

Abstract
N,C-capped dipeptides belong to a class of noncovalent proteasome inhibitors. Here we report that insertion of a β-amino acid in N,C-capped dipeptides markedly reduces the inhibitory potency against human constitutive proteasome, yet maintains potent inhibitory activity against human immunoproteasome, thus achieving thousands-fold selectivity for β5i over β5c. Structure-activity relationship studies reveal that β5c does not tolerate the β-amino acid based dipeptidomimetics as β5i does. In vitro, one such compound inhibited human T cell proliferation. Compounds of this class may have potential as therapeutics for autoimmune and inflammatory diseases with less mechanism-based cytotoxicity than agents that also inhibit the constitutive proteasome.
Keywords: β5i selective inhibitors, β-amino acid peptidomimetics, autoimmune, immunosuppression, T cell proliferation
Entry for the Table of Contents
Class-wide selective inhibitors for immunoproteasome β5i over constitutive proteasome β5c were serendipitously identified by incorporating β-amino acids into dipeptides, of which one compound inhibited T cell proliferation.
The ubiquitin-proteasome system is a highly regulated protein-degradation machinery that controls many cellular functions, including removal of damaged proteins to avoid toxic gain-of-function effects, timely removal of cyclins to allow progression of the cell cycle, and inactivation of endogenous inhibitors of transcription factors to allow their nuclear translocation in the cells’ response to intracellular or extracellular signals.[1] Products of the proteasome are also the major source of antigenic oligopeptides for major histocompatibility complex (MHC) class I antigen presentation.[2] The 20S core of eukaryotic proteasomes contains 2 copies each of 7 different α subunits and 7 different β subunits arranged in four stacked rings in α1–7β1–7β1–7α1–7 fashion.[3] Of the 7 β subunits in each of the inner rings, 3 are enzymatically active: β1 has caspase-like activity, while β2 is tryptic and β5 is chymotryptic. Four types of eukaryotic proteasome core particles have been identified. The constitutive proteasome (c-20S) is expressed in all types of cells. The immunoproteasome (i-20S) is constitutively expressed in immune cells at variable, cell-specific levels[4] and in other cells when induced by cytokines such as interferon- γ. The thymoproteasome (t-20S) is expressed only in the thymus. The spermatoproteasome (sp-20S) is expressed in spermatazoa. Three of the types are distinguished by their active subunits: β1cβ2cβ5c in c-20S, β1iβ2iβ5i in i-20S, and β1iβ2iβ5t in t-20S. The spermatoproteasome differs from c-20S only in α4, giving α4spβ1cβ2cβ5c in sp-20S. The proteasome is a validated drug target for multiple myeloma and mantle cell lymphoma. Several classes of proteasome inhibitors have been developed.[5] Because of the hypersensitivity of immunoglobulin-secreting plasma cells to proteasome inhibition,[6] the first-approved proteasome inhibitor drug, bortezomib, has also been used to control allograft rejection [7] and refractory systemic lupus erythematosus.[8]
Given its mechanism-based toxicity, bortezomib is generally used in suboptimal doses in patients with non-malignant disease. Suppression of unwanted immune responses might be achieved with less toxicity if inhibitors were available that target i-20S but spare c-20S. Encouraging this notion, mice with combined deficiency of β1i, β2i and β5i are viable, fertile and generally immune-competent.[9] Moreover, a relatively selective β5i inhibitor, ONX-0914 (formerly known as PR-957; Fig. 1A), has shown efficacy in animal models of rheumatoid arthritis, systemic lupus erythematosus, colitis and multiple sclerosis.[10] However, ONX0914 is an irreversibly-acting peptide epoxyketone. The combination of limited isoform selectivity and irreversibility raises questions about possible toxicity during long-term use of such an agent. Chronic use of a similar peptide sequence with other irreversibly acting warheads, such as sulfonyl fluoride (Fig. 1B), is subject to the same concern, as is use of a compound that reacts irreversibly with Cys48 of β5i (Fig. 1C).[11]
Figure 1.
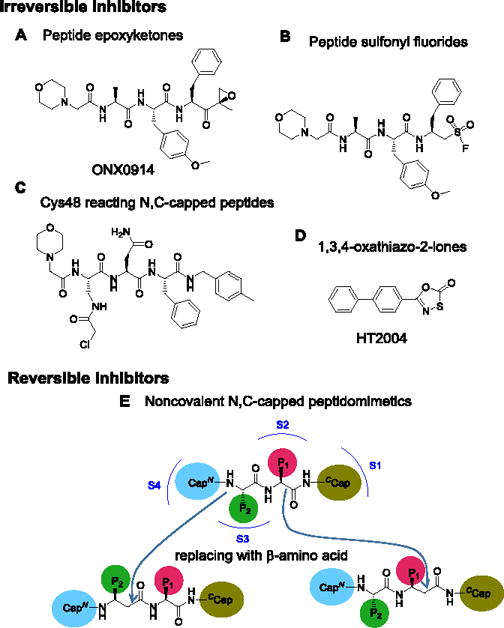
Structures of representative irreversible β5i selective inhibitors and schematic for β-amino acid based noncovalent N,C-capped peptidomimetics. A) Peptide epoxyketones; B) Peptide sulfonyl fluoride; C) β5i Cys48 reacting N,C-capped peptides; D) 1,3,4-oxathiazol-2-ones; E) Schematic for conversion of N,C-capped dipeptides to β-amino acid-based dipeptidomimetics. N- and C- caps bind to S4 and S1 pockets of the β5, respectively. P1 binds to S2 and P2 binds to S3.
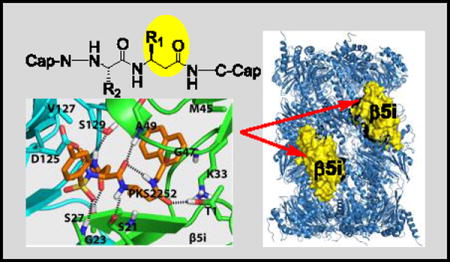
To identify reversible inhibitors with a high degree of selectivity for β5i over β5c, we took advantage of our ongoing effort to develop inhibitors that target the Mycobacterium tuberculosis proteasome (Mtb20S) but spare both hu c-20S and i-20S. We observed that Mtb20S and β5i both prefer bulky aromatic amino acids at the S1 pocket and both are inhibited by 1,3,4-oxathiazol-2-ones (Fig. 1D) that spare hu c-20S.[12] N,C-capped dipeptides are a versatile class of cell-permeable compounds that can be tailored for proteasomes of different species or forms.[13] This encouraged us to test if noncovalent N,C-capped dipeptides that we have reported to be selective for Mtb20S over both human β5i and β5c[13b] could be optimized to be selective for human β5i over β5c. Here we report achievement of this goal with β-amino acid based N,C-capped dipeptidomimetics.
Crystal structures of N,C-capped dipeptides with yeast proteasome (Ye20S) showed the C-cap in the S1 pocket, the P1 amino acid side chain in the S2 pocket, the P2 amino acid side chain in the S3 pocket and the N-cap in the S4 pocket.[14] For Mtb20S, both an S3 Asn side chain and an S1 naphthyl group are major determinants for potency and a diethyl amide chain of the Asn for selectivity.[13b] We hypothesized that because β5i but not β5c was inhibited by 1,3,4-oxathiazol-2-ones, and X-ray structures of Mtb20S with 1,3,4-oxathiazol-2-ones revealed marked conformational changes, β5i might be more plastic than β5c. This encouraged us to incorporate β-amino acids into the inhibitor while retaining the bulky 1-naphthalenemethyl C-cap. β-amino acids were incorporated separately at P1 or P2, or simultaneously at both P1 and P2. Synthetic routes are illustrated for mono-β amino acid dipeptidomimetic inhibitors (Scheme 1, S1) and dual-β amino acid dipeptidomimetics (Scheme S2). We confirmed all intermediates and final products by NMR and the final products by high-resolution MS.
Scheme 1.
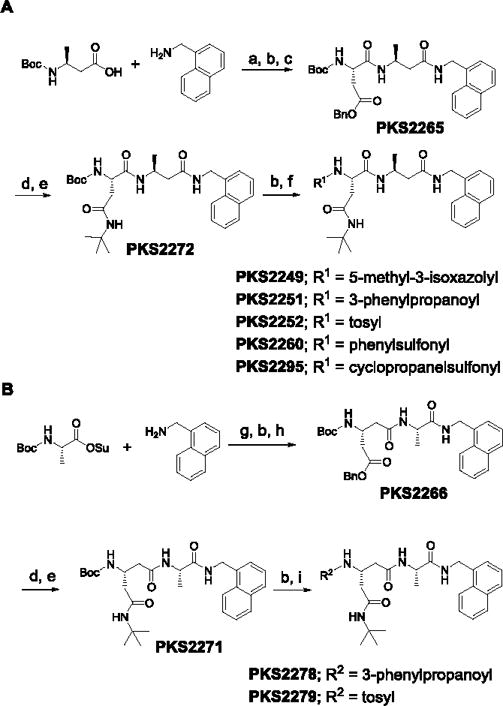
Synthetic route of P1-β-amino acid-based (A) and P2-β-amino acid-based (B) peptidomimetics. Reagents and conditions: a) HATU, HOAt, Hunig’s base, DMF; b) TFA in DCM (20%); c) Boc-Asp(OBn)OH, HATU, HOAt, Hünig’s base, DMF; d) Pd/C (10%), H2, methanol; e) tBuNH2, EDC, HOBt, Hünig’s base, DMF; f) R1COOH, HATU, HOAt, Hünig’s base, DMF or R1Cl, Et3N, DCM; g) Et3N, DCM, 0°C – RT; h) Boc-β-Glu(OBn)OH, HATU, HOAt, Hünig’s base, DMF; i) R2COOH, HATU, HOAt, Hünig’s base, DMF or R2Cl, Et3N, DCM.
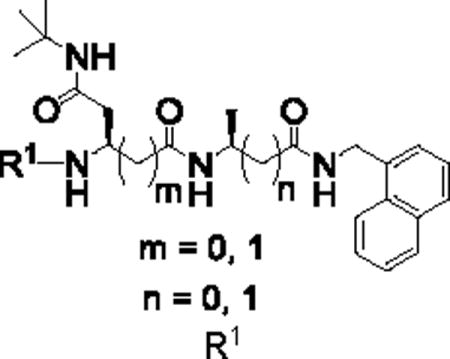
We first introduced (L)-β-Ala into the P1 position (Table 1). C-cap was fixed as 1-naphthalenemethyl group. P2 amino acid was Asn(t-Bu). For the N-cap, we used substituents that varied from hydrophobic phenylpropionate (PKS2251) to polar 5-methylisoxazole-3-carboxylate (PKS2249) and a tosyl moiety (PKS2252). Commercially available boc-(L)-β-homoalanine and 1-naphthalenemethylamine were first coupled using HATU to give boc-(L)-β-Ala-NHCH2–1-naphthyl. Boc-deprotection and coupling with Boc-Asp(OBn)-OH gave PKS2265. The O-benzyl protecting group of PKS2272 was removed by hydrogenolysis and the product converted to t-butyl amide PKS2272. Boc-deprotection of PKS2272 and subsequent reaction with a different N-cap gave the final compounds PKS2249, PKS2251, PKS2252, PKS2260 and PKS2295. PKS3054 was synthesized by coupling PKS2245 with PKS3052 (Fig. S1). Synthesis of P2-(L)-β-Gln-based dipeptidomimetics (PKS2278, PKS2279) was accomplished in a similar fashion (Fig. 1B). Boc-(L)-Ala-OSu was coupled with 1-naphthalenemethylamine and the product was mixed with TFA to remove boc protection group. The freed amine was coupled with (L)-β-Glu(OBn)-COOH, yielding PKS2266. After removal of the benzyl group by hydrogenolysis, the resulting product was converted to t-butyl amide PKS2271. Boc-deprotection of PKS2271 and subsequent reaction with phenylpropionate or tosyl gave PKS2278 and PKS2279, respectively. PKS2291 and PKS2292, in which both P1 and P2 are β-amino acids, were synthesized as depicted in Figure S2.
Table 1.
IC50s of dipeptidomimetics against β5i and β5c. Data were average of at least three independent experiments.
| ID |
|
IC50 (μM)* | SIapp | |
|---|---|---|---|---|
| β5i (ANW) | β5c (LLVY) | |||
| P1: β-AA | (m=0, n=1) | |||
| PKS2249 |
|
0.190 ± 0.018 | 88.38 ± 5.81 | 465 |
| PKS2251 |
|
0.057 ± 0.012 | 76.02 ± 25.61 | 1333 |
| PKS2252 |
|
0.0055 ± 0.0010 | 74.87 ± 10.52 | 13600 |
| PKS2260 |
|
0.012 ± 0.001 | 13.85 ± 3.79 | 1154 |
| PKS2272 |
|
0.135 ± 0.022 | >100 | >600 |
| PKS2295 |
|
0.148 ± 0.016 | 22.18 ± 5.77 | 150 |
| PKS3054 |
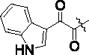
|
0.0053 ± 0.0003 | 0.30 ± 0.08 | 57 |
| P2: β-AA | (m=1, n=0) | |||
| PKS2278 |
|
3.35 ± 0.64 | >100 | >44 |
| PKS2279 |
|
0.014 ± 0.002 | 35.3 | 2521 |
| P1, P2: β-AAs | (m = 1, n = 1) | |||
| PKS2291 |
|
9.60 ± 0.66 | >100 | >11 |
| PKS2292 |
|
>100 | >100 | - |
AA: amino acid.
ANW: ac-ANW-AMC.
LLVY: suc-LLVY-AMC.
SIapp: apparent selectivity index = IC50β5c / IC50β5i.
Data were given as mean ± SEM.
IC50s against β5i and β5c were determined by a reported method (Table 1).[13b] Although the potency of these compounds varies with the N-caps, they were all markedly selective for β5i over β5c, with potency and selectivity indices ranging in tandem from high to low in the order of tosyl- (5.5 nM and 13,600-fold) > phenylpropionate (57 nM and 1,333-fold) > 5-methylisoxazole-3-carboxylate (190 nM and 465-fold). Further changes at N-cap on this scaffold with phenylsulfonamide, Boc, or cyclopropyl sulfonamide retained selectivity without further improvement in potency. N-cap (2-(1H-indol-3-yl)-2-oxoacetyl) improved potency to 5.3 nM, but reduced the selectivity index to 57-fold. Replacing the P1-β-(L)-Ala with (L)-Ala and P2-(L)-Asn with β-(L)-Gln in compound PKS2251 and PKS2252 resulted in compounds PKS2278 and PKS2279, whose potencies were reduced by 58-fold and 2.5-fold, respectively, indicating that the sulfonamide is a better N-cap than phenylpropionate to retain potency in P2 β-amino acid-based dipeptidomimetics. Furthermore, sulfonamide reserved largely unchanged selectivity. This dataset indicates that incorporation of β-amino acids in either position of this class of inhibitors is tolerated by β5i but not by β5c. However, incorporation of β-amino acids in both P1 and P2 at the same time markedly reduced the potency.
To begin to understand how the binding mode of the β-amino acid based dipeptidomimetics accounts for their marked selectivity, we created a homology model of a dimer of β5i and β6 of hu i-20S using the crystal structure of the mouse i-20S as template.[15] Assuming each moiety would similarly occupy S1–S4 pockets as described in Fig. 1, PKS2252 and PKS2279 were docked in silico in the active site (Figure 2). We found that inserting a carbon in either compound largely retains the hydrogen bond network between the backbone of the enzyme and the dipeptide ligands as shown by Blackburn et al.[14] Additionally, the N-cap sulfonamide is predicted to form a hydrogen bond with Gly23, and the side chain of P2 N-t-butyl amide is predicted to make two hydrogen bonds with Ser27 of the β5i and Ser129 of the β6, respectively. Ser27 is replaced by Ala27 in β5c. The one carbon extension in P1 of PKS2252 (Fig. 2A) is predicted to let the compound form a hydrogen bond between the carbonyl and the β-OH of the active site Thr1. In contrast, the one carbon extension in P2 of PKS2279 (Fig. 2B) is predicted not to lead to a hydrogen bond with the β-OH of the active site Thr1. Thus, P1-β-amino acid peptidomimetics and P2-β-amino acid peptidomimetics are likely to bind to β5i differently. As indicated in other reports,[15] S1 in β5i pocket is significantly bigger than that in β5c. Therefore, we postulate that the marked isoform selectivity displayed by the β-amino acid dipeptidomimetics may result from several factors: change of the key amino acids of the enzymes that are involved in the binding of the ligands, the enlarged S1 pocket in β5i over β5c and the plasticity of the β5i that allows it to undergo conformational changes to accommodate the one carbon extension in either P1 or P2 position.[12b]
Figure 2.
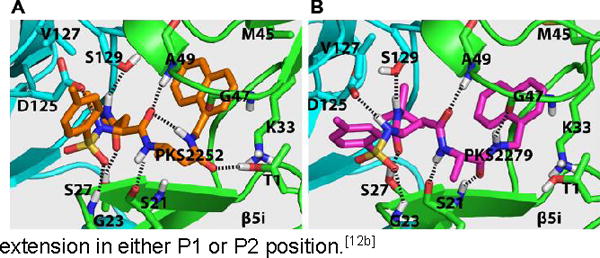
The predicted binding modes of PKS2252 (A) and PKS2279 (B) in β5iβ6 dimer of the human i-20S.
Next, we tested the impact of PKS3054 on the function of cells a substantial proportion of whose proteasomes contain i-20S. As shown in Table 2, PKS3054 inhibited the activation of human CD4-positive T lymphocytes by tetanus toxoid (EC50 0.20 μM); and the proliferation of T cells within PBMCs as stimulated by anti-CD3 antibody (EC50 0.57 μM). PKS3054 was not cytotoxic against PBMCs under these conditions (Figure S1).
Table 2.
Effects of PKS3054 in assays of adaptive immune responses
| Immunological assays1 | EC50 (μM)* |
|---|---|
|
|
|
| Hu CD4 T cell response to tetanus toxoid | 0.20 ± 0.04 |
| Hu PBMC response to anti-CD3 | 0.57 ± 0.62 |
Assays with PBMCs from 3 healthy donors, each in triplicate.
: Data were given as mean ± SEM.
In summary, we have developed a series of noncovalent β-amino acid-based peptidomimetic immunoproteasome-selective inhibitors. The surprising impact on isoform selectivity of incorporating a β-amino acid into N,C-capped dipeptides suggests a subtle yet distinct structural variation at the binding sites of the i-20S and c-20S β5 subunits. The SAR indicates that there is room to test further modifications in the interest of improving pharmacokinetic properties while retaining potency and selectivity. PKS3054 inhibits the activation of human T cells without apparent cytotoxicity, providing an early indication that such compounds may have utility in the control of immune disorders. Additionally, a recent survey found upregulated expression of i-20S over c-20S in lymphoblastic leukemia cells and multiple myeloma cell lines,[16] suggesting that selective inhibition of i-20S may have therapeutic potential in treatment of those malignancies.in Text Paragraph.
Experimental Section
Protocols of the biological experiments; docking studies; synthesis and compound characterization of all new compounds; Scheme S1 and S2; Figure S1; Table S1 and S2 are included in supporting information.
Supplementary Material
Acknowledgments
This work is supported by Alliance for Lupus Research (GL), Daedalus Fund for Innovation at Weill Cornell Medicine (GL) and by the Milstein Program in Chemical Biology and Translational Medicine (CN). We thank Ms. Rong Wang and Dr. George Sukenick at Memorial Sloan Kettering Cancer Center for assistance with high-resolution mass spectrometry. We thank Dr. J. David Warren at The Abby and Howard P. Milstein Synthetic Chemistry Core Facility at Weill Cornell Medicine for assistance. We thank Dr. Jane Salmon and Dr. Xiaoping Qing at Hospital for Special Surgery, New York, for discussions. The Department of Microbiology and Immunology is supported by the William Randolph Hearst Foundation.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Goldberg AL. Biochem Soc Trans. 2007;35:12–17. doi: 10.1042/BST0350012. [DOI] [PubMed] [Google Scholar]
- 2.Rock KL, York IA, Saric T, Goldberg AL. Adv Immunol. 2002;80:1–70. doi: 10.1016/s0065-2776(02)80012-8. [DOI] [PubMed] [Google Scholar]
- 3.Baumeister W, Walz J, Zuhl F, Seemuller E Cell. 1998;92:367–380. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 4.a) Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, Thomas JK, Muthusamy B, Leal-Rojas P, Kumar P, Sahasrabuddhe NA, Balakrishnan L, Advani J, George B, Renuse S, Selvan LD, Patil AH, Nanjappa V, Radhakrishnan A, Prasad S, Subbannayya T, Raju R, Kumar M, Sreenivasamurthy SK, Marimuthu A, Sathe GJ, Chavan S, Datta KK, Subbannayya Y, Sahu A, Yelamanchi SD, Jayaram S, Rajagopalan P, Sharma J, Murthy KR, Syed N, Goel R, Khan AA, Ahmad S, Dey G, Mudgal K, Chatterjee A, Huang TC, Zhong J, Wu X, Shaw PG, Freed D, Zahari MS, Mukherjee KK, Shankar S, Mahadevan A, Lam H, Mitchell CJ, Shankar SK, Satishchandra P, Schroeder JT, Sirdeshmukh R, Maitra A, Leach SD, Drake CG, Halushka MK, Prasad TS, Hruban RH, Kerr CL, Bader GD, Iacobuzio-Donahue CA, Gowda H, Pandey A. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wilhelm M, Schlegl J, Hahne H, Gholami A Moghaddas, Lieberenz M, Savitski MM, Ziegler E, Butzmann L, Gessulat S, Marx H, Mathieson T, Lemeer S, Schnatbaum K, Reimer U, Wenschuh H, Mollenhauer M, Slotta-Huspenina J, Boese JH, Bantscheff M, Gerstmair A, Faerber F, Kuster B. Nature. 2014;509:582–587. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 5.a) Huber EM, Groll M. Angew Chem Int Ed Engl. 2012;51:8708–8720. doi: 10.1002/anie.201201616. [DOI] [PubMed] [Google Scholar]; b) Groll M, Huber R. Biochim Biophys Acta. 2004;1695:33–44. doi: 10.1016/j.bbamcr.2004.09.025. [DOI] [PubMed] [Google Scholar]; c) Borissenko L, Groll M. Chem Rev. 2007;107:687–717. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]; d) Gallastegui N, Beck P, Arciniega M, Huber R, Hillebrand S, Groll M. Angew Chem Int Ed Engl. 2012;51:247–249. doi: 10.1002/anie.201106010. [DOI] [PubMed] [Google Scholar]
- 6.Neubert K, Meister S, Moser K, Weisel F, Maseda D, Amann K, Wiethe C, Winkler TH, Kalden JR, Manz RA, Voll RE. Nat Med. 2008;14:748–755. doi: 10.1038/nm1763. [DOI] [PubMed] [Google Scholar]
- 7.a) Raghavan R, Jeroudi A, Achkar K, Gaber AO, Patel SJ, Abdellatif A, Transplant J. 2010;2010 doi: 10.1155/2010/698594. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pai CC, Chen M, Mirsoian A, Grossenbacher SK, Tellez J, Ames E, Sun K, Jagdeo J, Blazar BR, Murphy WJ, Abedi M. Blood. 2014;124:1677–1688. doi: 10.1182/blood-2014-02-554279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frohlich K, Holle JU, Aries PM, Gross WL, Moosig F. Ann Rheum Dis. 2011;70:1344–1345. doi: 10.1136/ard.2010.133256. [DOI] [PubMed] [Google Scholar]
- 9.Kincaid EZ, Che JW, York I, Escobar H, Reyes-Vargas E, Delgado JC, Welsh RM, Karow ML, Murphy AJ, Valenzuela DM, Yancopoulos GD, Rock KL. Nat Immunol. 2012;13:129–135. doi: 10.1038/ni.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Muchamuel T, Basler M, Aujay MA, Suzuki E, Kalim KW, Lauer C, Sylvain C, Ring ER, Shields J, Jiang J, Shwonek P, Parlati F, Demo SD, Bennett MK, Kirk CJ, Groettrup M. Nat Med. 2009;15:781–787. doi: 10.1038/nm.1978. [DOI] [PubMed] [Google Scholar]; b) Basler M, Dajee M, Moll C, Groettrup M, Kirk CJ. Immunol J. 2010;185:634–641. doi: 10.4049/jimmunol.0903182. [DOI] [PubMed] [Google Scholar]; c) Muchamuel T, Jiang J, Lee SJ, Kirk CJ. Neurology. 2011;76:A459–A460. [Google Scholar]; d) Basler M, Mundt S, Muchamuel T, Moll C, Jiang J, Groettrup M, Kirk CJ. EMBO Mol Med. 2014;6:226–238. doi: 10.1002/emmm.201303543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dubiella C, Baur R, Cui H, Huber EM, Groll M. Angew Chem Int Ed Engl. 2015;54:15888–15891. doi: 10.1002/anie.201506631. [DOI] [PubMed] [Google Scholar]
- 12.a) Lin G, Li D, de Carvalho LP, Deng H, Tao H, Vogt G, Wu K, Schneider J, Chidawanyika T, Warren JD, Li H, Nathan C. Nature. 2009;461:621–626. doi: 10.1038/nature08357. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fan H, Angelo NG, Warren JD, Nathan CF, Lin G. ACS Med Chem Lett. 2014;5:405–410. doi: 10.1021/ml400531d. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sosic I, Gobec M, Brus B, Knez D, Zivec M, Konc J, Lesnik S, Ogrizek M, Obreza A, Zigon D, Janezic D, Mlinaric-Rascan I, Gobec S. Angew Chem Int Ed Engl. 2016;55:5745–5748. doi: 10.1002/anie.201600190. [DOI] [PubMed] [Google Scholar]
- 13.a) Blackburn C, Barrett C, Blank JL, Bruzzese FJ, Bump N, Dick LR, Fleming P, Garcia K, Hales P, Hu Z, Jones M, Liu JX, Sappal DS, Sintchak MD, Tsu C, Gigstad KM. Bioorg Med Chem Lett. 2010;20:6581–6586. doi: 10.1016/j.bmcl.2010.09.032. [DOI] [PubMed] [Google Scholar]; b) Lin G, Chidawanyika T, Tsu C, Warrier T, Vaubourgeix J, Blackburn C, Gigstad K, Sintchak M, Dick L, Nathan C. J Am Chem Soc. 2013;135:9968–9971. doi: 10.1021/ja400021x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blackburn C, Gigstad KM, Hales P, Garcia K, Jones M, Bruzzese FJ, Barrett C, Liu JX, Soucy TA, Sappal DS, Bump N, Olhava EJ, Fleming P, Dick LR, Tsu C, Sintchak MD, Blank JL. Biochem J. 2010;430:461–476. doi: 10.1042/BJ20100383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber EM, Basler M, Schwab R, Heinemeyer W, Kirk CJ, Groettrup M, Groll M. Cell. 2012;148:727–738. doi: 10.1016/j.cell.2011.12.030. [DOI] [PubMed] [Google Scholar]
- 16.de Bruin G, Xin BT, Kraus M, van der Stelt M, van der Marel GA, Kisselev AF, Driessen C, Florea BI, Overkleeft HS. Angew Chem Int Ed Engl. 2016;55:4199–4203. doi: 10.1002/anie.201509092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.