

Abstract
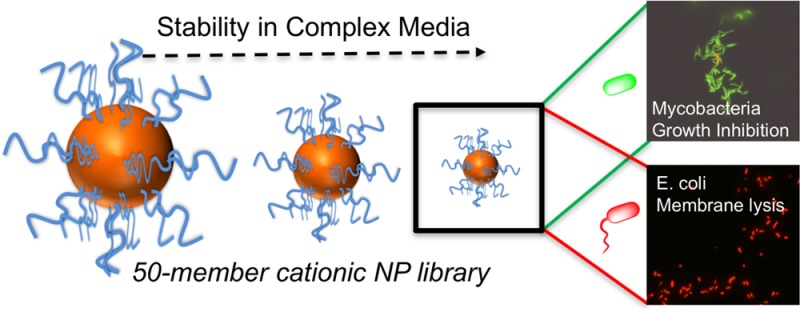
Because of the emergence of antimicrobial resistance to traditional small-molecule drugs, cationic antimicrobial polymers are appealing targets. Mycobacterium tuberculosis is a particular problem, with multi- and total drug resistance spreading and more than a billion latent infections globally. This study reports nanoparticles bearing variable densities of poly(dimethylaminoethyl methacrylate) and the unexpected and distinct mechanisms of action this multivalent presentation imparts against Escherichia coli versus Mycobacterium smegmatis (model of M. tuberculosis), leading to killing or growth inhibition, respectively. A convergent “grafting to” synthetic strategy was used to assemble a 50-member nanoparticle library, and using a high-throughput screen identified that only the smallest (2 nm) particles were stable in both saline and complex cell media. Compared with the linear polymers, the nanoparticles displayed two- and eight-fold enhancements in antimicrobial activity against M. smegmatis and E. coli, respectively. Mechanistic studies demonstrated that the antimicrobial particles were bactericidal against E. coli due to rapid disruption of the cell membranes. Conversely, against M. smegmatis the particles did not lyse the cell membrane but rather had a bacteriostatic effect. These results demonstrate that to develop new polymeric antituberculars the widely assumed, broad spectrum, membrane-disrupting mechanism of polycations must be re-evaluated. It is clear that synthetic nanomaterials can engage in more complex interactions with mycobacteria, which we hypothesize is due to the unique cell envelope at the surface of these bacteria.
Introduction
Infectious diseases are the second-leading cause of death worldwide and the third-leading cause of death in economically advanced countries.1 Microbial infection remains one of the most serious complications in medical equipment and instrumentation, food packaging, and food storage. Reisner and Woods found that 85–90% of all clinically reported infections are caused by only seven microorganisms with Escherichia coli and Staphylococcus aureus accountable for approximately half of all infections.2 Tuberculosis (TB) remains one of the top ten causes of death worldwide, causing an estimated 1.8 million deaths annually.3 Most worrying is the rise of drug resistance: In 2015 there was an estimated 480 000 new cases of multi-drug-resistant TB. Despite a steady stream of potential targets being discovered through developments in genomics and proteomics, antibiotic development in the industry has declined alarmingly.1,4,5 Because of the intense use and misuse of antibiotics, antibiotic resistance is now one of the most pressing global healthcare problems facing today’s society,4,6,7 and a lack of available treatments is becoming a very real possibility for some infections.8 There is, therefore, a pressing need for new and nontraditional solutions, which includes diagnostics, improved prescribing,9,10 control measures, and new drugs alongside a better understanding of the host–pathogen interactions.3
Antimicrobial peptides (AMPs) have had considerable attention as potential new lead antibiotics.11−13 It is hypothesized that their mode of action is to bind to negatively charged phospholipid head-groups present on the bacterial membrane, followed by the induction of membrane permeabilization, pore formation, and ultimately cell death,12,14 rather than targeting a specific enzyme, as small-molecule drugs typically do. While AMPs show potential as antimicrobial agents, the expense of large-scale peptide synthesis, protease susceptibility, biocompatibility issues, and toxicity need to be overcome. Cationic polymers have been explored as mimics of AMPs, particularly due to their resistance to proteases and the vast chemical and architectural space for synthetic polymers.15−23 Tew and coworkers have taken the design rules from natural AMPs and have shown that facially amphiphilic cationic materials can be tuned to be highly active toward Gram-negative or Gram-positive bacteria.24 They have also demonstrated that if the facial amphiphilicity is disrupted it switches off the activity toward a Gram-negative bacterial strain but is maintained against a Gram-positive strain.21
It has been shown that placing cationic polymers on multivalent scaffolds can dramatically enhance their affinity with the bacterial cytoplasmic membrane.25−29 By displaying multiple copies of the cationic polymer, a high density of positive charge is created, leading to an increase in adsorption onto negatively charged bacterial membranes. Qiao and coworkers have reported that 16- and 32-arm star branched polycations containing valine (isopropyl) and lysine (primary amine) functionalities were active under in vivo mimicking environments such as simulated body fluid against a range of Gram-negative, ESKAPE pathogens, E. coli, Klebsiella pneumoniae, and P. aeruginosa.30 The group went on to show that these polycations are effective against Acinetobacter baumannii in vivo. They also found that the bacteria did not grow resistant to the polycations over 600 generations. The star-branched polymers were found to disrupt the outer membrane of the bacteria and trigger cell-death pathways.31
Gold nanoparticles (AuNPs) have been used as an alternative to generate dense polycations. AuNPs are a versatile platform based on their useful optical and physical properties. Bunz et al.28 reported that cationic 2 and 6 nm AuNPs interact with cell membrane of Gram-positive (Bacillus subtilis) and Gram-negative (E. coli) bacteria, resulting in aggregation on the cell surface. Interestingly, 6 nm AuNPs were found not to lyse either of the strains, whereas 2 nm AuNPs rapidly lysed B. subtilis but not E. coli. Following on from this, Rotello et al.26 changed the quarternizing group on these 2 nm particles and found that activity increased as hydrophobicity of the quarternizing group increases. Feldheim et al.32 showed that ∼2 nm particles with specific mixtures of 4-mercaptobenzoic acid, cysteamine, 3-mercapto-1-propanesulfonic acid, and 2-diethylaminoethanethiol on their surface showed increased activity against M. smegmatis over E. coli. None of these particles lead to significant membrane disruption, though, implying that nonlytic mechanisms are an untapped route to modulate bacteria growth and infection with cationic polymers. Gibson and coworkers33 have observed that poly(2-dimethylamino)ethyl methacrylate (PDMAEMA) can selectively target M. smegmatis over Gram-negative strains such as E. coli and Pseudomonas putida and that PDMAEMA also did not lyse the mycobacterial cell membrane. Gram-negative bacteria have an inner and outer membrane, and hence these are typically harder to lyse than Gram-positive bacteria, which lack the outer membrane. However, mycobacteria are a unique subclass, which have a specialized thick layer of complex polysaccharides and lipids on their surface.34 This provides a physical barrier, enabling the pathogen to survive intracellularly in macrophages but also a physical barrier to drugs, which could include polycations. Therefore, disrupting this is likely to require design rules very different from, for example, E. coli.
Considering the above, this manuscript reports a detailed study into the use of “multicopy” multivalent nanoparticles and their antimicrobial activity against mycobacteria and also a Gram-negative strain. The particles are engineered to generate diversity in terms of size and cationic polymer density to ensure colloidal stability (preventing false-positives). The significant enhancements in antimicrobial activity upon changing from single polymer chains to polymer-immobilized nanoparticles are reported, along with discovery of very different modes of action between the different strains.
Experimental Section
Materials
2-(Dimethylamino)ethyl methacrylate (98%) 4,4′-azobis(4-cyanovaleric acid) (>98%) mesitylene (analytical standard) N-hydroxyethyl acrylamide (97%), dioxane, methanol, toluene, tetrahydrofuran, pentane, gold(III) chloride trihydrate (>99.9%), sodium citrate tribasic dihydrate (>99%), sodium borohydride, and propidium iodide (>94%) were purchased from Sigma-Aldrich and used as supplied unless otherwise stated. SYTO-9 green fluorescent nucleic acid stain was purchased from ThermoFischer Scientific. 2-(Dodecylthiocarbonothioylthio)-2-methylpropionic acid was synthesized as previously described.43 Ovine red blood cells were supplied by TCS Biosciences.
Physical and Analytical Methods
SEC analysis was performed using a Varian 390-LC MDS system equipped with a PL-AS RT/MT autosampler, a PL-gel 3 μm (50 Å∼ 7.5 mm) guard column, and two PL-gel 5 μm (300 Å∼ 7.5 mm) mixed-D columns using DMF with 5 mM NH4BF4 at 50 °C as eluent at a flow rate of 1.0 mL·min–1. The SEC system was equipped with ultraviolet (UV)/visible (set at 280 and 461 nm) and differential refractive index (DRI) detectors. Narrow-molecular-weight PMMA standard (200–1.0 Å∼ 106 g mol–1) was used for calibration using a second-order polynomial fit. NMR spectroscopy (1H, 13C) was conducted on a Bruker DPX-300 using deuterated chloroform or methanol as a solvent. Fluorescence microscopy was performed on a Nikon Eclipse Ti, inverted wide-field fluorescence microscope equipped with LED illumination, a 100× 1.45 NA objective, mCherry and GFP filter sets, and a 2k × 2k sCMOS Andor camera system. Transmission electron microscopy (TEM) was performed on a JEOL 2100 instrument at 200 kV, and images obtained were analyzed using ImageJ software.
Synthetic Section
Polymerization of Dimethylaminoethyl Methacrylate
In a typical reaction, 2-(dimethylamino)ethyl methyacrylate (0.5 g, 3.2 mmol), 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (23.2 mg, 0.64 mmol), and 4,4′-azobis(4-cyanovaleric acid) (3.6 mg, 0.13 mmol) were dissolved in dioxane (4 mL) in a glass vial. Mesitylene (100 μL) was added as an internal reference. A 25 μL aliquot was taken for 1H NMR analysis in CDCl3. The vial was fitted with a rubber septum and the solution was degassed by bubbling with nitrogen gas for 30 min. The vial was then placed in a thermostated water bath at 70 °C for 16 h. After this time, the reaction was opened to air and quenched in liquid nitrogen. A 25 μL aliquot was taken for 1H NMR analysis in CDCl3, and conversion was determined against mesitylene standard. The product was purified by precipitation from tetrahydrofuran into pentane and dried under vacuum. See Table 1 for analysis.
Table 1. Characterization of Polymers Synthesized by RAFT Polymerization Used in This Study.
| polymer | [M]:[CTA]a | conversion (%)b | Mn(th) (g mol–1)c | Mn(SEC) (g mol–1)d | Mw/Mnd |
|---|---|---|---|---|---|
| PHEA10 | 10 | 78 | 1600 | 2800 | 1.17 |
| PHEA25 | 25 | 81 | 3300 | 4900 | 1.21 |
| PHEA50 | 50 | 80 | 6200 | 7800 | 1.22 |
| PHEA100 | 100 | 88 | 11900 | 13600 | 1.17 |
| PDMAEMA10 | 10 | 79 | 2000 | 6300 | 1.48 |
| PDMAEMA25 | 25 | 84 | 4300 | 10100 | 1.63 |
| PDMAEMA50 | 50 | 86 | 8300 | 16300 | 1.82 |
| PDMAEMA100 | 100 | 89 | 16100 | 22500 | 1.71 |
Feed ratio of monomer to chain transfer agent.
Determined by 1H NMR spectroscopy.
Theoretical number-average molecular weight, calculated from the feed ratio and percent conversion.
Determined by size-exclusion chromatography in dimethylformamide (DMF) using poly(methyl methacrylate) standards. Mw, weight-average molecular weight; Mn, number-average molecular weight.
Polymerization of N-Hydroxyethyl Acrylamide
In a typical reaction, N-hydroxyethyl acrylamide (0.5 g, 4.3 mmol), 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (31.7 mg, 0.87 mmol), and 4,4′-azobis(4-cyanovaleric acid) (4.9 mg, 0.17 mmol) were dissolved in 1:1 methanol/toluene (4 mL) in a glass vial. Mesitylene (100 μL) was added as an internal reference. A 25 μL aliquot was taken for 1H NMR analysis in CDCl3. The vial was fitted with a rubber septum, and the solution was degassed by bubbling with nitrogen gas for 30 min. The vial was then placed in a thermostated water bath at 70 °C for 90 min. After this time, the reaction was opened to air and quenched in liquid nitrogen. A 25 μL aliquot was taken for 1H NMR analysis in MeOD, and conversion was determined against mesitylene standard. The product was purified by precipitation from methanol into diethyl ether and dried under vacuum. See Table 1 for analysis.
Synthesis of Citrate-Stabilized Gold Nanoparticles
HAuCl4 (114.4 mg, 0.29 mmol) in 350 mL of milli-Q H2O was heated to reflux under vigorous stirring. To this, sodium citrate tribasic dihydrate (299 mg, 1.02 mmol for ∼15 nm and 214 mg, 0.73 mmol for ∼30 nm) in 10 mL of milli-Q H2O was added in a single portion. The reaction was maintained at reflux for 30 min, during which a deep-red coloration formed. The reaction was allowed to cool to room temperature. Assuming complete reduction of the HAuCl4, the total gold concentration was 0.83 mM (0.16 mg·mL–1).
For ∼5 nm citrate-stabilized gold nanoparticles, to HAuCl4 (19.8 mg, 0.05 mmol) in 240 mL of milli-Q H2, sodium citrate tribasic dihydrate (13.8 mg, 0.05 mmol) was added. To this, NaBH4 (18.5 mg, 0.5 mmol) in ice-cold milli-Q H2O was added and stirred at room temperature overnight. Assuming complete reduction of the HAuCl4 the total gold concentration was 0.21 mM (0.04 mg·mL–1). Particles were concentrated using an Amicon Ultra-15 10 kDa MWC centrifugal dialysis unit.
Functionalization of Preformed Gold Nanoparticles with RAFT-Derived Polymers
100 μL of a 10 mg·mL–1 polymer solution in milli-Q water was added to a 1 mL solution of gold nanoparticles (0.83 mM) and left on a rolling stirrer for 30 min. Unattached polymer was removed by three centrifugation–resuspension cycles (10 000g, 15 min). Amicon-4 30 kDa centrifugal dialysis filters were required to remove unattached polymer from ∼5 nm particles.
Synthesis of 2 nm Particles
∼2 nm particles were synthesized using a modified procedure from Cameron et al.40 0.5 mM HAuCl4 in 25 mL of milli-Q H2O and 5.0 mM polymer (DP100 PDMAEMA, PHEA, or a mix of the two) in 0.5 mL of milli-Q H2O were combined under vigorous stirring. 50 mM sodium borohydride in 5 mL of milli-Q H2O was added in a single portion. An immediate color change from yellow to brown was observed. Stirring was continued for 3 h. Particles were concentrated, and unattached polymer was removed using an Amicon Ultra-15 10 kDa MWC centrifugal dialysis unit.
Microbiology Section
Bacterial Strains and Growth Conditions
Mycobacterium smegmatis MC2155 was grown in Middlebrook 7H9 media supplemented with 0.2% glycerol and 0.05% Tween 80. Escherichia coli Top10 were grown in Luria–Bertani (LB) media. For MBCs and CFUs bacteria were grown on LB agar containing no antibiotics.
Determination of Antibacterial Activities of 2 nm Particles
Minimum inhibitory concentration (MIC99) of the particles was determined against M. smegmatis and E. coli. The bacteria were cultured to mid-log phase and the inoculum was standardized to 1 × 105 CFU.mL–1 before addition to a 96-well microtiter plate in which the particles were serially diluted two-fold across the plate. Control wells contained culture controls and reference antibiotics (rifampicin for M. smegmatis and ampicillin for E. coli). The plates were incubated in a static incubator for 16 h for E. coli and 24 h for M. smegmatis. Following this incubation period, 25 μL of resazurin (one tablet (VWR) in 30 mL of sterile PBS) was added and left for a further incubation for 24 h for M. smegmatis and 2 h for E. coli. The MIC99 values were determined as the lowest concentration of particles at which the color did not change of resazurin (blue, no bacterial growth) to resorufin (pink, bacterial growth). The MIC99 values were determined in triplicate.
Minimal Bactericidal Concentration Determination
Minimum bactericidal concentration (MBC) of the particles were determined against M. smegmatis and E. coli. The bacteria were cultured to mid-log phase and the inoculum was standardized to 1 × 105 CFU.mL–1 before the addition to a 96-well microtiter plate in which the particles were serially diluted two-fold across the plate. The plates were incubated in a static incubator for 16 h for E. coli and 24 h for M. smegmatis. Following this incubation period, 100 μL of each culture was plated onto LB agar and incubated at 37 °C for 24 h for E. coli and 48 h for M. smegmatis. The MBC values were determined as the lowest concentration of particles that resulted in the observation of no bacterial growth. The MBC values were determined in triplicate.
Fluorescence Microscopy
LIVE/DEAD viability testing was carried out using a protocol from Molecular Probes. M. smegmatis and E. coli were grown to late-log phase (OD 0.8) before being harvested by centrifugation at 3300g for 15 min at room temperature. The cell pellet was resuspended in 0.85% NaCl, then aliquoted and incubated with 0.5 × MIC99 (3.90625 mg·mL–1 for E. coli and 1.953125 mg·mL–1 or M. smegmatis) and 2 × MIC99 (15.625 mg·mL–1 for E. coli and 7.8125 mg·mL–1 or M. smegmatis) of 100% PDMAEMA AuNPs for 15 min at room temperature. An aliquot was used as a live-cell control incubated with no particles, and a further aliquot was used as a heat-killed cells control, incubated at 80 °C for 30 min. After incubation, all samples were washed with 0.85% NaCl twice and resuspended in 0.85% NaCl. LIVE/DEAD bacterial viability dyes were used to determine membrane viability. In brief, SYTO-9 (3.34 mM in DMSO) and propidium iodide (20 mM in DMSO) were used in a 1:1 ratio, and 3 μL of the dye mixture was added for 1 mL of the bacterial suspension in 0.85% NaCl before incubating for 15 min in the dark. Five μL of the stained bacterial suspension was imaged using a Nikon Eclipse Ti inverted widefield fluorescence microscope equipped with LED illumination (at 100 × magnification) using GFP (excitation 470/40 nm, emission 525/50 nm) and mCherry (excitation 560/40 nm, emission 630/75 nm) filter sets to visualize the SYTO-9 and propidium iodide staining, respectively.
Hemolysis and Hemagglutination
Ovine red blood cells were pelleted by centrifugation at 6000g for 5 min, and the supernatant was removed and resuspended in PBS buffer. Particles were serially diluted two-fold in PBS, and 250 μL of particles was incubated with 250 μL of ovine red blood cells and incubated at room temperature for 1 h. For hemagglutination determination 25 μL of this mixture was added to 75 μL of PBS in a U-bottom 96-well plate and incubated for a further hour at room temperature. Polyethylenimine was used as a positive control for hemagglutination and cells in PBS alone as a negative control. Hemagglutination was determined visually by creation of a pellet (hemagglutination negative) or a blood film (hemagglutination positive). For hemolysis testing the rest of the particle–blood mixture was centrifuged at 6000g, and 10 μL of the supernatant was added to 90 μL of PBS buffer in a 96-well plate. The absorbance was measured at 450 nm and compared against deionized water as a positive control for lysis and PBS as a negative control to determine % hemolysis.
Results and Discussion
Antimicrobial polymers were prepared by reversible addition–fragmentation chain transfer (RAFT) polymerization of 2-dimethylaminoethyl methacrylate (DMAEMA).33 An uncharged, noninteracting polymer, poly(N-hydroxyethyl acrylamide) (PHEA), was also chosen to control charge density (vide infra) because it is an excellent stabilizing ligand for gold nanoparticles in biological media (Figure 1). Four different degrees of polymerization (DP) were targeted for each monomer, DP10, DP25, DP50, and DP100, to provide a range of molecular weights to screen for stability and activity. A crucial design criterion was to employ RAFT polymerization, which installs a trithiocarbonate end-group at the ω-terminus to enable direct immobilization on preformed gold nanoparticles (AuNPs) (vide infra) due to the high affinity of sulfur for gold surfaces. The produced polymers were characterized by 1H NMR and size exclusion chromatography (SEC) (Figure 1C,D). PHEA gave narrow molecular weight distributions, and the predicted molecular weights agreed with the feed ratio. PDMAEMAs had broader molecular weight distributions, as had been previously observed,33 but are suitable for the purposes of this investigation.
Figure 1.
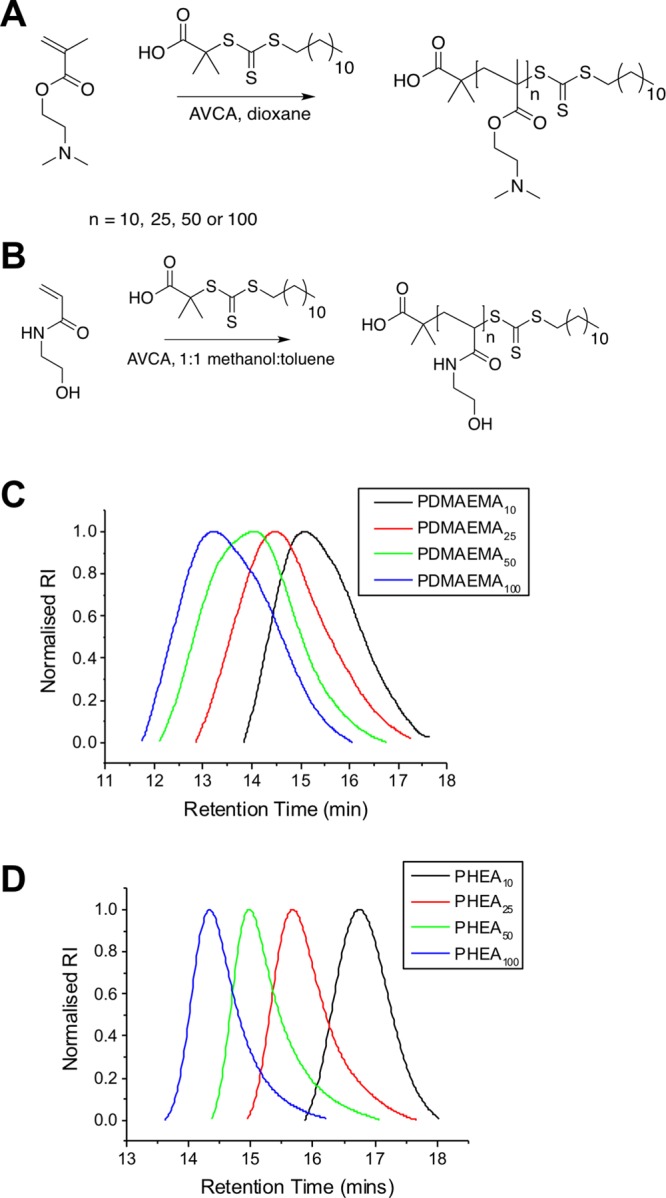
Synthesis of polymers. (A) PDMAEMA and (B) PHEA. SEC analysis of (C) PDMAEMA and (D) PHEA, as reported in Table 1.
A panel of citrate-stabilized AuNPs was synthesized using the established citrate/HAuCl4 reduction approach, with the additional reductant NaBH4 used to obtain the smallest nanoparticles.3,4Table 2 summarizes the produced AuNPs that were characterized by UV–visible spectroscopy and dynamic light scattering (DLS), giving diameters of 5–32 nm.
Table 2. Citrate-Stabilized Gold Nanoparticles Used in This Study.
| diameter (nm) |
||||||
|---|---|---|---|---|---|---|
| particlea | citrate:Au | NaBH4:Aub | λSPR (nm)c | ASPR/A450c | DLSd | UV–vise |
| Au5 | 1 | 10 | 510 | 1.26 | 4.4 ± 0.4 | 5 |
| Au16 | 3.5 | 521 | 1.65 | 22.8 ± 2.1 | 16 | |
| Au32 | 2.5 | 525 | N/A | 32.1 ± 8.3 | 32 | |
Aux:X = diameter of gold as determined by UV–vis spectroscopy.
NaBH4 used as reducing agent to obtain nanoparticles with diameter <10 nm.
SPR, surface plasmon resonance.
± standard error from three measurements.
Determined using the method of Haiss et al.35
These AuNPs were coated with polymers from Table 1 by a simple mixing strategy, followed by centrifugation/resuspension cycles to remove excess polymer.36 The DP10 PDMAEMA-coated nanoparticles were found to be unstable (rapid aggregation) in PBS; therefore, DP25, DP50, and DP100 polymers were coated onto the particles as statistical mixtures of PHEA and PDMEAMA (Figure 2A). Using three sizes of AuNPs, three polymer chain lengths, and five different ratios of each polymer (0–100% PHEA/100–0% PDMAEMA), a library of 45 distinct nanoparticles was obtained. These particles were characterized by UV–vis spectroscopy and DLS (SI). In each case there was a slight shift of the surface plasmon resonance (SPR) band to longer wavelengths (Figure 2C), consistent with polymer grafting, along with an increase in hydrodynamic diameter of the particles upon polymer coating (Figure 2B).
Figure 2.

(A) Synthetic procedure for the production of antimicrobial nanoparticle library by a simple mix-and-match strategy. (B) DLS and (C) UV–vis before (black) and after (red) functionalization of 32 nm particles with DP100 PHEA.
Initial screening of the antimicrobial activity of this nanoparticle library (data not shown) lead to inconsistent, irreproducible results, but these did suggest increased activity (lower MIC99 value) for the polymer-grafted nanoparticles compared with the polymers alone. In drug delivery, aggregation of nanoparticles in complex media is a common problem, with particles often being stable in simple solutions (water/PBS) but not in, for example, blood,37 which leads to irreproducible results and low stability upon storage: Therefore, this was studied in detail before progressing to antimicrobial testing. Gold nanoparticles are excellent scaffolds for studying aggregation as their optical properties change (red–blue color shift, decrease in SPRmax absorption) upon aggregation, enabling high-throughput screening by colorimetric assays.37−39 The nanoparticle library prepared here was evaluated for stability to aggregation in LB broth and Middlebrook 7H9 broth supplemented with 0.2% glycerol and 0.05% Tween 80, the media used for antimicrobial activity assays. In short, the nanoparticles were incubated for 16 h in the indicated media in a 96-well plate. After this time, the UV–vis spectra were recorded and stability evaluated by assessing the change in absorbance at 700 nm. Particles were classified as being colloidally unstable if this change reached a certain threshold (>0.2 shift in Abs700; see the SI for cut-offs and full data set). To simplify, the results are represented as heat maps in Figure 3, where green indicates a stable formulation and red unstable.
Figure 3.
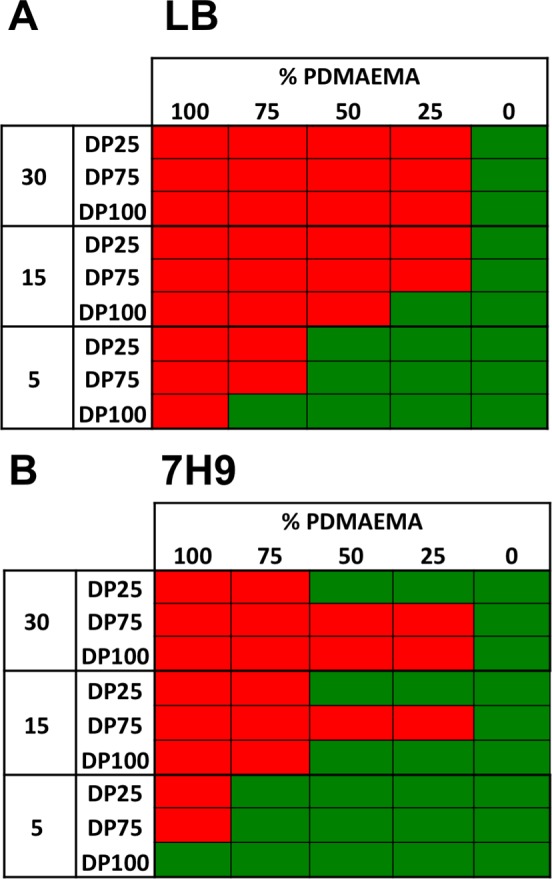
Heat maps showing stability of the 45 particle formulations in (A) LB and (B) 7H9 supplemented with 0.2% glycerol and 0.05% Tween 80, where red is unstable and green is stable.
Clearly, 30 nm particles are unstable in LB when any amount of PDMAEMA is included on their surface. Fifteen nm particles will only tolerate a small amount of PDMAEMA at the longest polymer chain length. In general, as particle size decreases the stability increases; however, even going to 5 nm particles there is not a combination of particle size and polymer length that is stable at all PDMAEMA inclusions. The introduction of PHEA increases stability, but for this study it was essential to be able to screen the impact of PDMAEMA content across all compositions. This highlights the importance of considering the stability of polymer–particle conjugates in complex media instead of just assuming stability due to being stable in PBS and suggests that other particle formations are probably less stable but do not generate the strong signal outputs of AuNPs, meaning it could go unnoticed.
With this information at hand, it was clear that smaller nanoparticles were essential to be able to obtain stable nanoparticles, which were suitable for testing. To achieve this, a direct reduction method employing polymers produced by the RAFT process as capping groups was used as reported by Cameron and coworkers40 to make ∼2 nm particles with the same PDMAEMA combinations (Figure 4A). Only DP100 polymers (PDMAEMA and PHEA) were used because the above screening indicated that this would give us stable nanoparticles across all compositions. Figure 4B shows a typical TEM image (see SI for all particle TEMs) of the nanoparticles, confirming that they are ∼2 nm in diameter, in agreement with DLS (SI) measurements showing that the particles are between 5 and 13 nm due to the hydrated polymer coating. Zeta potential of these particles shows the particles are more positive with more PDMAEMA incorporated as expected. Thermogravimetric analysis (TGA, SI) revealed that these hybrid materials are composed of 80% polymer:20% Au by mass. Full characterization of the library of particles made by this route is summarized in Table 3. Because of the lack of SPR band in the UV–vis spectra of these smaller particles, their increased stability was instead probed by dynamic light scattering in both water and in LB media (used for bacterial assays). In both cases there was no aggregation over an 18 h period (SI).
Figure 4.

(A) Synthetic scheme for direct particle formation. (B) Typical TEM of ∼2 nm particles formed by this method (100% PDMAEMA particles) (rest in SI). (C) Histogram of particle sizes determined by image analysis in ImageJ by measurement of >100 particles.
Table 3. Summary of Particles Synthesized by the Direct Reduction Route.
| AuPDMAEMA% | %PHEA100:%PDMAEMA100 | size DLS (nm)a | size TEM (nm)b | zeta potential (mV)c |
|---|---|---|---|---|
| Au0% | 100:0 | 13.7 ± 1.4 | 1.7 ± 0.05 | –3.4 ± 0.1 |
| Au25% | 75:25 | 13.6 ± 1.3 | 2.4 ± 0.09 | 11.8 ± 0.4 |
| Au50% | 50:50 | 8.1 ± 0.8 | 1.7 ± 0.06 | 15.7 ± 0.2 |
| Au75% | 25:75 | 6.5 ± 0.6 | 2.3 ± 0.06 | 19.2 ± 0.5 |
| Au100% | 0:100 | 5.3 ± 0.4 | 1.8 ± 0.04 | 23.2 ± 0.5 |
± standard error from three measurements.
± standard error from at least 100 measurements.
± standard error from five measurements.
This library of complex media-stable particles could then be screened for antimicrobial activity against M. smegmatis (a nonpathogenic model for M. tuberculosis) and E. coli (a Gram-negative bacterial strain). The minimum inhibitory concentration (MIC99) of the library was determined using a resazurin cell viability assay. In brief, the particles were serially diluted, bacterial cultures were added at 1 × 105 CFU·mL–1 and grown over an appropriate time scale (16 h for E. coli and 24 h for M. smegmatis), and resazurin (a blue, nonfluorescent, redox-active dye) was added after this time period and left for 1 h for E. coli and a further 24 h for M. smegmatis (standard method due to its slower growth rate). The MIC99 was reported as the lowest concentration at which resazurin was not reduced to resorufin (Figure 5 and Table 4).
Figure 5.
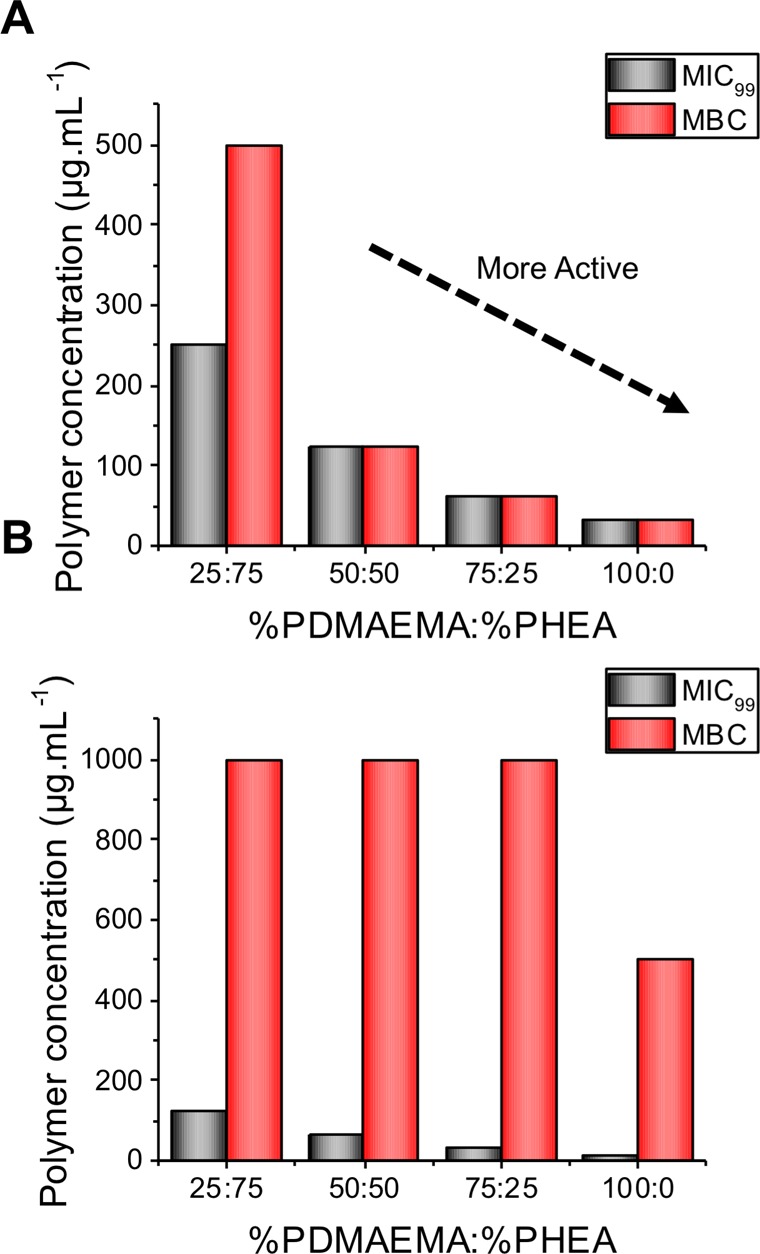
Comparison of MIC99 and MBC for (A) E. coli and (B) M. smegmatis. Values from three replicates, which each gave the same value, and hence no variance is shown.
Table 4. Minimum Inhibitory Concentrations and Minimum Bactericidal Concentrations of Particles against E. coli and M. smegmatis.
|
E. coli |
M. smegmatis |
|||||||
|---|---|---|---|---|---|---|---|---|
| particles | MIC99 (μg·mL–1 [Au]) | MIC99 (μg·mL–1 [polymer]) | MBC (μg·mL–1 [Au]) | MBC (μg·mL–1 [polymer]) | MIC99 (μg·mL–1 [Au]) | MIC99 (μg·mL–1 [polymer]) | MBC (μg·mL–1 [Au]) | MBC (μg·mL–1 [polymer]) |
| Au0% | >1000 | >4000 | N/Da | N/Da | >1000 | >4000 | N/Da | N/Da |
| Au25% | 62.5 | 250 | 125 | 500 | 31.25 | 125 | 250 | 1000 |
| Au50% | 31.3 | 125 | 31.3 | 125 | 15.6 | 62.5 | 250 | 1000 |
| Au75% | 15.6 | 62.5 | 15.6 | 62.5 | 7.8 | 31.3 | 250 | 1000 |
| Au100% | 7.8 | 31.3 | 7.8 | 31.25 | 3.9 | 15.6 | 125 | 500 |
| PHEA | >4000 | N/D | >4000 | N/D | ||||
| PDMAEMA | 250 | 250 | 31.3 | 62.5 | ||||
Not determined due to no MIC99 in the concentration range tested.
As would be expected, as the cationic character of the particles increased (increase in [PDMAEMA]), the MIC99 decreased (increase in antimicrobial activity) for both M. smegmatis and E. coli. The antimicrobial activity is greater for M. smegmatis over E. coli, which is the same trend as for the polymers alone;33 however, the difference in response between strains is significantly less. Interestingly, this corresponded to an eight-fold increase in activity (based on polymer concentration determined by TGA) compared with the MIC99 of the polymer alone for E. coli, whereas it is only a two-fold increase in activity against M. smegmatis. This confirms our hypothesis that the mycobacteria interact with PDMAEMA by a unique mechanism compared with Gram-negative bacteria.
These polymer–particle conjugates were further tested for their antimicrobial activity against the two bacterial genera to determine their minimum bactericidal concentration (MBC); this assay determines if new compounds are bactericidal or bacteriostatic. The minimum concentration to kill all of the bacteria is reported in Figure 5 and Table 3. The MIC99 and MBC correlate well for E. coli, as shown in Figure 5A, confirming that our method of presenting multiple copies of polycations on the gold nanoparticle surface leads to a drastic increase in bactericidal antimicrobial activity and offers an easy and practical route to enhancing activity. In contrast with the above, the MIC99 values for M. smegmatis are considerably lower than the MBCs with the MBC being up to eight times the MIC99. This suggests that the particles are bactericidal against E. coli but bacteriostatic against M. smegmatis, and hence the same polymer functions against the different genera by a distinct mechanism. Such observations have broad implications for the rational design of selective antimicrobials.
The core hypothesis of bactericidal activity by polycations involves mechanisms that all lead to permeability or disruption of the cell membrane.12 To probe this, LIVE/DEAD viability assays were employed to probe the nature of the particle–bacteria interaction and the ability of the particles to permeabilize the membranes. In this assay two dyes are used: a green SYTO 9 membrane permeable nucleic acid stain that indicates live cells and a red propidium iodide dye that is only able to cross damaged membrane and indicates dead cells. After incubation of the bacteria with particles below (0.5×) and above (2×) their MIC99, they were visualized by fluorescence microscopy. Figure 6A shows that even at 2× MIC M. smegmatis appear to have intact membranes, as judged by the green color of the individual bacteria and absence of red. However, E. coli are membrane-compromised even at 0.5× MIC (Figure 6B, red coloration), which agrees with the concept that the particles are acting with distinct modes of action against the two genera and appear to perturb the membrane of the E. coli and are bactericidal, whereas the particles inhibit the growth of M. smegmatis but do not appear to kill the bacteria or cause membrane damage. Growth inhibition but uncompromised bacterial membranes of M. smegmatis were also observed in the study by Feldheim et al.32 Fernandez-Trillo and coworkers demonstrated that primary amine-terminated dendrimers can induce clustering in Vibrio harveyi and that this process resulted in growth inhibition at higher concentrations as well as inducing membrane permeability,41 and they have shown that cationic polymers can effect quorum sensing in bacteria, and such mechanisms cannot be ruled out here.23,42
Figure 6.

Fluorescence microscopy of (A) M. smegmatis and (B) E. coli upon exposure to varying concentrations of 100% PDMAEMA AuNPs. Green channel is SYTO-9 nucleic acid stain and red channel is propidium iodide, which only enters membrane-compromised bacteria. Scale bar is 10 μm.
The increase in valency from single polymer chains to nanoparticles may also impact their unwanted interactions with host cells, and hence the particles were tested for their hemotoxicity against ovine red blood cells. It was determined that they were not hemolytic with <5% hemolysis observed at (6–200) × MIC99 of the particles (SI). For comparison, Rotello et al.26 found their quarternized particles to be up to 25% hemolytic in their MIC range (4–128 nM). The particles used here did, however, lead to some hemagglutination below the MIC99 (data in the SI), which is not observed using the polymers alone.33 This also correlated well with the study by Feldheim et al.,32 which suggested that there were hemocompatibility issues associated with their mixed particles that showed a similar mode of action against M. smegmatis, and the nanoparticle surface needs fine-tuning to resolve this.
Conclusions
We demonstrate the impact of the multivalent presentation of cationic antimicrobial polymers at nanoparticle surfaces on their antimicrobial activity against two different genera of bacteria and reveal distinct mechanisms of action that will help guide future design. Using PDMAEMA as the cationic polymer, a library (>50) nanoparticles ranging in diameter from 2 to 32 nm were synthesized, and the surface composition varied by the addition of a noninteracting, noncharged second polymer. It was found that only small (2 nm) particles lead to stable dispersions: Large nanoparticles did display antimicrobial activity, but the results were inconsistent and irreproducible due to the lack of colloidal stability in complex media (despite appearing stable in saline solutions alone). Against E. coli, nanoparticle formulation of PDMAEMA leads to dramatic increases in antimicrobial activity compared with the polymer alone, which was found to be due to increased membrane lysis and increases in both inhibitory and bactericidal activity. In stark contrast, against M. smegmatis (a surrogate for M. tuberculosis) there was a less dramatic increase in activity upon presenting polymers on the nanoparticle surface. Mechanistic investigations showed that against E. coli, membrane permeation and lysis was the primary mode of activity. Against M. smegmatis, a bacteriostatic mode of action dominated, but with no evidence of membrane lysis. These results are significant, as they show that there is not a universal strategy nor a “best polymer” against all bacteria but that the structural compositions of the cell wall have a dramatic impact on antimicrobial polymer function. These results will guide the development of increasingly active and potential therapeutics as alternatives to traditional small molecules and peptide-based antimicrobials.
Acknowledgments
Equipment was supported by the Innovative Uses for Advanced Materials in the Modern World (AM2), with support from Advantage West Midlands (AWM) and partial funding by the European Regional Development Fund (ERDF). Warwick Integrative Synthetic Biology (WISB) microscopy imaging facilities (grant ref: BB/M017982/1) are funded under the U.K. Research Councils’ Synthetic Biology for Growth programme. M.I.G. acknowledges the ERC for a starter grant (CRYOMAT 638661). E.F. holds a Sir Henry Dale Fellowship jointly funded by The Wellcome Trust and the Royal Society (grant number 104193/Z/14/Z). S.-J.R. thanks the University of Warwick and The Institute for Advanced Study for an Early Career Fellowship. The Leverhulme Trust is thanked for providing a studentship for L.E.W. (RPG – 2015 0 194). The Midlands Integrative Biosciences Doctoral Training Partnership (MIBTP) and BBSRC are thanked for providing a studentship for J.L. (BB/M01116X/1).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biomac.7b01561.
Figure S1: Particles before and after functionalization with DP10, 25, 50, and 100 polymers in PBS. Figure S2: Absorbance at 700 nm for each particle size and polymer combination after incubation in either LB or 7H9 media for 16 h. Figure S3: Transmission electron microscopy analysis and histograms of size distributions for particles. Figure S4: Distributions from dynamic light scattering of ∼2 nm particles with PHEA and PDMAEMA coatings. Figure S5. Thermogravimetric analysis of PHEA and PHEA-functionalized 2 nm AuNP. Figure S6: Particle size by DLS over time in water and LB media. Table S1: Percentage haemolysis caused by polymer-functionalized 2 nm particles. Figure S6: Hemagglutination of polymer-coated particles in 96-well plate format. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Nathan C. Nature 2004, 431 (7011), 899–902. 10.1038/431899a. [DOI] [PubMed] [Google Scholar]
- Reisner B. S.; Woods G. L. J. Clin. Microbiol. 1999, 37 (6), 2024–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. Global Tuberculosis Report 2016; World Health Organization, 2017. [Google Scholar]
- Opar A. Nat. Rev. Drug Discovery 2007, 6 (12), 943–944. 10.1038/nrd2477. [DOI] [PubMed] [Google Scholar]
- Fischbach M. A.; Walsh C. T. Science (Washington, DC, U. S.) 2009, 325 (5944), 1089–1093. 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein R. A. Emerging Infect. Dis. 2001, 7 (2), 188–192. 10.3201/eid0702.010206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu H. C. Science 1992, 257 (5073), 1064–1073. 10.1126/science.257.5073.1064. [DOI] [PubMed] [Google Scholar]
- O’Connell K. M. G.; Hodgkinson J. T.; Sore H. F.; Welch M.; Salmond G. P. C.; Spring D. R. Angew. Chem., Int. Ed. 2013, 52 (41), 10706–10733. 10.1002/anie.201209979. [DOI] [PubMed] [Google Scholar]
- Ventola C. L. P.T. 2015, 40 (4), 277–283. [PMC free article] [PubMed] [Google Scholar]
- Ventola C. L. P.T. 2015, 40 (5), 344–352. [PMC free article] [PubMed] [Google Scholar]
- Zasloff M. Nature 2002, 415 (6870), 389–395. 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- Brogden K. A. Nat. Rev. Microbiol. 2005, 3 (3), 238–250. 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- Hancock R. E. W.; Sahl H.-G. Nat. Biotechnol. 2006, 24 (12), 1551–1557. 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- Chan D. I.; Prenner E. J.; Vogel H. J. Biochim. Biophys. Acta, Biomembr. 2006, 1758 (9), 1184–1202. 10.1016/j.bbamem.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Ikeda T.; Yamaguchi H.; Tazuke S. Antimicrob. Agents Chemother. 1984, 26 (2), 139–144. 10.1128/AAC.26.2.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tew G. N.; Liu D.; Chen B.; Doerksen R. J.; Kaplan J.; Carroll P. J.; Klein M. L.; DeGrado W. F. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (8), 5110–5114. 10.1073/pnas.082046199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michl T. D.; Locock K. E. S.; Stevens N. E.; Hayball J. D.; Vasilev K.; Postma A.; Qu Y.; Traven A.; Haeussler M.; Meagher L.; Griesser H. J. Polym. Chem. 2014, 5 (19), 5813–5822. 10.1039/C4PY00652F. [DOI] [Google Scholar]
- Tew G. N.; Scott R. W.; Klein M. L.; Degrado W. F. Acc. Chem. Res. 2010, 43 (1), 30–39. 10.1021/ar900036b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda K.; DeGrado W. F. J. Am. Chem. Soc. 2005, 127 (12), 4128–4129. 10.1021/ja044205+. [DOI] [PubMed] [Google Scholar]
- Ilker M. F.; Nüsslein K.; Tew G. N.; Coughlin E. B. J. Am. Chem. Soc. 2004, 126 (48), 15870–15875. 10.1021/ja045664d. [DOI] [PubMed] [Google Scholar]
- Thaker H. D.; Cankaya A.; Scott R. W.; Tew G. N. ACS Med. Chem. Lett. 2013, 4 (5), 481–485. 10.1021/ml300307b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowery B. P.; Lee S. E.; Kissounko D. A.; Epand R. F.; Epand R. M.; Weisblum B.; Stahl S. S.; Gellman S. H. J. Am. Chem. Soc. 2007, 129 (50), 15474–15476. 10.1021/ja077288d. [DOI] [PubMed] [Google Scholar]
- Louzao I.; Sui C.; Winzer K.; Fernandez-Trillo F.; Alexander C. Eur. J. Pharm. Biopharm. 2015, 95 (Pt A), 47–62. 10.1016/j.ejpb.2015.05.026. [DOI] [PubMed] [Google Scholar]
- Lienkamp K.; Madkour A. E.; Musante A.; Nelson C. F.; Nüsslein K.; Tew G. N. J. Am. Chem. Soc. 2008, 130 (30), 9836–9843. 10.1021/ja801662y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei L.; Zhang X.; Wang Y.; Zhang W.; Lu Z.; Luo Y.; Zhao Y.; Li C. Polym. Chem. 2014, 5 (8), 3038. 10.1039/c3py01578e. [DOI] [Google Scholar]
- Li X.; Robinson S. M.; Gupta A.; Saha K.; Jiang Z.; Moyano D. F.; Sahar A.; Riley M. A.; Rotello V. M. ACS Nano 2014, 8 (10), 10682–10686. 10.1021/nn5042625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A.; Landis R. F.; Rotello V. M. F1000Research 2016, 5, 364. 10.12688/f1000research.7595.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden S. C.; Zhao G.; Saha K.; Phillips R. L.; Li X.; Miranda O. R.; Rotello V. M.; El-Sayed M. A.; Schmidt-Krey I.; Bunz U. H. F. J. Am. Chem. Soc. 2012, 134 (16), 6920–6923. 10.1021/ja301167y. [DOI] [PubMed] [Google Scholar]
- Feng Z. V.; Gunsolus I. L.; Qiu T. A.; Hurley K. R.; Nyberg L. H.; Frew H.; Johnson K. P.; Vartanian A. M.; Jacob L. M.; Lohse S. E.; Torelli M. D.; Hamers R. J.; Murphy C. J.; Haynes C. L. Chem. Sci. 2015, 6 (9), 5186–5196. 10.1039/C5SC00792E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam S. J.; Wong E. H. H.; O’Brien-Simpson N. M.; Pantarat N.; Blencowe A.; Reynolds E. C.; Qiao G. G. ACS Appl. Mater. Interfaces 2016, 8 (49), 33446–33456. 10.1021/acsami.6b11402. [DOI] [PubMed] [Google Scholar]
- Lam S. J.; O’Brien-Simpson N. M.; Pantarat N.; Sulistio A.; Wong E. H. H.; Chen Y.-Y.; Lenzo J. C.; Holden J. A.; Blencowe A.; Reynolds E. C.; Qiao G. G. Nat. Microbiol. 2016, 1 (11), 16162. 10.1038/nmicrobiol.2016.162. [DOI] [PubMed] [Google Scholar]
- Gifford J. C.; Bresee J.; Carter C. J.; Wang G.; Melander R. J.; Melander C.; Feldheim D. L. Chem. Commun. 2014, 50 (100), 15860–15863. 10.1039/C4CC06236A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips D. J.; Harrison J.; Richards S.-J.; Mitchell D. E.; Tichauer E.; Hubbard A. T. M.; Guy C.; Hands-Portman I.; Fullam E.; Gibson M. I. Biomacromolecules 2017, 18 (5), 1592–1599. 10.1021/acs.biomac.7b00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A. K.; Driessen N. N.; Appelmelk B. J.; Besra G. S. FEMS Microbiol. Rev. 2011, 35 (6), 1126–1157. 10.1111/j.1574-6976.2011.00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haiss W.; Thanh N. T. K.; Aveyard J.; Fernig D. G. Anal. Chem. 2007, 79 (11), 4215–4221. 10.1021/ac0702084. [DOI] [PubMed] [Google Scholar]
- Richards S.-J.; Gibson M. I. ACS Macro Lett. 2014, 3 (10), 1004–1008. 10.1021/mz5004882. [DOI] [PubMed] [Google Scholar]
- Gibson M. I.; Danial M.; Klok H.-A. ACS Comb. Sci. 2011, 13 (3), 286–297. 10.1021/co100099r. [DOI] [PubMed] [Google Scholar]
- Otten L.; Vlachou D.; Richards S.-J.; Gibson M. I. Analyst 2016, 141 (14), 4305–4312. 10.1039/C6AN00549G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S.-J.; Fullam E.; Besra G. S.; Gibson M. I. J. Mater. Chem. B 2014, 2 (11), 1490–1498. 10.1039/C3TB21821J. [DOI] [PubMed] [Google Scholar]
- Parry A. L.; Clemson N. A.; Ellis J.; Bernhard S. S. R.; Davis B. G.; Cameron N. R. J. Am. Chem. Soc. 2013, 135 (25), 9362–9365. 10.1021/ja4046857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leire E.; Amaral S. P.; Louzao I.; Winzer K.; Alexander C.; Fernandez-Megia E.; Fernandez-Trillo F. Biomater. Sci. 2016, 4 (6), 998–1006. 10.1039/C6BM00079G. [DOI] [PubMed] [Google Scholar]
- Lui L. T.; Xue X.; Sui C.; Brown A.; Pritchard D. I.; Halliday N.; Winzer K.; Howdle S. M.; Fernandez-Trillo F.; Krasnogor N.; Alexander C. Nat. Chem. 2013, 5 (12), 1058–1065. 10.1038/nchem.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips D. J.; Gibson M. I. Biomacromolecules 2012, 13 (10), 3200–3208. 10.1021/bm300989s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.