

Abstract
Single nucleotide polymorphisms in drug metabolizing Cytochrome P450 (CYP) enzymes are important contributors to inter-individual differences in drug metabolism leading to adverse drug reactions. Despite their extensive characterization and importance in pharmacogenetics of clinical drugs, the structural basis of CYP polymorphisms has remained scant. Here we report the crystal structures of human CYP2C9 and its polymorphic variants, *3 (I359L) and *30 (A477T), with an anti-hypertensive drug losartan. The structures show distinct interaction and occupation of losartan in the active site, the access channel and the peripheral binding site. The I359L substitution located far away from the active site remarkably altered the residue sidechains near the active site and the access channel. Whereas the T477 substitution illustrated hydrogen-bonding interaction with the reoriented sidechain of Q214. The results provide structural insights into reduced catalytic activity of the CYP2C9 variants and have important implications for understanding genetic polymorphisms in CYP-mediated drug metabolism.
Adverse drug reactions result in over half a million injuries and deaths and costs millions of dollars per year in the United States alone.1 Identifying human genetic polymorphisms responsible for pharmacokinetic and pharmacodynamic differences between individuals could lead to development of safer medication specific to an individual's DNA trait, often referred to as personalized medicine. Single nucleotide polymorphisms (SNP) in drug metabolizing cytochrome P450 (CYP) enzymes, which constitute the major enzyme family in drug metabolism with increasing importance in pharmacogenetics, are key determinants of variability in drug response.2-3 Over 700 alleles of CYPs in various ethnic population have been identified and examined via genetic testing, with many having a significant impact on enzyme activity and drug metabolism.4-5
The highly polymorphic CYP2C9 enzyme, metabolizes up to 15-18% of the drugs that include warfarin, tolbutamide and glimepiride,phenytoin, flurbiprofen and diclofenac, and losartan.6-8 Sixty CYP2C9 alleles have been reported until now, and many exhibited altered activities compared to the wild-type (WT) enzyme (Table S1).9 CYP2C9 *3 (I359L) was associated with reduced catalytic activities towards several substrates such as warfarin, tolbutamide, and losartan.10-11 Whereas CYP2C9 *30 (A477T) variant demonstrated loss of antihypertensive effects for losartan. 12-13 Both CYP2C9 *3 and *30 variants showed 77% and 99% reduced activity, respectively, with losartan compared to the WT enzyme in vitro.
Despite the huge amount of efforts to understand genetic variations and differences in drug response, and with over 100 structures of multiple human CYPs published in the presence or absence of various ligands, the structural basis of SNPs illustrating the effect of such amino acid substitutions is still lacking. Allelic variants are often difficult to purify and crystallize, largely due to lower expression, yield or stability than their WT counterparts. Moreover, structurally it might be difficult to comprehend the diminished in vitro activity of expressed variants since the amino acid changes in the protein occur in the region not directly located near the active site or the substrate recognition site. In this study, we report the crystal structures of CYP2C9 and its two important allelic variants, *3 and *30, in complex with an angiotensin II receptor antagonist losartan (Table S2). Losartan, a prodrug used mainly as an anti-hypertensive agent, is primarily oxidized in the liver by CYP2C9 and CYP3A4 to a more potent E-3174 (Figures S1 and S2).14
The CYP2C9 WT and the *30 variant demonstrated binding of three molecules of losartan, with one at the peripheral site, another in the active site and the third in the access channel. Whereas the *3 variant with only two molecules bound, lacked the losartan in the access channel and illustrated altered binding mode in the active site (Figure 1). The losartan binding site at the periphery in all the three complexes and the access channel in the WT and *30 complexes is located at the previously predicted substrate recognition site (SRS) 3, and 5 and 6, respectively, in the CYP2 family of enzymes.15-16
Figure 1.
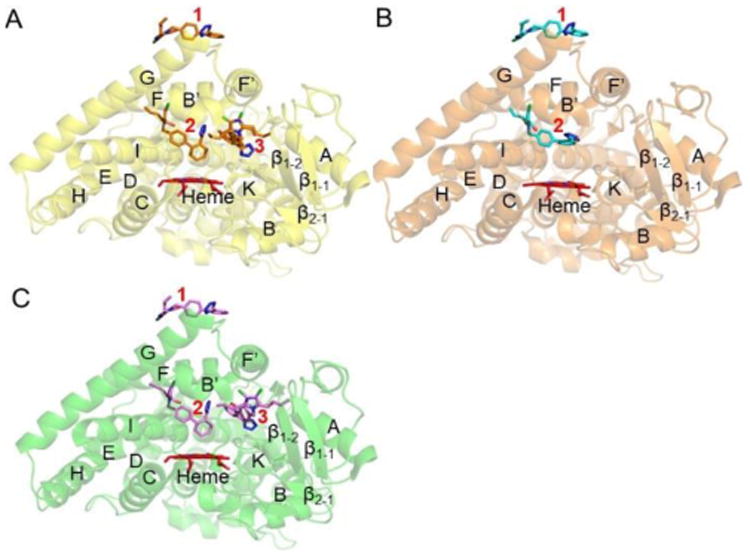
Structures of CYP2C9 and allelic variants in complex with losartan: (A) Structure of CYP2C9 WT (yellow) complexed with three molecules of losartan (orange sticks). (B) Structure of CYP2C9 *3 (orange) with two molecules of losartan (cyan sticks). (C) Structure of CYP2C9 *30 (green) in complex with three losartan molecules (pink sticks) in similar orientation and location to that in the WT complex. Heme is in red sticks.Location of each losartan is represented by red numbers: 1, peripheral site; 2, active site; and 3, access channel.
The structural overlay of all the three complexes (root-mean-square deviation, RMSD ∼ 0.2-0.25 Å) revealed the residues located within 5 Å of losartan bound on the periphery (Figure 2). The tetrazole ring of losartan is packed between the residues F226, P227, G228 and T229, whereas the Cl of the losartan interacts with the sidechain of K232. Interestingly, this peripheral site is similar to that observed in a CYP2C8 structure where a palmitic acid is hydrogen bonded in the same region comprising the cluster of amino acids from P227 to T229 as shown in Figure S3, A.17 The signature of conserved residues FPGT from 226 to 229 in the human CYP2C8, 2C9 and 2C19 and observation of ligands bound at this site in multiple CYP2C structures suggest a potential role of such site in substrate recognition. Across the larger family CYP2 enzymes, the residue at position 226 is variable hydrophobic, at 227 is conserved proline, at position 228 is either glycine or alanine, and position 229 includes either a hydrophobic or hydrophilic substitution. In addition, the peripheral binding site of losartan is close to the surface binding site of progesterone in CYP3A4, located in the Phe cluster of residues in the F-G cassette (Figure S3, B).18 The F and G helices are longer in the CYP2C9, which lacks such a hydrophobic cluster.
Figure 2.
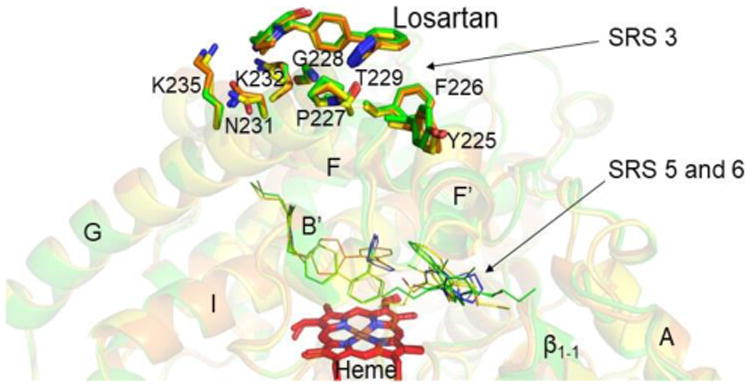
Structural overlay of CYP2C9-losartan complexes representing interactions of peripheral losatan (sticks) with the amino acid residue sidechains (sticks) located within 5 Å distance.
The active site losartan in all the CYP2C9 structures make two important polar interactions with the residue sidechains of R108 and N204. The presence of R108 in the active site is important for binding affinity of the potent CYP2C9 inhibitor benzbromarone and other ionized phenols.19 The R108, known to be crucial in the formation of 4′-hydroxydiclofenac,20-21 rotates by 90° into the active site in the current CYP2C9 complexes compared with the previously solved CYP2C9 structures (Figure S4, A).22 The guanidino group of R108 forms hydrogen bond with the tetrazole ring and the hydroxyl of the imidazole ring in losartan, thereby stabilizing substrate binding near the heme-iron. The active site losartan is significantly altered in the *3 complex, however, the polar contacts between losartan and R108/N204 sidechains are maintained in a similar fashion as observed with the WT and *30 complexes. The biphenyl rings of losartan in *3 active site flips by 90°, which is now 7 Å away from the heme-iron in place of 5 Å distance in the other two losartan complexes. In addition to these polar contacts, the residue sidechains of V113, F114, V237 and V292 were involved in hydrophobic interactions with the active site losartan in all the three structures as observed with other ligand bound structures of CYP2C9.22-24 Importantly, the site of hydroxylation of losartan in the active site of CYP2C9, irrespective of WT, *3 and *30, is positioned away from the heme-iron, in an orientation not consistent with demonstrated metabolism. Such altered pose of substrate has been observed in numerous crystallographic structures of CYP enzymes.25-26 Since substrate binding occurs during the first step in the catalytic cycle, it is possible for substrate to reorient in subsequent steps during the electron transfer from NADPH-cytochrome P450 reductase (CPR). Moreover, the crystallization experiments are performed in the absence of NADPH or CPR. Additional studies are warranted to elucidate the role of these redox partners in reorienting the ligand in a conformation suitable for metabolism. The water molecule is ordered by its interactions with the heme-iron in all the three structures.
In contrast to the *3 complex, the structures of CYP2C9 WT and *30 illustrated binding of an additional losartan in the substrate access channel. The third losartan in the WT and the *30 complex was observed in two similar orientations that were superimposable onto each other. In each of the two structures, the polar Q214 and N218 residue sidechains are positioned near the imidazole ring of losartan, while the tetrazole of losartan contacts the polar sidechain of T364 (Figure S4, B) and main chain oxygen of S365 and F476. Apart from the above residues, majority of residues surrounding the losartan are hydrophobic. In particular, the sidechain of F100 and F476 π-stacks with the imidazole and the phenyl ring, respectively, of the access channel losartan found in the WT and *30 complexes. Such π-stacking interactions by F100 and F476 sidechains are also seen in the warfarin complex,27 where the only molecule bound in the structure was in this access channel region.
Most importantly, the effect of SNP that results in amino acid substitution from isoleucine to leucine at position 359 located on the K-helix in the CYP2C9*3 complex is transduced on the I-helix residues from 307 to 311 (Figure 3). The sidechain of Y308 with the greatest influence is pushed towards the β4 loop by >3 Å, thereby affecting the secondary structural elements and the orientation of multiple residues. Such effect of I359L substitution more than 15 Å away from the active site significantly affects the important β4 loop involved in substrate interaction in the other losartan complexes. The disorder of the β4 loop leads to altered orientation of multiple residue side chains. Most importantly, the F476, demonstrated to be important in π-stacking with losartan and various substrates28-29 rotates 180° and protrude into the access channel where the third losartan is bound in the WT and *30 complexes. The *3 allele is the most functionally impaired of all the CYP2C9 genetic variants, and confers up to 80% and 77% decrease in catalytic efficiency toward glimepiride and losartan, respectively.12The significant structural change transduced from the I359L substitution on K-helix to the rotation of F476 on the β4 loop that blocks the access channel (Figure S5, A) may help us understand the loss of activity of not only losartan, but also many other important substrates including flurbiprofen and warfarin that are not metabolized by this defective allele. Furthermore, a recent study demonstrated that the loss of CYP2C9 activity in these individuals is not compensated for by switching to other CYP or metabolism pathway, further complicating clearance of the drug.30
Figure 3.
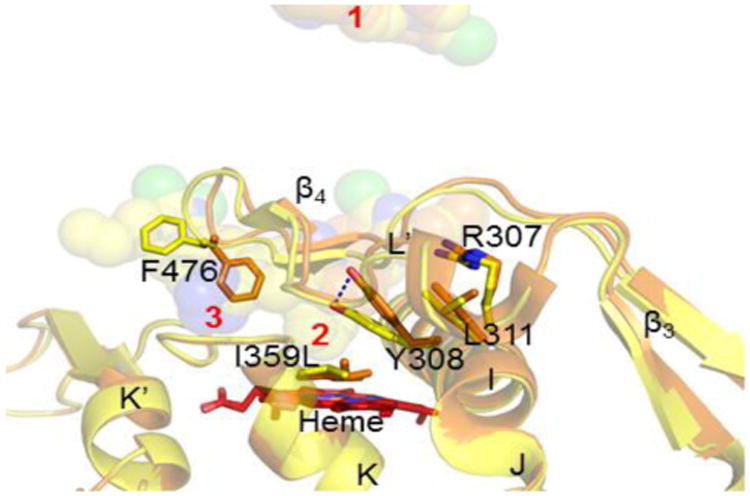
SNP in CYP2C9 *3 and its influence on substrate binding. Overlay of CYP2C9 WT (yellow) and *3 (orange) complexed with losartan. The effect of distal I359L is transduced to the neighboring I-helix residues. The Y308 sidechain is displaced by >3 Å (black dashes), subsequently affecting the β4 loop crucial for substrate interaction near the active site. Losartans shown as spheres are numbered in red. Part of the protein image isomitted for clarity.
Structural analysis of the CYP2C9 *30-losartan complex with a hydrophobic alanine to a polar threonine substitution at position 477 on the β4 loop revealed significant re-orientation of an adjacent Q214, which promotes the formation of a hydrogen bond between these two residue sidechains. (Figure 4).
Figure 4.
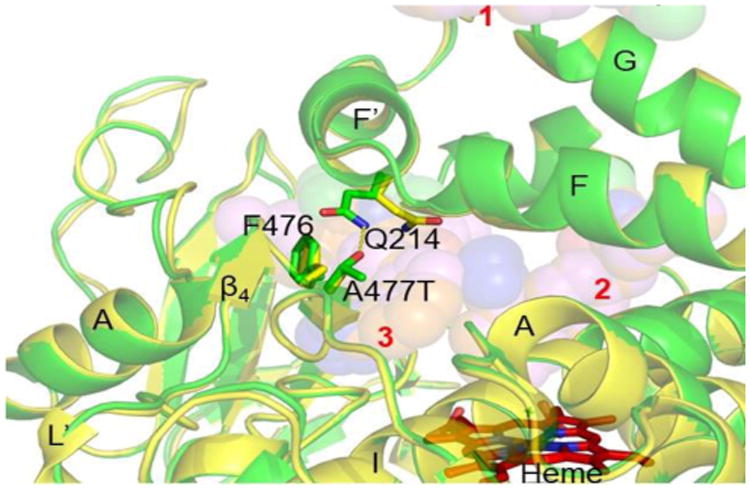
SNP in CYP2C9 *30. The *30 complex (green) associated with A477T substitution on the β4 loop is superimposed on the CYP2C9 WT complex (yellow) with losartan. The Q214 sidechain in the WT complex interacts with the imidazole ring of losartan, and rotates ∼180° in the *30 complex to hydrogen bond (yellow dashes) with the polar threonine substitution at 477. Losartans (spheres) are numbered in red.
The sidechain of Q214 in the CYP2C9 WT structure interacts with the imidazole ring of the access channel losartan near the hydroxyl moiety, the potential activation site of losartan. Interestingly, the same sidechain in the *30 variant rotates more than 90° from the losartan activation site to hydrogen bond with the hydrophilic substitution T477 (Figure S5, B). The results suggest the possible role of Q214 that may be crucial for activation of a prodrug losartan by CYP2C9. Indeed, CYP2C9*28 (Q214L) exhibited 87% decreased activity for losartan oxidation compared with WT.12 Of note, in the previous structures, the residue Q214 was either located in the region containing engineered amino acids for crystallization, or not modelled due to disordered electron density.22, 27, 31-32
Moreover, the structures of CYP2C9 in complex with losartan are significantly different than the previously reported CYP2C9 structures.22, 27, 31-32 All the published CYP2C9 structures until now (08/2017) either contained multiple stabilizing amino acid substitutions introduced in the F-G loop for crystallization purposes or had disordered or missing electron density of crucial amino acids in that region near the access channel (Figure S6). Although, these residues are outside the active site, it is evident that such engineering or poorly ordered residues affect ligand binding and protein conformations in this F-G region that was previously attributed to substrate specificity and enzymatic activity.33Furthermore, the structural overlay of the CYP2C9-losartan complex with the published flurbiprofen (RMSD ∼ 0.41 Å) and warfarin (RMSD ∼ 0.47 Å) complexes of CYP2C9 revealed differences and similarities of ligand binding. The active site flurbiprofen superimposes with the losartan in the active site, whereas warfarin that π-stacks with F476 overlays with the access channel losartan in the CYP2C9 complex (Figure S6, A and B). This is consistent with previous findings that the interactions of different ligands to CYP2C9 involve multiple substrate binding modes and sites.34-35
Overall, the structures are the first representation of allelic variants in CYP super family of enzymes that illustrate implications of polymorphism upon drug binding and provide a useful framework to infer altered drug metabolism by genetic variants. Not only do these structures elucidate the effects of *3and *30, but they also provide crucial insight in interpreting the *4 (I359T), *18 (I359L, D397A) and *28 (Q214L) variants that demonstrated reduced activity toward various substrates.36-38 It is likely that the I359T in *4 perturbs the residues on the I-helix and the effect is transduced to the active site in a similar fashion as *3 with I359L. The *18 variant includes amino acid substitution as in *3, whereas the *28 with the hydrophobic substitution of leucine at 214 may preclude or differ in interactions with the substrate compared to the polar glutamine as seen in the CYP2C9-losartan WT complex. Moreover, the findings of multiple substrate binding sites in each of these structures suggest the possible role of cooperativity or allosteric regulation by human drug metabolizing enzymes. It remains to be elucidated whether losartan bound at the peripheral site triggers the conformational change and move to the active site, or the binding occurs in sequential fashion with one losartan binding at the peripheral site followed by another in the active site via the access channel. Indeed, the structures will facilitate computational approaches for predictions of their effects in drug clearance. Such information would be very useful for not only obtaining mechanistic insights into how the allelic variations lead to altered catalytic activities toward one drug, but also evaluating their effects toward drug-drug interactions especially when designing candidates in drug discovery as part of the lead optimization process.
Supplementary Material
1) Experimental details - figures, tables, and methods.(PDF)
Acknowledgments
This work was supported by the grants KAKENHI22590054 and KAKENHI25293128 from Japan Society for the Promotion of Science to K.M., Japan, and the National Institute of Health R01 GM098538 to Q.Z, USA. The research was performed under the approval of the Photon Factory Program Advisory Committee (Proposal: 2013G597), Japan. The authors would like to thank Dr. James R. Halpert at the University of Connecticut for facilitating our collaboration.
Abbreviations
- WT
Wild-Type
- CYP
Cytochrome P450
- PDB
Protein Data Bank
- SRS
Substrate Recognition Site
- RMSD
Root Mean Square Deviation
- CPR
Cytochrome P450 Reductase
- SNP
Single Nucleotide Polymorphism
Footnotes
Supporting Information: The Supporting Information is available on the ACS website.
Notes: The authors declare no competing financial interests.
References
- 1.Classen DC, Pestotnik SL, Evans RS, Lloyd JF, Burke JP. JAMA. 1997;277:301–306. [PubMed] [Google Scholar]
- 2.Meyer UA, Zanger U, Skoda R, Grant DM. Psychopharmacol Ser. 1989;7:141–147. doi: 10.1007/978-3-642-74430-3_15. [DOI] [PubMed] [Google Scholar]
- 3.Daly AK. Curr Top Med Chem. 2004;4:1733–1744. doi: 10.2174/1568026043387070. [DOI] [PubMed] [Google Scholar]
- 4.Sim SC, Ingelman-Sundberg M. Hum Genomics. 2010;4:278–281. doi: 10.1186/1479-7364-4-4-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Leon J, Susce MT, Murray-Carmichael E. Mol Diagn Ther. 2006;10:135–151. doi: 10.1007/BF03256453. [DOI] [PubMed] [Google Scholar]
- 6.Rettie AE, Jones JP. Annu Rev Pharmacol Toxicol. 2005;45:477–494. doi: 10.1146/annurev.pharmtox.45.120403.095821. [DOI] [PubMed] [Google Scholar]
- 7.Zanger UM, Turpeinen M, Klein K, Schwab M. Anal Bioanal Chem. 2008;392:1093–1108. doi: 10.1007/s00216-008-2291-6. [DOI] [PubMed] [Google Scholar]
- 8.Miners JO, Birkett DJ. Br J Clin Pharmacol. 1998;45:525–538. doi: 10.1046/j.1365-2125.1998.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sim SC, Ingelman-Sundberg M. Methods Mol Biol. 2013;987:251–259. doi: 10.1007/978-1-62703-321-3_21. [DOI] [PubMed] [Google Scholar]
- 10.Goldstein JA. Br J Clin Pharmacol. 2001;52:349–355. doi: 10.1046/j.0306-5251.2001.01499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joy MS, Dornbrook-Lavender K, Blaisdell J, Hilliard T, Boyette T, Hu Y, Hogan SL, Candiani C, Falk RJ, Goldstein JA. Eur J Clin Pharmacol. 2009;65:947–953. doi: 10.1007/s00228-009-0707-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maekawa K, Harakawa N, Sugiyama E, Tohkin M, Kim SR, Kaniwa N, Katori N, Hasegawa R, Yasuda K, Kamide K, Miyata T, Saito Y, Sawada J. Drug Metab Dispos. 2009;37:1895–1903. doi: 10.1124/dmd.109.027003. [DOI] [PubMed] [Google Scholar]
- 13.Yin T, Maekawa K, Kamide K, Saito Y, Hanada H, Miyashita K, Kokubo Y, Akaiwa Y, Otsubo R, Nagatsuka K, Otsuki T, Horio T, Takiuchi S, Kawano Y, Minematsu K, Naritomi H, Tomoike H, Sawada J, Miyata T. Hypertens Res. 2008;31:1549–1557. doi: 10.1291/hypres.31.1549. [DOI] [PubMed] [Google Scholar]
- 14.Sica D, Gehr T, Ghosh S. Clin Pharmacokinet. 2005;44:797–814. doi: 10.2165/00003088-200544080-00003. [DOI] [PubMed] [Google Scholar]
- 15.Gotoh O. J Biol Chem. 1992;267:83–90. [PubMed] [Google Scholar]
- 16.Berka K, Hendrychova T, Anzenbacher P, Otyepka M. J Phys Chem A. 2011;115:11248–11255. doi: 10.1021/jp204488j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schoch GA, Yano JK, Wester MR, Griffin KJ, Stout CD, Johnson EF. J Biol Chem. 2004;279:9497–9503. doi: 10.1074/jbc.M312516200. [DOI] [PubMed] [Google Scholar]
- 18.Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- 19.Locuson CW, 2nd, Wahlstrom JL, Rock DA, Rock DA, Jones JP. Drug Metab Dispos. 2003;31:967–971. doi: 10.1124/dmd.31.7.967. [DOI] [PubMed] [Google Scholar]
- 20.Ridderstrom M, Masimirembwa C, Trump-Kallmeyer S, Ahlefelt M, Otter C, Andersson T. Biochem Biophys Res Commun. 2000;270:983. doi: 10.1006/bbrc.2000.2538. [DOI] [PubMed] [Google Scholar]
- 21.Dickmann LJ, Locuson CW, Jones JP, Rettie AE. Mol Pharmacol. 2004;65:842–850. doi: 10.1124/mol.65.4.842. [DOI] [PubMed] [Google Scholar]
- 22.Wester MR, Yano JK, Schoch GA, Yang C, Griffin KJ, Stout CD, Johnson EF. J Biol Chem. 2004;279:35630–35637. doi: 10.1074/jbc.M405427200. [DOI] [PubMed] [Google Scholar]
- 23.Haining RL, Jones JP, Henne KR, Fisher MB, Koop DR, Trager WF, Rettie AE. Biochemistry. 1999;38:3285–3292. doi: 10.1021/bi982161+. [DOI] [PubMed] [Google Scholar]
- 24.Tracy TS, Hutzler JM, Haining RL, Rettie AE, Hummel MA, Dickmann LJ. Drug Metab Dispos. 2002;30:385–390. doi: 10.1124/dmd.30.4.385. [DOI] [PubMed] [Google Scholar]
- 25.Ekroos M, Sjogren T. Proc Natl Acad Sci U S A. 2006;103:13682. doi: 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Vore NM, Scott EE. J Biol Chem. 2012;287:26576–26585. doi: 10.1074/jbc.M112.372813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams PA, Cosme J, Ward A, Angove HC, Vinkovic DM, Jhoti H. Nature. 2003;424:464–468. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- 28.Melet A, Assrir N, Jean P, Pilar Lopez-Garcia M, Marques-Soares C, Jaouen M, Dansette PM, Sari MA, Mansuy D. Arch Biochem Biophys. 2003;409:80–91. doi: 10.1016/s0003-9861(02)00548-9. [DOI] [PubMed] [Google Scholar]
- 29.Mosher CM, Hummel MA, Tracy TS, Rettie AE. Biochemistry. 2008;47:11725–11734. doi: 10.1021/bi801231m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flora DR, Rettie AE, Brundage RC, Tracy TS. J Clin Pharmacol. 2017;57:382–393. doi: 10.1002/jcph.813. [DOI] [PubMed] [Google Scholar]
- 31.Branden G, Sjogren T, Schnecke V, Xue Y. Drug Discov Today. 2014;19:905–911. doi: 10.1016/j.drudis.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 32.Skerratt SE, Andrews M, Bagal SK, Bilsland J, Brown D, Bungay PJ, Cole S, Gibson KR, Jones R, Morao I, Nedderman A, Omoto K, Robinson C, Ryckmans T, Skinner K, Stupple P, Waldron G. J Med Chem. 2016;59:10084–10099. doi: 10.1021/acs.jmedchem.6b00850. [DOI] [PubMed] [Google Scholar]
- 33.Wada Y, Mitsuda M, Ishihara Y, Watanabe M, Iwasaki M, Asahi S. J Biochem. 2008;144:323–333. doi: 10.1093/jb/mvn070. [DOI] [PubMed] [Google Scholar]
- 34.Korzekwa KR, Krishnamachary N, Shou M, Ogai A, Parise A, Rettie AE, Gonzalez FJ, Tracy TS. Biochemistry. 1998;37:4137–4147. doi: 10.1021/bi9715627. [DOI] [PubMed] [Google Scholar]
- 35.Locuson CW, Rock DA, Jones JP. Biochemistry. 2004;43:6948–6958. doi: 10.1021/bi049651o. [DOI] [PubMed] [Google Scholar]
- 36.Imai J, Ieiri I, Mamiya K, Miyahara S, Furuumi H, Nanba E, Yamane M, Fukumaki Y, Ninomiya H, Tashiro N, Otsubo K, Higuchi S. Pharmacogenetics. 2000;10:85–89. doi: 10.1097/00008571-200002000-00011. [DOI] [PubMed] [Google Scholar]
- 37.DeLozier TC, Lee SC, Coulter SJ, Goh BC, Goldstein JA. J Pharmacol Exp Ther. 2005;315:1085–1090. doi: 10.1124/jpet.105.091181. [DOI] [PubMed] [Google Scholar]
- 38.Maekawa K, Fukushima-Uesaka H, Tohkin M, Hasegawa R, Kajio H, Kuzuya N, Yasuda K, Kawamoto M, Kamatani N, Suzuki K, Yanagawa T, Saito Y, Sawada J. Pharmacogenet Genomics. 2006;16:497–514. doi: 10.1097/01.fpc.0000215069.14095.c6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1) Experimental details - figures, tables, and methods.(PDF)