving hepatocyte injury, pro‐inflammatory alterations and steatosis. BCP was administered daily during the course of the Liber‐DeCarli alcohol feeding protocol and at the end of the feeding period it was given 1 h prior to binge ethanol treatment (Figure 1A). BCP treatment significantly alleviated binge alcohol‐induced liver injury, as reflected by a decrease in serum ALT levels (Figure 1B), and normalized the histopathological signs of alcoholic steatohepatitis (ballooning of hepatocytes, microvesicular steatosis and neutrophil inflammatory infiltrates/loci) (Figure 1C), and attenuated oxidative tissue injury, as reflected by reduced amount of 4‐hydroxy‐nonenal accumulation (Figure 1D).
Figure 1.
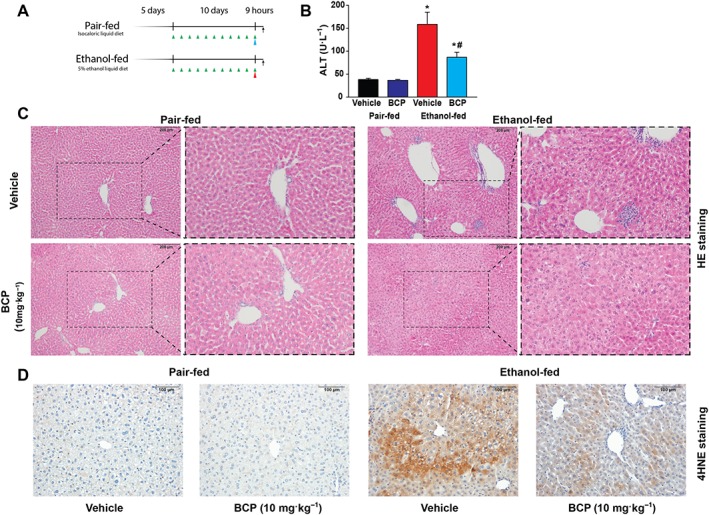
Protective effect of BCP treatment against chronic plus binge ethanol feeding induced hepatic injury. (A) Experimental alcohol‐ and isocaloric pair‐feeding protocol combined with single ethanol/maltodextrin binge at the end of the 10 days long diet. Green arrows indicate daily drug treatments (vehicle or 10 mg·kg−1 BCP). Red arrow represents single ethanol binge treatment, while the blue arrow represents an isocaloric maltodextrin gavage. (B) Determination of liver injury by measurement of ALT enzyme activity, and histological assessment of hepatic pathological alterations (C) on haematoxylin–eosin stained liver sections in vehicle or BCP treated pair‐fed or ethanol‐fed mice respectively. (D) Assessment of oxidative stress in vehicle or BCP treated pair‐fed or ethanol‐fed mice, by histological staining for 4‐hydroxy‐nonenal. Results are mean ± SEM, n = 8. *P < 0.05, significantly different from pair‐fed with vehicle treatment, #P < 0.05, significantly different from ethanol‐fed with vehicle pretreatment.
Treatment with BCP prevents the pro‐inflammatory phenotypic switch of hepatic macrophages upon chronic plus binge ethanol feeding
Hepatic macrophage population undergoes a major phenotypic switch during the course of alcohol feeding (Wang et al., 2014). In a healthy control liver, a mixed population of larger and smaller Kupffer cell population is present: larger, spindle‐shaped peri‐sinusoidal macrophages are involved in phagocytosis and are less prone to activation, while the smaller, round‐shaped peri‐central macrophage population is more prone to activation and pro‐inflammatory cytokine and reactive oxygen species production (Laskin et al., 2001). To characterize macrophage morphology, we employed the pan‐macrophage marker Iba‐1 (Rehg et al., 2012). As an actin cross‐linking protein, being involved in cytoskeletal reorganization, immunohistochemical detection of Iba‐1 gives a sharp and detailed picture of macrophage morphology (Figure 2A) (Kohler, 2007) in comparison to staining for cell membrane‐specific markers F4/80 or CD68 (Figure 2B and Supporting Information Fig. S1). Accordingly, we detected the presence of the large Kupffer cells along the hepatic sinuses with arborized morphology both in the vehicle and BCP treated pair‐fed groups (Figure 2A). However, upon ethanol‐feeding we detected small, round‐shaped macrophages with less arborized morphology, resembling `M1`‐type of morphology. BCP treatment partly prevented this morphological switch: large macrophages with arborization were present along the hepatic sinusoids (Figure 2A,B).
Figure 2.
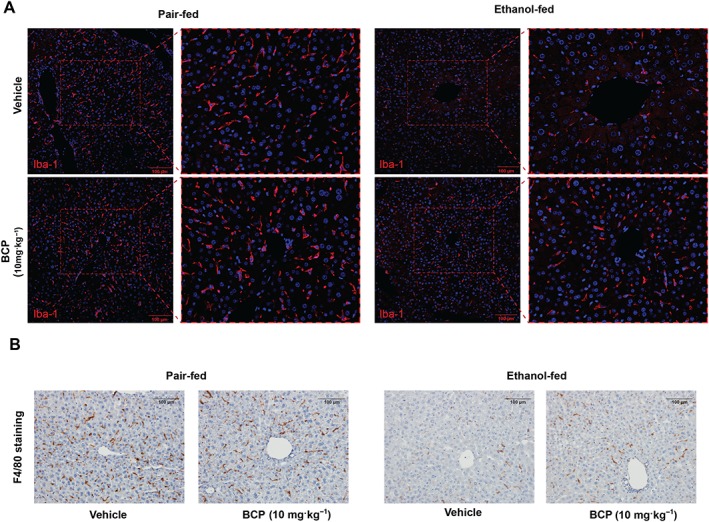
BCP prevents chronic plus binge ethanol feeding‐induced morphological changes in hepatic macrophages. (A) Representative confocal scanning microscopic images of Iba‐1 positive hepatic macrophages from pair‐fed and ethanol‐fed mice either treated with vehicle or BCP (10 mg·kg−1). (B) Representative light microscopic images of F4/80 positive hepatic macrophages from pair‐fed and ethanol‐fed mice either treated with vehicle or BCP (10 mg·kg−1).
In line with the above observations, mRNA markers of tissue‐resident macrophages, F4/80, CD68 and Iba‐1 were down‐regulated due to chronic plus binge ethanol feeding, an effect that was mitigated by BCP treatment (Figure 3A). In addition CD11b expression, a marker of pro‐inflammatory lymphocytes (mainly monocytes and macrophages) showed a striking increase in the ethanol‐fed groups and this effect was attenuated by BCP treatment (Figure 3A).
Figure 3.
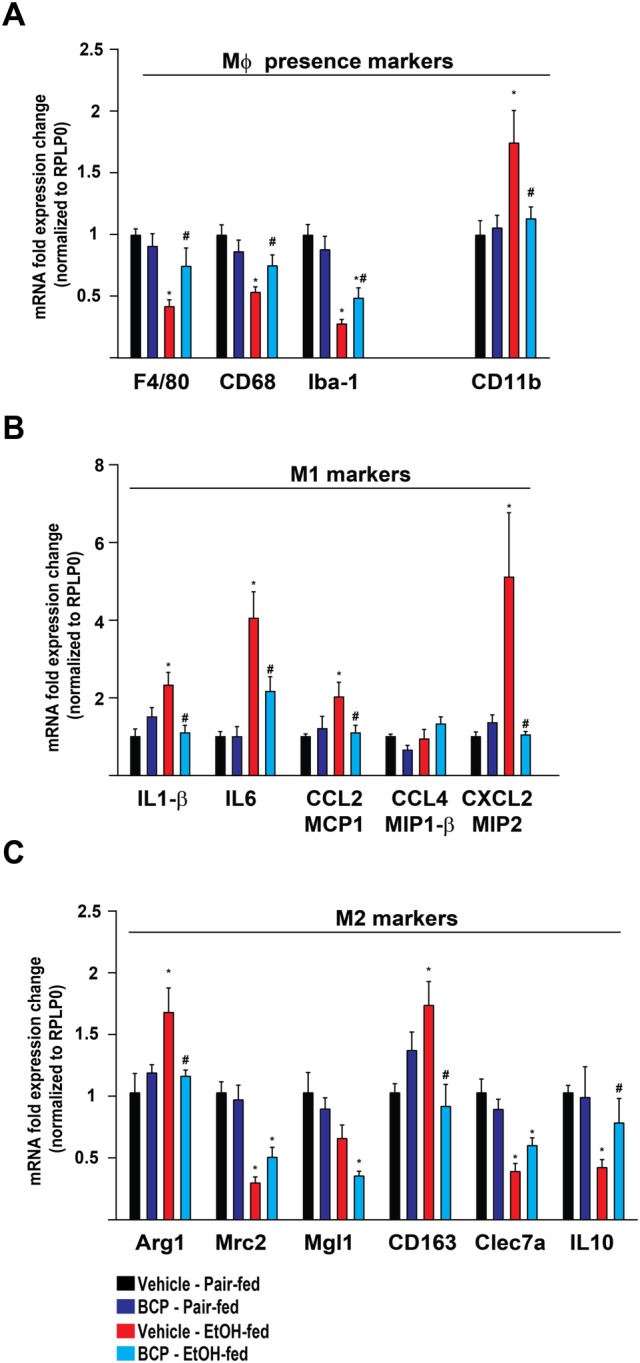
BCP prevents chronic plus binge ethanol feeding‐induced pro‐inflammatory phenotypic switch of hepatic macrophages. (A) Gene expression of classic macrophage markers, F4/80, CD68, Iba‐1 and CD11b in the liver of pair‐fed and ethanol‐fed mice either treated with vehicle or BCP (10 mg·kg−1). (B) Gene expression of pro‐inflammatory macrophage activation markers (M1 phenotype), IL‐1‐β, IL‐6, CCL2, CCL4 and CXCL2 in the liver of pair‐fed and ethanol‐fed mice either treated with vehicle or BCP (10 mg·kg−1). (C) Gene expression of alternative macrophage activation markers (M2 phenotype), Arg‐1, Mrc‐2, Mgl‐1, CD163, Clec7a and IL‐10 in the liver of pair‐fed and ethanol‐fed mice either treated with vehicle or BCP (10 mg·kg−1). Results are mean ± SEM, n = 6. *P < 0.05, significantly different from pair‐fed with vehicle treatment, #P < 0.05, significantly different from ethanol‐fed with vehicle pretreatment.
Ethanol‐fed mice displayed significant induction of `M1` markers of macrophage activation, including IL1ß, IL‐6 and the chemokines, CCL2 and CXCL2. In addition, there was also a parallel induction of genes characteristic of an alternative `M2` activation signature, such as up‐regulation of arginase 1 (Arg1) and CD163, both attenuated by BCP treatment (Figure 3B). Other markers of `M2` activation, like mannose receptor C type 2, macrophage galactose‐type C‐type lectin 1, C‐type lectin domain family 7 member A (Clec7a) and IL‐10 showed down‐regulation that was attenuated by BCP treatment only in the case of IL‐10 (Figure 3C). These findings show that chronic alcohol feeding promotes polarization of Kupffer cells towards a mixed M1/M2 phenotype, an effect that is minimized by BCP treatment mainly in the case of `M1` activation.
Treatment with BCP prevents vascular inflammation and subsequent hepatic neutrophil infiltration due to chronic plus binge ethanol feeding
Neutrophil infiltration is a key pathological finding in alcoholic hepatitis and has been shown to closely correlate with the severity of alcoholic hepatic injury (Dominguez et al., 2009; Bertola et al., 2013b). Immunohistochemical staining for the neutrophil marker Ly6G confirmed that a large number of neutrophils had infiltrated the livers of chronic plus binge ethanol‐fed mice, compared with control pair‐fed mice. This effect of ethanol feeding was largely attenuated by BCP treatment (Figure 4A,B). Chronic plus binge ethanol feeding also led to a marked up‐regulation of hepatic mRNA expression of the neutrophil marker, Ly6G (Figure 4C).
Figure 4.
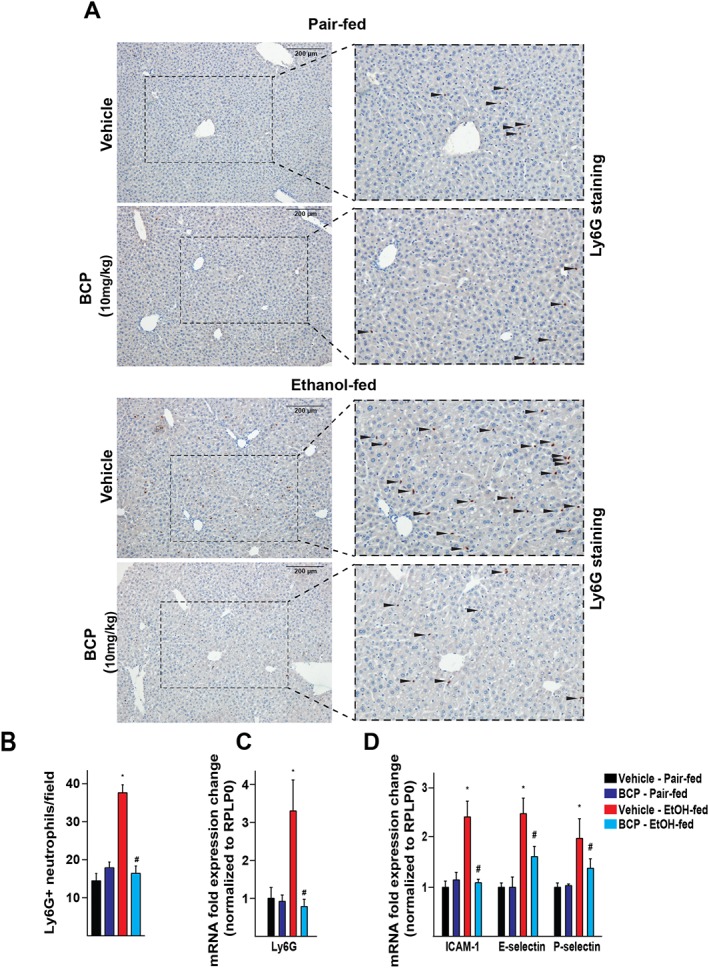
BCP prevents chronic plus binge ethanol feeding‐induced hepatic vascular inflammation and neutrophil infiltration. (A) Immunohistochemical detection of Ly6G positive neutrophil granulocytes from pair‐fed and ethanol‐fed mice either treated with vehicle or BCP (10 mg·kg−1). (B) Average number of neutrophil granulocytes on high power field microscopic images taken from histological sections of the livers of pair‐fed and ethanol‐fed mice either treated with vehicle or BCP (10 mg·kg−1). (C) Gene expression of the neutrophil marker Ly6G in the liver of pair‐fed and ethanol‐fed mice either treated with vehicle or BCP (10 mg·kg−1). (D) Gene expression of pro‐inflammatory vascular adhesion molecules, ICAM‐1, E‐selectin and P‐selectin in the liver of pair‐fed and ethanol‐fed mice either treated with vehicle or BCP (10 mg·kg−1). Results are mean ± SEM, n = 6. *P < 0.05, significantly different from pair‐fed with vehicle treatment, #P < 0.05, significantly different from ethanol‐fed with vehicle pretreatment.
To further investigate how chronic plus binge ethanol feeding leads to infiltration of neutrophils into the liver, hepatic expression of several vascular and tissue adhesion molecules were examined, reflecting vascular inflammatory processes. Chronic plus binge ethanol feeding resulted in the induction of ICAM‐1, E‐selectin and P‐selectin, which were significantly attenuated by BCP treatment (Figure 4D).
Treatment with BCP prevents alcoholic steatosis and preserves PPAR‐α‐dependent signalling
Daily administration of BCP prevented ethanol‐feeding‐induced development of microvesicular steatosis, as shown by Oil Red O staining on fresh frozen liver sections (Figure 5A). Quantification of liver triglycerol content confirmed the beneficial effect of BCP treatment on alcohol‐induced hepatic lipid accumulation (Figure 5B). As BCP and its metabolites have been proposed to affect COX2 and FAAH activity (Chicca et al., 2014), we aimed to measure endocannabinoids and related lipids in pair‐fed mice treated either with vehicle or BCP (10 mg·kg−1). None of the measured lipids showed changes due to BCP treatment (anandamide: 0.90 ± 0.06 fmol·mg−1 in vehicle vs. 1.12 ± 0.18 fmol·mg−1 in BCP treated livers; 2‐arachidonoylglycerol: 1.43 ± 0.23 pmol·mg−1 in vehicle vs. 1.41 ± 0.46 pmol·mg−1 in BCP treated livers; oleoylethanolamide: 13.66 ± 1.38 fmol·mg−1 in vehicle vs. 15.56 ± 1.98 fmol·mg−1 in BCP treated livers; arachidonic acid: 0.53 ± 0.03 pmol·mg−1 in vehicle vs. 0.41 ± 0.12 pmol·mg−1 in BCP treated livers), implying that the hepatoprotective effect of BCP was not related to COX2 or FAAH inhibition.
Figure 5.
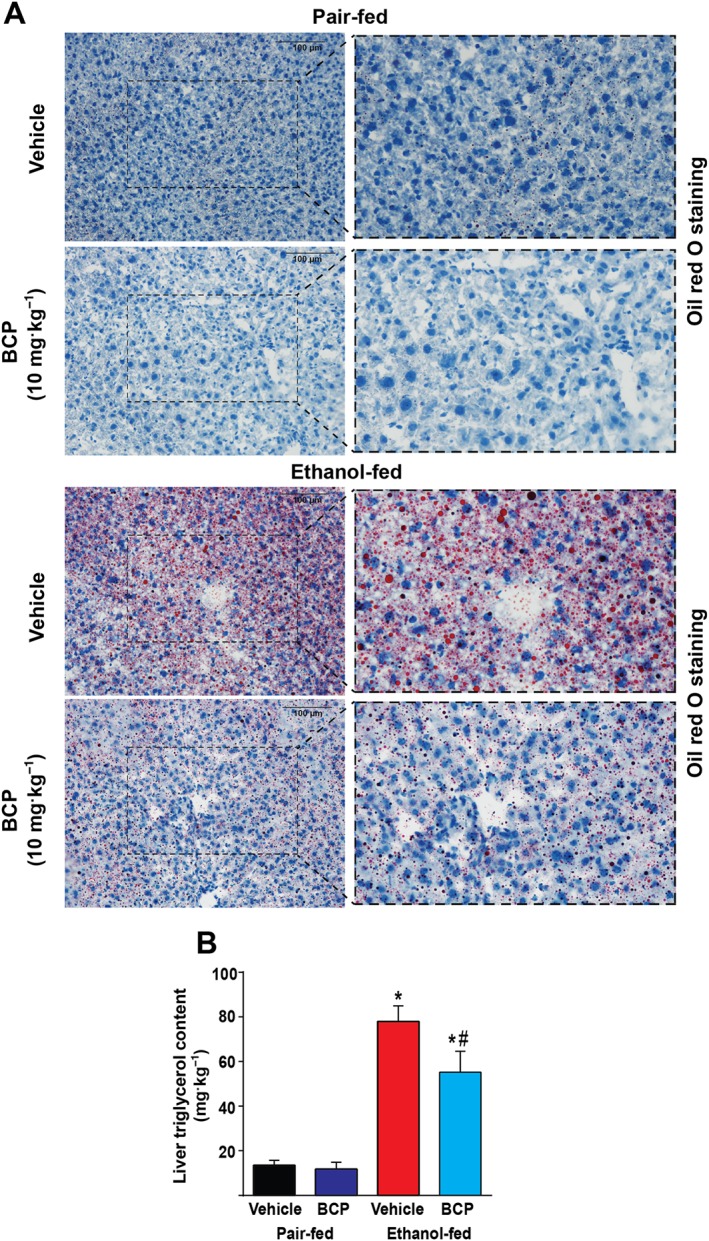
BCP prevents chronic plus binge ethanol feeding‐induced hepatic steatosis. (A) Histochemical detection of neutral lipids (Oil Red O staining) in fresh frozen liver sections of pair‐fed and ethanol‐fed mice either treated with vehicle or BCP (10 mg·kg−1). (B) Quantitative determination of triglycerol content in the livers of pair‐fed and ethanol‐fed mice either treated with vehicle or BCP (10 mg·kg−1). Results are mean ± SEM, n = 6.*P < 0.05, significantly different from pair‐fed with vehicle treatment, #P < 0.05, significantly different from ethanol‐fed with vehicle pretreatment.
Ethanol metabolism results in increased acetyl‐CoA formation, with concomitant protein‐acetylation and thereby leading to reprogramming of intermediate metabolism (Shepard and Tuma, 2009). Sirtuins (SIRT) are NAD+‐dependent protein deacetylase enzymes, capable of removing acetyl groups from proteins, and thereby preventing the development of alcoholic (Yin et al., 2014) and non‐alcoholic fatty liver disease (Li et al., 2014). In line with these, we detected an increase in overall lysine acetylation in chronic plus binge ethanol‐fed group that was attenuated by BCP treatment (Figure 6A). The increased acetylation pattern seen in ethanol‐fed livers was paralleled by a decrease in SIRT‐1 protein level; however, SIRT‐1 expression seems to be not influenced by BCP treatment.
Figure 6.
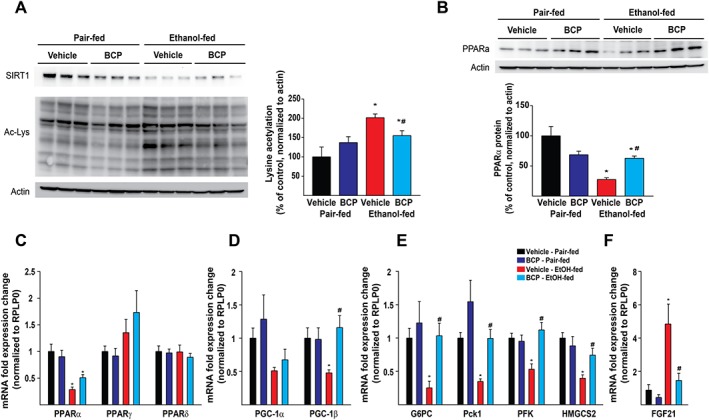
BCP prevents chronic plus binge ethanol feeding‐induced loss of PPAR‐α expression, decreases protein hyperacetylation and promotes PPAR‐α‐dependent signalling. Western blot analysis of hepatic protein lysine acetylation (A and B) and PPAR‐α protein expression (B) in the liver of pair‐fed and ethanol‐fed mice either treated with vehicle or BCP (10 mg·kg−1). (C) Gene expression of PPAR transcription factors, PPAR‐α, PPAR‐γ and PPAR‐δ in the liver of pair‐fed and ethanol‐fed mice either treated with vehicle or BCP (10 mg·kg−1). (D) Gene expression of PPAR coactivators, PPAR‐γ coactivator 1‐α (PGC‐1α), and PPAR‐γ coactivator 1‐β (PGC‐1β) in the liver of pair‐fed and ethanol‐fed mice either treated with vehicle or BCP (10 mg·kg−1). (E) Gene expression of PPAR‐α signalling‐related transcripts, glucose‐6‐phosphatase (G6PC), phosphoenolpyruvate carboxykinase 1 (Pck‐1), phosphofructokinase (PFK), 3‐hydroxy‐3‐methylglutaryl‐CoA synthase 2 (HMGCS2) and (F) FGF‐21 in the liver of pair‐fed and ethanol‐fed mice either treated with vehicle or BCP (10 mg·kg−1). Results are mean ± SEM, n = 6. *P < 0.05, significantly different from pair‐fed with vehicle treatment, #P < 0.05, significantly different from ethanol‐fed with vehicle pretreatment.
We detected a massive reduction both in protein (Figure 6B) and mRNA levels of PPAR‐α (Figure 6C) following ethanol feeding, which was mitigated by BCP treatment. In line with this observation, BCP treatment preserved the expression of PPAR‐α‐related mRNA targets (PPAR‐γ coactivator 1‐β, glucose‐6‐phosphatase, phosphoenolpyruvate carboxykinase 1, phosphofructokinase and 3‐hydroxy‐3‐methylglutaryl‐CoA synthase 2) upon chronic plus binge ethanol feeding (Figure 6D,E). Interestingly, we detected a massive ethanol‐feeding‐induced up‐regulation of a known PPAR‐α target gene, FGF21. However, this effect was markedly attenuated by BCP treatment (Figure 6F)
The beneficial effects of treatment with BCP against alcoholic steatosis are attenuated in CB2 receptor deficient mice
Because BCP has been proposed to act on potential targets (SIRT‐1, PPAR‐α and COX2) other than CB2 receptors, we tested the protective effects of BCP treatment in CB2 receptor deficient mice. We were not able to detect any marked effect of BCP treatment on macrophage morphology in ethanol‐fed CB2‐/‐ mice (Figure 7A). This was further substantiated by the analysis of expression of F4/80 and CD11b, which were not affected by BCP treatment in CB2‐/‐ mice (Figure 7B).
Figure 7.
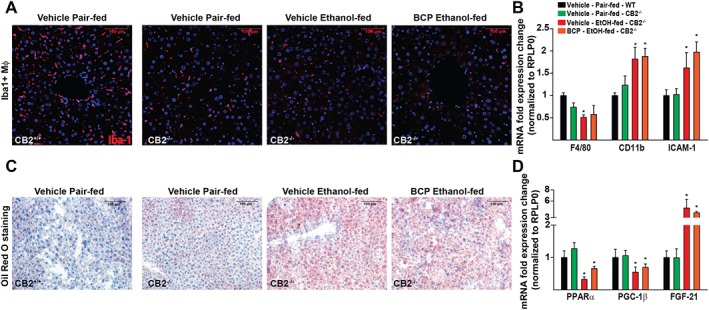
BCP‐induced protection against alcoholic steatohepatitis is attenuated in CB2‐/‐ mice. (A) Confocal scanning microscopic images of Iba‐1 positive hepatic macrophages from pair‐fed wild type mice and from ethanol‐fed CB2‐/‐ mice either treated with vehicle or BCP (10 mg·kg−1). (B) Gene expression of classic macrophage markers, F4/80, and the pro‐inflammatory vascular adhesion molecule ICAM‐1 and the monocyte marker CD11b in the liver of pair‐fed and ethanol‐fed CB2 receptor deficient mice either treated with vehicle or BCP (10 mg·kg−1). (C) Histochemical detection of neutral lipids (Oil Red O staining) in fresh frozen liver sections of pair‐fed wild type mice and from ethanol‐fed CB2‐/‐ mice either treated with vehicle or BCP (10 mg·kg−1). (D) Gene expression of PPAR‐α, PPAR‐γ coactivator 1‐β (PGC1‐β) and FGF21 in the liver of pair‐fed and ethanol‐fed CB2‐/‐ mice either treated with vehicle or BCP (10 mg·kg−1). Results are mean ± SEM, n = 6. *P < 0.05, significantly different from sham with vehicle pretreatment.
We observed mild steatosis even in control pair‐fed CB2‐/‐ mice. There was a significant induction of microvesicular steatosis in ethanol‐fed CB2‐/‐ mice; however, this was not affected by BCP treatment (Figure 7C). On examining PPAR‐α expression and PPAR‐α‐related targets, we saw that effects observed in wild type ethanol‐fed and BCP treated mice were lost in CB2‐/‐ mice (Figure 7D).
Pharmacokinetic properties of BCP
BCP reached comparable serum (Figure 8A), hepatic (Figure 8B) and brain levels (Figure 8C) in both ethanol‐fed and pair‐fed animals. BCP tissue concentrations indicated a 50‐fold higher distribution in liver compared to brain. In addition BCP showed good oral bioavailability in comparison to the used intraperitoneal administration (Figure 8D).
Figure 8.
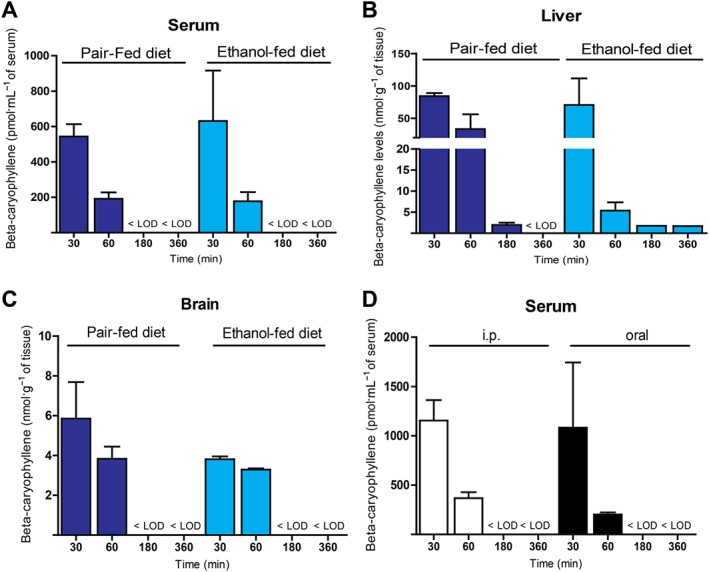
Pharmacokinetic properties of BCP. Determination of serum (A), hepatic (B), and brain (C) tissue levels of BCP 30, 60, 180 and 360 min after chronic administration (10 mg·kg−1 daily for 10 days) to either pair‐fed or ethanol‐fed animals. (D) Determination of serum levels of BCP 30, 60, 180 and 360 min after BCP administration (single dose of 10 mg·kg−1 given i.p. or orally). Results are mean ± SEM, n = 3.
Discussion
In the present study, we have demonstrated that treatment with the dietary phytochemical BCP exerted marked hepatoprotective effects in the setting of chronic liver injury induced by chronic and binge alcohol feeding in mice. We show that (i) in the chronic and binge alcohol‐induced liver injury model BCP attenuates the pro‐inflammatory phenotypic `M1` switch of Kupffer cells and (ii) BCP treatment reduces expression of tissue and vascular adhesion molecules ICAM‐1, E‐Selectin and P‐Selectin, as well as consequent neutrophil infiltration; (iii) BCP beneficially influences alcohol‐induced hepatic metabolic dysregulation (steatosis, protein hyperacetylation and PPAR‐α signalling); (iv) these protective effects of BCP involve activation of CB2 receptors.
The CB2 receptors are primarily expressed on immune and immune derived cells, including the Kupffer cells of the liver (Teixeira‐Clerc et al., 2010; Pacher and Mechoulam, 2011; Cao et al., 2013). Activation of CB2 receptors mediate anti‐inflammatory actions, limiting inflammatory and subsequent oxidative/nitrative tissue injury and organ damage (Pacher and Mechoulam, 2011).
Resident macrophages of the liver (Kupffer cells), account approximately for 10–15% of all liver cells. These cells are localized along the sinusoidal space of Disse anchored to endothelial cells by long cytoplasmic processes, being involved in maintaining tissue homeostasis and providing immunosurveillance. In addition, they also play a protective role in promoting liver regeneration (Meijer et al., 2000). Kupffer cells may become activated by a variety of stimuli that trigger innate immunity, involving endogenous molecules released during hepatocyte injury (e.g. hyaluronic acid, heparin sulfate, high‐mobility group box 1 or heat shock proteins) (Tsung et al., 2005). In addition, Kupffer cell activation might be triggered by environmental toxins, as in the case of alcoholic steatohepatitis by ethanol and its metabolites or by endotoxin, absorbed due to increased gastrointestinal permeability. Pathologically over‐activated macrophages, as seen in alcoholic‐, and in non‐alcoholic fatty liver disease, may further aggravate tissue injury and inflammation, leading to liver failure. As BCP has been proposed to protect the colonic mucosa against dextran sulfate‐induced colitis (Cho et al., 2007; Bento et al., 2011), it is possible that BCP could exert similar protection against ethanol‐induced mucosal damage and thereby also attenuate endotoxin translocation from the gut to the portal circulation (Bode et al., 1987).
Kupffer cells consist of three major subsets; the CD11b+ cells, and CD68+ cells overlapping with CD32+ cells, representing precursors of CD68+ cells (expressing stem cell markers), such as c‐kit (CD117) and CD34 (Kinoshita et al., 2010). Detailed flow cytometric and immunohistochemical studies also revealed that the CD68+ cells are large and spindle‐shaped, while the CD11b+ cells are small and round or oval‐shaped (Ikarashi et al., 2013). While large macrophages are more phagocytic and generate increased quantities of lysosomal enzymes, the smaller macrophages release more reactive oxygen species, and appear to be more susceptible to `M1`‐type activation and cytokine production. Interestingly, we found that chronic and binge alcohol feeding resulted in pro‐inflammatory phenotypic switch in liver Kupffer cells, which was attenuated by BCP treatment.
Experimental CB2 receptor agonists, such as JWH‐133, HU910 and HU308, have been shown to favourably affect overall hepatic macrophage activation upon chronic alcohol exposure (Louvet et al., 2011) and hepatocyte injury (Batkai et al., 2007; Rajesh et al., 2008; Horvath et al., 2012a). However, not a single CB2 receptor agonist has yet been approved for clinical testing in liver disease. In this study, we demonstrated that the FDA approved food additive BCP, which is present in various food and medicinal plants, as well as in cannabis essential oil, exerted hepatoprotective effects in a model of alcohol‐induced chronic liver injury. The observed protective effects were attenuated in CB2‐/‐ mice indicating involvement of CB2 receptors. To increase the translational potential of our study, we also tested the pharmacokinetic properties of oral and i.p. BCP administration both in healthy and in ethanol‐fed mice. BCP was orally bioavailable, and ethanol‐feeding did not interfere with its kinetics (e.g. absorption or metabolism). The BCP dose used in our study in mice (10 mg·kg−1) is convertible to a human equivalent dose of approximately 0.8–1 mg·kg−1·day−1 for adult human subjects (Nair and Jacob, 2016). Based on the mouse pharmacokinetics data, repeated oral dosing of BCP might be desirable, the benefit of which remains to be tested.
Our results also suggest that BCP has beneficial properties on intermediary metabolism in ethanol‐fed animals, potentially by decreasing pro‐inflammatory cytokine expression in a CB2 receptor‐dependent manner and thereby preserving PPAR‐α‐related signalling that is characteristic for the healthy liver.
An intriguing field of investigation is how pro‐inflammatory alterations influence hepatic intermediary metabolism. It is now well established that pro‐inflammatory cytokines (IL‐1ß or TNFα) may profoundly affect hepatic lipid and glucose metabolism in a paracrine manner (Stienstra et al., 2010; Louvet et al., 2011). IL‐1ß suppressed human and mouse PPAR‐α promoter activity and in parallel interfered with the ability of PPAR‐α to activate transcription of target genes (Stienstra et al., 2010). We found that alcohol markedly impaired PPAR‐α signalling and increased lipid accumulation in the liver, which were significantly attenuated by BCP treatment; however, the effects seem to relate to BCP‐induced activation of CB2 receptors rather than to a direct PPAR‐α mediated effect.
During hepatic ethanol metabolism, liver mitochondria convert acetate into acetyl‐CoA that is further processed in the citric acid cycle. However, due to ethanol metabolism, there is an increased level of NADH that inhibits further metabolism of the acetyl‐CoA by the citric acid cycle. In addition, excess NADH inhibits gluconeogenesis by preventing the oxidation of lactate to pyruvate, leading to accumulation of lactate (lactic acidosis) and causing hypoglycemia (Tsai et al., 2015). Further consequences of hepatic acetyl‐CoA accumulation involves increased protein acetylation (histones, transcription factors – sterol regulatory element‐binding protein 1, forkhead box protein O1, forkhead box O3 and PPAR‐γ coactivator 1‐α; Shepard and Tuma, 2009), ketone body formation, increased fatty acid synthesis and fat storage, and buildup of acetaldehyde. Increased production of acetaldehyde forms covalent bonds with many important functional groups in proteins, impairing protein function. It may also react with phospholipids and arachidonic acid, which triggers lipid peroxidation reactions promoting hepatocyte cell death. Consistently with the above, we found increased protein acetylation and lipid peroxidation (4‐HNE formation) in livers of ethanol‐fed groups, which were attenuated by BCP treatment. Collectively, our results demonstrate that BCP treatment exerts beneficial effects against liver injury induced by chronic plus binge ethanol feeding by attenuating the Kupffer cell‐mediated pro‐inflammatory response (activation and/or pro‐inflammatory phenotypic `M1` switch), neutrophil‐mediated oxidative/nitrative stress/injury, vascular inflammation (expression of vascular adhesion molecules), and hepatic metabolic dysregulation (steatosis, protein hyperacetylation, and PPAR‐α signalling). Our results also indicate that these in vivo protective effects of BCP against alcohol‐induced hepatic injury may involve, at least in part, mechanisms mediated by CB2 receptors. Our study may have immediate translational potential in liver disease as BCP is an FDA approved food additive for humans.
Author contributions
Z.V.V. and P.P. conception and design of research; Z.V.V., C.M., K.E., R.C., D.N., A.C., B.T.N., J.P., T.L. and L.C. performed experiments; Z.V.V., C.M., A.C., J.P. and B.T.N. analysed data; Z.V.V., R.C., G.H., A.C., B.G., G.K., J.G. and P.P. interpreted results of experiments; Z.V.V. and P.P. prepared figures; Z.V.V and P.P. drafted manuscript; Z.V.V., A.C., G.H., B.G., G.K., J.G. and P.P. edited and revised manuscript; Z.V.V., C.M., K.E., R.C., D.N., A.C., B.T.N., J.P., T.L., L.C., G.H., B.G.,G.K., J.G. and P.P. approved final version of manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Primer sequences used in the present study.
Figure S1 Supplementary Figure 1 shows co‐localization of the two utilized macrophage markers, EGF‐like module‐containing mucin‐like hormone receptor‐like 1 (F4/80) and ionized calcium‐binding adapter molecule 1 ((Iba‐1). F4/80 stains the cell membrane of macrophages, while Iba‐1 as an actin‐binding protein gives a detailed picture of cytoskeletal organization and cell shape. The two markers are highly co‐localized.
Acknowledgements
The recent work was supported by the Intramural Research Program of NIAAA/NIH (to P. Pacher). C. Matyas was supported by the scholarship of the Hungarian‐American Enterprise Scholarship Fund/Council on International Educational Exchange. Z. V. Varga was supported by the Rosztoczy Foundation.
Varga, Z. V. , Matyas, C. , Erdelyi, K. , Cinar, R. , Nieri, D. , Chicca, A. , Nemeth, B. T. , Paloczi, J. , Lajtos, T. , Corey, L. , Hasko, G. , Gao, B. , Kunos, G. , Gertsch, J. , and Pacher, P. (2018) β‐Caryophyllene protects against alcoholic steatohepatitis by attenuating inflammation and metabolic dysregulation in mice. British Journal of Pharmacology, 175: 320–334. doi: 10.1111/bph.13722.
This article has been contributed to by US Government employees and their work is in the public domain in the USA.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Mansouri S, Ojha S, Al Maamari E, Al Ameri M, Nurulain SM, Bahi A (2014). The cannabinoid receptor 2 agonist, beta‐caryophyllene, reduced voluntary alcohol intake and attenuated ethanol‐induced place preference and sensitivity in mice. Pharmacol Biochem Behav 124: 260–268. [DOI] [PubMed] [Google Scholar]
- Batkai S, Osei‐Hyiaman D, Pan H, El‐Assal O, Rajesh M, Mukhopadhyay P et al. (2007). Cannabinoid‐2 receptor mediates protection against hepatic ischemia/reperfusion injury. FASEB J 21: 1788–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bento AF, Marcon R, Dutra RC, Claudino RF, Cola M, Leite DF et al. (2011). beta‐Caryophyllene inhibits dextran sulfate sodium‐induced colitis in mice through CB2 receptor activation and PPARgamma pathway. Am J Pathol 178: 1153–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertola A, Mathews S, Ki SH, Wang H, Gao B (2013a). Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc 8: 627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertola A, Park O, Gao B (2013b). Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E‐selectin. Hepatology 58: 1814–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode C, Kugler V, Bode JC (1987). Endotoxemia in patients with alcoholic and non‐alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J Hepatol 4: 8–14. [DOI] [PubMed] [Google Scholar]
- Cao Z, Mulvihill MM, Mukhopadhyay P, Xu H, Erdelyi K, Hao E et al. (2013). Monoacylglycerol lipase controls endocannabinoid and eicosanoid signaling and hepatic injury in mice. Gastroenterology 144: 808–817 .e815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicca A, Caprioglio D, Minassi A, Petrucci V, Appendino G, Taglialatela‐Scafati O et al. (2014). Functionalization of beta‐caryophyllene generates novel polypharmacology in the endocannabinoid system. ACS Chem Biol 9: 1499–1507. [DOI] [PubMed] [Google Scholar]
- Cho HI, Hong JM, Choi JW, Choi HS, Kwak JH, Lee DU et al. (2015). beta‐Caryophyllene alleviates D‐galactosamine and lipopolysaccharide‐induced hepatic injury through suppression of the TLR4 and RAGE signaling pathways. Eur J Pharmacol 764: 613–621. [DOI] [PubMed] [Google Scholar]
- Cho JY, Chang HJ, Lee SK, Kim HJ, Hwang JK, Chun HS (2007). Amelioration of dextran sulfate sodium‐induced colitis in mice by oral administration of beta‐caryophyllene, a sesquiterpene. Life Sci 80: 932–939. [DOI] [PubMed] [Google Scholar]
- Choi IY, Ju C, Anthony Jalin AM, Lee DI, Prather PL, Kim WK (2013). Activation of cannabinoid CB2 receptor‐mediated AMPK/CREB pathway reduces cerebral ischemic injury. Am J Pathol 182: 928–939. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez M, Miquel R, Colmenero J, Moreno M, Garcia‐Pagan JC, Bosch J et al. (2009). Hepatic expression of CXC chemokines predicts portal hypertension and survival in patients with alcoholic hepatitis. Gastroenterology 136: 1639–1650. [DOI] [PubMed] [Google Scholar]
- Gertsch J, Leonti M, Raduner S, Racz I, Chen JZ, Xie XQ et al. (2008). Beta‐caryophyllene is a dietary cannabinoid. Proc Natl Acad Sci U S A 105: 9099–9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertsch J, Pertwee RG, Di Marzo V (2010). Phytocannabinoids beyond the Cannabis plant ‐ do they exist? Br J Pharmacol 160: 523–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath B, Magid L, Mukhopadhyay P, Batkai S, Rajesh M, Park O et al. (2012a). A new cannabinoid CB2 receptor agonist HU‐910 attenuates oxidative stress, inflammation and cell death associated with hepatic ischaemia/reperfusion injury. Br J Pharmacol 165: 2462–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath B, Mukhopadhyay P, Kechrid M, Patel V, Tanchian G, Wink DA et al. (2012b). beta‐Caryophyllene ameliorates cisplatin‐induced nephrotoxicity in a cannabinoid 2 receptor‐dependent manner. Free Radic Biol Med 52: 1325–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikarashi M, Nakashima H, Kinoshita M, Sato A, Nakashima M, Miyazaki H et al. (2013). Distinct development and functions of resident and recruited liver Kupffer cells/macrophages. J Leukoc Biol 94: 1325–1336. [DOI] [PubMed] [Google Scholar]
- Jeong WI, Osei‐Hyiaman D, Park O, Liu J, Batkai S, Mukhopadhyay P et al. (2008). Paracrine activation of hepatic CB1 receptors by stellate cell‐derived endocannabinoids mediates alcoholic fatty liver. Cell Metab 7: 227–235. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita M, Uchida T, Sato A, Nakashima M, Nakashima H, Shono S et al. (2010). Characterization of two F4/80‐positive Kupffer cell subsets by their function and phenotype in mice. J Hepatol 53: 903–910. [DOI] [PubMed] [Google Scholar]
- Kohler C (2007). Allograft inflammatory factor‐1/Ionized calcium‐binding adapter molecule 1 is specifically expressed by most subpopulations of macrophages and spermatids in testis. Cell Tissue Res 330: 291–302. [DOI] [PubMed] [Google Scholar]
- Laskin DL, Weinberger B, Laskin JD (2001). Functional heterogeneity in liver and lung macrophages. J Leukoc Biol 70: 163–170. [PubMed] [Google Scholar]
- Li Y, Wong K, Giles A, Jiang J, Lee JW, Adams AC et al. (2014). Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology 146: 539–549 .e537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvet A, Teixeira‐Clerc F, Chobert MN, Deveaux V, Pavoine C, Zimmer A et al. (2011). Cannabinoid CB2 receptors protect against alcoholic liver disease by regulating Kupffer cell polarization in mice. Hepatology 54: 1217–1226. [DOI] [PubMed] [Google Scholar]
- Mahmoud MF, Swefy SE, Hasan RA, Ibrahim A (2014). Role of cannabinoid receptors in hepatic fibrosis and apoptosis associated with bile duct ligation in rats. Eur J Pharmacol 742: 118–124. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer C, Wiezer MJ, Diehl AM, Schouten HJ, Schouten HJ, Meijer S et al. (2000). Kupffer cell depletion by CI2MDP‐liposomes alters hepatic cytokine expression and delays liver regeneration after partial hepatectomy. Liver 20: 66–77. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay B, Cinar R, Yin S, Liu J, Tam J, Godlewski G et al. (2011). Hyperactivation of anandamide synthesis and regulation of cell‐cycle progression via cannabinoid type 1 (CB1) receptors in the regenerating liver. Proc Natl Acad Sci U S A 108: 6323–6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair AB, Jacob S (2016). A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 7: 27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojha S, Javed H, Azimullah S, Haque ME (2016). beta‐Caryophyllene, a phytocannabinoid attenuates oxidative stress, neuroinflammation, glial activation, and salvages dopaminergic neurons in a rat model of Parkinson disease. Mol Cell Biochem 418: 59–70. [DOI] [PubMed] [Google Scholar]
- Osei‐Hyiaman D, DePetrillo M, Pacher P, Liu J, Radaeva S, Batkai S et al. (2005). Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet‐induced obesity. J Clin Invest 115: 1298–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Mechoulam R (2011). Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog Lipid Res 50: 193–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Hasko G, Huffman JW, Mackie K, Pacher P (2008). CB2 cannabinoid receptor agonists attenuate TNF‐alpha‐induced human vascular smooth muscle cell proliferation and migration. Br J Pharmacol 153: 347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehg JE, Bush D, Ward JM (2012). The utility of immunohistochemistry for the identification of hematopoietic and lymphoid cells in normal tissues and interpretation of proliferative and inflammatory lesions of mice and rats. Toxicol Pathol 40: 345–374. [DOI] [PubMed] [Google Scholar]
- Shepard BD, Tuma PL (2009). Alcohol‐induced protein hyperacetylation: mechanisms and consequences. World J Gastroenterol 15: 1219–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestri C, Di Marzo V (2013). The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metab 17: 475–490. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054‐1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JE, van Rooijen N et al. (2010). Kupffer cells promote hepatic steatosis via interleukin‐1beta‐dependent suppression of peroxisome proliferator‐activated receptor alpha activity. Hepatology 51: 511–522. [DOI] [PubMed] [Google Scholar]
- Tam J, Cinar R, Liu J, Godlewski G, Wesley D, Jourdan T et al. (2012). Peripheral cannabinoid‐1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab 16: 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam J, Liu J, Mukhopadhyay B, Cinar R, Godlewski G, Kunos G (2011). Endocannabinoids in liver disease. Hepatology 53: 346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira‐Clerc F, Belot MP, Manin S, Deveaux V, Cadoudal T, Chobert MN et al. (2010). Beneficial paracrine effects of cannabinoid receptor 2 on liver injury and regeneration. Hepatology 52: 1046–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira‐Clerc F, Julien B, Grenard P, Tran Van Nhieu J, Deveaux V, Li L et al. (2006). CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Nat Med 12: 671–676. [DOI] [PubMed] [Google Scholar]
- Tsai WW, Matsumura S, Liu W, Phillips NG, Sonntag T, Hao E et al. (2015). ATF3 mediates inhibitory effects of ethanol on hepatic gluconeogenesis. Proc Natl Acad Sci U S A 112: 2699–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH et al. (2005). Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol 175: 7661–7668. [DOI] [PubMed] [Google Scholar]
- Wang M, You Q, Lor K, Chen F, Gao B, Ju C (2014). Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J Leukoc Biol 96: 657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Jia Y, Lee JH, Jun HJ, Lee HS, Hwang KY et al. (2014). trans‐Caryophyllene is a natural agonistic ligand for peroxisome proliferator‐activated receptor‐alpha. Bioorg Med Chem Lett 24: 3168–3174. [DOI] [PubMed] [Google Scholar]
- Yin H, Hu M, Liang X, Ajmo JM, Li X, Bataller R et al. (2014). Deletion of SIRT1 from hepatocytes in mice disrupts lipin‐1 signaling and aggravates alcoholic fatty liver. Gastroenterology 146: 801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Sun T, Wang X (2013). Activation of type 2 cannabinoid receptors (CB2R) promotes fatty acid oxidation through the SIRT1/PGC‐1alpha pathway. Biochem Biophys Res Commun 436: 377–381. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primer sequences used in the present study.
Figure S1 Supplementary Figure 1 shows co‐localization of the two utilized macrophage markers, EGF‐like module‐containing mucin‐like hormone receptor‐like 1 (F4/80) and ionized calcium‐binding adapter molecule 1 ((Iba‐1). F4/80 stains the cell membrane of macrophages, while Iba‐1 as an actin‐binding protein gives a detailed picture of cytoskeletal organization and cell shape. The two markers are highly co‐localized.