

Abstract
Purpose
Resistance to anti-HER2 therapies in HER2+ breast cancer can occur through activation of alternative survival pathways or reactivation of the HER signaling network. Here we employed BT474 parental and treatment-resistant cell line models to investigate a mechanism by which HER2+ breast cancer can reactivate the HER network under potent HER2-targeted therapies.
Experimental Design
Resistant derivatives to lapatinib (L), trastuzumab (T), or the combination (LR/TR/LTR) were developed independently from two independent estrogen receptor ER+/HER2+ BT474 cell lines (AZ/ATCC). Two derivatives resistant to the L-containing regimens (BT474/AZ-LR and BT474/ATCC-LTR lines) that showed HER2 reactivation at the time of resistance were subjected to massive parallel sequencing and compared to parental lines. Ectopic expression and mutant-specific siRNA interference were applied to analyze the mutation functionally. In vitro and in vivo experiments were performed to test alternative therapies for mutant HER2 inhibition.
Results
Genomic analyses revealed that the HER2L755S mutation was the only common somatic mutation gained in the BT474/AZ-LR and BT474/ATCC-LTR lines. Ectopic expression of HER2L755S induced acquired L resistance in the BT474/AZ, SK-BR-3, and AU565 parental cell lines. HER2L755S-specific siRNA knockdown reversed the resistance in BT474/AZ-LR and BT474/ATCC-LTR lines. The HER1/2 irreversible inhibitors afatinib and neratinib substantially inhibited both resistant cell growth and the HER2 and downstream AKT/MAPK signaling driven by HER2L755S in vitro and in vivo.
Conclusion
HER2 reactivation through acquisition of the HER2L755S mutation was identified as a mechanism of acquired resistance to L-containing HER2-targeted therapy in preclinical HER2-amplified breast cancer models, which can be overcome by irreversible HER1/2 inhibitors.
Keywords: HER2 L755S mutation, HER2-positive breast cancer, acquired resistance, lapatinib, trastuzumab
Introduction
The HER2 gene is amplified and/or overexpressed in about 15% of breast cancers, which clinically defines the HER2+ breast cancer subtype. HER2 overexpression has been shown to result in activation of downstream AKT and MAPK signaling through either homo- or hetero-dimerization with other HER family members. HER2+ breast cancers have higher proliferation rates and have been shown to be associated with poorer prognosis prior to the advent of HER2-targeted treatments (1). Currently, the U.S. Food and Drug Administration (FDA)-approved HER2-targeted therapies include the monoclonal antibodies trastuzumab (T) and pertuzumab (P), the small molecule HER1/2 tyrosine kinase inhibitor (TKI) lapatinib (L), and the antibody-drug conjugate trastuzumab emtansine (T-DM1), all of which have greatly improved the outcome of HER2+ breast cancer patients (2–7). Our group and others have shown that anti-HER2 drug combinations, such as L+T, can more completely block the HER receptor layer than each single agent alone, and, thereby, achieve tumor regression and eradication in preclinical models (8–10). In the NeoALTTO trial, the L+T combination therapy showed superior effect over L or T therapy alone when combined with chemotherapy (11). In our 12-week neoadjuvant L+T trial (TBCRC006/NCT00548184) in patients with stages II and III HER2+ breast cancer, a high pathological complete response (pCR) rate (27%) was achieved with L+T combination even without the addition of chemotherapy (8, 9, 11, 12). Despite the benefit of HER2-targeted therapy, de novo and acquired resistance to L, T, or the combination commonly occurs (12–15).
We and others have shown that acquired resistance to anti-HER2 therapies is a convergent phenotype (15). Resistance can occur through a multitude of mechanisms that result in HER pathway reactivation (15) or activation of alternative survival pathways such as upregulation of ER signaling (10), upregulation of the PI3K pathway via PIK3CA mutations or reduced PTEN expression (16–18), and upregulation of β1-integrin signaling (19). Therefore, germane to the development of fit-for-purpose biomarkers and optimal alternative therapies for HER2+ breast cancer patients is the elucidation of resistance mechanisms predicting resistance to L, T, and combination of anti-HER treatments.
Recent massive parallel sequencing studies have revealed that HER2 can drive breast cancer growth not only by amplification in HER2+ breast cancer but also through HER2-activating mutations preferentially in breast cancers lacking HER2 overexpression and/or gene amplification (20, 21). HER2 mutations occur in about ~3% of breast cancer patients, among which the HER2L755S mutation is the most common (results based on the Cancer Genome Atlas Study (TCGA; cBioPortal) (22, 23). This mutation has been associated with L resistance when overexpressed in HER2-negative cells (20, 24, 25). Yet it is not clear whether it is an activating mutation (20), and little is known about its role in activating HER2 and driving acquired resistance to HER2-targeted therapies in HER2+ breast cancer. Here we demonstrate that the HER2L755S mutation, which was detected in the BT474/AZ-LR and BT474/ATCC-LTR lines, can induce resistance to potent anti-HER2 therapies by reactivating HER2 signaling in HER2+ breast cancer models. This resistance can be pharmacologically overcome by irreversible dual HER1/2 inhibitors. Treatment of HER2+ breast cancer patients harboring activating L-resistant HER2 mutations such as the L755S mutation with irreversible HER1/2 inhibitors may improve their clinical outcome.
Materials and Methods
Chemicals
Lapatinib and trastuzumab were purchased from LC Laboratories and Mckesson Specialty Health, respectively. Stocks of L and T were prepared as described previously (10, 19). Afatinib (Afa) and neratinib (Nrb) were purchased from LC Laboratories and Selleck Chemicals, respectively. Stocks of Afa and Nrb were prepared with DMSO.
Cell lines
Source, culture medium, and conditions of the BT474/AZ parental (P), SK-BR-3 and AU565 lines were described previously (10). The BT474/ATCC-P line was purchased from ATCC and cultured in the same medium and conditions as the BT474/AZ-P line. Resistant (R) derivatives of both BT474-P lines to HER2-targeted therapies were derived independently: cells were treated with gradually increasing doses until they resumed growth in the presence of 1 μM L (LR), 50 μg/ml T (TR), or the combination (LTR) as previously described (Suppl. Table S1) (10). All cell lines were authenticated at the MD Anderson Characterized Cell Line Core Facility within 6 months of performing the experiments. All cell lines were tested to be mycoplasma-free by MycoAlert™ Mycoplasma Detection Kit (Lonza).
Whole-exome sequencing
Cell line genomic DNA (gDNA) was isolated using the Wizard Genomic DNA Kit (Promega). Exome libraries of the BT474/AZ-P, BT474/AZ-LR, BT474/ATCC-P, and BT474/ATCC-LTR cell line genomic DNA were generated using the Agilent SureSelect XT kit and Agilent Automation Systems NGS system per the manufacturer’s instructions. Paired-end 101 bp sequencing was performed on a Illumina HiSeq 2000 sequencer, achieving a median coverage of 52.96x (range 21.22–91.96x) (Suppl. Table S2). Sequence reads have been deposited to the NCBI Sequence Read Archive under the accession SRP076305.
Paired-end whole-exome sequencing reads were aligned to the human reference genome GRCh37 using the Burrow-Wheeler Aligner (26). Local realignment, duplicate removal, and quality score recalibration were performed using the Genome Analysis Toolkit (GATK) (27). Mutations in the derivative cell lines that were not detected in the BT474/ATCC parental cell line (BT474/ATCC-P) were defined using MuTect (28) for the single nucleotide variants, and a combination of Strelka and Varscan2 (29, 30) for insertions and deletions, with the BT474/ATCC-P line as the reference. SNVs and indels with mutant allelic fraction of less than 1% and/or supported by fewer than 5 reads were disregarded (31). Variants found with >5% global minor allele frequency in dbSNP (Build 137) or that were covered by <10 reads in the tumor or <5 reads in the BT474/ATCC-P cell line were disregarded. Variants for which the tumor variant allele fraction was <5 times than that of the variant allele fraction found in the BT474/ATCC-P cell line were disregarded. The cancer cell fraction (CCF) of each mutation was inferred using ABSOLUTE (v1.0.6) (32) and mutations were classified according to pathogenicity (details see Supplementary Methods).
Single nucleotide polymorphism (SNP) array
SNP array analysis of the BT474/AZ-P, BT474/AZ-LR, BT474/ATCC-P, and BT474/ATCC-LTR cell lines was performed with the Human Omni2.5–8 BeadChip Kit (Illumina) following the manufacturer’s instructions. Log2 ratios and B-allele frequencies were exported from GenomeStudio. Allele-specific copy number alterations were estimated using ASCAT (33) as previously described. SNP array data have been deposited to the NCBI Gene Expression Omnibus under the accession GSE83608.
RNA sequencing
Total RNA of the BT474/AZ-P, BT474/AZ-LR, BT474/ATCC-P, and BT474/ATCC-LTR cell lines was extracted with RNeasy Mini Kit (Qiagen). The RNA-seq libraries of these lines were prepared using the TruSeq RNA Sample Preparation Kit (Illumina) and the Agilent Automation NGS system per manufacturers’ instructions. Samples were sequenced on an Illumina HiSeq platform with paired-end 100 bp reads.
For the analysis of the expression of novel mutations (i.e. found in the BT474 derivative cell lines but not in the BT474/ATCC-P cell line), paired-end sequence reads aligned using Tophat (34) were interrogated from pileup files generated using Samtools (35) from the aligned RNA sequencing data. Mutations with MAF of at least 0.01% in the RNA sequencing data were considered to be expressed. The expression of the HER2L755S mutation was visualized using the Integrative Genomics Viewer (IGV) (36). Sequence reads have been deposited to the NCBI Sequence Read Archive under the accession number SRP076300.
Ectopic expression of wildtype (WT) and mutant HER2
HA-tagged WT-, G572V-, L755S-, and G572V/L755S-HER2 were expressed in the BT474/AZ-P line using a doxycycline-inducible lentiviral system (37). WT and HER2L755S were also expressed in SK-BR-3 and AU565 lines. Mutant HER2 cDNA constructs were generated through site-directed mutagenesis (Stratagene). Primers used for the mutagenesis were:
G572V-F: GTGTCAGCCCCAGAATGTCTCAGTGACCTGTTTTG;
G572V-R: CAAAACAGGTCACTGAGACATTCTGGGGCTGACAC;
L755S-F: GTGGCCATCAAAGTGTCGAGGGAAAACACATCC;
L755S-R: GGATGTGTTTTCCCTCGACACTTTGATGGCCAC.
WT and mutant HER2 cDNA were shuffled into doxycycline (Dox)-inducible pHAGE-Ubc-DEST-HA expression plasmids (from Dr. Thomas Westbrook, BCM, Houston, TX) (37). Lentiviral supernatants were generated by transient transfection of 293T cells using TransIT 293 transfection reagent (Mirus Bio LLC) and harvested 48 hours post transfection. BT474/AZ-P cells were infected with lentiviral supernatant and selected with 800μg/ml Geneticin (Invitrogen) 48 hours after infection. Transduced cell lines (BT474/AZ-P-WT, BT474/AZ-P-G572V, BT474/AZ-P-L755S, and BT474/AZ-P-G572V/L755S) as well as the transduced SK-BR-3 and AU565 lines were further selected with 1μg/ml Dox + 1μM L for 3–5 weeks (or, where indicated, continuously) as previously described (38).
Mutant-specific RNA interference
Small interfering RNAs (siRNAs) were designed to selectively knock down the HER2L755S mutation (C>T mismatch was placed at position 16 (Seq1) or position 17 (Seq2) of the 19mer siRNA (39)): Seq1: 5’-UGGCCAUCAAAGUGUCGAGdTdT-3’, Seq2: 5’-GUGGCCAUCAAAGUGUCGAdTdT-3’ (Sigma). The non-targeting control (Ctrl) siRNA sequence was: 5’-UUCUCCGAACGUGUCACGUdTdT-3’ (40). 5000 cells/well of the BT474 parental or resistant lines were plated in 96-well plates in six replicates for siRNA transfection (zero day). Cells were transfected with transfection reagent alone (mock), Ctrl siRNA, or HER2L755S-specific siRNAs by reverse transfection using RNAiMAX Lipofectamine per manufacturer’s instructions (Invitrogen). Medium was replaced the next day with original treatments of each line. Cell growth was assessed 7 days after transfection by cell growth assay (10).
Cell growth assay
5000 cells/well of the BT474/AZ-P, BT474/AZ-LR, BT474/AZ-LTR, BT474/ATCC-P, BT474/ATCC-LTR cells were plated in 96-well plates in quadruplicate to measure the drug response or siRNA interference effect (zero days). Medium was replaced the next day with regular medium or drug-containing medium, and replaced again at 4 days. Cell growth was assessed at six days by methylene blue assay as described previously (10). Relative growth percentage was determined by ((O.D. 655 nm at six days − O.D. 655 nm at zero days) Treatment)/((O.D. 655 nm at six days − O.D. 655 nm at zero days) Control), as previously described (10).
Immunoblotting assay
Protein lysates were extracted as described previously (10). Twenty μg of each protein sample were separated by electrophoresis on NuPAGE Novex 4–12% Bis-Tris Gels (Invitrogen) and transferred by electroblotting onto nitrocellulose membranes using the iBlot® 2 Dry Blotting System (Invitrogen). Blots were blocked with 5% milk for 1h and then reacted at 4°C overnight with respective primary antibodies (see Supplementary Methods) diluted in 5% bovine serum albumin (BSA)+0.05% Tween-20 at dilutions as per manufacturer’s directions. Blots were washed 3 times in PBS with 0.05% Tween-20 (PBST) and then incubated with matching HRP-linked secondary antibody (Cell Signaling) diluted in 5% BSA+0.05% Tween-20 at 1:2000 dilutions for one hour. The blots were then washed 3 times with PBST, visualized by chemiluminescence on a ChemiDoc™ Touch Imaging System, and analyzed using the Image Lab Software Version 5.2.1 (BioRad). Experiments were repeated at least twice. Details of immunoblotting see Supplementary Methods.
Xenograft studies
BT474/AZ-LR cells were maintained as described previously (10). Animal care was in accordance with institutional guidelines. Ovariectomized 5–6-week-old athymic mice (Harlan Sprague Dawley) supplemented with estrogen pellets (E2) (41) received subcutaneous injection of 5×106 BT474/AZ-LR cells (ER+/HER2+) as described previously (8). Starting next day, mice were treated with 100mg/kg L once daily orally, 7d/week. After two weeks, 16 mice bearing xenografts that reached ~150mm3 were randomized to 2 treatment groups (8 mice/group): continued L (E2+L, 100mg/kg once daily orally, 7d/week) or switched to Afa (E2+Afa, 20mg/kg once daily orally, 7d/week). The rest of the mice remained on E2+L treatment until tumors reached ~350mm3 and were then randomized to 3 groups (12 mice/group): estrogen deprivation by removal of estrogen pellets (ED) plus vehicle (0.5% hydroxypropyl-methylcellulose, 0.1% Tween 80) (ED+Veh), ED+L, or ED+Afa. Tumor volumes were measured twice per week as described previously (8, 10, 42). Mice were sacrificed and tumors were harvested when the tumors reached 1000mm3 or at completion of the experiment (Day 16 for E2 arms and Day 85 for ED arms except for mice which reached complete tumor eradication). Each tumor analyzed was from a different mouse; tumor tissue was harvested from each individual mouse and preserved in liquid nitrogen or formalin-fixed and paraffin embedded (FFPE) for later analyses.
Immunohistochemistry (IHC)
BT474 tumor xenografts were excised and tumor slices were immediately placed into formalin and fixed overnight. The next day, fixed tissue slices were washed with 70% ethanol before processing and paraffin embedding. IHC was performed as described previously (10) using the same primary antibodies against phospho proteins as in the western blots.
Statistical analyses
For the L755S mutant silencing experiment, data analyses were performed for BT474/AZ and BT474/ATCC models separately. Cell growth was compared using a general linear model with cell lines (parental/resistant derivative), treatment (siRNA transfected/mock, drug treatments/DMSO), and their interaction. Results from two independent experiments were combined for analysis and each experiment was considered a categorical blocking factor. For purposes of plotting, model–estimated group means and 95% confidence limits were used. Plots show data from two experiments combined. For the drug response experiments, IC50 values were generated through GraphPad Prism (version 6.05) using the Log (inhibitor) vs. response-variable slope model (GraphPad).
For the xenograft experiment, Kaplan-Meier survival plots were generated from survival analysis performed for progression (tumor size tripling/doubling from day of randomization in the E2/ED treatment groups, respectively), and regression (tumor size halving from day of randomization). Time to doubling/tripling was estimated by linear interpolation. Complete response was defined as complete tumor disappearance for at least 3 consecutive weeks and complete response rates were calculated based on the total number of animals treated in each group. Tumor size halving required two consecutive observations of the regression; time to regression was defined by the 2nd observation time. If the tumor size had not reached an event threshold by the last observation, then it was considered as censored at the last time point. Difference of time to tumor progression/regression between groups was analyzed by generalized Wilcoxon test and pairwise comparison with p-value adjustment using simulated method. Differences of IHC H-score, relative expression of HER2L755S and total HER2 RNA, and relative cell growth and colony formation upon drug treatments were compared using one-way ANOVA.
Results
The HER2L755S mutation is associated with acquired resistance to L-containing regimens and reactivates HER2 signaling in BT474 models
As previously described by our group, the BT474/AZ cell line is a subline of the BT474/ATCC cell line that can be effectively grown in vivo as xenografts (10). Both BT474 models are ER+/HER2+ and harbor HER2 gene amplification. We have previously shown that in the BT474/AZ cell model, during the development of L resistance, HER2 signaling was inhibited at the early stage and then reactivated after prolonged L treatment (BT474/AZ-late LR as described previously (10), hereafter termed the BT474/AZ-LR line, Fig. 1A, Suppl. Fig. S1A). HER2 signaling was not reactivated in the BT474/AZ-LTR derivative (Suppl. Fig. S1A), or in derivatives resistant to L-containing regimens of five additional HER2+ breast cancer cell models (HCC202, AU565, MDA-MB-361, UACC812 and HCC1954) we have previously described (10, 19). Here we focused on the phenomenon of HER2 reactivation in the L-containing regimen since it has been previously shown that L has a stronger inhibitory effect on HER2 kinase activity than T (43, 44). To understand whether HER2 reactivation during L resistance is found only in the specific BT474/AZ subline of the BT474 cell model, we next investigated its parental line BT474/ATCC. Importantly, in the BT474/ATCC model, we also found HER2 and its downstream signaling to be reactivated in its LT-resistant derivative (BT474/ATCC-LTR line) (Fig. 1A, Suppl. Fig. S1A) but not in the LR derivative. Therefore, we observed HER2 reactivation in certain derivatives resistant to L-containing regimens in both the BT474/AZ (LR) and BT474/ATCC (LTR) models.
Fig. 1. The HER2L755S mutation reactivates HER2 signaling in BT474 models and is associated with acquired resistance to L-containing regimens.
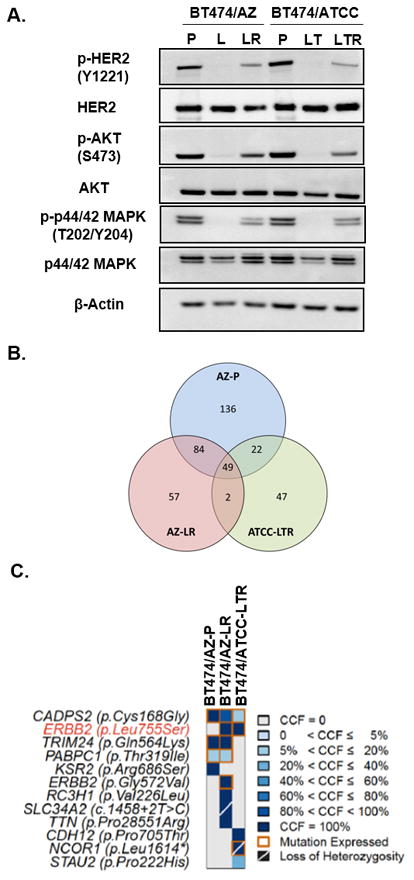
A, Western blot analyses of L/LT-treated BT474 parental and resistant cells. BT474/AZ-P were treated 6h with DMSO or 1μM L. BT474/ATCC-P cells were treated with DMSO or 1μM L + 10μg/ml T. BT474/AZ-LR cells were cultured in the presence of 1μM L and BT474/ATCC-LTR cells were cultured in the presence of 1μM L+ 10μg/ml T. B, Number of mutations detected using whole exome sequencing identified in single samples or multiple samples in the BT474/AZ-P, BT474/AZ-LR, and BT474/ATCC-LTR lines using the BT474/ATCC-P line as reference. C, Pathogenic mutations identified by whole-exome sequencing (supplementary methods) in the BT474/AZ-P, BT474/AZ-LR, and BT474/ATCC-LTR lines using the BT474/ATCC-P line as reference are shown. The shade of blue in each block represents the cancer cell fraction (CCF) of each mutation in each line. Orange squares represent mutations that were found to be expressed using RNA sequencing. Mutations associated with the loss of the wild-type allele are indicated by a diagonal bar. The HER2 (encoded by the gene ERBB2) L755S mutation is highlighted in red.
Previously we have shown that HER2 knockdown can significantly inhibit growth of the BT474/AZ-LR line, suggesting that the resistant growth of this line is dependent on the reactivated HER2 signaling in the presence of HER2-targeted therapy (10). HER2 activating mutations have been shown to be associated with resistance to L in HER2-negative breast cancer (20). Therefore, we hypothesized that HER2 reactivation in the two HER2+ BT474 resistant lines could result from HER2 activating mutations. To test this, we subjected both BT474 parental lines (BT474/AZ-P and BT474/ATCC-P) and their two HER2-reactivated L-resistant derivatives (BT474/AZ-LR and BT474/ATCC-LTR) to whole exome sequencing and RNA sequencing (Suppl. Table S3). Using the BT474/ATCC-P line as reference for mutation calling (i.e. the sequencing results of the BT474/ATCC-P cells were used as the ‘germline’ reference for the analysis of the resistant models), we identified a HER2 mutation (L755S) that was present in both L-resistant cell line derivatives but was absent in the BT474/ATCC-P line (Fig. 1B, Suppl. Fig. S1B). Importantly, the HER2L755S mutation was identified as the only pathogenic mutation shared by the two resistant lines, but not present in the BT474/AZ-P line (Fig. 1C, Suppl. Fig. S1B). Furthermore, analysis of the CCF of the mutations using ABSOLUTE (32) revealed that the HER2L7 55S mutation was clonally present in the two BT474 resistant lines (i.e. bioinformatically inferred to be present in virtually all cells, Fig. 1C). Using digital PCR, we could confirm the presence of this mutation in BT474/AZ-LR and BT474/ATCC-LTR, but not in the BT474/AZ-P and BT474/ATCC-P cells at the sensitivity of 1/10000 (Suppl. Fig. S1H). In addition, cDNA Sanger sequencing also confirmed that the HER2L755S mutation was clonally expressed only in the two HER-reactivated LR/LTR lines (BT474/AZ-LR and BT474/ATCC-LTR, Suppl. Table S3) but not in the BT474 parental lines, BT474/AZ-LTR and TR, BT474/ATCC-LR and TR derivative lines (Suppl. Figs. S1B, S1C, S1D, S1F, S1G). Additional sequencing also confirmed that the HER2L755S mutation was present only in the two HER-reactivated LR/LTR lines of BT474 model but not in any of the derivatives resistant to L-containing regimens of five additional HER2+ breast cancer cell models mentioned above (10, 19).
Apart from the L755S mutation, whole exome and RNA sequencing of BT474 and its derivative cell line models identified a second HER2 mutation (G572V), which was in the BT474/AZ-LR but not in the BT474/AZ-P, BT474/ATCC-P, BT474/ATCC-LTR, BT474/AZ-LTR, BT474/ATCC-LR, BT474/AZ-TR, or BT474/ATCC-TR derivative lines (Suppl. Table S3, Suppl. Figs. S1B, S1E). There have been no reports of the HER2G572V mutation in cell line or clinical sequencing data. Given that the L755S but not the G572V mutation was found to be shared by the two resistant lines, we hypothesized that the HER2L755S mutation but not the G572V mutation may reactivate HER2 signaling and drive resistance to HER2-targeted therapies in the BT474/AZ-LR and BT474/ATCC-LTR lines.
The HER2L755S mutation is the driver of acquired resistance in the two BT474 L/LT-resistant derivatives with HER2 reactivation
To investigate whether the HER2L755S mutation can induce L resistance in the HER2+ breast cancer preclinical models and test if the HER2G572V mutation has any function in inducing L resistance, we ectopically expressed HA-tagged WT-, G572V-, L755S-, and G572V/L755S-HER2 in the BT474/AZ-P cells. Expression of the Dox-inducible HA-tagged HER2 constructs was verified by western blot analysis (Fig. 2A). To avoid the effect of the overexpressed endogenous WT HER2, we selected the exogenous-HER2 expressing cells with Dox+L for 5 weeks. After 5 weeks of selection, only the L755S HER2- and G572/L755S HER2-expressing cells, but not the WT HER2- and G572V- HER2 expressing cells, survived the selection (Fig. 2B). No additive survival benefit was observed comparing the G572/L755S HER2-expressing cells versus the L755S HER2-expressing cells. In these surviving L-resistant L755S HER2- and G572/L755S HER2-transduced cells, HER2 and downstream signaling was reactivated in the presence of L (Fig. 2C). This suggests that the HER2L755S mutation can induce L resistance in BT474/AZ-P cells and that the HER2G572V mutation can neither induce resistance nor enhance resistance driven by the HER2L755S mutation in this model system. Exogenous overexpression of the HER2L755S but not WT HER2 also conferred L resistance in two additional HER2+ breast cancer models, SK-BR-3 and AU565 (Suppl. Fig. S2). This suggests that the HER2L755S mutation can confer L resistance in HER2+ breast cancer models irrespective of their genetic background.
Fig. 2. The HER2L755S mutation but not the G572V mutation is the driver of acquired resistance in the two BT474 LR/LTR derivatives with HER2 reactivation.

A, Dox-inducible ectopic expression of C-terminal HA-tagged WT and mutant HER2 constructs were validated by western blot. B, WT-, G572V-, L755S- and G572V/L755S-HER2-expressing BT474/AZ-P cells were selected with Dox+L for 5 weeks. Pictures were taken at 4X magnification using an Olympus IX70 microscope with a RETIGA 1300R Fast 1394 camera and analyzed with Image-pro plus software (version 5.0). Scale Bar: 50μ. C, WT-, G572V-, L755S-, and G572V/L755S-HER2-expressing BT474/AZ-P cells were treated with or without 1μM L for 6h followed by western blot. L755S- and G572V/L755S-HER2-expressing BT474/AZ-P cells which survived the Dox+L selection for >5 weeks (L755S-LR and G572V/L755S-LR) were analyzed by western blot for HER2 and downstream signaling. D, BT474 parental and HER2-reactivated LR/LTR cells were transfected with siRNA (seq1) targeting the HER2L755S mutant. Culture medium was replaced the next day with regular medium or drug-containing medium, and replaced again at 4 days. Cell growth was assessed at six days by methylene blue assay. Relative percent (%) growth was normalized to mock transfection. Statistical analyses were performed for AZ and ATCC separately. Model–estimated group means and 95% confidence limits were plotted combining two independent experiments.
To confirm that the HER2L755S mutation is the driver of acquired resistance in the BT474/AZ-LR and BT474/ATCC-LTR cells, several siRNAs specifically targeting the HER2L755S mutation were designed (Supplementary Methods) and applied to the resistant derivatives and their parental lines with two siRNAs tested effective and specific. Mutant-specific Q-PCR assays (Suppl. Fig. S3A) confirmed effective and selective knockdown of the mutation in the two resistant lines (Suppl. Fig. S3B). The mutant-specific siRNA seq1 significantly inhibited growth of the two resistant lines (Fig. 2D). Modest growth inhibition was observed in the BT474 parental cells by mutant-specific siRNA, which could be attributed to the partial target effect of mutant-specific siRNA on total HER2 (Fig. 2D, Suppl. Fig. S3B). Growth inhibition of the two resistant lines by mutant-specific siRNA, however, was markedly greater than that of their parental lines (Fig. 2D). To further confirm, we tested a second HER2L755S mutant-specific siRNA, seq2 (Suppl. Fig. S3C), which showed similar substantially higher growth inhibition in the BT474/AZ-LR cells over BT474/AZ-P cells (Suppl. Fig. S4A). Furthermore, western blot analysis following HER2L755S knockdown using siRNA seq2 showed a selective inhibition of phospho-HER2 as well as phospho-AKT levels in BT474/AZ-LR cells as opposed to BT474/AZ-P cells (Suppl. Fig. S4B). This suggests that the HER2 signaling reactivation observed in the LR derivatives is indeed driven by HER2L755S. Collectively, the results suggest that the HER2L755S mutation is the driver of acquired L and LT resistance in BT474/AZ-LR and BT474/ATCC-LTR lines respectively, with HER2 reactivation.
Next, we asked whether the HER2L755S mutation can also confer resistance to more potent and commonly used anti-HER2 drug regimens. As shown in Suppl. Fig. S5, we found that the HER2L755S mutation conferred complete resistance not only to the dual regimen L+T but also T+P and partial resistance to the antibody-drug conjugate trastuzumab emtansine (T-DM1), both when expressed endogenously in the BT474/AZ-LR cell line (Suppl. Fig. S5A) as well as when expressed exogenously by Dox induction in the BT474/AZ-P cells (Suppl. Fig. S5B).
Irreversible HER1/2 inhibitors overcome acquired resistance to L-containing HER2-targeted therapy conferred by the HER2L755S mutation in the BT474 models in vitro
It has been suggested by structural modeling that the HER2L755S mutation disrupts the inactive conformation of the kinase domain, which is required for L binding (24, 25). The HER1/2 irreversible TKIs such as Afa and Nrb were designed to covalently bind and irreversibly block enzymatically active HER1/2 receptors (45). Afatinib has been shown to effectively inhibit Ba/F3 cells ectopically expressing HER2L755S (24, 25), and Nrb has shown great efficacy in inhibiting growth of MCF10A cells ectopically expressing HER2L755S (20). Therefore, we hypothesized that the irreversible HER1/2 inhibitors can overcome acquired resistance to L-containing HER2-targeted therapy in our HER2-amplified BT474 resistant lines endogenously harboring the HER2L755S mutation. Cell growth assays showed that the BT474/AZ-LR and BT474/ATCC-LTR cells, which are highly resistant to L treatment (IC50≥3μM), can be effectively inhibited by Afa and Nrb in a dose-dependent manner (IC50<25nM) (Fig. 3A). Furthermore, Western blot analyses confirmed that reactivated HER2 signaling in the two resistant lines was significantly inhibited by 6h of 50nM Afa or Nrb treatment, which is lower than the clinically relevant concentrations of these agents (100nM) (Fig. 3B). Importantly, in the BT474/AZ model, the activity of Afa was observed only in the LR cells that were dependent on active HER2 signaling for resistant growth, and not in the LTR cells where HER signaling remained inhibited under anti-HER2 treatment (Suppl. Fig. S6) (10).
Fig. 3. The irreversible HER1/2 inhibitors overcome acquired resistance to HER2-targeted therapy conferred by the HER2L755S mutation in BT474 models in vitro.
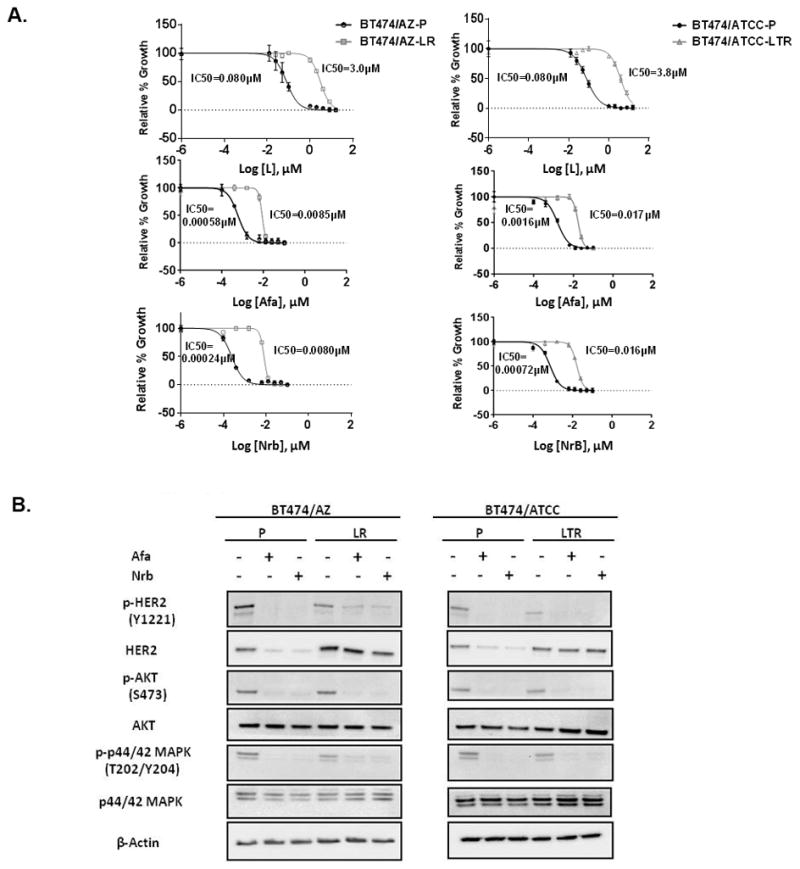
A, Responses of BT474/AZ-LR and BT474/ATCC-LTR lines and their relative parental lines (BT474/AZ-P and BT474/ATCC-P) to lapatinib (L), afatinib (Afa), and neratinib (Nrb) were measured by cell growth assay. Data was analyzed by GraphPad Prism (version 6.05) to generate drug response curves and relative IC50 values using the Log (inhibitor) vs. response-variable slope model (Bars=SEM) with normalization of data defining the biggest number in each dataset as 100% and the smallest number in the same dataset as 0%. B, BT474/AZ-P, BT474/AZ-LR, BT474/ATCC-P, and BT474/ATCC-LTR lines were treated with or without 6h of 50nM Afa or Nrb followed by Western blot.
Afatinib overcomes acquired resistance to HER2-targeted therapy in the BT474/AZ-LR xenografts in vivo
To investigate whether Afa can serve as an effective treatment to overcome resistance to HER2-targeted therapy induced by the HER2L755S mutation in HER2-amplified breast cancer, we tested its effect in vivo using the BT474/AZ-LR line grown as xenografts. Mice bearing BT474/AZ-LR xenografts at ~150mm3 that were developed in the presence of E2-supplemented L treatment were randomized to E2+L and E2+Afa treatment groups (Fig. 4A). Tumor progression was completely inhibited in the E2+Afa arm compared to E2+L arm (p=0.0038) (Fig. 4B). Harvested on Day 16 post randomization, the E2+Afa tumors were markedly smaller compared to the E2+L tumors (Fig. 4B, Suppl. Fig. S7).
Fig. 4. Afatinib effectively overcomes acquired resistance in the BT474/AZ-LR xenografts.

A, Mice prepped with estrogen (E2) pellets were injected with 5×106 BT474/AZ-LR cells and treated with L until randomization to 5 groups: E2+L, E2+Afa, ED+Veh, ED+L, and ED+Afa (for details see Materials and Methods). B, Kaplan-Meier analyses of progression-free survival within 16 days of treatment of E2+Afa or E2+L. Tumor progression was defined as tumor size tripling since randomization. TTP: time to tumor progression. C, Kaplan-Meier analyses of progression-free survival and tumor regression (graph showing change of proportion of non-regressing tumors) within 85 days of ED+Veh, ED+L, or ED+Afa treatment. Tumor regression was defined as tumor size halving since the day of randomization. TTR: time to tumor regression. D, p-HER2 (Y1221), p-AKT (S473), and p-p44/42 MAPK (T202/Y204) levels of xenografts in each treatment arm was assessed by IHC and scored as H-score by a pathologist. Two tumors (Suppl. Fig. S8B and S8C) that were resistant to the ED+Afa regimen were not included in the analysis. *: p<0.05, **: p<0.01, ***: p<0.001.
We have previously shown that in both preclinical and clinical HER2+/ER+ tumors, blockade of both HER2 and ER signaling is required for long-term tumor regression to be achieved (10, 12). Therefore, endocrine deprivation (ED) treatment was added to the experiment to mimic the clinical scenario of treating ER+/HER2+ breast cancer patients. Mice bearing the BT474/AZ-LR xenografts at an average size of ~350mm3 that had developed in the presence of E2+L were randomized to ED+Veh, ED+L, and ED+Afa arms. The tumors continued to grow in both ED and ED+L groups but regressed in the ED+Afa group (Suppl. Fig. S8A), except one mouse in the ED+Afa group that showed de novo resistance (Suppl. Fig. S8B) and so has been excluded from the analysis of the growth curves. With ED treatment, median time to tumor progression (TTP) was numerically, though not significantly, increased in the ED+L group compared to the ED+Veh group (33.7 and 8.5 days, respectively, p= 0.0928), and not yet achieved in the ED+Afa group at Day 85 (p=0.0014) (Fig. 4C, Suppl. Table S4). Only two mice of the ED+Afa group progressed with the treatment (Suppl. Fig. S8B and S8C). Conversely, median time to tumor regression (TTR) was not achieved in the ED+Veh and ED+L groups at Day 85 (Suppl. Table S4). Compared to ED+Veh group, response (regression) rate was significantly improved in the ED+Afa group (p=0.0094) but not the ED+L group (Suppl. Table S4). Importantly, complete regression (CR) was achieved in 5/12 of the mice in the ED+Afa arm after 71 days of treatment, and no tumor regrowth was observed after removal of Afa and resupplementing E2 pellets for >100 days (Suppl. Fig. S8D). We next examined the effect of Afa on HER2 and key downstream signaling in the HER2-reactivated BT474/AZ-LR xenografts. Levels of p-HER2/p-AKT/p-MAPK were immunohistochemically assessed, and p-HER2/p-AKT/p-MAPK levels were significantly reduced in tumors which switched to Afa treatment compared to those continued with L treatment in both E2 and ED settings (Fig. 4D). In the ED+Afa arm, we focused on analyzing signaling changes within the 10/12 mice that were not resistant to the therapy.
Discussion
Acquired resistance to HER2-targeted therapies in HER2+ breast cancer can occur through reactivating the HER pathway or switching to alternative survival pathways (10, 15). In this study, we identified HER2 reactivation by acquisition of the HER2L755S mutation as a mechanism of acquired resistance to HER2-targeted therapy in our two HER2+ BT474 cell line models. In previous pre-clinical studies, forced expression of this mutation in HER2-negative cells was found to result in L resistance (20, 24, 25), and HER2 activating mutations were reported as a mechanism other than HER2 amplification to drive breast tumor growth (20). Here we report on the novel observation that in HER2-amplified breast cancer cells the HER2 mutation (L755S) can be gained during HER2-targeted therapy, and that this mutation may result in reactivation of the HER signaling network and induction of resistance to potent L-containing anti-HER2 therapy models. Interestingly, Zabransky et al. (46) reported in preclinical in vitro and mouse models that HER2 missense mutations as single copies may require additional oncogenic input to induce tumorigenesis and may not by themselves predict response to HER2-targeted therapies. Our study, however, demonstrates that the HER2+ BT474/AZ-LR xenografts endogenously expressing the HER2L755S mutation are tumorigenic even under L treatment, and that this mutation can restore HER2 activity and cause resistance to therapy in HER2+ breast cancer.
Since we have identified the HER2L755S mutation as the mechanism of acquired resistance in two of the BT474 derivatives resistant to L-containing regimens, we postulated that the resistant clone could have originated from clonal selection of a preexisting subclone present prior to HER2-targeted therapy, or it could have appeared de novo during the course of treatment. Although direct evidence of the presence of the HER2L755S mutation in the BT474/AZ-P and BT474/ATCC-P cells could not be obtained through digital droplet PCR analysis, we cannot rule out the possibility that the methods employed here lacked the sensitivity to detect a minor resistant subclone (<0.01%) in the parental lines.
It is worth mentioning that in the BT474/AZ-LR line, we found the HER2 L755S mutation was clonally present in all cells of the resistant line based on bioinformatics inferences made using ABSOLUTE and on single cell cloning. Importantly, however, this mutation was expressed at approximately ~30% of the alleles at the transcriptomic level and displayed a variant allelic fraction of approximately 30%. Thus, in the case of simultaneous HER2 amplification and L755S mutation, the most parsimonious explanation for these findings is that the HER2L755S mutation likely developed later in the evolution of the cell line.
The clinical relevance of the HER2 mutations stems from their presence in intrinsic/post-treatment clinical HER2+ breast cancers. To date, HER2 missense mutations have been reported by TCGA in ~3% of breast cancer patients with primary HER2+ tumors (results based on cBioPortal) (22, 23). A similar rate of HER2 mutations (2.3%) was also recently reported in the study by Zuo et al. in a large cohort of 910 HER2+ primary breast tumors (47). The role of HER2 mutations in acquired resistance to HER2-targeted therapies in HER2+ breast cancer, however, still remains unclear. Importantly, the HER2L755S mutation, which is the most common HER2 mutation in breast cancer, has been reported in 3/40 (7.59%) metastatic patients who had received prior T treatment in the adjuvant setting (48). Additionally, the study by Zuo et al. analyzed 18 pairs of primary and metastatic lesions, 16 of which had received 1 year of adjuvant T treatment, and observed that the drug-resistant HER2L755S mutation was present in 3/18 metastatic lesions but not in any of the paired primary tumors. Interestingly, a second close mutation, HER2K753E, which also confers preclinical resistance to L and T, also emerged in 2 of these 18 metastatic lesions (47). This suggests that these two HER2 mutations are associated with clinically acquired resistance to T. Through targeted sequencing of 76 HER2+ primary invasive carcinomas, Boulbes et al. (49) identified 12 missense mutations in the HER family kinase domain, including 3 in the HER2 kinase domain (but excluding the L755S variant) that are associated with aggressiveness of the tumor and resistance to T-based therapy in the metastatic setting. Overall, these studies suggest the occurrence and role of the HER2L755S mutation, amongst other mutations, in intrinsic and acquired HER2 therapy resistance in breast cancer.
Importantly, we have demonstrated that dual HER1/2 irreversible kinase inhibitors such as Afa and Nrb can effectively overcome acquired resistance to HER2-targeted therapy in breast cancer cells harboring simultaneous HER2 amplification and L755S somatic mutation. In our ER+/HER2+ BT474 models, we have shown that reactivated HER2 and downstream signaling (such as AKT and MAPK) by the HER2L755S mutation can be significantly inhibited by low and clinically relevant concentrations of these inhibitors in vitro and in vivo. These cells, in vitro, were resistant not only to the dual regimen L+T but also T+P and were less sensitive to T-DM1. We have further shown that Afa combined with endocrine therapy can achieve stable complete tumor regression in the ER+/HER2+ BT474/AZ-LR xenografts harboring the L755S mutation. This suggests that treating ER+/HER2+ breast cancer patients harboring the HER2L755S mutation with irreversible HER1/2 inhibitors such as Afa or Nrb instead of L might improve clinical outcome. The ExteNet trial has demonstrated a small additional benefit from one year of Nrb after completion of adjuvant T in high risk HER2+ breast cancers (50). Whether this added benefit is related to the presence of HER2 mutations remains to be determined. The therapeutic potential of the HER1/2 irreversible TKIs in HER2-negative breast cancer patients where HER2 signaling is activated by HER2 mutations is being investigated by Ma et al. (NCT01670877) (51). The results of that study so far indicate that in 16 heavily-pretreated HER2-negative metastatic breast cancer patients (14/16 with known activating HER2 mutations, 2/16 with HER2 mutations of unknown significance), single-agent Nrb treatment resulted in a 36% clinical benefit rate in the patients whose tumors harbored known activating HER2 mutations. Yet the majority of the patients with those HER2 mutations did not benefit from Nrb treatment alone. A recent preclinical study in ER+ MCF7 cells expressing HER2 kinase domain mutations (52), as well as early results from an additional clinical trial investigating the efficacy of Nrb + fulvestrant in ER+ metastatic breast cancer patients with HER2 mutations (SUMMIT, NCT01953926) (53), further support the notion that a simultaneous inhibition of ER (fulvestrant) and the mutant HER2 by irreversible TKIs is needed. However, a significant portion of patients did not benefit even with the combination of Nrb+fulvestrant while other patients developed acquired resistance. Interestingly, we observed acquired resistance to ED+Afa treatment in our BT474/AZ-LR xenografts (Suppl. Fig. S8C). It is important to understand the mechanism of intrinsic and acquired resistance to Afa and Nrb in breast tumors expressing wildtype or mutant HER2. More Afa/Nrb-resistant models are currently under development through in vitro and in vivo approaches to understand the involvement of additional acquired HER2 mutations, as has been recently suggested (54), as well as the role of additional pathways.
To summarize, here we have identified HER2 reactivation through acquisition of the HER2L755S mutation as a mechanism of acquired resistance to L-containing HER2-targeted therapy in preclinical HER2+ breast cancer models. This resistance can be overcome by treating the tumors harboring the L755S mutation with irreversible HER1/2 inhibitors. Our findings warrant further studies investigating the role of this mutation and other HER2 mutations in acquired resistance in HER2+ breast cancer through sequencing analyses of larger numbers of tumor pairs pre- and post HER2-targeted treatment in the clinical setting. Likewise, additional clinical trials are needed to identify the subset of breast cancer patients whose tumors harbor HER2 mutations who might benefit from irreversible HER1/2 TKIs such as Nrb and Afa.
Supplementary Material
Translational relevance.
Despite the efficacy of first-generation HER2-targeted therapy such as trastuzumab and lapatinib in HER2-positive breast cancer, acquired resistance to these drugs, alone or in combination, frequently occurs in patients. HER2 pathway reactivation by the HER2L755S mutation was found to constitute a mechanism of acquired resistance to lapatinib-containing HER2-targeted therapies in HER2+ breast cancer models. This mutation also conferred resistance to the dual HER2 blockade trastuzumab+pertuzumab and less sensitivity to the antibody-drug conjugate trastuzumab emtansine (T-DM1). Our findings support the contention that second-generation irreversible HER1/2 inhibitors, such as afatinib and neratinib, may offer therapeutic approaches for HER2+ breast cancer patients whose tumors harbor the HER2L755S mutation.
Acknowledgments
Research support: This study was supported in part by the NCI Specialized Programs of Research Excellence (SPORE) grants P50 CA058183 and CA186784-01 and the Dan L. Duncan Comprehensive Cancer Center grant P30CA125123, by the National Cancer Institute grant U54 CA 112970, and by the Breast Cancer Research Foundation, and a Stand Up To Cancer Dream Team Translational Research Grant (SU2C-AACR-DT0409). Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research, the scientific partner of SU2C.
The authors thank the Biostatistics and Informatics shared resource at the Dan L. Duncan Comprehensive Cancer Center for biostatistics analysis, the Smith Breast Center Pathology Core for IHC staining, Dr. Thomas Westbrook for providing the pHAGE-Ubc-DEST-HA expression plasmids and Dr. John Belmont at the Laboratory for Translation Genomics, Children’s Nutrition Research Center, Baylor College of Medicine, for performing the SNP arrays of the BT474 cell lines. STR DNA fingerprinting (cell line authentication) was done by the CCSG-funded Characterized Cell Line Core, NCI # CA016672. D.J.Z. is funded in part by NIH GM007309. K.L.S is supported by the NIH (UO1CA168394). B.H.P is funded in part by the Breast Cancer Research Foundation, and the Avon Foundation. S.G.H is partly supported by the NCI Cancer Center Support Grant (P30CA125123). JSR-F is funded in part by the Breast Cancer Research Foundation. Research reported in this publication was supported in part by a Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute (Grant No. P30CA008748). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
- Thirty-Seventh Annual CTRC-AACR San Antonio Breast Cancer Symposium, San Antonio, TX, USA, December 9–13, 2014. Xiaowei Xu et al. Clonal evolution of the HER2 L755S mutation leads to acquired HER-targeted therapy resistance that can be reversed by the irreversible HER1/2 inhibitor afatinib. Abstract P5-05-03.
- The annual meeting of the American Association for Cancer Research, Philadelphia, PA, USA, April 18–22, 2015. Xiaowei Xu et al. Clonal evolution of the HER2 L755S mutation as a mechanism of acquired HER-targeted therapy resistance. Abstract 737.
- Experimental Biology Annual Meeting, San Diego, CA, USA, April 2 – 6, 2016. Xiaowei Xu et al. Clonal Evolution of the HER2 L755S Mutation Leads to Acquired HER-targeted Therapy Resistance That Can Be Reversed by the Irreversible HER1/2 Inhibitor Afatinib. Abstract 1107.5.
Conflicts of Interest
R. S. has received in the past 3 years research grants from AstraZeneca and Gilead and has served on an advisory board of Eli Lilly.
Under licensing agreements between Horizon Discovery, Ltd. and The Johns Hopkins University, D. J. Z and B. H. P are entitled to a share of royalties received by the university on sales of products.
M. F. R receives funding from GlaxoSmithKline.
Abbott has licensed technology of which J. W. G is an inventor and which is used in this research. This potential conflict of interest has been reviewed and managed by OHSU.
C. K. O is on the advisory boards of Genentech, Perkin Elmer, Pfizer, Ventana/Roche, and AstraZeneca, and receives book royalties from Wolter Kluwer. C. K. O is also an expert witness consultant for O’Melveny and Myers.
Authors’ Contributions
Conception and design: X. Xu, A. Nardone, H. Hu, C. De Angelis, B. Weigelt, J. S. Reis-Filho, C. K. Osborne, R. Schiff
Development of methodology: X. Xu, C. De Angelis, A. Nardone, H. Hu, J. S. Reis-Filho, B. Weigelt, R. Schiff
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): X. Xu, L. Qin, C. De Angelis, A. Nardone, H. Hu, L. Heiser, N. Wang, J. Veeraraghavan, V. Sethunath, S. Nanda, M. Shea, T. Mitchell, I. Waters, D. J. Zabransky, C. Gutierrez, M. F. Rimawi, C. Nagi, B. H. Park, J. W. Gray, R. Schiff
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): X. Xu, C. De Angelis, V. Sethunath, J. Veeraraghavan, A. Nardone, H. Hu, K. Burke, C. K. Y. Ng, E. Chen, A. Renwick, T. Wang, C. Shaw, S. G. Hilsenbeck, R. Schiff
Writing, review, and/or revision of the manuscript: X. Xu, C. De Angelis, J. Veeraraghavan, K. A. Burke, A. Nardone, V. Sethunath, L. Qin, H. Hu, L. M. Heiser, N. Wang, C. K. Y. Ng, E. S. Chen, A. Renwick, T. Wang, S. Nanda, M. Shea, T. Mitchell, I. Waters, D. J. Zabransky, C. Gutierrez, C. Nagi, F. C. Geyer, G. C. Chamness, K. L. Scott, B. H. Park, C. A. Shaw, S. G. Hilsenbeck, M. F. Rimawi, J. W. Gray, B. Weigelt, J. S. Reis-Filho, C. K. Osborne, R. Schiff.
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): X. Xu, C. De Angelis, R. Schiff
Study supervision: R. Schiff, C. K. Osborne
References
- 1.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 2.Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. The New England journal of medicine. 2006;355(26):2733–43. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 3.Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE, Jr, Davidson NE, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. The New England journal of medicine. 2005;353(16):1673–84. doi: 10.1056/NEJMoa052122. [DOI] [PubMed] [Google Scholar]
- 4.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. The New England journal of medicine. 2001;344(11):783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 5.Smith I, Procter M, Gelber RD, Guillaume S, Feyereislova A, Dowsett M, et al. 2-year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: a randomised controlled trial. Lancet. 2007;369(9555):29–36. doi: 10.1016/S0140-6736(07)60028-2. [DOI] [PubMed] [Google Scholar]
- 6.Gianni L, Pienkowski T, Im YH, Roman L, Tseng LM, Liu MC, et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(1):25–32. doi: 10.1016/S1470-2045(11)70336-9. [DOI] [PubMed] [Google Scholar]
- 7.Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. The New England journal of medicine. 2012;367(19):1783–91. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arpino G, Gutierrez C, Weiss H, Rimawi M, Massarweh S, Bharwani L, et al. Treatment of human epidermal growth factor receptor 2-overexpressing breast cancer xenografts with multiagent HER-targeted therapy. Journal of the National Cancer Institute. 2007;99(9):694–705. doi: 10.1093/jnci/djk151. [DOI] [PubMed] [Google Scholar]
- 9.Rimawi MF, Wiechmann LS, Wang YC, Huang C, Migliaccio I, Wu MF, et al. Reduced dose and intermittent treatment with lapatinib and trastuzumab for potent blockade of the HER pathway in HER2/neu-overexpressing breast tumor xenografts. Clin Cancer Res. 2011;17(6):1351–61. doi: 10.1158/1078-0432.CCR-10-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang YC, Morrison G, Gillihan R, Guo J, Ward RM, Fu X, et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers--role of estrogen receptor and HER2 reactivation. Breast Cancer Res. 2011;13(6):R121. doi: 10.1186/bcr3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, Aura C, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet. 2012;379(9816):633–40. doi: 10.1016/S0140-6736(11)61847-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rimawi MF, Mayer IA, Forero A, Nanda R, Goetz MP, Rodriguez AA, et al. Multicenter phase II study of neoadjuvant lapatinib and trastuzumab with hormonal therapy and without chemotherapy in patients with human epidermal growth factor receptor 2-overexpressing breast cancer: TBCRC 006. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31(14):1726–31. doi: 10.1200/JCO.2012.44.8027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blackwell KL, Pegram MD, Tan-Chiu E, Schwartzberg LS, Arbushites MC, Maltzman JD, et al. Single-agent lapatinib for HER2-overexpressing advanced or metastatic breast cancer that progressed on first- or second-line trastuzumab-containing regimens. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2009;20(6):1026–31. doi: 10.1093/annonc/mdn759. [DOI] [PubMed] [Google Scholar]
- 14.Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol. 2006;3(5):269–80. doi: 10.1038/ncponc0509. [DOI] [PubMed] [Google Scholar]
- 15.Rimawi MF, Schiff R, Osborne CK. Targeting HER2 for the treatment of breast cancer. Annu Rev Med. 2015;66:111–28. doi: 10.1146/annurev-med-042513-015127. [DOI] [PubMed] [Google Scholar]
- 16.Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 17.Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008;68(22):9221–30. doi: 10.1158/0008-5472.CAN-08-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Contreras AHS, Wang T, Mayer I, Forero A, Nanda R, et al., editors. PIK3CA mutations and/or low PTEN predict resistance to combined anti-HER2 therapy with lapatinib and trastuzumab and without chemotherapy in TBCRC006, a neoadjuvant trial of HER2-positive breast cancer patients. Cancer Res; San Antonio Breast Cancer Symposium; Dec 10–14; San Antonio, TX. 2013. [Google Scholar]
- 19.Huang C, Park CC, Hilsenbeck SG, Ward R, Rimawi MF, Wang YC, et al. beta1 integrin mediates an alternative survival pathway in breast cancer cells resistant to lapatinib. Breast cancer research : BCR. 2011;13(4):R84. doi: 10.1186/bcr2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer discovery. 2013;3(2):224–37. doi: 10.1158/2159-8290.CD-12-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrari A, Vincent-Salomon A, Pivot X, Sertier AS, Thomas E, Tonon L, et al. A whole-genome sequence and transcriptome perspective on HER2-positive breast cancers. Nature communications. 2016;7:12222. doi: 10.1038/ncomms12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trowe T, Boukouvala S, Calkins K, Cutler RE, Jr, Fong R, Funke R, et al. EXEL-7647 inhibits mutant forms of ErbB2 associated with lapatinib resistance and neoplastic transformation. Clin Cancer Res. 2008;14(8):2465–75. doi: 10.1158/1078-0432.CCR-07-4367. [DOI] [PubMed] [Google Scholar]
- 25.Kancha RK, von Bubnoff N, Bartosch N, Peschel C, Engh RA, Duyster J. Differential sensitivity of ERBB2 kinase domain mutations towards lapatinib. PLoS One. 2011;6(10):e26760. doi: 10.1371/journal.pone.0026760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20(9):1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology. 2013;31(3):213–9. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome research. 2012;22(3):568–76. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics (Oxford, England) 2012;28(14):1811–7. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 31.De Mattos-Arruda L, Weigelt B, Cortes J, Won HH, Ng CK, Nuciforo P, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2014;25(9):1729–35. doi: 10.1093/annonc/mdu239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nature biotechnology. 2012;30(5):413–21. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Loo P, Nordgard SH, Lingjaerde OC, Russnes HG, Rye IH, Sun W, et al. Allele-specific copy number analysis of tumors. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(39):16910–5. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics (Oxford, England) 2009;25(9):1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England) 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nature biotechnology. 2011;29(1):24–6. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, et al. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(9):3665–70. doi: 10.1073/pnas.1019736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rexer BN, Chanthaphaychith S, Dahlman K, Arteaga CL. Direct inhibition of PI3K in combination with dual HER2 inhibitors is required for optimal antitumor activity in HER2+ breast cancer cells. Breast cancer research : BCR. 2014;16(1):R9. doi: 10.1186/bcr3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwarz DS, Ding H, Kennington L, Moore JT, Schelter J, Burchard J, et al. Designing siRNA that distinguish between genes that differ by a single nucleotide. PLoS Genet. 2006;2(9):e140. doi: 10.1371/journal.pgen.0020140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474(7350):179–83. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 41.Ariazi EA, Lewis-Wambi JS, Gill SD, Pyle JR, Ariazi JL, Kim HR, et al. Emerging principles for the development of resistance to antihormonal therapy: implications for the clinical utility of fulvestrant. J Steroid Biochem Mol Biol. 2006;102(1–5):128–38. doi: 10.1016/j.jsbmb.2006.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morrison G, Fu X, Shea M, Nanda S, Giuliano M, Wang T, et al. Therapeutic potential of the dual EGFR/HER2 inhibitor AZD8931 in circumventing endocrine resistance. Breast Cancer Res Treat. 2014;144(2):263–72. doi: 10.1007/s10549-014-2878-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scaltriti M, Verma C, Guzman M, Jimenez J, Parra JL, Pedersen K, et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene. 2009;28(6):803–14. doi: 10.1038/onc.2008.432. [DOI] [PubMed] [Google Scholar]
- 44.Nahta R, Yuan LX, Du Y, Esteva FJ. Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: effects on insulin-like growth factor I signaling. Mol Cancer Ther. 2007;6(2):667–74. doi: 10.1158/1535-7163.MCT-06-0423. [DOI] [PubMed] [Google Scholar]
- 45.Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther. 2012;343(2):342–50. doi: 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- 46.Zabransky DJ, Yankaskas CL, Cochran RL, Wong HY, Croessmann S, Chu D, et al. HER2 missense mutations have distinct effects on oncogenic signaling and migration. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(45):E6205–14. doi: 10.1073/pnas.1516853112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zuo WJ, Jiang YZ, Wang YJ, Xu XE, Hu X, Liu GY, et al. Dual Characteristics of Novel HER2 Kinase Domain Mutations in Response to HER2-Targeted Therapies in Human Breast Cancer. Clin Cancer Res. 2016;22(19):4859–69. doi: 10.1158/1078-0432.CCR-15-3036. [DOI] [PubMed] [Google Scholar]
- 48.Wagle NLN, Richardson AL, Leshchiner I, Mayer IA, Forero-Torres A, Hobday TJ, Dees E, Nanda R, Rimawi MF, Guo H, Barry WT, Wolff AC, Gabriel SB, Garraway LA, Winer EP, Krop IE, editors. 2014 ASCO Annual Meeting. 2014. Whole exome sequencing (WES) of HER2+ metastatic breast cancer (MBC) from patients with or without prior trastuzumab (T): A correlative analysis of TBCRC003. [Google Scholar]
- 49.Boulbes DR, Arold ST, Chauhan GB, Blachno KV, Deng N, Chang WC, et al. HER family kinase domain mutations promote tumor progression and can predict response to treatment in human breast cancer. Mol Oncol. 2015;9(3):586–600. doi: 10.1016/j.molonc.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chan A, Delaloge S, Holmes FA, Moy B, Iwata H, Harvey VJ, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17(3):367–77. doi: 10.1016/S1470-2045(15)00551-3. [DOI] [PubMed] [Google Scholar]
- 51.Ma CXBR, Gao F, Freedman RA, Pegram MD, Blackwell K, et al., editors. Phase II trial of neratinib for HER2 mutated, non-amplified metastatic breast cancer (HER2mut MBC). J Clin Oncol.2016 ASCO annual meeting; 2016 Jun 2–6; Chicago, IL. 2016. [Google Scholar]
- 52.Croessmann S, Cutler RE, Jr, Lalani AS, Park BH, Arteaga CL. Inhibition of mutant HER2 results in synthetic lethality when combined with ER antagonists in ER+/HER2 mutant human breast cancer cells. Thirty-Ninth Annual CTRC-AACR San Antonio Breast Cancer Symposium; San Antonio, TX, USA. December 6–10, 2016. [Google Scholar]
- 53.Hyman D, Saura C, Arteaga CL, Mayer I, Shapiro G, Loi S, Lalani A, Xu F, Cutler R, Butturini A, Bryce R, Meric-Bernstam F, Baselga J, Solit D. Neratinib + fulvestrant in ERBB2-mutant, HER2–non-amplified, estrogen receptor (ER)-positive, metastatic breast cancer (MBC): Preliminary analysis from the phase II SUMMIT trial. Thirty-Ninth Annual CTRC-AACR San Antonio Breast Cancer Symposium; San Antonio, TX, USA. December 6–10, 2016. [Google Scholar]
- 54.Hanker AB, Sheehan JH, Koch JP, Lanman R, Hyman DM, Cutler RE, Jr, Lalani AS, Cross D, Lovly CM, Meiler J, Arteaga CL. An acquired HER2 T798I gatekeeper mutation induces resistance to neratinib in a patient with HER2 mutant-driven breast cancer. Thirty-Ninth Annual CTRC-AACR San Antonio Breast Cancer Symposium; San Antonio, TX, USA. December 6–10, 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.