

Abstract
Astrocyte swelling occurs after CNS injury and contributes to brain swelling, which can increase mortality. Mechanisms proffered to explain astrocyte swelling emphasize the importance of either aquaporin-4 (AQP4), an astrocyte water channel, or of Na+-permeable channels, which mediate cellular osmolyte influx. However, the spatio-temporal functional interactions between AQP4 and Na+-permeable channels that drive swelling are poorly understood. We hypothesized that astrocyte swelling after injury is linked to an interaction between AQP4 and Na+-permeable channels that are newly upregulated. Here, using co-immunoprecipitation and Förster resonance energy transfer, we report that AQP4 physically co-assembles with the sulfonylurea receptor 1 – transient receptor potential melastatin 4 (SUR1-TRPM4) monovalent cation channel to form a novel heteromultimeric water/ion channel complex. In vitro cell-swelling studies using calcein fluorescence imaging of COS-7 cells expressing various combinations of AQP4, SUR1 and TRPM4 showed that the full tripartite complex, comprised of SUR1-TRPM4-AQP4, was required for fast, high-capacity transmembrane water transport that drives cell swelling, with these findings corroborated in cultured primary astrocytes. In a murine model of brain edema involving cold-injury to the cerebellum, we found that astrocytes newly upregulate SUR1-TRPM4, that AQP4 co-associates with SUR1-TRPM4, and that genetic inactivation of the solute pore of the SUR1-TRPM4-AQP4 complex blocked in vivo astrocyte swelling measured by diolistic labeling, thereby corroborating our in vitro functional studies. Together, these findings demonstrate a novel molecular mechanism involving the SUR1-TRPM4-AQP4 complex to account for bulk water influx during astrocyte swelling. These findings have broad implications for the understanding and treatment of AQP4-mediated pathological conditions.
Keywords: Astrocyte, Ion Channels, Ions, Osmosis, Water
Graphical abstract
INTRODUCTION
Astrocyte swelling occurs after many types of central nervous system (CNS) injury and contributes importantly to brain swelling (Stokum et al. 2016). In large hemispheric infarction, brain swelling is associated with up to 80% mortality (Berrouschot et al. 1998; Heron et al. 2009). Current treatments for brain swelling are reactive, and are limited to osmotic therapies and surgical decompression.
Aquaporin-4 (AQP4) is a major water channel that is expressed in the CNS exclusively by astrocytes (Nielsen et al. 1997). AQP4 mediates bulk water influx during astrocyte swelling after ischemia (Manley et al. 2000). AQP4 is the main route for transmembrane water flux in astrocytes (Solenov et al. 2004), and has roles in a variety of physiological and pathological processes (Saadoun et al. 2005; Iliff et al. 2012). These observations have led to the hypothesis that AQP4 promotes astrocyte swelling primarily by increasing the water permeability of the astrocyte plasmalemma (Verkman et al. 2006). However, without an identified driving force, this hypothesis is not sufficient to explain how AQP4, a passive water channel, can mediate water influx and cell swelling.
In brain cells, where transmembrane hydrostatic pressure is negligible, the transport of water through aquaporins is determined mostly by transmembrane osmotic gradients, which are generated by osmolyte transporters. A large number of Na+-permeable channels have been implicated in astrocyte swelling (Staub et al. 1990; Su et al. 2002; Chen et al. 2003), consistent with Na+ being the primary osmolyte that drives astrocyte swelling. These observations have led to the hypothesis that the osmotic gradient generated by Na+ influx through membrane channels is the major determinant of astrocyte swelling. However, since astrocytes that lack AQP4 still swell when exposed to osmotic gradients (Solenov et al. 2004), this hypothesis does not sufficiently explain why the expression of AQP4 is critical (Manley et al. 2000). Moreover, for Na+ influx to result in increased intracellular osmotic pressure, cytoplasmic Na+ concentration ([Na+]) must rise but, after injury, astrocytes with preserved levels of adenosine triphosphate (ATP) quickly correct changes in cytoplasmic [Na+] (Cotrina et al. 1998; Floyd et al. 2005) through the activity of ion exchangers (Rose and Ransom 1997b) and gap junctions (Rose and Ransom 1997a).
The above discordant hypotheses underscore the need for a more refined model of ion and water channel interactions to account for astrocyte swelling. All prior studies of astrocyte swelling assumed that the relative spatial distribution of water and Na+ channels are incidental to their functional interaction. However, this assumption is challenged by a group of channels that contain pores that are permeable to both water and solute. In these “water co-transporters”, osmolyte influx is immediately translated to water influx via the generation of small localized osmotic pressures (Duquette et al. 2001; Lapointe et al. 2002). Water co-transporters are highly efficient, mediating ~65% of intestinal water uptake (Zeuthen et al. 2016), and have been shown to be associated with the majority of perisynaptic glutamate clearance (MacAulay et al. 2004). Interestingly, certain water co-transporters, including the Na+/K+/2Cl− co-transporter-1 (NKCC1) and volume-regulated anion channels (VRACs), mediate cell volume changes in astrocytes (Nilius 2004; Kimelberg et al. 2006; Jayakumar and Norenberg 2010; Zeuthen 2010).
The concept of water/ion transport coupling is utilized outside of water co-transporters, notably by AQP4 itself. Under normal physiological conditions, the physical co-assembly of AQP4 with transient receptor potential vanilloid 4 (TRPV4) is critical to the role of AQP4 in astrocyte volume homeostasis (Benfenati et al. 2011). Under normal conditions, AQP4 also physically co-assembles with Na+-K+-ATPase, although the functional implications of this interaction are unknown (Illarionova et al. 2010). After CNS injury, AQP4 becomes “dysregulated”, wherein its subcellular localization shifts from its normal polarized location at the astrocyte endfoot to widespread expression throughout the plasmalemma (Frydenlund et al. 2006; Steiner et al. 2012). However, the fate of AQP4’s binding partner(s) under conditions of CNS injury, when astrocyte swelling dominates, is unknown.
After CNS injury, astrocytes exhibit de novo upregulation of sulfonylurea receptor 1 (SUR1) – transient receptor potential melastatin 4 (TRPM4) channels (Chen and Simard 2001; Chen et al. 2003; Simard et al. 2006; Mehta et al. 2015). TRPM4, the pore-forming subunit, is a nonselective monovalent cation channel activated by intracellular calcium (Cai2+) (Vennekens and Nilius 2007). SUR1, an ATP-binding cassette (ABC) transporter that regulates pore-forming subunits, physically co-associates with TRPM4 and doubles its Cai2+ sensitivity (Chen and Simard 2001; Chen et al. 2003; Woo et al. 2013). TRPM4 alone can form functional plasmalemmal ion channels in the absence of SUR1, whereas SUR1 does not traffic to the membrane without a pore-forming subunit (Sharma et al. 1999; Zerangue et al. 1999). Previous work implicated SUR1-TRPM4 in oncotic astrocyte swelling (Chen and Simard 2001; Chen et al. 2003) and brain edema formation (Simard et al. 2006). Furthermore, clinical trials indicate that pharmacological blockade of SUR1-TRPM4 reduces brain swelling after ischemic stroke (Sheth et al. 2016). Unlike most other ion channels and transporters implicated in astrocyte swelling, SUR1-TRPM4 is only expressed after injury (Chen and Simard 2001; Chen et al. 2003; Simard et al. 2006; Mehta et al. 2015), when astrocyte swelling predominates. However, the role of SUR1-TRPM4, if any, in AQP4-mediated water transport is unknown.
We hypothesized that, after injury, AQP4’s normal homeostatic partner(s) may be replaced with newly-expressed Na+-permeable channels, accounting for astrocyte swelling. Here, we report that the SUR1-TRPM4 ion channel physically co-assembles with AQP4 to form a novel heteromultimeric water/ion channel complex. In vitro functional studies showed that SUR1-TRPM4 and AQP4 synergize to mediate fast, high-capacity transmembrane water influx and astrocyte swelling. Studies of astrocyte volume changes in a murine model of cerebellar edema demonstrated a key role for the SUR1-TRPM4-AQP4 complex in astrocyte swelling. These findings have important implications for CNS conditions marked by brain swelling.
MATERIALS AND METHODS
Cell culture
COS-7 cells and HEK 293 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 4.5 g/L glucose (Invitrogen) supplemented with 10% fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin. HEK 293 cell lines with stable expression of SUR1 or empty vector were generated as previously described (Woo et al. 2013).
Primary C57B6 mouse astrocytes were isolated from P<5 mouse pups using a modified version of the method described by McCarthy and de Vellis (McCarthy and de Vellis 1980) or were commercially obtained (M-ASM-330; Lonza, Basel, Switzerland). GFAP immunolabeling demonstrated that cultures were >95% GFAP+. Astrocyte cultures were maintained for experiments <2 months. Prior to experiments, astrocytes were activated overnight with a cocktail of TNFα (20 ng/mL), IFNγ (20 ng/mL), and LPS (1 μg/mL), or with vehicle alone. Quantitative polymerase chain reaction (qPCR) analysis was used to quantify mRNA upregulation upon astrocyte activation, using previously described methods (Kurland et al. 2016).
For small interfering RNA (siRNA) knockdown of TRPM4, primary mouse astrocytes were treated for 2 days with 47 pmol siRNA per 1 mL DMEM complexed with 1 μl oligofectamine (112252011; Thermo Fisher Scientific Inc. Waltham, MA) per 1 mL DMEM. The TRPM4 siRNA sequence (Loh et al. 2014) knockdown efficiency was validated by qPCR. A universal siRNA negative control (AM4611; Thermo Fisher Scientific Inc.) served as control.
Co-immunoprecipitation and immunoblot analysis
Cell or tissue lysates were isolated with pH 8 0.5% CHAPS lysis buffer (CIB-1; FIVEphoton Biochemicals, San Diego, CA). Immunocomplexes were formed by incubating lysates with anti-AQP4 antibody (sc-9888; Santa Cruz Biotechnology, Dallas, TX) or anti-TRPM4 antibody (custom antibody). Immunocomplexes were then isolated with either protein G-Sepharose or anti-chicken IgY antibody agarose, washed 3× with lysis buffer, and eluted by boiling 5 min in 2× NuPAGE LDS sample buffer (NP0007; Thermo Fisher Scientific Inc.). The isolated proteins were analyzed with immunoblot assay. For immunoassay, proteins were detected with anti-AQP4 (AB3594; Millipore, Billerica, MA), anti-TRPM4 (custom antibody (Woo et al. 2013)), anti-SUR1 (custom antibody (Woo et al. 2013)), or anti-Myc (2278S; Cell Signaling Technology, Danvers, MA). Surface biotinylation assay was performed as previously described (Woo et al. 2013).
Förster resonance energy transfer
COS-7 cells were transfected with plasmids encoding AQP4 M1 or M23, SUR1, TRPM4, TRPV4, or KIR2.1, each fused with citrine or cerulean at the N or C terminus. Positive controls included citrine-AQP4 M1 with cerulean-AQP4 M1, or with TRPV4. For negative control, AQP4 M1-cerulean was co-expressed with citrine-KIR2.1. Cells were incubated for 48 hr after transfection, fixed with 4% paraformaldehyde for 5 min, and coverslipped prior to Förster resonance energy transfer (FRET) imaging. FRET fluorescence imaging and FRET efficiency experiments were performed as previously described (Woo et al. 2013). FRET efficiency was analyzed over the cellular margins to exclude the nucleus and endoplasmic reticulum
Calcein imaging
COS-7 cells or primary mouse astrocytes were plated on perfusion chambers (80606; Ibidi, Planegg, Germany). For COS-7 experiments, COS-7 cells were transfected with plasmids encoding different combinations of AQP4 M1 or M23, TRPM4, and SUR1. COS-7 cells also were co-transfected with plasmids encoding cerulean fluorescent protein (CFP) to identify transfected cells. Prior to the experiment, COS-7 cells or astrocytes were loaded with calcein by incubating with 5 μM calcein-AM (C3100MP; Thermo Fisher Scientific Inc.) for 5 min in HBSS.
Prior to the experiment, cells were incubated at 36 °C; imaging was conducted at room temperature. During experiments, cultured cells were continuously perfused with HBSS; the chamber exchange time was 4.36±0.38 sec. Fluorescence images were acquired every 60 sec with an epifluorescence microscope (AE31E; Motic, Richmond, British Columbia) with a GFP filter (49001 ET CFP; Chroma Technology Corporation, Bellows Falls, VT). Baseline fluorescence measurements were obtained for 40 sec (COS-7 cells), or 60 sec (primary astrocytes), whereupon the control solution was switched to a treatment solution containing either 200 mOsm HBSS (for hypotonic stress) or 10 μM A23187 to induce TRPM4 currents (Vennekens and Nilius 2007). Experiments were conducted using either 0.003% dimethyl sulfoxide (DMSO) (control), or 30 μM glibenclamide or 10 μM 9-phenanthrol in baseline and treatment solutions.
Calcein fluorescence time series were calculated by normalizing calcein fluorescence at each time point (Ft) by the calcein fluorescence at time 0 (F0). Steady-state swelling magnitude was calculated as Fend/F0 where Fend is the calcein fluorescence at the final time point and F0 is the calcein fluorescence at time 0. Net water influx was calculated by integrating the area under Ft/F0 traces between the time of A23187 treatment and the end of the experiment. Swelling rate, which was only calculated for experiments that led to volume increases, was calculated as the reciprocal of the time a trace took to move from the 25th percentile value of Ft/F0 to the 90th percentile value of Ft/F0, for t = 1…140. Calcein fluorescence “heat maps” were calculated by dividing the final calcein image by the initial calcein image, and processing the result with the ImageJ HeatMap Histogram plugin (Péan 2012).
Patch clamp electrophysiology
Patch clamp electrophysiology was used to study vehicle-treated or activated primary mouse astrocytes. A nystatin perforated patch technique was used to minimize disturbance of the cytoplasm, which can cause rapid rundown of TRPM4 current. For these experiments,16.5 μl of nystatin stock solution (50 mg of nystatin was dissolved in 1 mL DMSO) was added to 5 mL of the pipette solution to yield 165 μg/mL nystatin and 3.3 μg/mL DMSO. To record whole cell currents independent of K+ channels such as KATP, the 7.2 pH extracellular solution contained 145 mM CsCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 165 μg/mL nystatin. To elicit membrane currents, the holding potential was set to −50 mV, and ramp pulses were from −100 to +10 mV, 4 mV/msec, every 15 seconds.
Cerebellar cold injury
Animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Maryland and were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Male ~30g wild-type (WT) C57B6 mice and TRPM4−/− mice on a C57B6 background were anesthetized with 60 mg/kg ketamine and 7.5 mg/kg xylazine (Gerzanich et al. 2009). Body temperature was maintained with a homeothermic blanket controlled by a rectal thermometer. A 5 × 5 mm craniectomy was performed over the right cerebellum, with the dura left intact. A 2.5 mm diameter liquid-nitrogen-cooled copper rod was placed in contact with the exposed dura for 45 seconds, after which the cerebellar cold injury was repeated a total of 3×. A random number generator was used to assign animals to groups.
Immunolabeling and TUNEL assay
Coronal cryosections (12 μm) were obtained at approximately 12 mm posterior to bregma, a location that was centered over the anterior-posterior axis of the cold injury. Immunolabeling was performed as previously described (Stokum et al. 2015b) with primary antibodies against GFAP (C9205; Sigma-Aldrich, St. Louis, MO), AQP4 (AB3594; Millipore), TRPM4 (sc-27540; Santa Cruz Biotechnology), or SUR1 (custom antibody (Woo et al. 2013)). For TUNEL assay, cryosections were processed according to kit instructions (C10617; Thermo Fischer Scientific Inc.). Imaging was conducted with standard epifluorescence and confocal techniques.
For quantification, images were obtained of cerebellar granule cell layer ipsilateral to cold injury. The contralateral side served as control. To quantify SUR1 and TRPM4 immunolabeling in granule cell layer astrocytes, AQP4 images were thresholded at 2x the background immunofluorescence, and were used to mask the SUR1 and TRPM4 channels. SUR1 and TRPM4 % region of interest (ROI) was calculated by dividing the number of positive (>2× background immunofluorescence) SUR1 or TRPM4 pixels in the AQP4+ granule cell layer ROI by the total AQP4+ granule cell layer ROI area.
In situ astrocyte volume quantification
At 3 days after sham surgery or cerebellar cold injury, mice were euthanized by pentobarbital overdose and intracardially perfused with normal saline and 1.5% paraformaldehyde. Brains were dissected and post-fixed for 1 hr in 1.5% paraformaldehyde. The cerebellum was sectioned into 150 μm sections with a vibratome (1000 plus; The Vibratome Co., St. Louis, MO) and stored in PBS for up to 1 week. Sections were submitted to diolistic labeling with the Helios gene gun system (Bio-Rad; Hercules, CA), wherein tungsten particles (1.1 μm) (165-2267-MSDS; Bio-Rad, Hercules, CA) coated with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) dye were delivered to tissue sections (Gan et al. 2000). Brain slices were immunolabeled for GFAP (SAB5201116; Sigma-Aldrich) and coverslipped. Z-stacks (slice thickness: 0.5 μm; pixel size: 0.26 × 0.26 μm) were obtained with the LSM510 meta (Zeiss, Jena, Germany) laser scanning microscope system that covered the entire DiI-labeled arborization of GFAP+ cerebellar granule cell layer astrocytes. A 3-dimensional (3D) region-growing algorithm (Kroon 2008) was used to segment the brightly fluorescent intracellular astrocyte volume from the dark extracellular space. The intracellular volume was obtained by multiplying the number of intracellular voxels by the volume of a single voxel.
Data Analysis
Data are presented as mean ± SE. Analysis was performed with Origin Pro (V8; OriginLab Corp, North Hampton MA). Student t-test and 1-way ANOVA with Tukey’s HSD post-hoc comparisons were used where appropriate.
RESULTS
SUR1-TRPM4 co-immunoprecipitates with AQP4
We postulated that, following CNS injury, changes in AQP4 ion channel binding partners might account for astrocyte swelling. Initial experiments were conducted wherein the AQP4 M1 or M23 isoforms (Jung et al. 1994), SUR1, TRPM4, and a SUR1-TRPM4 fusion protein were variously expressed in a COS-7 heterologous expression system, and tested for possible pair-wise interactions using co-immunoprecipitation (co-IP). In each experiment, the specificity of the immunoprecipitating antibody was verified in COS-7 cells that did not express the protein targeted for immunoprecipitation. Additionally, the specificity of the assay was verified by omitting the immunoprecipitation antibody.
Immunoprecipitation of TRPM4 was performed in COS-7 cells expressing TRPM4 and AQP4 M1 or AQP4 M23. Immunoblot for AQP4 demonstrated co-IP of AQP4 M1 (Figure 1A, lane 2) and AQP4 M23 (Figure 1B, lane 2) dimers, trimers, and tetramers. After omission of TRPM4 expression, or omission of the anti-TRPM4 immunoprecipitation antibody, AQP4 was not observed, confirming specificity (Figure 1A and 1B, lanes 3 and 4). This experiment indicated co-association of AQP4 with TRPM4.
Figure 1. SUR1-TRPM4 co-immunoprecipitates with AQP4.
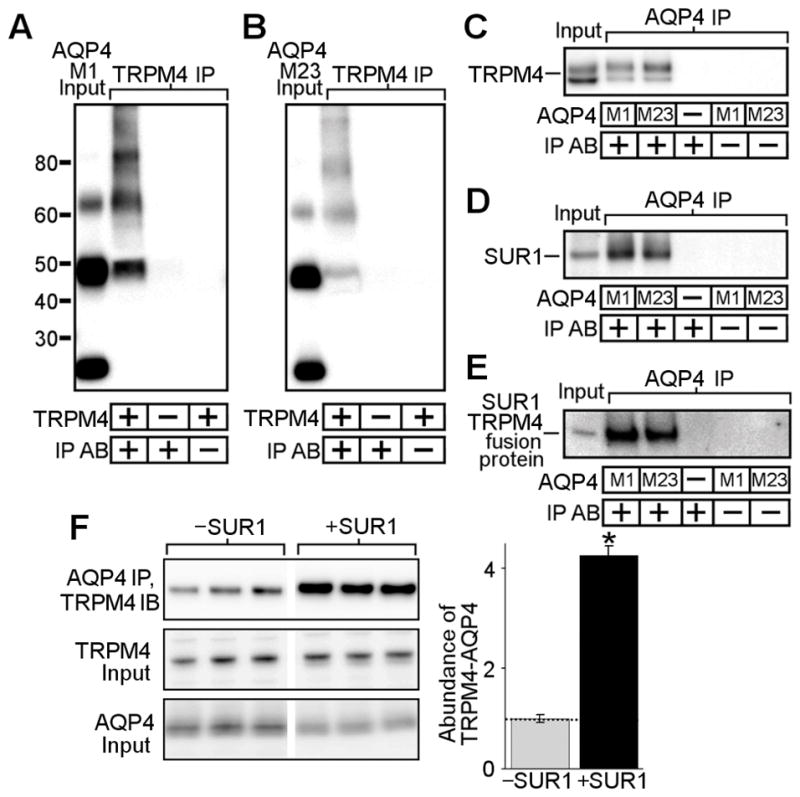
(A–E) Co-immunoprecipitation experiments in COS-7 cells transfected with AQP4 M1 or M23 isoforms, TRPM4, SUR1, or an SUR1-TRPM4 fusion protein; AQP4 immunoblots showing AQP4 M1 or M23 in total lysates (A, B lane 1), AQP4 upon TRPM4 immunoprecipitation (IP) (A, B lane 2), no AQP4 upon TRPM4 IP in cells that express AQP4 but not TRPM4 (A, B lane 3), and no AQP4 when the anti-TRPM4 IP antibody (IP AB) is omitted (A, B lane 4); TRPM4 immunoblot showing TRPM4 in total lysates (C lane 1), TRPM4 upon IP of AQP4 M1 (C lane 2) or M23 (C lane 3), no TRPM4 upon AQP4 IP in cells that express TRPM4 but lack AQP4 (C lane 4), and no TRPM4 when the anti-AQP4 IP antibody is omitted (C lanes 5 and 6); SUR1 immunoblot showing SUR1 in total lysates (D lane 1), SUR1 upon IP of AQP4 M1 (D lane 2) or M23 (D lane 3), no SUR1 upon AQP4 IP in cells that express SUR1 but lack AQP4 (D lane 4), and no SUR1 when the anti-AQP4 IP antibody is omitted (D lanes 5 and 6); SUR1 immunoblot showing the SUR1-TRPM4 fusion protein in total lysates (E lane 1), the SUR1-TRPM4 fusion protein upon IP of AQP4 M1 (E lane 2) or M23 (E lane 3), no SUR1-TRPM4 fusion protein upon AQP4 IP in cells that express SUR1-TRPM4 but lack AQP4 (E lane 4), and no SUR1-TRPM4 fusion protein when the IP antibody is omitted (E lanes 5 and 6); all results represent n=5 replicates. (F) Co-immunoprecipitation experiment in HEK 293 cells stably expressing either empty vector (−SUR1) or SUR1 (+SUR1) and transfected with AQP4 M1 and TRPM4, showing 4-fold greater TRPM4 abundance upon AQP4 IP in SUR1+ cells versus SUR1− cells (top row); TRPM4 and AQP4 in total lysates (middle, bottom rows) do not show differences in expression; n=6; *p<0.05 in t-test; densities were normalized to average SUR1− band density (dotted line).
To verify AQP4-TRPM4 interactions, the above co-IP experiments were performed in reverse. Immunoprecipitation of AQP4 was performed in COS-7 cells expressing TRPM4 and AQP4 M1 or AQP4 M23. Immunoblot for TRPM4 demonstrated co-IP of TRPM4 (Figure 1C, lanes 2 and 3). After omission of AQP4 expression, or omission of the anti-AQP4 immunoprecipitation antibody, TRPM4 was not observed, confirming specificity (Figure 1C, lanes 4 – 6). This experiment confirmed co-association of AQP4 with TRPM4.
To investigate AQP4 and SUR1 interactions, immunoprecipitation of AQP4 was performed in COS-7 cells expressing SUR1 and AQP4 M1 or AQP4 M23. Immunoblot for SUR1 demonstrated co-IP of SUR1 (Figure 1D, lanes 2 and 3). After omission of AQP4 expression, or omission of the anti-AQP4 immunoprecipitation antibody, SUR1 was not observed, confirming specificity (Figure 1D, lanes 4 – 6). This experiment indicated co-association of AQP4 with SUR1.
Lastly, to confirm that AQP4 interacts with the assembled SUR1-TRPM4 channel, Immunoprecipitation of AQP4 was performed in COS-7 cells expressing AQP4 M1 or AQP4 M23 and a functional SUR1-TRPM4 fusion protein (Woo et al. 2013). Immunoblot for SUR1 demonstrated co-IP of the SUR1-TRPM4 fusion protein (Figure 1E, lanes 2 and 3). After omission of AQP4 expression, or omission of the anti-AQP4 immunoprecipitation antibody, the SUR1-TRPM4 fusion protein was not observed, confirming specificity (Figure 1E, lanes 4 – 6).
Together, the above experiments indicated that AQP4 M1 and M23 co-associate with the SUR1-TRPM4 heteromer through physical co-associations with both TRPM4 and SUR1. Since direct interaction of SUR1 with TRPM4 has been validated in previous reports (Chen et al. 2003; Woo et al. 2013; Mehta et al. 2015), these experiments were not repeated here.
SUR1 increases co-association of TRPM4 with AQP4
The above co-IP experiments showed that AQP4 independently co-associates with TRPM4 and SUR1. Thus, we hypothesized that the octomeric SUR1-TRPM4 channel may recruit more AQP4 than the tetrameric TRPM4 channel.
AQP4 M1 and TRPM4 were co-expressed in HEK 293 cells stably transfected with either empty vector (SUR1−) or an SUR1 expression plasmid (SUR1+). Immunoprecipitation of AQP4 was performed. Immunoblot for TRPM4 demonstrated greater co-IP of TRPM4 by AQP4 in SUR1+ cells versus SUR1− cells (Figure 1F, top row). Densitometric quantification revealed a ~4-fold increase in AQP4-TRPM4 complex formation upon SUR1 expression (Figure 1F). Control immunoblots of direct lysates did not show differences in TRPM4 or AQP4 expression between SUR1− versus SUR1+ cells (Figure 1F, middle and lower row). These data strongly suggest that 4 additional AQP4 binding domains are provided by the 4 SUR1 monomers that co-associate with TRPM4 tetramers to form the octomeric SUR1-TRPM4 channel.
AQP4 co-associates more with TRPM4 than with TRPV4
It was previously shown that AQP4 co-assembles with TRPV4 in astrocytes from normal brain tissues (Benfenati et al. 2011). We assessed the relative abundance of the AQP4-TRPM4 versus the AQP4-TRPV4 complex in an in vitro expression system.
Myc-tagged TRPM4 and Myc-tagged TRPV4 were co-expressed with either AQP4 M1 or AQP4 M23 in COS-7 cells. Myc-tagging allows for direct immunoblot comparison of TRPM4 versus TRPV4 protein abundance. Anti-Myc immunoblot of total lysate showed two bands at ~120 kDa that corresponded to TRPM4 and two bands at ~100 kDa that corresponded to TRPV4 (Figure 2A, top panel). AQP4 was immunoprecipitated, followed by anti-Myc immunoblot to assess TRPM4 and TRPV4 pulldown. After IP with anti-AQP4 antibody, the TRPM4 bands were markedly denser than the TRPV4 bands (Figure 2A, bottom panel), indicating that the AQP4-TRPM4 complex is more abundant than the AQP4-TRPV4 complex.
Figure 2. AQP4 co-associates more with TRPM4 than with TRPV4.

(A) Anti-myc immunoblots of direct lysate input and AQP4 immunoprecipitation (IP) in COS-7 cells expressing AQP4, myc-TRPM4, and myc-TRPV4, showing equivalent abundance of myc-TRPM4 and myc-TRPV4 in direct lysate, but markedly greater abundance of myc-TRPM4 versus myc-TRPV4 upon IP of AQP4 M1 or M23 isoforms; displayed immunoblot includes all (n=3) independent experimental replicates. (B) Densitometric quantification of the data in panel (A), showing that the AQP4/myc-TRPM4 complex is 5–14 times more abundant than the AQP4/myc-TRPV4 complex; *p<0.05 in t-test between groups; n=3 independent experiments.
The results in Figure 2A were quantified. First, the co-IP efficiencies of TRPM4 and TRPV4 with AQP4 were calculated by normalizing the immunoprecipitated TRPM4 and TRPV4 band densities in Figure 2A, bottom panel to the input TRPM4 and TRPV4 band densities in Figure 2A, top panel. Next, the co-IP efficiencies of TRPM4 and TRPV4 with AQP4 were normalized to the TRPV4 efficiency. This analysis showed that when TRPM4, TRPV4, and AQP4 are co-expressed, the AQP4-TRPM4 complex is ~5-fold (M1) and ~14.7-fold (M23) more abundant than the AQP4-TRPV4 complex (Figure 2B). These results suggest that the AQP4-TRPM4 interaction, and hence the SUR1-TRPM4-AQP4 complex, predominates when AQP4, TRPM4, and TRPV4 are co-expressed.
SUR1-TRPM4 and AQP4 co-assemble to form a heteromultimeric complex
While co-IP experiments indicated AQP4 co-association with the SUR1-TRPM4 ion channel, co-IP cannot discriminate between direct and indirect intermolecular interactions, which may occur over relatively large distances. Thus, to determine whether AQP4 directly co-associates with SUR1-TRPM4 to form a heteromultimeric channel complex, we performed intermolecular FRET analysis.
FRET analysis was conducted by expressing fluorophore-tagged constructs in COS-7 cells. C-terminus cerulean fused AQP4 M1 or M23 was co-expressed with N-terminus citrine fused TRPM4. C-terminus citrine fused AQP4 M1 or M23 was co-expressed with N-terminus cerulean fused SUR1. Biotin pull-down experiments confirmed that fluorophore-tagged proteins were expressed at the plasmalemma (Figure 3A). Surface expressed biotinylated proteins exhibited slightly greater molecular weight (Figure 3A).
Figure 3. Intermolecular FRET analysis of AQP4 with TRPM4 and SUR1.

(A) Immunoblots of total protein lysate (lane 1) and surface lysate from surface biotinylation assay (lane 2) showing surface expression of fluorophore-tagged AQP4, TRPM4, and SUR1 in COS-7 cells; results represent n=3 replicates. (B–E) Fluorescence micrographs of COS-7 cells transfected with citrine (Ci)-TRPM4 and AQP4 M1-cerulean (Ce) (B), Ci-TRPM4 and AQP4 M23-Ce (C), AQP4 M1-Ci and SUR1-Ce (D), and AQP4 M23-Ci and SUR1-Ce (E), showing expression of Ci- and Ce-tagged constructs in cytoplasmic organelles, and at the plasmalemma cellular margin; FRET signal (white) was present at the cellular margins (arrows), indicating AQP4-TRPM4 and AQP4-SUR1 interactions at the plasmalemma; results represent n>35 cells per construct combination.
Expression of fluorophore-tagged AQP4 resulted in punctate membrane and intracellular fluorescence (Figure 3B), as reported previously (Smith et al. 2014). This expression pattern likely reflects the formation of orthogonal arrays of particles (OAP), and vesicular localization of AQP4. Expression of fluorophore-tagged TRPM4 (Figure 3B) and SUR1 (Figure 3D) resulted in vesicular and membrane localized fluorescence, consistent with previously reported trafficking patterns (Crnich et al. 2008).
FRET images of transfected COS-7 cells were captured with cerulean excitation and citrine detection. When TRPM4 was co-expressed with AQP4 M1 (Figure 3B) and M23 (Figure 3C), a FRET signal was detected on the cellular margins, indicative of interactions at the plasmalemma. Even though SUR1 alone does not normally traffic to the plasmalemma (Sharma et al. 1999; Zerangue et al. 1999), FRET also was detected on the cellular margins when SUR1 was co-expressed with AQP4 M1 (Figure 3D) and M23 (Figure 3E). This result may reflect low-level endogenous expression of TRPM4 in COS-7 cells. Alternatively, AQP4 may have shielded the SUR1 endoplasmic reticulum retention domain (Zerangue et al. 1999).
To quantify FRET efficiency, acceptor photobleaching (Woo et al. 2013) was performed in COS-7 cells transfected with fluorophore-tagged constructs. ROIs for photobleaching were defined over the cellular margins to select for membrane-localized proteins (Figure 4A).
Figure 4. Quantification of FRET efficiency.

(A) Example plasmalemmal (arrows) ROI used in photobleaching quantification of FRET efficiency. (B) Quantification of FRET efficiency in COS-7 cells with acceptor photobleaching showing that the negative control (AQP4 M1-Ce with Ci-KIR2.1) exhibited ~3% FRET efficiency, positive controls (Ce-M1 with Ci-M1) (Ce-M1 and TRPV4-Ci) exhibited ~8–10% FRET efficiency, and combinations of AQP4 M1 and M23 with TRPM4 and SUR1 exhibited ~8–12% FRET efficiency; n>35 independent cells/condition; *p<0.05 t-test versus M1-Ce/Ci-Kir6.2 negative control.
AQP4 and KIR K+ channels are not known to form direct protein-protein interactions. Thus, for a negative control, AQP4 M1 was co-expressed with the KIR2.1 K+ channel. The negative control exhibited ~3% FRET efficiency. Since AQP4 monomers form tetramers, AQP4 M1 was co-expressed with AQP4 M1 for a positive control. For a second positive control, AQP4 M1 was co-expressed with TRPV4. Positive controls exhibited FRET efficiencies of 8–10%. For combinations of AQP4 M1 and M23 with TRPM4 and SUR1, FRET efficiencies of 8.5–11.5% were recorded, which were in the range of positive controls (Figure 4B).
Together, Co-IP and FRET data indicated that SUR1-TRPM4 and AQP4 physically co-assemble to form a novel heteromultimeric channel complex.
AQP4 synergizes with SUR1-TRPM4 to mediate cellular water uptake and swelling
Na+-permeable channels are the major routes for osmolyte influx during astrocyte swelling, while AQP4 has a major role in associated water influx (see Introduction). The SUR1-TRPM4-AQP4 complex contains both Na+-permeable and water-permeable pores. Thus, we reasoned that the constituent proteins of the SUR1-TRPM4-AQP4 complex synergize to mediate water influx and cell swelling.
Possible interacting roles of SUR1, TRPM4, and AQP4 in generating transmembrane water flux were investigated in COS-7 cells transfected with combinations of AQP4, TRPM4, and SUR1. Changes in cell volume were continuously monitored with calcein, a self-quenching dye that exhibits fluorescence proportional to cytoplasmic volume (Hamann et al. 2002).
First, to verify that transfection with plasmids encoding AQP4 M1 and M23 resulted in plasmalemmal expression of functional water channels, empty vector, AQP4 M1 or AQP4 M23 were expressed. COS-7 cells were exposed to control HBSS (295 mOsm) and then stimulated with hypotonic stress (200 mOsm HBSS) to promote cellular water uptake. In COS-7 cells transfected with empty vector, hypotonic stress resulted in a gradual increase in calcein fluorescence, indicating cell swelling (Figure 5A). In contrast, in COS-7 cells that expressed AQP4 M1 or M23, hypotonic stress resulted in an immediate sharp increase in calcein fluorescence, followed by a slow return towards baseline (Figure 5A). This response indicates fast cell swelling followed by regulatory volume decrease, as previously demonstrated (Benfenati et al. 2011). Overall, these results showed that functional AQP4 channels were expressed at the cell membrane.
Figure 5. SUR1-TRPM4 synergizes with AQP4 to mediate cellular water uptake and swelling.

(A) Traces of calcein fluorescence (Ft/F0) in COS-7 cells showing that, compared to expression of empty vector (CTR), expression of AQP4 M1 or M23 in COS-7 cells results in hypotonic stress-induced swelling. (B, C) Traces of calcein fluorescence in COS-7 cells showing that, compared to control conditions (green, pink, and blue), expression of TRPM4 (orange) sensitizes COS-7 cells to Ca2+ ionophore A23187-induced water influx, which is increased in rate upon co-expression of AQP4 M1 (black) or M23 (red) (B) and blocked by treatment with the TRPM4 antagonist 9-phenanthrol (C). (D, E) Traces of calcein fluorescence in COS-7 cells showing that, compared to control conditions (blue, pink, grey), A23187-induced water influx in TRPM4-expressing COS-7 cells (orange) is increased in magnitude upon co-expression of SUR1 (green), further increased in both magnitude and rate upon co-expression of SUR1 and AQP4 M1 (black) or M23 (red) (D), and inhibited by the SUR1 antagonist glibenclamide to levels similar to TRPM4-expressing cells (E); n=3 independent experiments with 3 cells/experiment; *p<0.05 in ANOVA with Tukey tests versus leftmost column or between groups denoted with brackets.
To determine whether TRPM4 and AQP4 interact to generate cell swelling, TRPM4 and AQP4 were co-expressed in COS-7 cells. Transfected COS-7 cells were treated with the calcium ionophore, A23187, to activate TRPM4 by raising Cai2+ (Woo et al. 2013). Expression of empty vector, AQP4 M1, or M23 alone did not result in cell swelling following A23187, as shown by calcein fluorescence (Figure 5B). These results were unsurprising, given that no osmolyte carrier was expressed. Expression of TRPM4 alone resulted in statistically significant swelling upon A23187 treatment (Figure 5B), indicating that TRPM4 can generate osmotic gradients that result in transmembrane water influx through non-aquaporin routes. Co-expression of AQP4 with TRPM4, and formation of the TRPM4-AQP4 complex, resulted in marginally faster swelling rate (Figure 5B). However, co-expression of AQP4 did not increase steady-state swelling magnitude (Figure 5B), nor alter net water influx (t-test; p= 0.74). Treatment with 9-phenanthrol, a TRPM4 antagonist (Guinamard et al. 2014), blocked A23187-induced swelling (Figure 5C). Together, these results indicate that TRPM4 and AQP4 can cooperate to mediate relatively low-capacity water influx.
Upon activation, TRPM4 current is quickly desensitized (Vennekens and Nilius 2007), limiting its ability to mediate influx of osmolytes. Since AQP4 water flux is primarily determined by osmolyte flux, TRPM4 desensitization may limit water flux, perhaps accounting for the unimpressive traces in Figure 5B. SUR1 physically co-associates with TRPM4, doubles its Ca2+ sensitivity, and reduces TRPM4 desensitization (Woo et al. 2013). We reasoned that co-expression of SUR1 may boost water flux and promote cell swelling.
To characterize the role of SUR1 in modulating TRPM4- and AQP4-dependent water flux, SUR1, TRPM4, and AQP4 were variously co-expressed in COS-7 cells. Expression of only SUR1, AQP4 M1, or AQP4 M23 did not result in COS-7 swelling following A23187 treatment (Figure 5D). As before, expression of TRPM4 alone resulted in moderate swelling following A23187 treatment (Figure 5D). When SUR1 was co-expressed with TRPM4, A23187 triggered a greater increase in calcein fluorescence steady-state magnitude compared to COS-7 cells that expressed TRPM4 alone (Figure 5D), indicating increased solute influx compared to cells expressing TRPM4 alone.
Impressively, co-expression of AQP4 with SUR1 and TRPM4, and formation of the SUR1-TRPM4-AQP4 complex, resulted in markedly greater increases in calcein fluorescence steady-state magnitude upon A23187 treatment, and the increases were faster than in COS-7 cells expressing TRPM4 or SUR1 and TRPM4 (Figure 5D). As estimated from calcein fluorescence dequenching, expression of SUR1-TRPM4-AQP4 M1 resulted in ~7.6-fold greater net water influx than expression of TRPM4 alone (t-test, p = 0.0073), and ~3.2-fold greater net water influx than expression of SUR1 and TRPM4 (t-test, p = 9.3×10−5). Treatment with glibenclamide, a SUR1 antagonist (Gopalakrishnan et al. 2000; Woo et al. 2013), inhibited A23187-triggered changes in calcein fluorescence (Figure 5E) to levels observed in COS-7 cells that co-expressed AQP4 and TRPM4, consistent with blockade of SUR1. Overall, these results indicate that (i) co-expression of SUR1 with TRPM4, and formation of the SUR1-TRPM4 channel, enhances solute influx; (ii) addition of a passive water channel to SUR1-TRPM4 substantially boosts water influx magnitude and rate; (iii) the SUR1-TRPM4-AQP4 complex is a potent mediator of transmembrane water flux.
The SUR1-TRPM4-AQP4 complex mediates swelling of activated primary astrocytes
Possible roles of the SUR1-TRPM4-AQP4 complex in astrocyte swelling were studied in cultured primary murine astrocytes. To stimulate upregulation of SUR1, TRPM4, and AQP4, astrocytes were activated overnight with TNFα, IFNγ, and LPS, a cocktail that mimics the pro-inflammatory extracellular milieu after CNS injury (Li et al. 2001; Caso et al. 2007; Lambertsen et al. 2012). Compared with control (vehicle), activation resulted in ~9-fold increased AQP4 mRNA, ~5.4-fold increased SUR1 mRNA, and ~2.5-fold increased TRPM4 mRNA (Figure 6A). Patch clamp experiments were performed to verify expression of functional SUR1-TRPM4 channels in activated astrocytes. Control astrocytes showed no inward current upon stimulation with diazoxide, an SUR1-specific agonist (Figure 6B). In contrast, activated astrocytes exhibited large, inward, and glibenclamide-sensitive currents at physiological potentials upon diazoxide treatment (Figure 6B).
Figure 6. The SUR1-TRPM4-AQP4 complex mediates swelling of activated primary astrocytes.

(A, B) Activation (ACT) of primary mouse astrocytes with overnight TNFα, IFNγ, and LPS (20 ng/mL, 20 ng/mL, 1 μg/mL, respectively) resulted in upregulation of AQP4, TRPM4, and SUR1 mRNA versus control (CTR) astrocytes (A), and expression of functional SUR1-TRPM4 channels that mediate inward glibenclamide-sensitive current upon exposure to the SUR1 activator, diazoxide (100 μM) (B); *p<0.05 in t-test versus control; n=5 replicates for qPCR and n>15 cells per condition for electrophysiology. (C, D) Fluorescence immunocytochemistry micrographs of activated astrocytes showing that AQP4 closely co-localizes with TRPM4 (C) and SUR1 (D) on the plasmalemma cell margin (arrows). (E, F) Calcein fluorescence micrographs of astrocytes at time 0 seconds (F0) and at time 300 seconds (F300) in experiments with A23187 calcium ionophore, showing increased membrane calcein fluorescence in activated, but not in control astrocytes upon A23187 application; F300/F0 heat maps show change in calcein fluorescence with A23187 treatment. (G) Traces of calcein fluorescence (Ft/F0) in primary astrocytes showing that, compared to control astrocytes (grey), activation (red) sensitizes astrocytes to A23187-induced water influx, which is inhibited with glibenclamide (Glib.) (blue) and blocked with 9-phenanthrol (9-Phen.) (green). (H, I) Compared to control (CTR), ~70% knockdown of TRPM4 mRNA with siRNA (H) resulted in ~65% reduction in activated astrocyte swelling upon A23187 application; n=3 independent experiments with 3 cells/experiment; *p<0.05 in t-test or ANOVA with Tukey tests versus leftmost column; #p<0.05 in ANOVA with Tukey tests versus ACT.
Immunocytochemistry was used to determine possible co-localization of AQP4, TRPM4, and SUR1 in activated primary astrocytes. AQP4 and TRPM4 (Figure 6C), and AQP4 and SUR1 (Figure 6D) immunolabeling was present in the intracellular compartment, and along the plasmalemmal margin of the cell, indicating membrane localization. Co-localization analysis was performed using ROIs defined at the plasmalemma. Co-localization analysis between AQP4 and TRPM4 at the cellular margin yielded a Pearson’s coefficient of 0.90±0.03. Co-localization analysis of AQP4 and SUR1 at the cellular margin yielded a Pearson’s coefficient of 0.81±0.01. These experiments indicate membrane expression of the SUR1-TRPM4-AQP4 complex in activated primary astrocytes.
To determine the role of the SUR1-TRPM4-AQP4 complex in astrocyte swelling, primary astrocytes were stimulated with A23187 to activate TRPM4 current as above, while volume changes were measured with calcein imaging. Parenthetically, sustained increased Cai2+ is prominent in astrocytes after CNS injury (Duffy and MacVicar 1996; Ding et al. 2009; Dong et al. 2013). Calcein fluorescence micrographs and calcein fluorescence traces showed increased membrane calcein fluorescence upon A23187 treatment in activated (Figure 6F, 6G), but not in control (Figure 6E, 6G) astrocytes. Interestingly, increases in calcein fluorescence occurred primarily at the cellular margins of activated astrocytes (Figure 6F), consistent with the subcellular localization of SUR1-TRPM4-AQP4 (Figure 6C, 6D). A23187-induced swelling in activated astrocytes was blocked by 9-phenanthrol and attenuated by glibenclamide (Figure 6G).
To confirm specificity, primary astrocytes were treated with small interfering RNA (siRNA) against TRPM4, or with a control sequence. TRPM4 knockdown disrupts the ion pore-forming subunit of the SUR1-TRPM4-AQP4 channel, preventing the SUR1-TRPM4-AQP4 complex from mediating solute flux (Figure 5). Compared to control, TRPM4 siRNA resulted in ~70% knockdown of TRPM4 mRNA (Figure 6H) and ~65% attenuation of swelling in activated astrocytes following A23187 treatment (Figure 6I).
After cerebellar cold injury, granule cell layer astrocytes express SUR1-TRPM4-AQP4
To verify the foregoing molecular findings in vivo, we studied swelling of astrocytes in the cerebellar granule cell layer in a cold injury model of brain edema (Murakami et al. 1999; van den Bos et al. 2005). Granule cell layer astrocytes, which are exquisitely sensitive to swelling after cerebellar ischemic injury (Friede 1963; Chan-Palay and Palay 1972), are an ideal system to study mechanisms of astrocyte swelling.
Male C57B6 mice were submitted to cerebellar cold injury, a model of brain edema (Murakami et al. 1999) that generates lesions that replicate many of the molecular, histopathological and functional changes that occur following arterial occlusion (Pollay and Stevens 1980; Darby et al. 1993; Itabashi et al. 2001; O’Quinn et al. 2011; Remond et al. 2011; Ongstad and Gourdie 2013; Strungs et al. 2013). When examined at 3 days after injury, cold injury resulted in a TUNEL+ necrotic lesion core surrounded by hypertrophic GFAP+ astrocytes (Figure 7A).
Figure 7. De novo expression of the SUR1-TRPM4-AQP4 complex in granule cell layer astrocytes after cold injury.

(A) Montage of micrographs of mouse cerebellum labeled for TUNEL (white), GFAP (red), and DAPI (blue) showing TUNEL+ cold injury with surrounding upregulation of GFAP in the cerebellar granule cell layer (dotted lines); boxes show ROIs for ipsilateral injured (INJ) tissue and contralateral control (CTR) tissue; the results shown are representative of 5 mice. (B, C) Micrographs of mouse cerebellum immunolabeled for AQP4 and TRPM4 (B) or SUR1 (C) showing ~3-fold (p=0.004) upregulation of SUR1 and ~3.6-fold (p=0.008) upregulation of TRPM4 in injured AQP4+ cerebellar granule cell layer astrocytes versus control; co-localization analysis showed Pearson’s correlation coefficient of ~0.84 for AQP4 and TRPM4 (B), and ~0.77 for AQP4 and SUR1 (C) in injured tissues; *p<0.05 in t-test versus CTR. (D, E) Immunoblots of co-IP experiments from cold injured and sham injured mouse cerebellum, showing AQP4 immunoblot detecting AQP4 upon TRPM4 immunoprecipitation (IP) in injured tissues with low (low ex) and higher exposures (high ex) (D), and SUR1 immunoblot detecting SUR1 upon AQP4 IP in injured tissues (E); the results shown are representative of 3 replicates.
Normally, astrocytes constitutively express high levels of AQP4 (Nielsen et al. 1997), but do not express SUR1 or TRPM4 (Chen and Simard 2001; Chen et al. 2003; Simard et al. 2006; Mehta et al. 2015). Thus, immunohistochemistry was used to determine if TRPM4 and SUR1 were upregulated in cerebellar astrocytes following cold injury. Sections immunolabeled for TRPM4 or SUR1 were co-immunolabeled for AQP4, an astrocyte-specific marker. Cerebellar granule cell layer AQP4 labeling was uniformly present across the astrocyte plasmalemma (Figure 7B, 7C), as previously reported (Hubbard et al. 2015). In the injured granule cell layer, AQP4 closely co-localized with TRPM4 (Figure 7B) and SUR1 (Figure 7C). Co-localization analysis of AQP4 and TRPM4 in the injured cerebellar granule cell layer revealed a Pearson’s coefficient of ~0.84 (Figure 7B). A similar analysis of AQP4 and SUR1 revealed a Pearson’s coefficient of ~0.77 (Figure 7C).
To quantify TRPM4 and SUR1 expression specifically in cerebellar astrocytes, the AQP4 channel was used to mask the TRPM4 and SUR1 channels prior to quantification. Compared to control tissues, TRPM4 and SUR1 immunoreactivities were increased in injured cerebellar granule cell layer astrocytes (Figure 7B, C). Quantification showed that TRPM4 immunoreactivity increased by ~3.6-fold (p=0.008) (Figure 7B), and SUR1 immunoreactivity by ~3.0-fold (p=0.004) (Figure 7C).
To evaluate for possible interactions between AQP4 and TRPM4 or SUR1, cerebellar tissues from mice submitted to sham injury or cerebellar cold injury were studied by co-immunoprecipitation (co-IP). After IP using anti-TRPM4 antibody, immunoblot showed AQP4 in cold injured cerebellum, but not sham injured control cerebellum (Figure 7D). After IP with anti-AQP4 antibody, immunoblot showed SUR1 in cold injured cerebellum, but not sham injured control cerebellum (Figure 7E).
Together, these data are consistent with the de novo formation of a 3-partner macromolecular complex in vivo after CNS injury, in accord with results from in vitro co-IP and FRET studies (Figure 1, 3, 4).
The SUR1-TRPM4-AQP4 complex mediates astrocyte swelling after cerebellar cold injury
We showed that the SUR1-TRPM4-AQP4 complex mediates fast, high-capacity transmembrane water flux and astrocyte swelling in vitro (Figure 5, 6). To confirm the relevance of these findings to pathological astrocyte swelling after CNS injury, we studied granule cell layer astrocyte swelling following a cerebellar cold injury. Astrocyte swelling was compared between wild-type and TRPM4−/− knockout mice, which lack the ion pore-forming subunit of the SUR1-TRPM4-AQP4 channel, and thus cannot mediate SUR1-TRPM4-AQP4 solute flux (Figure 5, 6).
Measurement of astrocyte volume is technically challenging, with conventional GFAP immunolabeling resulting in marked underestimation of intracellular volume (Bushong et al. 2002). To accurately measure astrocyte volume, brain tissue sections were processed for diolistic labeling, which results in a sparse fluorescent cellular labeling that fully stains the plasmalemma of labeled cells (Gan et al. 2000). Astrocytes were identified with GFAP immunolabeling. Z-stacks of diolistic labeled astrocytes were obtained with confocal microscopy. An automated 3-D region-growing algorithm was used to segment the bright intracellular volume from the dark extracellular space (Figure 8A). Co-labeling with GFAP and TUNEL confirmed that astrocytes selected for analysis were viable (Figure 8B).
Figure 8. SUR1-TRPM4 mediates astrocyte swelling after cerebellar cold injury.
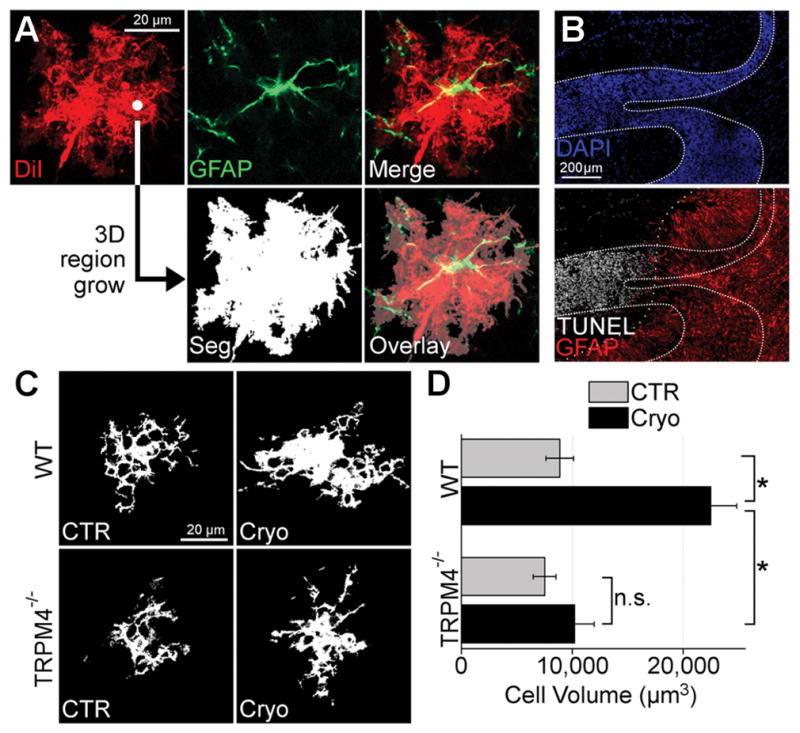
(A) Image processing pipeline for astrocyte volume quantification; DiI-stained (red), GFAP+ (green) astrocytes were imaged; 3D region growing of DiI image outputs a binary image segmentation (Seg) of intracellular voxels (white) from extracellular voxels (black); overlay demonstrates segmentation coverage of astrocyte arborization. (B) Montage of micrographs of murine cerebellum with granule cell layer defined with DAPI (dense dotted lines) and co-labeled for GFAP (red) and TUNEL (white) showing TUNEL+ granule cell layer tissues do not overlap (sparse dotted line) with GFAP+ granule cell layer tissues; the results shown are representative of n=4 mice. (C) Segmented images of cerebellar granule cell layer astrocytes from wild-type (WT) and TRPM4−/− mice submitted to control sham surgery (CTR) or cerebellar cold injury (Cryo) showing that in WT mice, granule cell layer astrocytes in cold injured cerebellum exhibited swelling of somata and processes, whereas astrocytes from TRPM4−/− mice were protected from astrocyte swelling after cerebellar cold injury; the results shown are representative of n>15 cells from 3 independent mice. (D) Quantification of granule cell layer astrocytic volume in WT and TRPM4−/− mice submitted to sham surgery (CTR) or cerebellar cold injury (Cryo) showing that after cerebellar cold injury, WT astrocytes increased in volume from 8.86 × 104 μm3 to 22.47 × 104 μm3; TRPM4−/− astrocytes increased in volume from 7.5 × 104 μm3 to only 10.2 × 104 μm3; TRPM4 knockout led to significant reduction in astrocyte swelling after cold injury; *p<0.05 in ANOVA with Tukey tests between groups denoted with brackets; n.s.=non-significant; n>15 cells from 3 different mice.
In sham injured WT mice, granule cell layer astrocytes exhibited characteristic veil-like processes that formed acini (Figure 8C) (Chan-Palay and Palay 1972). Volumetric analysis showed that in sham injured WT mice, mean astrocyte volume was 8.86 × 104 μm3 (Figure 8D), a value comparable to previous reports (Halassa et al. 2007). After cold injury, WT astrocytes exhibited swollen somata and processes (Figure 8C) and mean astrocyte volume in WT increased ~2.5-fold to 22.47 × 104 μm3 (Figure 8D).
Neither astrocyte morphology (Figure 8C) nor volume (Figure 8D) were different between sham-injured TRPM4−/− and sham-injured WT mice. However, deletion of TRPM4 largely blocked the formation of astrocyte swelling at 3 days after cold injury (Figure 8C and 8D). These results, together with findings from calcein imaging (Figure 5, 6), indicate that the SUR1-TRPM4-AQP4 complex is a key mediator of transmembrane water flux and astrocyte swelling following CNS injury.
DISCUSSION
Here, we report that AQP4, SUR1 and TRPM4 physically co-assemble to form a heteromultimeric water/ion channel complex that mediates transmembrane water flux and astrocyte swelling in vitro. Moreover, we demonstrate that upregulation of SUR1-TRPM4 and co-assembly with AQP4 mediates astrocyte swelling in vivo after CNS injury. Our findings indicate that the physical co-association of AQP4 with a newly expressed Na+-permeable channel plays a heretofore unrecognized role in transmembrane water flux during pathological astrocyte swelling.
To study astrocyte swelling in vivo, we utilized a cold injury model of brain edema. Cold injury is a well-characterized model of brain edema (Murakami et al. 1999) that creates highly reproducible lesions (Strungs et al. 2013) that closely resemble many of the molecular, histopathological and functional changes that occur following arterial occlusion (Martins and Doyle 1978; Pollay and Stevens 1980; Darby et al. 1993; Itabashi et al. 2001; O’Quinn et al. 2011; Remond et al. 2011; Ongstad and Gourdie 2013). Cold injury results in an area of frozen tissue immediately below the probe, with a surrounding area of reduced cerebral blood flow (<15/100g min) (Darby et al. 1993), blood-brain barrier disruption, and brain edema formation (Murakami et al. 1999). Moreover, we applied this injury model to the cerebellum, in order to capitalize on the exquisite sensitivity of astrocytes in the cerebellar granule cell layer to ischemia-induced swelling (Friede 1963; Chan-Palay and Palay 1972).
To examine the role of the SUR1-TRPM4-AQP4 complex in astrocyte swelling in vivo, we studied TRPM4−/− mice, which lack the osmolyte channel of the SUR1-TRPM4-AQP4 complex. Our studies of astrocyte swelling in WT versus TRPM4−/− mice showed that genetic ablation of TRPM4 blocked astrocyte swelling at 3 days after cold injury, indicating a key role for SUR1-TRPM4-AQP4 in astrocyte swelling in vivo. Given that knockout of TRPM4 largely blocked astrocyte swelling, an AQP4-dependent phenomenon (Manley et al. 2000), we infer that the contribution of TRPM4 to astrocyte swelling is upstream of AQP4.
Functional role of the SUR1-TRPM4-AQP4 complex
After CNS injury, AQP4 shifts from its normal polarized location at the astrocyte endfoot to widespread expression throughout the plasmalemma (Frydenlund et al. 2006; Steiner et al. 2012). Here, we show that this phenomenon, termed AQP4 dysregulation, may be accompanied by a switch in AQP4 binding partners from TRPV4 to TRPM4. This switch appears to be facilitated by a greater affinity of AQP4 for its new binding partner, TRPM4, as we found here (Figure 2).
As a Ca2+-sensitive, non-selective monovalent cation channel, TRPM4 functions normally as a negative regulator of Ca2+ influx – channel opening, triggered by an increase in Ca2+, causes cell depolarization that reduces the inward driving force for Ca2+ (Vennekens and Nilius 2007). Co-assembly of TRPM4 with SUR1 doubles TRPM4’s affinity for Ca2+/calmodulin, and thus its sensitivity to Ca2+, thereby amplifying TRPM4’s function as negative regulator of Ca2+ influx (Chen and Simard 2001; Chen et al. 2003; Woo et al. 2013). Based on our findings in the present report, we propose that co-assembly of SUR1-TRPM4 with AQP4 may further amplify this fundamental role of TRPM4 as negative regulator of Ca2+ influx, since the influx of water via AQP4 would serve to dilute any residual Ca2+ influx. After injury, astrocytes commonly exhibit elevated levels of intracellular Ca2+ (Duffy and MacVicar 1996; Ding et al. 2009; Dong et al. 2013). The SUR1-TRPM4-AQP4 complex appears to be well suited to oppose any further rise in intracellular Ca2+ by the dual complementary functions of cell depolarization and Ca2+ dilution.
AQP4 co-assembly with SUR1-TRPM4: structure and function
Based on our co-IP and FRET data (Figure 1–4), as well as previous structural studies (Ho et al. 2009; Liao et al. 2013; Li et al. 2017), we propose the structural model of the SUR1-TRPM4-AQP4 complex illustrated in Figure 9. Based on analogous structures (Mikhailov et al. 2005), the SUR1-TRPM4 channel is believed to be comprised of a central pore formed by four TRPM4 subunits, which are surrounded by four equidistantly spaced SUR1 subunits. Since FRET data indicate that AQP4 is closely co-associated (<10 nm) with both TRPM4 and SUR1, we speculate that AQP4 may intercalate between SUR1 subunits (Figure 9A). Interestingly, while AQP4 co-associates with TRPM4 alone (Figure 1A, 1B, 1C), co-expression of SUR1 with TRPM4, and formation of the SUR1-TRPM4 channel, increases TRPM4 co-association with AQP4 by ~4-fold (Figure 1F). This result is consistent with SUR1 providing four additional AQP4 binding sites in the SUR1-TRPM4 channel versus the TRPM4 channel (Figure 9). Notably, the close apposition of SUR1-TRPM4 and AQP4 is similar to the arrangement of ion and water channels observed in water co-transporters (MacAulay et al. 2004; Zeuthen et al. 2016). Structural studies are currently underway to examine the proposed model in Figure 9.
Figure 9. Model of SUR1-TRPM4-AQP4 structure and function.
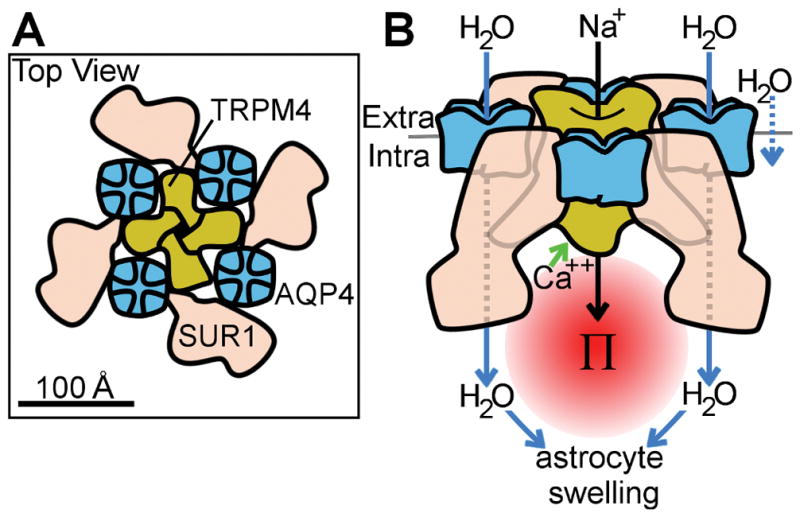
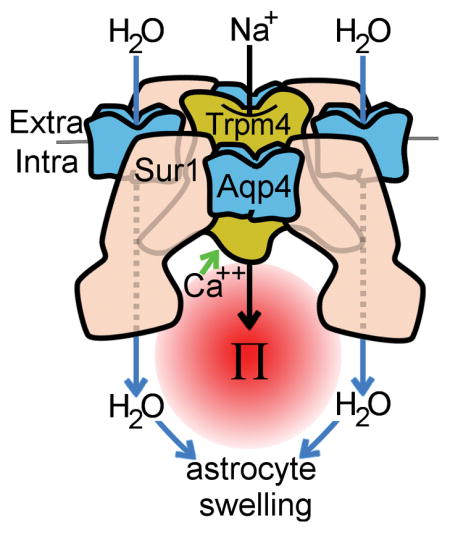
(A) Putative model of SUR1-TRPM4-AQP4 derived from co-immunoprecipitation experiments in Figure 1 and FRET experiments in Figure 3 and Figure 4, showing AQP4 tetramers (blue) intercalated between SUR1 monomers (pink), and interacting with the central tetrameric TRPM4 (yellow); protein profiles and relative protein sizes are based upon previously published structures for AQP4 (Ho et al. 2009), SUR1 (Li et al. 2017), and TRP channels (Liao et al. 2013); overall channel topology is based on the structure of the KATP channel (Li et al. 2017). (B) Model of SUR1-TRPM4-AQP4 water flux derived from calcein imaging experiments in Figure 5 and Figure 6, and astrocyte swelling experiments in Figure 8, showing that raised intracellular Ca2+ triggers SUR1-TRPM4 channel activation and Na+ influx, resulting in raised local osmotic pressure (Π) which, in turn, drives water influx (blue arrows) through AQP4, resulting in astrocyte swelling; some water flux occurs through AQP4-independent routes, including through the plasmalemma (rightmost arrow).
In our functional studies, the expression of AQP4 alone was not sufficient to mediate cell swelling, an unsurprising result given that AQP4 is a purely passive water channel. By contrast, when TRPM4 or SUR1-TRPM4 were expressed and activated without AQP4, some cell swelling did occur, likely due to transmembrane water influx through non-aquaporin routes. Although cells that expressed TRPM4 and SUR1-TRPM4 swelled, water influx developed slowly over ~2 min and was relatively small in magnitude. In contrast, co-expression of AQP4 with SUR1 and TRPM4, and formation of the SUR1-TRPM4-AQP4 complex increased steady-state swelling magnitude by ~1.5-fold, net water uptake by ~7.6-fold, and nearly quadrupled the rate of water uptake (Figure 5D). Notably, co-expression of AQP4 with TRPM4 did not result in similar gains in water flux (Figure 5B), indicating that SUR1-TRPM4 and AQP4 uniquely interact to generate bulk water flux. Given that AQP4 physically co-associates with SUR1-TRPM4, these results indicate that the addition of a passive water pore to the SUR1-TRPM4 channel greatly boosts water transport rate and magnitude.
The physical co-assembly of ion and water channels may accelerate water flux by strengthening osmotic coupling. Osmotic pressures generated by ion influx are eroded in a spatially-dependent manner by (i) intracellular diffusion barriers such as a relatively viscous cytoplasm, tortuous cellular morphology, and the intracellular protein lattice (Baumgartner et al. 1999; Ovádi and Saks 2004), and by (ii) compensatory mechanisms that resist changes in cytoplasmic ion concentrations (Rose and Ransom 1997b; Ho 2006). Thus, osmotic coupling between randomly distributed ion and water channels might not allow the specific spatial targeting of water influx that is needed to support the proposed function of diluting Ca2+ influx. By contrast, by directly coupling ion flux with water flux, the translation of osmotic pressure to water influx is spatially optimized for the proposed function (Figure 9B). This model, which emphasizes the cooperative importance of ion and water flux, helps explain the interdependent, complementary roles of AQP4 and Na+-permeable channels in astrocyte swelling (Staub et al. 1990; Manley et al. 2000; Su et al. 2002; Chen et al. 2003). Interestingly, in the present report, swelling in activated primary astrocytes occurred primarily at the cell margins, a specific plasmalemma domain that expressed the SUR1-TRPM4-AQP4 complex (Figure 6F). This result is consistent with the above model, and indicates that specialized water transport membrane domains may account for transmembrane water flux. Future experiments will be needed to directly address the impact of spatial proximity on water transport efficiency, and to determine if other mediators of high-capacity water influx utilize similar strategies.
Pathological role of SUR1-TRPM4-AQP4 water flux in astrocyte swelling
Whereas the adaptive consequence of formation of the SUR1-TRPM4-AQP4 complex may be the improved regulation of intracellular Ca2+, its pathological consequence is astrocyte swelling. Our data show that, in vitro after activation by pro-inflammatory cytokines, and in vivo after injury, astrocytes de novo upregulate the SUR1-TRPM4-AQP4 complex, accounting for cell swelling (Figure 6, 7, 8). We demonstrate that SUR1-TRPM4-AQP4 transmembrane water influx is triggered by increased Cai2+, which is prominent in astrocytes after brain injury (Duffy and MacVicar 1996; Ding et al. 2009; Dong et al. 2013). Astrocyte swelling (i) reduces the extracellular space, which impairs clearance of toxic metabolites (Sykova and Chvatal 2000); (ii) triggers dysregulated release of glutamate, which contributes to neuronal death (Kimelberg et al. 1990); (iii) exacerbates cerebral edema (Friede 1963; Stokum et al. 2016), which together may worsen outcome after CNS injury.
Given the numerous Na+-permeable channels implicated in astrocyte swelling (Stokum et al. 2015a; Stokum et al. 2016), it is unlikely that the SUR1-TRPM4-AQP4 complex is the sole mediator of astrocyte swelling. However, to our knowledge, none of these other Na+ permeable channels has been shown to co-assemble with AQP4. Our findings indicate that the SUR1-TRPM4-AQP4 complex is particularly important in the subacute phase (~3 days), which coincides with peak brain swelling after ischemic stroke.
The divergent time scales examined in our calcein swelling experiments (minutes after stimulation) and in vivo swelling experiments (3 days after injury) complicate a direct comparison between these results. Future in vivo experiments involving two-photon imaging of astrocyte swelling, or TMA+ quantification of extracellular space could contribute to our understanding of the temporal role of the SUR1-TRPM4-AQP4 complex in astrocyte volume changes after injury.
Therapeutic implications of AQP4/ion channel interactions
In the normal healthy brain, the AQP4-TRPV4 complex plays a major role in astrocyte volume homeostasis (Benfenati et al. 2011). However, our data show that, following injury, SUR1-TRPM4 binds with AQP4, and drives astrocyte swelling. These findings suggest that AQP4 function may be fundamentally determined by context-specific interactions with various ion channels. It is possible that other AQP4-dependent processes, including brain edema formation (Manley et al. 2000), vasogenic edema clearance (Papadopoulos et al. 2004), glymphatic system function (Iliff et al. 2012), astrocytic migration (Saadoun et al. 2005), and neuroinflammation (Fukuda and Badaut 2012) are similarly dependent on interactions between AQP4 and various ion channels.
If AQP4 is dependent on ion channel function, ion channel antagonists could be used to indirectly modulate AQP4. Given that AQP4-specific inhibition has not yet become practical, this is a tempting prospect. In light of our findings, indirect attenuation of AQP4 function may partially underlie the beneficial effects of glibenclamide following ischemic stroke (Chen et al. 2003; Simard et al. 2006; Sheth et al. 2016).
Main Points.
AQP4 co-assembles with SUR1-TRPM4 to form a water/ion channel complex.
Ion flux through SUR1-TRPM4 generates osmotic pressure that drives AQP4 water flux
The SUR1-TRPM4-AQP4 complex mediates astrocyte swelling after CNS injury.
Acknowledgments
The authors thank Dr. Joseph Maubin for support with confocal microscopy, Dr. Scott Thompson for the use of the Helios Gene Gun System, and Dr. Joseph Kao for careful review of the manuscript. This work was supported by grants to J.M.S. from NHLBI (R01 HL082517) and NINDS (R01 NS060801, R01 NS102589) and to V.G. from NINDS (R01 NS061934).
Footnotes
POTENTIAL CONFLICT OF INTEREST
J.M.S. holds a US patent (7,285,574) and is a member of the scientific advisory board and holds shares in Remedy Pharmaceuticals. The authors have no additional competing financial interests.
References
- Baumgartner W, Islas L, Sigworth F. Two-microelectrode voltage clamp of Xenopus oocytes: voltage errors and compensation for local current flow. Biophysical journal. 1999;77(4):1980–1991. doi: 10.1016/S0006-3495(99)77039-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati V, Caprini M, Dovizio M, Mylonakou MN, Ferroni S, Ottersen OP, Amiry-Moghaddam M. An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control in astrocytes. Proc Natl Acad Sci U S A. 2011;108(6):2563–8. doi: 10.1073/pnas.1012867108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrouschot J, Sterker M, Bettin S, Köster J, Schneider D. Mortality of space-occupying (‘malignant’) middle cerebral artery infarction under conservative intensive care. Intensive care medicine. 1998;24(6):620. doi: 10.1007/s001340050625. [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. The Journal of neuroscience. 2002;22(1):183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115(12):1599–608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- Chan-Palay V, Palay SL. The form of velate astrocytes in the cerebellar cortex of monkey and rat: high voltage electron microscopy of rapid Golgi preparations. Z Anat Entwicklungsgesch. 1972;138(1):1–19. doi: 10.1007/BF00519921. [DOI] [PubMed] [Google Scholar]
- Chen M, Dong Y, Simard JM. Functional coupling between sulfonylurea receptor type 1 and a nonselective cation channel in reactive astrocytes from adult rat brain. J Neurosci. 2003;23(24):8568–77. doi: 10.1523/JNEUROSCI.23-24-08568.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Simard JM. Cell swelling and a nonselective cation channel regulated by internal Ca2+ and ATP in native reactive astrocytes from adult rat brain. Journal of Neuroscience. 2001;21(17):6512–6521. doi: 10.1523/JNEUROSCI.21-17-06512.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotrina ML, Kang J, Lin JH, Bueno E, Hansen TW, He L, Liu Y, Nedergaard M. Astrocytic gap junctions remain open during ischemic conditions. Journal of Neuroscience. 1998;18(7):2520–2537. doi: 10.1523/JNEUROSCI.18-07-02520.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crnich RC, Gonzales AL, Earley S. Membrane Trafficking of TRPM4 in Smooth Muscle Cells. The FASEB Journal. 2008;22(1 Supplement):937.4–937.4. [Google Scholar]
- Darby JM, Nemoto EM, Yonas H, Yao L, Melick JA, Boston JR. Local cerebral blood flow measured by xenon-enhanced CT during cryogenic brain edema and intracranial hypertension in monkeys. J Cereb Blood Flow Metab. 1993;13(5):763–72. doi: 10.1038/jcbfm.1993.97. [DOI] [PubMed] [Google Scholar]
- Ding S, Wang T, Cui W, Haydon PG. Photothrombosis ischemia stimulates a sustained astrocytic Ca2+ signaling in vivo. Glia. 2009;57(7):767–76. doi: 10.1002/glia.20804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong QP, He JQ, Chai Z. Astrocytic Ca(2+) waves mediate activation of extrasynaptic NMDA receptors in hippocampal neurons to aggravate brain damage during ischemia. Neurobiol Dis. 2013;58:68–75. doi: 10.1016/j.nbd.2013.05.005. [DOI] [PubMed] [Google Scholar]
- Duffy S, MacVicar BA. In vitro ischemia promotes calcium influx and intracellular calcium release in hippocampal astrocytes. J Neurosci. 1996;16(1):71–81. doi: 10.1523/JNEUROSCI.16-01-00071.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquette PP, Bissonnette P, Lapointe JY. Local osmotic gradients drive the water flux associated with Na(+)/glucose cotransport. Proc Natl Acad Sci U S A. 2001;98(7):3796–801. doi: 10.1073/pnas.071245198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd CL, Gorin FA, Lyeth BG. Mechanical strain injury increases intracellular sodium and reverses Na+/Ca2+ exchange in cortical astrocytes. Glia. 2005;51(1):35–46. doi: 10.1002/glia.20183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friede RL. Cerebellar edema. A metabolic and cellstatistical analysis. Arch Neurol. 1963;8:67–81. doi: 10.1001/archneur.1963.00460010083010. [DOI] [PubMed] [Google Scholar]
- Frydenlund DS, Bhardwaj A, Otsuka T, Mylonakou MN, Yasumura T, Davidson KG, Zeynalov E, Skare O, Laake P, Haug FM, et al. Temporary loss of perivascular aquaporin-4 in neocortex after transient middle cerebral artery occlusion in mice. Proc Natl Acad Sci U S A. 2006;103(36):13532–6. doi: 10.1073/pnas.0605796103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda AM, Badaut J. Aquaporin 4: a player in cerebral edema and neuroinflammation. J Neuroinflammation. 2012;9:279. doi: 10.1186/1742-2094-9-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan WB, Grutzendler J, Wong WT, Wong RO, Lichtman JW. Multicolor “DiOlistic” labeling of the nervous system using lipophilic dye combinations. Neuron. 2000;27(2):219–25. doi: 10.1016/s0896-6273(00)00031-3. [DOI] [PubMed] [Google Scholar]
- Gerzanich V, Woo SK, Vennekens R, Tsymbalyuk O, Ivanova S, Ivanov A, Geng Z, Chen Z, Nilius B, Flockerzi V, et al. De novo expression of Trpm4 initiates secondary hemorrhage in spinal cord injury. Nat Med. 2009;15(2):185–91. doi: 10.1038/nm.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan M, Molinari EJ, Shieh CC, Monteggia LM, Roch JM, Whiteaker KL, Scott VE, Sullivan JP, Brioni JD. Pharmacology of human sulphonylurea receptor SUR1 and inward rectifier K(+) channel Kir6.2 combination expressed in HEK-293 cells. Br J Pharmacol. 2000;129(7):1323–32. doi: 10.1038/sj.bjp.0703181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinamard R, Hof T, Del Negro CA. The TRPM4 channel inhibitor 9-phenanthrol. Br J Pharmacol. 2014;171(7):1600–13. doi: 10.1111/bph.12582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Takano H, Dong JH, Haydon PG. Synaptic islands defined by the territory of a single astrocyte. J Neurosci. 2007;27(24):6473–7. doi: 10.1523/JNEUROSCI.1419-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann S, Kiilgaard JF, Litman T, Alvarez-Leefmans FJ, Winther BR, Zeuthen T. Measurement of cell volume changes by fluorescence self-quenching. Journal of Fluorescence. 2002;12(2):139–145. [Google Scholar]
- Heron M, Hoyert DL, Murphy SL, Xu J, Kochanek KD, Tejada-Vera B. Deaths: final data for 2006. Natl Vital Stat Rep. 2009;57(14):1–134. [PubMed] [Google Scholar]
- Ho JD, Yeh R, Sandstrom A, Chorny I, Harries WE, Robbins RA, Miercke LJ, Stroud RM. Crystal structure of human aquaporin 4 at 1.8 A and its mechanism of conductance. Proc Natl Acad Sci U S A. 2009;106(18):7437–42. doi: 10.1073/pnas.0902725106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SN. Intracellular water homeostasis and the mammalian cellular osmotic stress response. Journal of cellular physiology. 2006;206(1):9–15. doi: 10.1002/jcp.20445. [DOI] [PubMed] [Google Scholar]
- Hubbard JA, Hsu MS, Seldin MM, Binder DK. Expression of the Astrocyte Water Channel Aquaporin-4 in the Mouse Brain. ASN Neuro. 2015;7(5) doi: 10.1177/1759091415605486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4(147):147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illarionova N, Gunnarson E, Li Y, Brismar H, Bondar A, Zelenin S, Aperia A. Functional and molecular interactions between aquaporins and Na, K-ATPase. Neuroscience. 2010;168(4):915–925. doi: 10.1016/j.neuroscience.2009.11.062. [DOI] [PubMed] [Google Scholar]
- Itabashi Y, Prado GL, Abo M, Miura H, Abe Y. Dissociation of brain edema induced by cold injury in rat model: MR imaging and perfusion studies with 14C-iodo-antipyrine. Ann Nucl Med. 2001;15(4):337–42. doi: 10.1007/BF02988240. [DOI] [PubMed] [Google Scholar]
- Jayakumar AR, Norenberg MD. The Na-K-Cl Co-transporter in astrocyte swelling. Metab Brain Dis. 2010;25(1):31–8. doi: 10.1007/s11011-010-9180-3. [DOI] [PubMed] [Google Scholar]
- Jung JS, Bhat RV, Preston GM, Guggino WB, Baraban JM, Agre P. Molecular characterization of an aquaporin cDNA from brain: candidate osmoreceptor and regulator of water balance. Proc Natl Acad Sci U S A. 1994;91(26):13052–6. doi: 10.1073/pnas.91.26.13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelberg HK, Goderie SK, Higman S, Pang S, Waniewski RA. Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J Neurosci. 1990;10(5):1583–91. doi: 10.1523/JNEUROSCI.10-05-01583.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelberg HK, Macvicar BA, Sontheimer H. Anion channels in astrocytes: biophysics, pharmacology, and function. Glia. 2006;54(7):747–57. doi: 10.1002/glia.20423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroon D. Region Growing. [Accessed August 3, 2016];MathWorks File Exchange. 2008 http://www.mathworks.com/matlabcentral/fileexchange/19084-region-growing?focused=5098324&tab=functionUpdated March 6, 2008.
- Kurland DB, Gerzanich V, Karimy JK, Woo SK, Vennekens R, Freichel M, Nilius B, Bryan J, Simard JM. The Sur1-Trpm4 channel regulates NOS2 transcription in TLR4-activated microglia. Journal of neuroinflammation. 2016;13(1):130. doi: 10.1186/s12974-016-0599-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertsen KL, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab. 2012;32(9):1677–98. doi: 10.1038/jcbfm.2012.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe JY, Gagnon M, Poirier S, Bissonnette P. The presence of local osmotic gradients can account for the water flux driven by the Na+–glucose cotransporter. The Journal of physiology. 2002;542(1):61–62. doi: 10.1113/jphysiol.2002.013328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HL, Kostulas N, Huang YM, Xiao BG, van der Meide P, Kostulas V, Giedraitas V, Link H. IL-17 and IFN-gamma mRNA expression is increased in the brain and systemically after permanent middle cerebral artery occlusion in the rat. J Neuroimmunol. 2001;116(1):5–14. doi: 10.1016/s0165-5728(01)00264-8. [DOI] [PubMed] [Google Scholar]
- Li N, Wu JX, Ding D, Cheng J, Gao N, Chen L. Structure of a Pancreatic ATP-Sensitive Potassium Channel. Cell. 2017;168(1):101–110. e10. doi: 10.1016/j.cell.2016.12.028. [DOI] [PubMed] [Google Scholar]
- Liao M, Cao E, Julius D, Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504(7478):107–112. doi: 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh KP, Ng G, Yu CY, Fhu CK, Yu D, Vennekens R, Nilius B, Soong TW, Liao P. TRPM4 inhibition promotes angiogenesis after ischemic stroke. Pflugers Arch. 2014;466(3):563–76. doi: 10.1007/s00424-013-1347-4. [DOI] [PubMed] [Google Scholar]
- MacAulay N, Hamann S, Zeuthen T. Water transport in the brain: role of cotransporters. Neuroscience. 2004;129(4):1029–1042. doi: 10.1016/j.neuroscience.2004.06.045. [DOI] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6(2):159–63. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- Martins AN, Doyle TF. Cerebral blood flow in the monkey after focal cryogenic injury. Stroke. 1978;9(5):509–13. doi: 10.1161/01.str.9.5.509. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85(3):890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta RI, Tosun C, Ivanova S, Tsymbalyuk N, Famakin BM, Kwon MS, Castellani RJ, Gerzanich V, Simard JM. Sur1-Trpm4 Cation Channel Expression in Human Cerebral Infarcts. J Neuropathol Exp Neurol. 2015;74(8):835–49. doi: 10.1097/NEN.0000000000000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov MV, Campbell JD, de Wet H, Shimomura K, Zadek B, Collins RF, Sansom MS, Ford RC, Ashcroft FM. 3-D structural and functional characterization of the purified KATP channel complex Kir6.2-SUR1. EMBO J. 2005;24(23):4166–75. doi: 10.1038/sj.emboj.7600877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K, Kondo T, Yang G, Chen SF, Morita-Fujimura Y, Chan PH. Cold injury in mice: a model to study mechanisms of brain edema and neuronal apoptosis. Progress in neurobiology. 1999;57(3):289–299. doi: 10.1016/s0301-0082(98)00047-1. [DOI] [PubMed] [Google Scholar]
- Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17(1):171–80. doi: 10.1523/JNEUROSCI.17-01-00171.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B. Is the volume-regulated anion channel VRAC a “water-permeable” channel? Neurochem Res. 2004;29(1):3–8. doi: 10.1023/b:nere.0000010430.23137.be. [DOI] [PubMed] [Google Scholar]
- O’Quinn MP, Palatinus JA, Harris BS, Hewett KW, Gourdie RG. A Peptide Mimetic of the Connexin43 Carboxyl Terminus Reduces Gap Junction Remodeling and Induced Arrhythmia Following Ventricular Injury. Circulation Research. 2011;108(6):704–715. doi: 10.1161/CIRCRESAHA.110.235747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongstad EL, Gourdie RG. A Connexin43 CT-Mimetic Peptide Increases Fibroblast Migration in an in vitro Model of the Injury Border Zone. Circulation. 2013;128(22) [Google Scholar]
- Ovádi J, Saks V. On the origin of intracellular compartmentation and organized metabolic systems. Molecular and cellular biochemistry. 2004;256(1–2):5–12. doi: 10.1023/b:mcbi.0000009855.14648.2c. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Manley GT, Krishna S, Verkman AS. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004;18(11):1291–3. doi: 10.1096/fj.04-1723fje. [DOI] [PubMed] [Google Scholar]
- Péan S. [Accessed June 10];HeatMap Histogram (ImageJ plugin) 2012 http://www.samuelpean.com/heatmap-histogram/. Updated.
- Pollay M, Stevens FA. Blood-brain barrier restoration following cold injury. Neurol Res. 1980;1(3):239–45. doi: 10.1080/01616412.1980.11739556. [DOI] [PubMed] [Google Scholar]
- Remond MC, Iaffaldano G, O’Quinn MP, Mezentseva NV, Garcia V, Harris BS, Gourdie RG, Eisenberg CA, Eisenberg LM. GATA6 reporter gene reveals myocardial phenotypic heterogeneity that is related to variations in gap junction coupling. American Journal of Physiology-Heart and Circulatory Physiology. 2011;301(5):H1952–H1964. doi: 10.1152/ajpheart.00635.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR, Ransom BR. Gap junctions equalize intracellular Na+ concentration in astrocytes. Glia. 1997a;20(4):299–307. doi: 10.1002/(sici)1098-1136(199708)20:4<299::aid-glia3>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Rose CR, Ransom BR. Regulation of intracellular sodium in cultured rat hippocampal neurones. The Journal of physiology. 1997b;499(Pt 3):573. doi: 10.1113/jphysiol.1997.sp021951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadoun S, Papadopoulos MC, Watanabe H, Yan D, Manley GT, Verkman AS. Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. J Cell Sci. 2005;118(Pt 24):5691–8. doi: 10.1242/jcs.02680. [DOI] [PubMed] [Google Scholar]
- Sharma N, Crane A, Clement JPt, Gonzalez G, Babenko AP, Bryan J, Aguilar-Bryan L. The C terminus of SUR1 is required for trafficking of KATP channels. J Biol Chem. 1999;274(29):20628–32. doi: 10.1074/jbc.274.29.20628. [DOI] [PubMed] [Google Scholar]
- Sheth KN, Elm JJ, Molyneaux BJ, Hinson H, Beslow LA, Sze GK, Ostwaldt AC, Del Zoppo GJ, Simard JM, Jacobson S, et al. Safety and efficacy of intravenous glyburide on brain swelling after large hemispheric infarction (GAMES-RP): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2016;15(11):1160–9. doi: 10.1016/S1474-4422(16)30196-X. [DOI] [PubMed] [Google Scholar]
- Simard JM, Chen M, Tarasov KV, Bhatta S, Ivanova S, Melnitchenko L, Tsymbalyuk N, West GA, Gerzanich V. Newly expressed SUR1-regulated NC(Ca-ATP) channel mediates cerebral edema after ischemic stroke. Nat Med. 2006;12(4):433–40. doi: 10.1038/nm1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJ, Jin BJ, Ratelade J, Verkman AS. Aggregation state determines the localization and function of M1- and M23-aquaporin-4 in astrocytes. J Cell Biol. 2014;204(4):559–73. doi: 10.1083/jcb.201308118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solenov E, Watanabe H, Manley GT, Verkman A. Sevenfold-reduced osmotic water permeability in primary astrocyte cultures from AQP-4-deficient mice, measured by a fluorescence quenching method. American Journal of Physiology-Cell Physiology. 2004;286(2):C426–C432. doi: 10.1152/ajpcell.00298.2003. [DOI] [PubMed] [Google Scholar]
- Staub F, Baethmann A, Peters J, Weigt H, Kempski O. Effects of lactacidosis on glial cell volume and viability. Journal of Cerebral Blood Flow & Metabolism. 1990;10(6):866–876. doi: 10.1038/jcbfm.1990.143. [DOI] [PubMed] [Google Scholar]
- Steiner E, Enzmann GU, Lin S, Ghavampour S, Hannocks MJ, Zuber B, Rüegg MA, Sorokin L, Engelhardt B. Loss of astrocyte polarization upon transient focal brain ischemia as a possible mechanism to counteract early edema formation. Glia. 2012;60(11):1646–1659. doi: 10.1002/glia.22383. [DOI] [PubMed] [Google Scholar]
- Stokum JA, Gerzanich V, Simard JM. Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab. 2016;36(3):513–38. doi: 10.1177/0271678X15617172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokum JA, Kurland DB, Gerzanich V, Simard JM. Mechanisms of astrocyte-mediated cerebral edema. Neurochem Res. 2015a;40(2):317–28. doi: 10.1007/s11064-014-1374-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokum JA, Mehta RI, Ivanova S, Yu E, Gerzanich V, Simard JM. Heterogeneity of aquaporin-4 localization and expression after focal cerebral ischemia underlies differences in white versus grey matter swelling. Acta Neuropathol Commun. 2015b;3(61) doi: 10.1186/s40478-015-0239-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strungs EG, Ongstad EL, O’Quinn MP, Palatinus JA, Jourdan LJ, Gourdie RG. Cryoinjury Models of the Adult and Neonatal Mouse Heart for Studies of Scarring and Regeneration. Wound Regeneration and Repair: Methods and Protocols. 2013;1037:343–353. doi: 10.1007/978-1-62703-505-7_20. [DOI] [PubMed] [Google Scholar]
- Su G, Kintner DB, Flagella M, Shull GE, Sun D. Astrocytes from Na+-K+-Cl− cotransporter-null mice exhibit absence of swelling and decrease in EAA release. American Journal of Physiology-Cell Physiology. 2002;282(5):C1147–C1160. doi: 10.1152/ajpcell.00538.2001. [DOI] [PubMed] [Google Scholar]
- Sykova E, Chvatal A. Glial cells and volume transmission in the CNS. Neurochem Int. 2000;36(4–5):397–409. doi: 10.1016/s0197-0186(99)00131-x. [DOI] [PubMed] [Google Scholar]
- van den Bos EJ, Mees BM, de Waard MC, de Crom R, Duncker DJ. A novel model of cryoinjury-induced myocardial infarction in the mouse: a comparison with coronary artery ligation. Am J Physiol Heart Circ Physiol. 2005;289(3):H1291–300. doi: 10.1152/ajpheart.00111.2005. [DOI] [PubMed] [Google Scholar]
- Vennekens R, Nilius B. Insights into TRPM4 function, regulation and physiological role. Handb Exp Pharmacol. 2007;(179):269–85. doi: 10.1007/978-3-540-34891-7_16. [DOI] [PubMed] [Google Scholar]
- Verkman A, Binder DK, Bloch O, Auguste K, Papadopoulos MC. Three distinct roles of aquaporin-4 in brain function revealed by knockout mice. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2006;1758(8):1085–1093. doi: 10.1016/j.bbamem.2006.02.018. [DOI] [PubMed] [Google Scholar]
- Woo SK, Kwon MS, Ivanov A, Gerzanich V, Simard JM. The sulfonylurea receptor 1 (Sur1)-transient receptor potential melastatin 4 (Trpm4) channel. J Biol Chem. 2013;288(5):3655–67. doi: 10.1074/jbc.M112.428219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerangue N, Schwappach B, Jan YN, Jan LY. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron. 1999;22(3):537–48. doi: 10.1016/s0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]
- Zeuthen T. Water-transporting proteins. J Membr Biol. 2010;234(2):57–73. doi: 10.1007/s00232-009-9216-y. [DOI] [PubMed] [Google Scholar]
- Zeuthen T, Gorraitz E, Her K, Wright EM, Loo DD. Structural and functional significance of water permeation through cotransporters. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1613744113. [DOI] [PMC free article] [PubMed] [Google Scholar]