

Abstract
Diabetes is the result of having inadequate supply of functional insulin-producing β cells. Two possible approaches for replenishing the β cells are: 1) replacement by transplanting cadaveric islets or β cells derived from human embryonic stem (hESC)/induced pluripotent stem (iPSC) cells and 2) induction of endogenous regeneration. This review focuses on endogenous regeneration, which can follow two pathways: enhanced replication of existing β cells and formation of new β cells from cells not expressing insulin, either by conversion from a differentiated cell type (transdifferentiation) or differentiation from progenitors (neogenesis). Exciting progress on both pathways suggest that regeneration may have therapeutic promise.
eTOC BLURB
Aguayo-Mazzucato and Bonner-Weir review the current approaches toward inducing endogenous β-cell regeneration. The authors highlight exciting progress in our understanding and enhancement of regenerative pathways (proliferation, transdifferentiation and neogenesis) which may hold therapeutic promise in alleviating some of the disease burden by overcoming the inadequate β-cell supply in diabetes.
INTRODUCTION
Diabetes, both type 1 and type 2, is a disease characterized by an absolute or relative deficiency of β cells (Weir et al., 1990). Therefore, replenishing the lost functional or absolute β cell mass is a strategy that can alleviate some of the burdens of the disease. There are two general approaches to replenish β cells: 1) replacement therapy by transplanting cadaveric islets or β cells derived from human embryonic stem (hESC)/induced pluripotent stem (iPSC) cells and 2) induction of endogenous regeneration. The latter approach is the topic of this review.
Replacement therapy using cadaveric islets has been shown as a proof of principle (Shapiro et al., 2000) to reverse diabetes in 88% of patients at 1 year and 71% at 2 years (Hering et al., 2016) with 10% maintaining insulin independence at 5 years but 80% with detectable C-peptide at the same time point (Ryan et al., 2005). However, the availability of healthy islets is insufficient for widespread application of this. The recent breakthroughs in deriving glucose responsive β-like cells from human pluripotent stem cells (Korytnikov and Nostro, 2016; Pagliuca et al., 2014; Rezania et al., 2014; Russ et al., 2015) have given encouragement for β cell replacement therapy. However, besides the need to gain fully functional insulin responses, this approach has other major challenges in becoming a therapy, including recurrent autoimmune attacks in type 1 diabetes, the inherent risks of placing foreign tissue in the body and potential tumor formation from not fully differentiated cells. Encapsulation of cell aggregates within immunobarriers, either microcapsules or macrocapsules, is the main strategy to protect the transplanted cells as well as containing potentially undifferentiated cell that can later be removed. However, encapsulation brings substantial issues of insufficient oxygen/nutrient access, foreign body response and impaired insulin kinetics (O’Sullivan et al., 2011; Weir, 2013). Even so, some clinical trials with macroencapsulation of hESC-derived cells have started (Clinicaltrials.gov NCT02939118; NCT02239354).
Therefore, there is a continued interest in understanding the processes of endogenous expansion of β cells in order to regenerate their endogenous mass. β cell mass is defined as the total weight of β cells within a pancreas and is determined by the balance between death (apoptosis/necrosis) and birth (replication of existing cells and neogenesis/transdifferentiation) of β cells as well as individual cell volume (atrophy/hypertrophy). The endocrine pancreas is a slow turnover tissue with relatively long lifespan (rodents 2–3 months)(Finegood et al., 1995). The renewal capacity of the pancreatic β cell pool is lower than tissues with well-characterized adult stem cell niches such as blood, skin and gut. Even so, the low frequency of both proliferation and apoptosis in the adult allows sustained β cell mass expansion over the first 7 months in rats (Montanya et al., 2000). Many rodent studies, often using genetically modified mice, have examined pathways involved in development or postnatal growth. In this review, we will combine studies on neogenesis and transdifferentiation. Even though these terms are often used differently, they both represent new β cells derived from a cell not expressing insulin. Neogenesis is usually considered as newly formed β cells by differentiation from stem/progenitor cells; these progenitors may have arisen from dedifferentiated duct cells (Bonner-Weir et al., 2004). On the other hand, transdifferentiation has been defined (Shen et al., 2003) as the direct conversion of a terminally differentiated cell type into another cell type-β cells in this case.
Islet regeneration has mainly been studied in rodent models and only more recently isolated human islet, acinar or duct cells are being used in vitro. While the concept of islet regeneration by any means other than replication of preexisting cells suffered disfavor for about a decade, neogenesis/transdifferentiation has regained favor. It is likely that both proliferation and new formation of β cells are mechanisms of islet regeneration, with different emphasis depending on the injury and on the species. We will discuss the state of our knowledge of β cell/islet regeneration in rodents and humans with highlights of some exciting new studies.
CLASSIC ISLET REGENERATION MODELS
The classic rodent models of pancreatic regeneration, partial pancreatectomy (Bonner-Weir et al., 1993) and partial duct ligation (Wang et al., 1995), have both increased β-cell replication and neogenesis reported. A third model becoming a classic model of regeneration is that of diphtheria toxin-targeted cell ablation.
Partial pancreatectomy (Px), consisting of the removal of 60–90% of the pancreas in adult rats and 50–80% in adult mice, has demonstrated the substantial regenerative capacity of the adult pancreas with whole new lobes being formed and enhanced proliferation of pre-existing β and acinar cells (Bonner-Weir et al., 1993; Bonner-Weir et al., 1983; Brockenbrough et al., 1988; Li et al., 2010; Peshavaria et al., 2006). We showed that after partial pancreatectomy in adult rats, pancreatic-duct cells undergo a reproducible dedifferentiation and expansion forming transient areas composed of proliferating ductules; these foci of regeneration subsequently recapitulate aspects of embryonic pancreatic differentiation to form new pancreatic lobes with new islets (Bonner-Weir et al., 1993; Li et al., 2010). Recent data has shown that by 1 year of age there was a loss of this plasticity of the pancreatic ducts resulting in less neogenesis (Tellez et al., 2016). In these older rats, without the neogenesis, the β cell mass did not increase after Px even though β cell replication and apoptosis frequency did not differ with that of 1 month old and individual cell size had similar increases. At least two groups using 60% Px in adult mice also reported both neogenesis and enhanced replication (Ackermann Misfeldt et al., 2008; Peshavaria et al., 2006) while others reported only enhanced replication (Dor et al., 2004; Lee et al., 2006; Teta et al., 2007). Using genetically modified mice and this model, Ackermann Misfeldt et al. showed that the regulation of β cell proliferation by the transcription factor FoxM1 differed between β cells formed by neogenesis, which did not need the transcription factor to proliferate, and the mature adult ones that depended on FoxM1 to divide. This difference in the dependency of a transcription factor had been previously reported between embryonic and adult β cells (Zhang et al., 2006). In summary, partial pancreatectomy provides a model to study both neogenesis and replication in rodents.
Partial duct ligation involves the surgical ligation of the pancreatic duct at the level of the pylorus, resulting in an obstruction of drainage of exocrine secretions from the distal tail region and subsequent loss of acinar cells by death and dedifferentiation. This model has been used for several decades to study mechanisms involved in β cell formation (Edstrom, 1971a, b; Hultquist et al., 1979; Lardon et al., 2004; Rooman et al., 2002; Wang et al., 1995). Two distinct features of this model are 1) that regeneration is limited to the pancreatic portion distal to the ligation allowing an internal control and 2) there is no change in glucose homeostasis. In a comprehensive study in rats Wang et al. found a doubling of β cell population that could not be accounted for by a low BrdU incorporation. Finding an increased population of small clusters or singlet insulin positive cells suggested to them that neogenesis also occurred. One of the first studies (Xu et al., 2008) to use this model in mice showed expansion of β cell mass from endogenous multipotent progenitor cells that depended partly on activation of neurogenin 3 (Ngn3), a transcription factor that is necessary for pancreatic endocrine cell development. They identified and sorted GFP-expressing cells from the ligated portion of the pancreas of Ngn3:GFP mice and showed that when transplanted into Ngn3null pancreas in vitro these cells could become islet cells including insulin-expressing β cells. With knockdown of Ngn3 using an intraductal injection of lentivirus, they showed that the activation of Ngn3 in cells within the ductal complexes was involved. However, subsequently considerable controversy has arisen over this model with most other groups finding no increase in β–cells. The question of whether the PDL-induced changes in tissue composition might skew the results was raised (see (Kopp et al., 2011; Van de Casteele et al., 2014). A rigorous study using pancreatic acinar-specific transcription factor 1a (Ptf1a) promoter-driven lineage tracing (Pan et al., 2013) provided evidence of a small contribution of β cells from acinar cells; there was a rapid dedifferentiation of acinar cells to a ductal phenotype and then endocrine differentiation over several weeks. Hyperglycemia due to partial ablation of the β cells by streptozocin accelerated this process.
The diphtheria toxin-targeted cell ablation was originally developed by introducing the diphtheria toxin A chain as a transgene into mice such that only cells that transcribed the specific promoter of choice would be killed(Herrera et al., 1994). An inducible version directing the toxin to the insulin-expressing cells was used to show the regenerative capacity of 20–30% residual β cells and that the immunosuppressive drugs used in islet transplantation blocked the recovery (Nir et al., 2007). Another inducible variation, first used in hepatocytes (Saito et al., 2001) and then in islet cells by Herrera’s group (Thorel et al., 2010), has targeted transgene expression of the human heparin-binding epidermal growth factor-like precursor (HB-EGF), which acts as the diphtheria toxin receptor (DTR). Primates are 1000 fold more sensitive to diphtheria toxin (DT) than mice so the human receptor as transgene confers sensitivity; expressing cells are only ablated when DT is injected. The studies of the Herrera group are described below in the Transdifferentiation section. Using crosses of ROSA26 LSL:DTR mice with either Elastase (acinar-specific) or pancreatic and duodenal homeobox 1 (Pdx1)(global pancreas)-driven expression of Cre and injections of DT, Criscimanna et al. (Criscimanna et al., 2011) showed that the severity and type of pancreatic injury determined the regenerative mechanism. In the PDXcreROSADTR mice in which both the acinar and islet tissue were ablated, both regenerated from the pancreatic duct cells (which for an unexplained reason had not been ablated) but in ElacreROSADTR in which only the acinar cells were ablated, only new acinar regenerated from the ductal cells. Overall, the diphtheria toxin β-cell ablation model may induce less inflammation than the partial pancreatectomy and PDL and therefore may have less confounding effects introduced by edema or inflammation.
PHYSIOLOGICAL MODELS OF COMPENSATORY GROWTH IN RODENTS AND HUMANS: PREGNANCY AND INSULIN RESISTANCE
Pregnancy and high fat diets are not really regeneration models but rather models of physiological compensatory growth that help identify and study relevant pathways that can then be applied in islet regenerative strategies. The concept of compensatory growth to meet increased demand due to obesity, pregnancy or solely insulin resistance has been well studied in rodents and seen mainly as enhanced replication rather than neogenesis. Both models have provided insights into pathways involved with enhanced β cell growth in the adult. Interestingly, comparison of rodent pancreas and autopsied or surgically resected human pancreas has shown differences in compensatory growth between the species and between physiological compensatory models.
In pregnant rodents, proliferation of preexisting β cells and their enhanced function (Parsons et al., 1992) seem to be the main mechanisms of compensating although at least one lineage tracing study reported neogenesis in addition (Toselli et al., 2014). Signaling through the prolactin receptor by placental lactogen or prolactin was shown stimulate β cell proliferation. The mechanism of this includes the repression of the expression of menin, which as a tumor suppressor blocked β cell replication (Karnik et al., 2007; Kim et al., 2010), and induced serotonin production (Schraenen et al., 2010); (Kim et al., 2010) which in turn stimulated proliferation in an autocrine and/or paracrine manner (Kim et al., 2010). In pregnant humans, while there is a 40% increase in the relative volume of β cells (%β cell/pancreas), there was no observed change in replication nor apoptosis (Butler et al., 2010). However there were increased proportion of small islets, increased number of insulin+ cells with the ducts (1.2±0.02 % vs 0.4±0.1%) and a 3-fold increase in singlet insulin+ cells. Even so, one must be cautious about concluding the expansion was all neogenesis since the lack of measurable increase in replication may be due to the suppression of protein expression of the marker of proliferation Ki67 by autopsy conditions of warm and cold ischemia (Sullivan et al., 2015).
Another model of physiological β cell compensation is insulin resistance or obesity that can result in 30 fold increased β cell mass in mice (Bruning et al., 1997) but only about 30% in humans (Kloppel et al., 1985; Rahier et al., 2008). Moreover, while this compensatory growth is mainly by replication in mice, neogenesis rather than enhanced replication has been suggested in two studies of humans. Using surgical specimens Yoneda et al (Yoneda et al., 2013) reported increased number of single or small clusters of insulin+ cells, bihormone-expressing cells and percentage of insulin+ cells within ducts in patients with impaired glucose tolerance or newly diagnosed T2D compared to non diabetic patients; no difference in proliferation (Ki67+) was seen. Similarly Mezza et al. reported in surgical specimens from insulin-resistant subjects an increased proportion of scattered insulin+ cells and small islets and 3-fold increase of cells co-expressing insulin and the duct marker cytokeratin 19 compared to insulin-sensitive subjects; proliferation as judged by KI67 staining was not detectable (Mezza et al., 2014). A third study (Hanley et al., 2010), using organ donor pancreas, reported overall increased neogenesis (clusters of 3 or less insulin+ cells) with obesity or T2D. Additionally increased PCNA+ insulin cells were found in the non-diabetic obese but decreased PCNA+ cells in obese T2D.
We have learned from these physiological β-cell compensation models that the predominant compensatory mechanism may vary by species and condition, reinforcing the need to look into both strategies as a way to regenerate β-cells as a treatment of diabetes.
ENHANCED PROLIFERATION AS A STRATEGY
Inducing β cell proliferation is, in theory, a straightforward and effective way to increase absolute β cell mass; however, in human islets it has been quite difficult. Over the years there has been much controversy as to whether adult human β cells could proliferate, but in 2001 (Tyrberg et al., 2001) showed that adult human islets incorporated H3 thymidine when transplanted in mice and this incorporation increased in response to hyperglycemia. In both rodents and human there is a decline in the percentage of replicating β cells after the neonatal period (Gregg et al., 2012; Meier et al., 2008; Scaglia et al., 1997; Teta et al., 2005), but with a low stable frequency well in to adulthood. However, as often ignored but pointed out by Chintinne et al. (Chintinne et al., 2010), the actual pool of replicating β cells is six fold greater in adults than in neonates even with the lower frequency because the overall population is substantially larger.
One of the goals of the field has been to understand what regulated the age-related decrease in replication and strategies to enhance β cell proliferation (Table 1) both in rodents as proof of principle and in humans. In rodents many of the different strategies effective in increasing β cell proliferation have been loss or gain of function studies of different cell cycle molecules. For example, knockout of the nuclear regulator of cyclin-dependent kinase cyclin D2 showed it was necessary to regulate the transition of β cells to a replicative state (Georgia et al., 2010). Consistent with an age-related decrease in the proliferative capacity of β cells there are increases of expression of cell cycle inhibitors. The accumulation of p16Ink4a in cells has been seen as both an effector of senescence and an indicator of cellular senescence in β cells (Krishnamurthy et al., 2006). P16Ink4a blocks replication but its expression is repressed by a histone-lysine N/methyltransferase enzyme encoded by the enhancer of zeste 2 polycomb repressive complex 2 (Ezh2) (Zhou et al., 2013). Ezh2 levels decrease with age, in parallel p16Ink4a increases. Replenishing Ezh2 in young mice increased β cell replication, but after 8 months of age this strategy was no longer effective due to an enrichment of a trithorax group protein complex at the Ink4a locus (Zhou et al., 2013). Others showed that the transgenic maintenance of platelet-derived growth factor receptor a (Pdgfra) expression led to sustained Ezh2 expression and β cell replication in 14 month old mice (Chen et al., 2011). Senescent (proliferative-arrested) cells secrete an array of cytokines and chemokines (known as senescence-associated secreted proteins) that can impair proliferation of neighboring cells (Rodier et al., 2009). Senolytic therapies (Palmer et al., 2015) that specifically eliminate senescent cells may allow the remaining β cells to respond to physiological stimuli of proliferation such as pregnancy, hyperglycemia or weight gain.
Table 1.
Strategies to induce beta cell replication
| Rodents | Human islets | References | |
|---|---|---|---|
| KO Cyclin D2 | Effective | NA | Georgia et al. 2010 |
| Increase Ezh2 | Effective | NA | Zhou et al. 2013 |
| Senolytic therapies | TBD | TBD | Palmer et al. 2015 |
| Glycolytic flux and glucokinase activator. | Effective | Led to DNA damage, growth arrest and apoptosis. | Salpeter et al. 2010, 2011, 2013; Porat et al. 2011; Rieck et al. 2012; Tornovsky-Babeay et al. 2014 |
| Adenosine kinase inhibitors | Effective | Not effective | Annes et al. 2012 |
| Silencing of CDKN2C/p18 or CDKN1a/p21 | TBD | Effective | Robitaille et al. 2016 |
| Harmine | Effective | Effective (also induces α-cell proliferation) | Wang P et al. 2015, Wang YJ et al 2016 |
| Aminopyrazine | Effective | Effective | Shen et al. 2015 |
| SerpinB1 | Effective | Effective | El Ouaamari et al. 2016 |
A series of papers from the Dor lab support the concept that glucose metabolism is more important than cell cycle components in regulating β cell proliferation and can overcome cell cycle inhibitors. Cyclin D2, shown to be high in quiescent β cells, did not differ in β cells of mice between 1 and 6 months old. Yet it became downregulated in replicating cells, and regulation of its mRNA was glucose-dependent (Salpeter et al., 2010; Salpeter et al., 2011). Moreover, glucose metabolism (glycolytic flux) within the β cell was shown to be the main regulator of their compensatory growth, with the rate-limiting enzyme of glycolysis glucokinase being a potential target for inducing proliferation (Porat et al., 2011). In parabiosis and transplantation experiments examining replication in islets of old (8 month) and young (1 month) mice, they found β cell replication in old islets increased in a young environment, without changes in levels of cell cycle inhibitors p16, p18, p27 or p21 mRNA (Salpeter et al., 2013). Interestingly, they noted that the blood glucose levels were lower in their old mice and speculated that in the young environment, glucose might be driving the replication. This ability to induce compensatory growth in old mice is consistent with their experiments in which old mice (1–2y) showed 2–3 fold increase in β cell proliferation using either inducible DTA ablation of β cells or a small molecule glucokinase activator (Stolovich-Rain et al., 2012).
However, the translation of these findings into human cells is complicated by the fact that there are some differences in cell cycle proteins in human islets compared to rodent as laid out in the “roadmap” of the cell cycle molecules in human islets by Stewart’s group (Fiaschi-Taesch et al., 2013). One difference seems to be the cytoplasmic location of G1/S cell cycle molecules in human β cells in vitro. With adenoviral overexpression, Cdk6 plus cyclin D3 became nuclear and drove replication; however, in parallel, the cell cycle inhibitors p16, p21 and p27 also underwent cytoplasmic-to-nuclear localization (Fiaschi-Taesch et al., 2013). There is a concern that a substantial proportion of the β cells that entered the cell cycle showed accumulation of double-stranded DNA damage, which may result in apoptosis and not in expansion of functional β cells (Rieck et al., 2012). Such lack of completion of cell cycle was also seen with mitogenic push from overexpression of hepatocyte nuclear factor 4 (Hnf4) (Rieck et al., 2012). Similarly, long-term over activation of glucokinase or hyperglycemia in T2D was associated with DNA strand breaks, DNA damage response and activation of tumor suppressor protein p53 leading to growth arrest and apoptosis (Tornovsky-Babeay et al., 2014).
Even so, work proceeds on trying to enhance human β-cell proliferation. The most exciting results come from the use of high throughput screens to identify new pathways and compounds that can drive human β cell replication. Adenosine kinase inhibitors promoted β cell replication in mice, rats and pigs without affecting proliferation of α, PP, fibroblasts, exocrine or hepatic cells but, unfortunately the initial report found no effect on isolated human islets (Annes et al., 2012). Since then others have shown 5-iodotubercidin (Dirice et al., 2016) and harmine (Wang et al., 2015), both of which target tyrosine-regulated kinases DYRK1A and NFAT, stimulated human β proliferation. Harmine also stimulated α cell proliferation (Wang et al., 2016). Targeting the same pathway, aminopyrazine compounds stimulated β cell proliferation in dispersed adult human and mouse islets by inhibiting DYRK1a and glycogen synthase kinase 3 β (GSK3β) (Shen et al., 2015). Silencing of CDKN2C/p18 or CDKN1a/p21, both cell cycle inhibitors, was able to drive proliferation of human β cells (Robitaille et al., 2016). Using the LIRKO mouse in which the ablation of the insulin receptor in hepatocytes led to massive β cell hyperplasia, proteinase inhibitor SerpinB1 was identified as a hepatocyte-secreted protein that enhanced β-cell proliferation of human, mice and zebrafish (El Ouaamari et al., 2016). All of these represent potential β cell proliferation strategies but a major potential issue is whether other cell types within the body also respond with enhanced replication; unfortunately, most studies have not examined this question of specificity.
TRANSDIFFERENTIATION/NEOGENESIS AS A STRATEGY
A different, and complementary, strategy is to replenish the β cell mass by differentiation of new β cells. There is not convincing evidence of a true adult pancreatic stem cell, even though embryonically multipotent progenitors give rise to ductal, endocrine and exocrine lineages. While considerable work supports new β cell formation either from progenitors or facultative progenitors within the pancreas or by transdifferentiation of differentiated pancreatic cells into functional β cells, the presence of facultative progenitors after birth is not universally accepted. However, the concept of potential plasticity of the different pancreatic cell types has become somewhat accepted. The main questions studied are which cell type is involved, what factors trigger the process and whether these findings translate to human. The cell types suggested are pancreatic duct cells (or some particular cell within the ducts), centroacinar cells, acinar cells and islet cells particularly glucagon-expressing α, and somatostatin-expressing δ cells (Figure 1). With the recent publications of single cell RNAseq data from human islet preparations, there is an increasing awareness of heterogeneities within a particular cell type (Baron et al., 2016; Dorrell et al., 2016) as well as potentially intermediate stages (Segerstolpe et al., 2016; Xin et al., 2016). Whether such intermediates are artifacts of the technique or show transition states remains to be determined. It may well be that only some of these phenotypes have potential plasticity. This methodology of single cell analysis should be useful in furthering our understanding of β cell regeneration.
Figure 1.
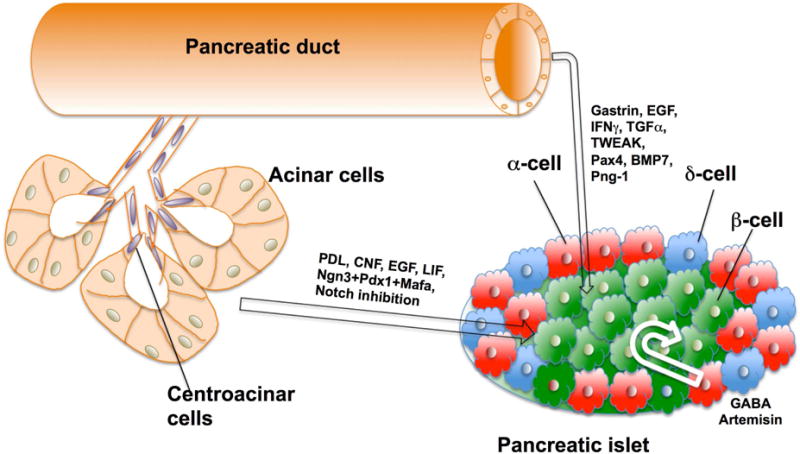
Cell sources within the pancreas that have been shown to differentiate into β-cells. The strategies used to induce this transdifferentiation are indicated within the arrows. For a complete description of these approaches refer to the text.
DUCTS
The pancreatic ductal tree serves as the conduit for the digestive enzymes to get from acinar cells to the duodenum, but we had been proponents that, in addition, its epithelium itself serves as a pool for progenitors for both islet and acinar tissues after birth and into adulthood (Bonner-Weir et al., 2016; Bonner-Weir et al., 2004; Inada et al., 2008). Our studies with the partial pancreatectomy rat model (Bonner-Weir et al., 1993; Li et al., 2010; Sharma et al., 1999) support the idea that there is a massive dedifferentiation of the ductal epithelium of the largest ducts (common pancreatic duct) and proliferation across these ducts giving rise to new lobes rapidly; within 1 week after surgery these new lobes were difficult to distinguish from the old ones. The treatment with gastrin in the 95% pancreatectomy induced further duct dedifferentiation and expression of Ngn3 and the β cell specific transcription factor Nkx6.1, followed by increased population of β cells (Tellez and Montanya, 2014). The papers on PDL from the Heimberg lab (mentioned above) also indicated that pancreatic ducts where the source of the cells with activated Ngn3. In vitro expansion of human ductal tissue and its subsequent differentiation to islet cells was observed after 3–4 weeks culture, when there was a significant increase in insulin as well as formation of functional islet-like structures (Bonner-Weir et al., 2000; Gao et al., 2005). In addition to replication in grafts of human isolated islet preparations under the kidney capsule of mice, duct cells proliferated and had induction of Nkx6.1 expression (Tyrberg et al., 2001). Similarly, insulin-positive cells expressing duct markers were found in grafts of purified human duct cells (Yatoh et al., 2007).
Several groups have identified markers that potentially identify progenitor populations within the ductal tree (Table 2). The most widely accepted marker is CD133 expressed by the progenitor populations from mouse or human pancreas (Gomez et al., 2015; Jin et al., 2016; Lee et al., 2013; Sugiyama et al., 2007b). Yet, others identified CD133+ cells in adult human duct cells as characterized by their expression of cytokeratin 19 (CK19) and carbonic anhydrase 2 (CAR2) (Lee et al., 2013). However, the expression of NGN3 was found in CD133high CD49fhigh cells from fetal mice and human pancreas (Sugiyama et al., 2007b) and as CD133+ cells from adult human, being 2 % of acinar and duct cells in surgical biopsies and 10% in organ donor pancreas (Gomez et al., 2015). Another subpopulation of cells, those expressing double cortin-like kinase 1 (Dclk1), has also been suggested in mouse and human to be a quiescent but long-lived pancreatic progenitor (Westphalen et al., 2016). These cells were found in both ducts and acini, responded to injury and may give rise to pancreatic cancer, but their role in endocrine pancreas regeneration has not been clarified. The recent identification by single cell RNAseq of at least 4 different gene expression profiles of human duct cells (Baron et al., 2016) does not clarify whether there is a separate progenitor population within the ductal tree or just a generalized potential plasticity.
Table 2.
Markers of ductal progenitors
| Markers | Rodent | Humans | References |
|---|---|---|---|
| CD133 | ✔ | ✔ | (Gomez et al., 2015; Jin et al., 2016; Lee et al., 2013; Sugiyama et al., 2007a) |
| CD49fhigh | ✔ | ✔ | Sugiyama et al., 2007 |
| Dclk1 | ✔ | ✔ | Westphalen et al., 2016 |
| ALDH1+ CAC* | ✔ | TBD | Rovira et al., 2010 |
CAC-centroacinar cells
Centroacinar cells (CAC) are a unique cell type at the junction of the terminal ducts and the acini. In zebrafish, they have been shown to contribute to pancreatic regeneration (Delaspre et al., 2015). In human pancreas they have strong CD133 expression (Immervoll et al., 2011). In mice, isolated aldehyde dehydrogenase isoform 1 (ALDH1)+ CACs/terminal ductal cells were shown to generate endocrine cells in vitro in either pancreatospheres or dorsal bud explants (Rovira et al., 2010). However the lack of transcription factor Hes1 in these ALDH1+ cells has led to questions of whether they were actually CAC cells (Delaspre et al., 2015) since in the adult pancreas Hes1 expression is expected to persists in this cell population.
The use of Cre-lox based lineage tracing did not resolve the question of whether pancreatic ducts contain a pool of β-cell progenitors. Such experiments using duct transcription factors hepatocyte nuclear factor-1-β (Hnf1β) (Solar et al., 2009) or SRY-Box 9 (Sox9) (Kopp et al., 2011) as promoters found no labeling of either islets or acinar cells after birth nor after pancreatic duct ligation in contrast to studies using the carbonic anhydrase 2 promoter (Inada et al., 2008), the inducible insulin promoter (Nakamura et al., 2011; Zhang et al., 2016) and the Sox9 promoter (Zhang et al., 2016). Using Sox9 promoter line of mice from the Sander lab but with a founder with higher recombination efficiency, this latter study found that moderate hyperglycemia was needed to drive SOX9+ ductal cells to β cells and that a long-term low dose of gastrin/epidermal growth factor enhanced the neogenesis. One remaining issue with these discrepant lineage-tracing experiments is that those that do not find neogenesis also did not find any postnatal ductal contribution to the acinar population (Kopp et al., 2011; Solar et al., 2009). This lack seems unexplained but the massive growth of the pancreas in the neonatal period, particularly between 2 and 4 weeks when there is a 5-fold increase in pancreatic weight in mice, could not be solely from replication of preexisting acinar cells or their change in cell volume as these would not generate new lobes. Both the ductal branching system that continues to add pancreatic lobes to the growing pancreas after birth (Bonner-Weir et al., 2016) and the labeled pancreatic lobes at 4 weeks age seen with CAII lineage tracing (Inada et al., 2008) support the postnatal pancreas expansion from expanding and differentiating ductal cells. Further evidence that ducts can serve as β-cell progenitors in the adult mouse comes from a series of papers from Collombat in which mice ectopically expressing paired box 4 (Pax4) in glucagon-expressing cells could repeatedly recover from toxin-induced diabetes via duct epithelial cells continuously forming new α cells that then converted to β cells (Al-Hasani et al., 2013; Collombat et al., 2009; Courtney et al., 2013; Pfeifer et al., 2013). The ductal origin of the new glucagon-expressing cells was lineage-traced using the Hfn1βCreER driver (Al-Hasani et al., 2013). These experiments are discussed further under islet transdifferentiation.
The process of transdifferentiation from duct cells into β cells can be promoted through a number of strategies (Figure 1). Transgenic over-expression of interferon-γ in the β cells (Gu and Sarvetnick, 1993), over-expression of transforming growth factor (TGF)-α in pancreatic ducts (Wang et al., 1993), pancreatic or ductal deletion of Fbw7 component of the SCF type ubiquitin ligase (Sancho et al., 2014) and Pax4 ectopic expression in glucagon-positive cells (Collombat et al., 2009) all showed duct-to-β cell progression. Inflammatory cytokines have been shown to stimulate epithelial -to-mesenchymal transition as well as endocrine differentiation program in duct cells through NGN3 activation in a STAT3-dependent manner (Valdez et al., 2016). Transgenic hepatic overexpression of the cytokine TWEAK (TNF-like weak inducer of apoptosis, TNFSF12) promoted pancreatic ductal proliferation and expression of Ngn3 and resulted in focal regions of proliferating ductules and β cells (Wu et al., 2013). In vitro culture of human islet-depleted exocrine tissue (both duct and acinar cells) or duct cell lines with BMP7 (Klein et al., 2015), gastrin and epithelial growth factor (EGF) (Suarez-Pinzon et al., 2005), or preadipocyte factor 1 (Pref-1) (Rhee et al., 2016) have had varying success with differentiating cells that when transplanted could reverse diabetes in mice.
On the basis of the above data the authors conclude that at least some, if not most, of the pancreatic duct cells in rodents and in humans can serve postnatally as β-cell progenitors although this process may not be robust
ACINAR
Acinar cells comprise the most abundant pancreatic cell type and therefore constitute an attractive source of reprogrammable tissue that could generate β cells. Much of the earlier work was on acinar cells in vitro and was led by the Brussels groups. After several days of suspension culture, rat exocrine tissue was seen to transdifferentiate/dedifferentiate to ductal cells (Rooman et al., 2000) but if cultured with growth factor EGF and leukemia inhibitory factor, newly formed β cells were seen that could restore normoglycemia in alloxan-diabetic mice (Baeyens et al., 2005). Further work showed that inhibiting notch signaling lead to 30 % of the acinar cells forming β cells (Baeyens et al., 2009). Then a transient cytokine mixture of EGF and ciliary neurotrophic factor (CNTF) that activated Stat3 signaling in mice that led to in vivo conversion of acinar-to-β cells could reverse alloxan-induced diabetes (Baeyens et al., 2014). Some of these findings were also found with in vitro using human pancreatic tissue (Houbracken et al., 2011), including the activation of STAT3 and MAPK-induced NGN3 expression in transduced cells with some resultant insulin-positive cells (Lemper et al., 2015).
Additional in vivo work used modification of transcription factor expression within the acinar compartment. Pioneering work introduced a viral vector driving key developmental transcription factors Ngn3, Pdx1, and Mafa into the pancreatic parenchyma to reprogram the differentiated exocrine cells in adult mice into β-like cells. These induced β cells were indistinguishable from endogenous islet β cells in size, shape and ultrastructure, and they expressed genes essential for β-cell function and ameliorated hyperglycemia (Zhou et al., 2008). More recently Pdx1 expression induced in acinar cells in transgenic mice resulted in reprogramming into endocrine precursor cells that migrated into islets and differentiated into insulin, somatostatin or PP cells; these cells improved STZ-induced diabetes (Miyazaki et al., 2016). As mentioned above, the study on pancreatic duct ligation with Ptf1a promoter-driven lineage tracing showed some β cell formation from acinar cells via a dedifferentiation to a ductal phenotype (Pan et al., 2013).
ISLET CELLS
Pancreatic islets are non-random organized microorgans of several endocrine cell types, with insulin-producing β-cells being the predominant one. Recently there has been surprising evidence using genetically manipulated mice that β cells could come from transdifferentiation of other islet cell types, namely glucagon-producing α-cells and somatostatin-producing δ-cells. The recent identification of a “virgin beta cell subpopulation” that are urocortin 3negative, MAFAnegative and insulinpositive subpopulation in the periphery of the islet, adds to the potential list of progenitors that could contribute towards a functional beta cell pool (van der Meulen et al., 2017). Moreover, other studies have shown β cells can lose their phenotype and become dedifferentiated and dysfunctional but could perhaps be redifferentiated.
First, the Herrera lab used their model of near total β cell ablation by injections of diphtheria toxin in RIP:DTR (β-cell expression of diphtheria toxin receptor HB-EGF) transgenic mice. Over 10 months with the first 5 months animals were treated with insulin to maintain blood glucose levels below 20 mM, β cells regenerated to about 10% of mass of the controls (Thorel et al., 2010). All islets were involved, and there were no extrainsular β cells that would suggest neogenesis. Surprisingly, lineage tracing showed when the ablation occurred in adult mice there was α,-to-β cell conversion rather than an expansion of the few residual β cells. Most of the β cells also expressed glucagon. However, in juvenile mice (before puberty) no α,-to-β cell conversion was seen but rather β cells derived from δ cells involving dedifferentiation, proliferation and re-expression of islet developmental genes in a forkhead box protein O1 (FoxO1)-dependent process (Chera et al., 2014). These rigorous experiments were the first to clearly show the plasticity of the pancreatic endocrine cells. The hope of such α- to-β cell conversion occurring in humans has led to studies searching for bihormonal cells in human pancreas. In the several autopsy studies published, only 3–4 % islet cells coexpress both insulin and glucagon were found in T2D pancreas with some aberrant transcription factor expression (Butler et al., 2013) ;(Spijker et al., 2015). In surgical specimens from non diabetic subjects, insulin-resistant subjects had higher frequency of bihormonal cells than in insulin-sensitive subjects (Mezza et al., 2014). Yet these human findings may reflect a dedifferentiation of the β cells rather than their transdifferentiation from another endocrine cell type; dedifferentiation of β cell will be discussed below.
Secondly, a series of papers from Collombat (Collombat et al., 2009);(Al-Hasani et al., 2013) (Courtney et al., 2013) showed that by in vivo ectopic expression of Pax4 or inhibition of transcription factor Arx- expression in α cells resulted in their conversion to β cells. Importantly, these neo-generated β-like cells were functional and could reverse chemically-induced diabetes even after repetitive ablations of the β cells. Moreover, a recent study identified GABA as an inducer of this α-to-β-like cell conversion in mice and in human islet transplanted under the kidney capsule of immuno-incompetent mice (Ben-Othman et al., 2017). The role of GABA had been suggested in expansion of the β cells earlier in a provocative study (Soltani et al., 2011) in which GABA treatment led to improved glucose levels in autoimmune diabetic NOD mice and the reversal of diabetes in a multiple low dose streptozocin diabetes mouse model. A particularly exciting complementary finding was that artemisins, already used for malaria treatment, caused both the translocation of ARX from the nucleus to the cytoplasm, thus inhibiting its function for maintenance of the α cell identity, and the stabilization of gephyrin which enhances GABA receptor signaling (Li et al., 2017). Treatment in vitro of human islets with artemisins, led to improved glucose-stimulated insulin release and changes in gene profiles that suggest the α-to-β transdifferentiation seen in mice. These data provide a possible unprecedented β-cell regeneration strategy using a known and approved therapeutic agent; we expect clinical trials will be coming.
A provocative proposal was made that dedifferentiation of β cells was quantitatively more important than actual β cell death/loss in diabetes and that redifferentiation of these β cells might be a reasonable therapy (Talchai et al., 2012). This proposal from the Accili group was based on their findings that β-cell ablation of the transcription factor FoxO1 in mice resulted in chronic hyperglycemia and a reversion of β cells to a progenitor state with a subset adopting an α cell phenotype (Talchai et al., 2012). Yet the term “dedifferentiation” can be more than the loss of hormone protein expression (Weir et al., 2013). In a series of papers in rats we showed that chronic hyperglycemia, even moderate (10–20 mg/dl), resulted in β cell dysfunction and change of phenotype (Jonas et al., 1999; Laybutt et al., 2003; Laybutt et al., 2002). The gene expression changes, including the loss of β cell identity gene expression and the gain of disallowed gene expression, were reversed with normalization of the glycemic levels after 2 weeks of hyperglycemia and partially after 12 weeks. Similarly, after induced transgene expression of a mutated KATP channel in β cells, hyperglycemia rapidly ensued leading to dedifferentiation and degranulation of the β cells (Brereton et al., 2014; Wang et al., 2014). Treatment with insulin or glibenclamide to maintain normoglycemia prevented the dedifferentiation, and if treatment was started after 4 wks, the β cells recovered function and granulation. Together these data would suggest that the maintenance of the glycemic levels in vivo may preserve the functionality of the β cells, but it is not clear yet if there is a limit to the length of time of dedifferentiation of β cells after which reversal is not possible. The Accili group extended this concept to humans with type 2 diabetes (T2D), reporting 16.8% hormonenegativesynaptophysinpositive (ie. dedifferentiated) islet cells in the pancreas of type 2 diabetes subjects, with a large number of these having misexpression of key transcription factors and “no loss of cells with general endocrine features”(Cinti et al., 2016). (Synaptophysin and chromogranin are pan-endocrine makers that have overall equivalent expression in the different islet endocrine cells and can identify an endocrine cell that has lost its hormone expression). They concluded that the 30–50% deficit of β cells seen in T2D (Butler et al., 2003; Rahier et al., 2008; Yoon et al., 2003) was not due to death but dedifferentiation or transdifferentiation of β cells to other islet types. In contrast, the Butlers found that chromograninpositivehormonenegative (ie. dedifferentiated) islet cells could account for no more than 2% of the deficit of β cells in T2D and suggested that instead these hormonenegative cells may reflect an attempted regeneration (Butler et al., 2016; Md Moin et al., 2016).
CONCLUSIONS
There have been great advances in our understanding and enhancement of regenerative strategies of β cells. The major pancreatic cell types (islet, acinar and duct) have been shown to have a degree of plasticity that could lend them to being expanded and differentiated to new β cells. There are recent encouraging findings that suggest endogenous replenishment may be possible in humans. Yet, it is unclear how much enhanced replication or transdifferentiation/neogenesis could be achieved in vivo in humans; it is likely that a combination of both processes will be needed to restore normoglycemia. Mechanistic and developmental studies in rodents have laid the foundation for identifying small molecules and drugs already approved for their medical use that could enhance both regenerative pathways. Even so, a major question remains as how the interventions to expand β cell mass could be specifically targeted to β cells.
There are other major obstacles that need to be overcome in type 1 and type 2 diabetes. Of foremost importance, the autoimmune attack characteristic of type 1 diabetes needs to be addressed so that regenerative strategies can have a long lasting beneficial effect. There have only been a few studies that have tried to replenish β cells in autoimmune diabetic NOD mice. One successful study combined anti-CD3/CD8 conditioning regimen with treatment with gastrin and EGF to reverse diabetes in NOD mice. Both β cell neogenesis and replication were enhanced (Wang et al., 2012; Zhang et al., 2016). For type 2 diabetes, it remains to be determined whether the pancreatic cells are still receptive to growth, expansion and differentiation techniques. If the cells have already become senescent, it is unlikely that they could be activated to regenerate unless the senescence pathway is directly targeted.
Acknowledgments
This study was supported by grants from the NIH DK110390, P30 DK036836 Joslin Diabetes Research Center [DRC] and P30 DK057521 BADRC P&F PI (CAM), the Diabetes Research and Wellness Foundation, and an important group of private donors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
No conflicts of interest relevant to this article are reported.
References
- Ackermann Misfeldt A, Costa RH, Gannon M. Beta-cell proliferation, but not neogenesis, following 60% partial pancreatectomy is impaired in the absence of FoxM1. Diabetes. 2008;57:3069–3077. doi: 10.2337/db08-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hasani K, Pfeifer A, Courtney M, Ben-Othman N, Gjernes E, Vieira A, Druelle N, Avolio F, Ravassard P, Leuckx G, et al. Adult duct-lining cells can reprogram into beta-like cells able to counter repeated cycles of toxin-induced diabetes. Dev Cell. 2013;26:86–100. doi: 10.1016/j.devcel.2013.05.018. [DOI] [PubMed] [Google Scholar]
- Annes JP, Ryu JH, Lam K, Carolan PJ, Utz K, Hollister-Lock J, Arvanites AC, Rubin LL, Weir G, Melton DA. Adenosine kinase inhibition selectively promotes rodent and porcine islet beta-cell replication. Proc Natl Acad Sci USA. 2012;109:3915–3920. doi: 10.1073/pnas.1201149109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeyens L, Bonne S, Bos T, Rooman I, Peleman C, Lahoutte T, German M, Heimberg H, Bouwens L. Notch signaling as gatekeeper of rat acinar-to-beta-cell conversion in vitro. Gastroenterology. 2009;136:1750–1760. doi: 10.1053/j.gastro.2009.01.047. [DOI] [PubMed] [Google Scholar]
- Baeyens L, De Breuck S, Lardon J, Mfopou JK, Rooman I, Bouwens L. In vitro generation of insulin-producing beta cells from adult exocrine pancreatic cells. Diabetologia. 2005;48:49–57. doi: 10.1007/s00125-004-1606-1. [DOI] [PubMed] [Google Scholar]
- Baeyens L, Lemper M, Leuckx G, De Groef S, Bonfanti P, Stange G, Shemer R, Nord C, Scheel DW, Pan FC, et al. Transient cytokine treatment induces acinar cell reprogramming and regenerates functional beta cell mass in diabetic mice. Nat Biotechnol. 2014;32:76–83. doi: 10.1038/nbt.2747. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, Ryu JH, Wagner BK, Shen-Orr SS, Klein AM, et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell systems. 2016;3:346–360. doi: 10.1016/j.cels.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Othman N, Vieira A, Courtney M, Record F, Gjernes E, Avolio F, Hadzic B, Druelle N, Napolitano T, Navarro-Sanz S, et al. Long-Term GABA Administration Induces Alpha Cell-Mediated Beta-like Cell Neogenesis. Cell. 2017;168:73–85 e11. doi: 10.1016/j.cell.2016.11.002. [DOI] [PubMed] [Google Scholar]
- Bonner-Weir S, Aguayo-Mazzucato C, Weir GC. Dynamic development of the pancreas from birth to adulthood. Ups J Med Sci. 2016;121:155–158. doi: 10.3109/03009734.2016.1154906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE. A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes. 1993;42:1715–1720. doi: 10.2337/diab.42.12.1715. [DOI] [PubMed] [Google Scholar]
- Bonner-Weir S, Taneja M, Weir GC, Tatarkiewicz K, Song KH, Sharma A, O’Neil JJ. In vitro cultivation of human islets from expanded ductal tissue. Proc Natl Acad Sci USA. 2000;97:7999–8004. doi: 10.1073/pnas.97.14.7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner-Weir S, Toschi E, Inada A, Reitz P, Fonseca SY, Aye T, Sharma A. The pancreatic ductal epithelium serves as a potential pool of progenitor cells. Pediatr Diabetes. 2004;5(Suppl 2):16–22. doi: 10.1111/j.1399-543X.2004.00075.x. [DOI] [PubMed] [Google Scholar]
- Bonner-Weir S, Trent DF, Weir GC. Partial pancreatectomy in the rat and subsequent defect in glucose-induced insulin release. J Clin Invest. 1983;71:1544–1553. doi: 10.1172/JCI110910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brereton MF, Iberl M, Shimomura K, Zhang Q, Adriaenssens AE, Proks P, Spiliotis II, Dace W, Mattis KK, Ramracheya R, et al. Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nature communications. 2014;5:4639. doi: 10.1038/ncomms5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockenbrough JS, Weir GC, Bonner-Weir S. Discordance of exocrine and endocrine growth after 90% pancreatectomy in rats. Diabetes. 1988;37:232–236. doi: 10.2337/diab.37.2.232. [DOI] [PubMed] [Google Scholar]
- Bruning JC, Winnay J, Bonner-Weir S, Taylor SI, Accili D, Kahn CR. Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell. 1997;88:561–572. doi: 10.1016/s0092-8674(00)81896-6. [DOI] [PubMed] [Google Scholar]
- Butler AE, Campbell-Thompson M, Gurlo T, Dawson DW, Atkinson M, Butler PC. Marked expansion of exocrine and endocrine pancreas with incretin therapy in humans with increased exocrine pancreas dysplasia and the potential for glucagon-producing neuroendocrine tumors. Diabetes. 2013;62:2595–2604. doi: 10.2337/db12-1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AE, Cao-Minh L, Galasso R, Rizza RA, Corradin A, Cobelli C, Butler PC. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia. 2010;53:2167–2176. doi: 10.1007/s00125-010-1809-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AE, Dhawan S, Hoang J, Cory M, Zeng K, Fritsch H, Meier JJ, Rizza RA, Butler PC. beta-Cell Deficit in Obese Type 2 Diabetes, a Minor Role of beta-Cell Dedifferentiation and Degranulation. The Journal of clinical endocrinology and metabolism. 2016;101:523–532. doi: 10.1210/jc.2015-3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- Chen H, Gu X, Liu Y, Wang J, Wirt SE, Bottino R, Schorle H, Sage J, Kim SK. PDGF signalling controls age-dependent proliferation in pancreatic beta-cells. Nature. 2011;478:349–355. doi: 10.1038/nature10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chera S, Baronnier D, Ghila L, Cigliola V, Jensen JN, Gu G, Furuyama K, Thorel F, Gribble FM, Reimann F, et al. Diabetes recovery by age-dependent conversion of pancreatic delta-cells into insulin producers. Nature. 2014;514:503–507. doi: 10.1038/nature13633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintinne M, Stange G, Denys B, In’t Veld P, Hellemans K, Pipeleers-Marichal M, Ling Z, Pipeleers D. Contribution of postnatally formed small beta cell aggregates to functional beta cell mass in adult rat pancreas. Diabetologia. 2010;53:2380–2388. doi: 10.1007/s00125-010-1851-4. [DOI] [PubMed] [Google Scholar]
- Cinti F, Bouchi R, Kim-Muller JY, Ohmura Y, Sandoval PR, Masini M, Marselli L, Suleiman M, Ratner LE, Marchetti P, et al. Evidence of beta-Cell Dedifferentiation in Human Type 2 Diabetes. The Journal of clinical endocrinology and metabolism. 2016;101:1044–1054. doi: 10.1210/jc.2015-2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat P, Xu X, Ravassard P, Sosa-Pineda B, Dussaud S, Billestrup N, Madsen OD, Serup P, Heimberg H, Mansouri A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell. 2009;138:449–462. doi: 10.1016/j.cell.2009.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney M, Gjernes E, Druelle N, Ravaud C, Vieira A, Ben-Othman N, Pfeifer A, Avolio F, Leuckx G, Lacas-Gervais S, et al. The inactivation of Arx in pancreatic alpha-cells triggers their neogenesis and conversion into functional beta-like cells. PLoS Genet. 2013;9:e1003934. doi: 10.1371/journal.pgen.1003934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscimanna A, Speicher JA, Houshmand G, Shiota C, Prasadan K, Ji B, Logsdon CD, Gittes GK, Esni F. Duct cells contribute to regeneration of endocrine and acinar cells following pancreatic damage in adult mice. Gastroenterology. 2011;141:1451–1462. doi: 10.1053/j.gastro.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaspre F, Beer RL, Rovira M, Huang W, Wang G, Gee S, Vitery Mdel C, Wheelan SJ, Parsons MJ. Centroacinar Cells Are Progenitors That Contribute to Endocrine Pancreas Regeneration. Diabetes. 2015;64:3499–3509. doi: 10.2337/db15-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirice E, Walpita D, Vetere A, Meier BC, Kahraman S, Hu J, Dancik V, Burns SM, Gilbert TJ, Olson DE, et al. Inhibition of DYRK1A Stimulates Human beta-Cell Proliferation. Diabetes. 2016;65:1660–1671. doi: 10.2337/db15-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- Dorrell C, Schug J, Canaday PS, Russ HA, Tarlow BD, Grompe MT, Horton T, Hebrok M, Streeter PR, Kaestner KH, et al. Human islets contain four distinct subtypes of beta cells. Nature communications. 2016;7:11756. doi: 10.1038/ncomms11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edstrom C. Alloxan sensitivity of rats at various intervals following ligation of the pancreatic ducts. Acta Soc Med Ups. 1971a;76:77–86. [PubMed] [Google Scholar]
- Edstrom C. Further quantitative structural studies of the pancreatic islet parenchyma in rats with duct ligation. Acta Soc Med Ups. 1971b;76:127–138. [PubMed] [Google Scholar]
- El Ouaamari A, Dirice E, Gedeon N, Hu J, Zhou JY, Shirakawa J, Hou L, Goodman J, Karampelias C, Qiang G, et al. SerpinB1 Promotes Pancreatic beta Cell Proliferation. Cell metabolism. 2016;23:194–205. doi: 10.1016/j.cmet.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiaschi-Taesch NM, Kleinberger JW, Salim FG, Troxell R, Wills R, Tanwir M, Casinelli G, Cox AE, Takane KK, Srinivas H, et al. Cytoplasmic-nuclear trafficking of G1/S cell cycle molecules and adult human beta-cell replication: a revised model of human beta-cell G1/S control. Diabetes. 2013;62:2460–2470. doi: 10.2337/db12-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finegood DT, Scaglia L, Bonner-Weir S. Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes. 1995;44:249–256. doi: 10.2337/diab.44.3.249. [DOI] [PubMed] [Google Scholar]
- Gao R, Ustinov J, Korsgren O, Otonkoski T. In vitro neogenesis of human islets reflects the plasticity of differentiated human pancreatic cells. Diabetologia. 2005;48:2296–2304. doi: 10.1007/s00125-005-1935-8. [DOI] [PubMed] [Google Scholar]
- Georgia S, Hinault C, Kawamori D, Hu J, Meyer J, Kanji M, Bhushan A, Kulkarni RN. Cyclin D2 is essential for the compensatory beta-cell hyperplastic response to insulin resistance in rodents. Diabetes. 2010;59:987–996. doi: 10.2337/db09-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez DL, O’Driscoll M, Sheets TP, Hruban RH, Oberholzer J, McGarrigle JJ, Shamblott MJ. Neurogenin 3 Expressing Cells in the Human Exocrine Pancreas Have the Capacity for Endocrine Cell Fate. PLoS One. 2015;10:e0133862. doi: 10.1371/journal.pone.0133862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg BE, Moore PC, Demozay D, Hall BA, Li M, Husain A, Wright AJ, Atkinson MA, Rhodes CJ. Formation of a human beta-cell population within pancreatic islets is set early in life. The Journal of clinical endocrinology and metabolism. 2012;97:3197–3206. doi: 10.1210/jc.2012-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu D, Sarvetnick N. Epithelial cell proliferation and islet neogenesis in IFN-g transgenic mice. Development. 1993;118:33–46. doi: 10.1242/dev.118.1.33. [DOI] [PubMed] [Google Scholar]
- Hanley SC, Austin E, Assouline-Thomas B, Kapeluto J, Blaichman J, Moosavi M, Petropavlovskaia M, Rosenberg L. {beta}-Cell mass dynamics and islet cell plasticity in human type 2 diabetes. Endocrinology. 2010;151:1462–1472. doi: 10.1210/en.2009-1277. [DOI] [PubMed] [Google Scholar]
- Hering BJ, Clarke WR, Bridges ND, Eggerman TL, Alejandro R, Bellin MD, Chaloner K, Czarniecki CW, Goldstein JS, Hunsicker LG, et al. Phase 3 Trial of Transplantation of Human Islets in Type 1 Diabetes Complicated by Severe Hypoglycemia. Diabetes Care. 2016;39:1230–1240. doi: 10.2337/dc15-1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera PL, Huarte J, Zufferey R, Nichols A, Mermillod B, Philippe J, Muniesa P, Sanvito F, Orci L, Vassalli JD. Ablation of islet endocrine cells by targeted expression of hormone-promoter-driven toxigenes. Proc Natl Acad Sci USA. 1994;91:12999–13003. doi: 10.1073/pnas.91.26.12999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houbracken I, de Waele E, Lardon J, Ling Z, Heimberg H, Rooman I, Bouwens L. Lineage tracing evidence for transdifferentiation of acinar to duct cells and plasticity of human pancreas. Gastroenterology. 2011;141:731–741. doi: 10.1053/j.gastro.2011.04.050. [DOI] [PubMed] [Google Scholar]
- Hultquist GT, Karlsson U, Hallner AC. The regenerative capacity of the pancreas in duct-ligated rats. Exp Pathol (Jena) 1979;17:44–52. doi: 10.1016/s0014-4908(79)80009-5. [DOI] [PubMed] [Google Scholar]
- Immervoll H, Hoem D, Steffensen OJ, Miletic H, Molven A. Visualization of CD44 and CD133 in normal pancreas and pancreatic ductal adenocarcinomas: non-overlapping membrane expression in cell populations positive for both markers. J Histochem Cytochem. 2011;59:441–455. doi: 10.1369/0022155411398275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada A, Nienaber C, Katsuta H, Fujitani Y, Levine J, Morita R, Sharma A, Bonner-Weir S. Carbonic anhydrase II-positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc Natl Acad Sci USA. 2008;105:19915–19919. doi: 10.1073/pnas.0805803105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Gao D, Feng T, Tremblay JR, Ghazalli N, Luo A, Rawson J, Quijano JC, Chai J, Wedeken L, et al. Cells with surface expression of CD133highCD71low are enriched for tripotent colony–forming progenitor cells in the adult murine pancreas. Stem Cell Res. 2016;16:40–53. doi: 10.1016/j.scr.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas JC, Sharma A, Hasenkamp W, Ilkova H, Patane G, Laybutt R, Bonner-Weir S, Weir GC. Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. The Journal of biological chemistry. 1999;274:14112–14121. doi: 10.1074/jbc.274.20.14112. [DOI] [PubMed] [Google Scholar]
- Karnik SK, Chen H, McLean GW, Heit JJ, Gu X, Zhang AY, Fontaine M, Yen MH, Kim SK. Menin controls growth of pancreatic beta-cells in pregnant mice and promotes gestational diabetes mellitus. Science (New York, NY) 2007;318:806–809. doi: 10.1126/science.1146812. [DOI] [PubMed] [Google Scholar]
- Kim H, Toyofuku Y, Lynn FC, Chak E, Uchida T, Mizukami H, Fujitani Y, Kawamori R, Miyatsuka T, Kosaka Y, et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med. 2010;16:804–808. doi: 10.1038/nm.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein D, Alvarez-Cubela S, Lanzoni G, Vargas N, Prabakar KR, Boulina M, Ricordi C, Inverardi L, Pastori RL, Dominguez-Bendala J. BMP-7 Induces Adult Human Pancreatic Exocrine-to-Endocrine Conversion. Diabetes. 2015;64:4123–4134. doi: 10.2337/db15-0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloppel G, Lohr M, Habich K, Oberholzer M, Heitz PU. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res. 1985;4:110–125. doi: 10.1159/000156969. [DOI] [PubMed] [Google Scholar]
- Kopp JL, Dubois CL, Schaffer AE, Hao E, Shih HP, Seymour PA, Ma J, Sander M. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development. 2011;138:653–665. doi: 10.1242/dev.056499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korytnikov R, Nostro MC. Generation of polyhormonal and multipotent pancreatic progenitor lineages from human pluripotent stem cells. Methods (San Diego, Calif) 2016;101:56–64. doi: 10.1016/j.ymeth.2015.10.017. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- Lardon J, Huyens N, Rooman I, Bouwens L. Exocrine cell transdifferentiation in dexamethasone-treated rat pancreas. Virchows Arch. 2004;444:61–65. doi: 10.1007/s00428-003-0930-z. [DOI] [PubMed] [Google Scholar]
- Laybutt DR, Glandt M, Xu G, Ahn YB, Trivedi N, Bonner-Weir S, Weir GC. Critical reduction in beta-cell mass results in two distinct outcomes over time. Adaptation with impaired glucose tolerance or decompensated diabetes. The Journal of biological chemistry. 2003;278:2997–3005. doi: 10.1074/jbc.M210581200. [DOI] [PubMed] [Google Scholar]
- Laybutt DR, Sharma A, Sgroi DC, Gaudet J, Bonner-Weir S, Weir GC. Genetic regulation of metabolic pathways in beta-cells disrupted by hyperglycemia. The Journal of biological chemistry. 2002;277:10912–10921. doi: 10.1074/jbc.M111751200. [DOI] [PubMed] [Google Scholar]
- Lee CS, De Leon DD, Kaestner KH, Stoffers DA. Regeneration of pancreatic islets after partial pancreatectomy in mice does not involve the reactivation of neurogenin-3. Diabetes. 2006;55:269–272. [PubMed] [Google Scholar]
- Lee J, Sugiyama T, Liu Y, Wang J, Gu X, Lei J, Markmann JF, Miyazaki S, Miyazaki J, Szot GL, et al. Expansion and conversion of human pancreatic ductal cells into insulin-secreting endocrine cells. Elife. 2013;2:e00940. doi: 10.7554/eLife.00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemper M, Leuckx G, Heremans Y, German MS, Heimberg H, Bouwens L, Baeyens L. Reprogramming of human pancreatic exocrine cells to beta-like cells. Cell Death Differ. 2015;22:1117–1130. doi: 10.1038/cdd.2014.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Casteels T, Frogne T, Ingvorsen C, Honore C, Courtney M, Huber KV, Schmitner N, Kimmel RA, Romanov RA, et al. Artemisinins Target GABAA Receptor Signaling and Impair alpha Cell Identity. Cell. 2017;168:86–100 e115. doi: 10.1016/j.cell.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WC, Rukstalis JM, Nishimura W, Tchipashvili V, Habener JF, Sharma A, Bonner-Weir S. Activation of pancreatic-duct-derived progenitor cells during pancreas regeneration in adult rats. J Cell Sci. 2010;123:2792–2802. doi: 10.1242/jcs.065268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Md Moin AS, Dhawan S, Cory M, Butler PC, Rizza RA, Butler AE. Increased Frequency of Hormone Negative and Polyhormonal Endocrine Cells in Lean Individuals With Type 2 Diabetes. The Journal of clinical endocrinology and metabolism. 2016;101:3628–3636. doi: 10.1210/jc.2016-2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, Butler PC. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57:1584–1594. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezza T, Muscogiuri G, Sorice GP, Clemente G, Hu J, Pontecorvi A, Holst JJ, Giaccari A, Kulkarni RN. Insulin resistance alters islet morphology in nondiabetic humans. Diabetes. 2014;63:994–1007. doi: 10.2337/db13-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki S, Tashiro F, Miyazaki J. Transgenic Expression of a Single Transcription Factor Pdx1 Induces Transdifferentiation of Pancreatic Acinar Cells to Endocrine Cells in Adult Mice. PLoS One. 2016;11:e0161190. doi: 10.1371/journal.pone.0161190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montanya E, Nacher V, Biarnes M, Soler J. Linear correlation between beta-cell mass and body weight throughout the lifespan in Lewis rats: role of beta-cell hyperplasia and hypertrophy. Diabetes. 2000;49:1341–1346. doi: 10.2337/diabetes.49.8.1341. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Minami K, Tamura K, Iemoto K, Miki T, Seino S. Pancreatic beta-cells are generated by neogenesis from non-beta-cells after birth. Biomed Res. 2011;32:167–174. doi: 10.2220/biomedres.32.167. [DOI] [PubMed] [Google Scholar]
- Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by beta cell regeneration. J Clin Invest. 2007;117:2553–2561. doi: 10.1172/JCI32959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan ES, Vegas A, Anderson DG, Weir GC. Islets transplanted in immunoisolation devices: a review of the progress and the challenges that remain. Endocrine reviews. 2011;32:827–844. doi: 10.1210/er.2010-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, Melton DA. Generation of functional human pancreatic beta cells in vitro. Cell. 2014;159:428–439. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AK, Tchkonia T, LeBrasseur NK, Chini EN, Xu M, Kirkland JL. Cellular Senescence in Type 2 Diabetes: A Therapeutic Opportunity. Diabetes. 2015;64:2289–2298. doi: 10.2337/db14-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan FC, Bankaitis ED, Boyer D, Xu X, Van de Casteele M, Magnuson MA, Heimberg H, Wright CV. Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development. 2013;140:751–764. doi: 10.1242/dev.090159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JA, Brelje TC, Sorenson RL. Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology. 1992;130:1459–1466. doi: 10.1210/endo.130.3.1537300. [DOI] [PubMed] [Google Scholar]
- Peshavaria M, Larmie BL, Lausier J, Satish B, Habibovic A, Roskens V, Larock K, Everill B, Leahy JL, Jetton TL. Regulation of pancreatic beta-cell regeneration in the normoglycemic 60% partial-pancreatectomy mouse. Diabetes. 2006;55:3289–3298. doi: 10.2337/db06-0017. [DOI] [PubMed] [Google Scholar]
- Pfeifer A, Courtney M, Ben-Othman N, Al-Hasani K, Gjernes E, Vieira A, Druelle N, Avolio F, Faurite B, Mansouri A, et al. Induction of multiple cycles of pancreatic beta-cell replacement. Cell cycle (Georgetown, Tex) 2013;12:3243–3244. doi: 10.4161/cc.26357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porat S, Weinberg-Corem N, Tornovsky-Babaey S, Schyr-Ben-Haroush R, Hija A, Stolovich-Rain M, Dadon D, Granot Z, Ben-Hur V, White P, et al. Control of pancreatic beta cell regeneration by glucose metabolism. Cell metabolism. 2011;13:440–449. doi: 10.1016/j.cmet.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes, obesity & metabolism. 2008;10(Suppl 4):32–42. doi: 10.1111/j.1463-1326.2008.00969.x. [DOI] [PubMed] [Google Scholar]
- Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O’Dwyer S, Quiskamp N, Mojibian M, Albrecht T, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol. 2014;32:1121–1133. doi: 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- Rhee M, Lee SH, Kim JW, Ham DS, Park HS, Yang HK, Shin JY, Cho JH, Kim YB, Youn BS, et al. Preadipocyte factor 1 induces pancreatic ductal cell differentiation into insulin-producing cells. Sci Rep. 2016;6:23960. doi: 10.1038/srep23960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieck S, Zhang J, Li Z, Liu C, Naji A, Takane KK, Fiaschi-Taesch NM, Stewart AF, Kushner JA, Kaestner KH. Overexpression of hepatocyte nuclear factor-4alpha initiates cell cycle entry, but is not sufficient to promote beta-cell expansion in human islets. Mol Endocrinol. 2012;26:1590–1602. doi: 10.1210/me.2012-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille K, Rourke JL, McBane JE, Fu A, Baird S, Du Q, Kin T, Shapiro AM, Screaton RA. High-throughput Functional Genomics Identifies Regulators of Primary Human Beta Cell Proliferation. The Journal of biological chemistry. 2016;291:4614–4625. doi: 10.1074/jbc.M115.683912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nature cell biology. 2009;11:973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooman I, Heremans Y, Heimberg H, Bouwens L. Modulation of rat pancreatic acinoductal transdifferentiation and expression of PDX-1 in vitro. Diabetologia. 2000;43:907–914. doi: 10.1007/s001250051468. [DOI] [PubMed] [Google Scholar]
- Rooman I, Lardon J, Bouwens L. Gastrin stimulates beta-cell neogenesis and increases islet mass from transdifferentiated but not from normal exocrine pancreas tissue. Diabetes. 2002;51:686–690. doi: 10.2337/diabetes.51.3.686. [DOI] [PubMed] [Google Scholar]
- Rovira M, Scott SG, Liss AS, Jensen J, Thayer SP, Leach SD. Isolation and characterization of centroacinar/terminal ductal progenitor cells in adult mouse pancreas. Proc Natl Acad Sci USA. 2010;107:75–80. doi: 10.1073/pnas.0912589107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ HA, Parent AV, Ringler JJ, Hennings TG, Nair GG, Shveygert M, Guo T, Puri S, Haataja L, Cirulli V, et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. The EMBO journal. 2015;34:1759–1772. doi: 10.15252/embj.201591058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan EA, Paty BW, Senior PA, Bigam D, Alfadhli E, Kneteman NM, Lakey JR, Shapiro AM. Five-year follow-up after clinical islet transplantation. Diabetes. 2005;54:2060–2069. doi: 10.2337/diabetes.54.7.2060. [DOI] [PubMed] [Google Scholar]
- Saito M, Iwawaki T, Taya C, Yonekawa H, Noda M, Inui Y, Mekada E, Kimata Y, Tsuru A, Kohno K. Diphtheria toxin receptor-mediated conditional and targeted cell ablation in transgenic mice. Nat Biotechnol. 2001;19:746–750. doi: 10.1038/90795. [DOI] [PubMed] [Google Scholar]
- Salpeter SJ, Khalaileh A, Weinberg-Corem N, Ziv O, Glaser B, Dor Y. Systemic regulation of the age-related decline of pancreatic beta-cell replication. Diabetes. 2013;62:2843–2848. doi: 10.2337/db13-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salpeter SJ, Klein AM, Huangfu D, Grimsby J, Dor Y. Glucose and aging control the quiescence period that follows pancreatic beta cell replication. Development. 2010;137:3205–3213. doi: 10.1242/dev.054304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salpeter SJ, Klochendler A, Weinberg-Corem N, Porat S, Granot Z, Shapiro AM, Magnuson MA, Eden A, Grimsby J, Glaser B, et al. Glucose regulates cyclin D2 expression in quiescent and replicating pancreatic beta-cells through glycolysis and calcium channels. Endocrinology. 2011;152:2589–2598. doi: 10.1210/en.2010-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho R, Gruber R, Gu G, Behrens A. Loss of Fbw7 reprograms adult pancreatic ductal cells into alpha, delta, and beta cells. Cell Stem Cell. 2014;15:139–153. doi: 10.1016/j.stem.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaglia L, Cahill CJ, Finegood DT, Bonner-Weir S. Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology. 1997;138:1736–1741. doi: 10.1210/endo.138.4.5069. [DOI] [PubMed] [Google Scholar]
- Schraenen A, Lemaire K, de Faudeur G, Hendrickx N, Granvik M, Van Lommel L, Mallet J, Vodjdani G, Gilon P, Binart N, et al. Placental lactogens induce serotonin biosynthesis in a subset of mouse beta cells during pregnancy. Diabetologia. 2010;53:2589–2599. doi: 10.1007/s00125-010-1913-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segerstolpe A, Palasantza A, Eliasson P, Andersson EM, Andreasson AC, Sun X, Picelli S, Sabirsh A, Clausen M, Bjursell MK, et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell metabolism. 2016;24:593–607. doi: 10.1016/j.cmet.2016.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro AM, Lakey JR, Ryan EA, Korbutt GS, Toth E, Warnock GL, Kneteman NM, Rajotte RV. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med. 2000;343:230–238. doi: 10.1056/NEJM200007273430401. [DOI] [PubMed] [Google Scholar]
- Sharma A, Zangen DH, Reitz P, Taneja M, Lissauer ME, Miller CP, Weir GC, Habener JF, Bonner-Weir S. The homeodomain protein IDX-1 increases after an early burst of proliferation during pancreatic regeneration. Diabetes. 1999;48:507–513. doi: 10.2337/diabetes.48.3.507. [DOI] [PubMed] [Google Scholar]
- Shen CN, Horb ME, Slack JM, Tosh D. Transdifferentiation of pancreas to liver. Mech Dev. 2003;120:107–116. doi: 10.1016/s0925-4773(02)00337-4. [DOI] [PubMed] [Google Scholar]
- Shen W, Taylor B, Jin Q, Nguyen-Tran V, Meeusen S, Zhang YQ, Kamireddy A, Swafford A, Powers AF, Walker J, et al. Inhibition of DYRK1A and GSK3B induces human beta-cell proliferation. Nature communications. 2015;6:8372. doi: 10.1038/ncomms9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solar M, Cardalda C, Houbracken I, Martin M, Maestro MA, De Medts N, Xu X, Grau V, Heimberg H, Bouwens L, et al. Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev Cell. 2009;17:849–860. doi: 10.1016/j.devcel.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Soltani N, Qiu H, Aleksic M, Glinka Y, Zhao F, Liu R, Li Y, Zhang N, Chakrabarti R, Ng T, et al. GABA exerts protective and regenerative effects on islet beta cells and reverses diabetes. Proc Natl Acad Sci USA. 2011;108:11692–11697. doi: 10.1073/pnas.1102715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spijker HS, Song H, Ellenbroek JH, Roefs MM, Engelse MA, Bos E, Koster AJ, Rabelink TJ, Hansen BC, Clark A, et al. Loss of beta-Cell Identity Occurs in Type 2 Diabetes and Is Associated With Islet Amyloid Deposits. Diabetes. 2015;64:2928–2938. doi: 10.2337/db14-1752. [DOI] [PubMed] [Google Scholar]
- Stolovich-Rain M, Hija A, Grimsby J, Glaser B, Dor Y. Pancreatic beta cells in very old mice retain capacity for compensatory proliferation. J Biol Chem. 2012;287:27407–27414. doi: 10.1074/jbc.M112.350736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Pinzon WL, Lakey JR, Brand SJ, Rabinovitch A. Combination therapy with epidermal growth factor and gastrin induces neogenesis of human islet b-cells from pancreatic duct cells and an increase in functional b-cell mass. The Journal of clinical endocrinology and metabolism. 2005:3401–3409. doi: 10.1210/jc.2004-0761. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Rodriguez RT, McLean GW, Kim SK. Conserved markers of fetal pancreatic epithelium permit prospective isolation of islet progenitor cells by FACS. Proc Natl Acad Sci U S A. 2007a;104:175–180. doi: 10.1073/pnas.0609490104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Rodriguez RT, McLean GW, Kim SK. Conserved markers of fetal pancreatic epithelium permit prospective isolation of islet progenitor cells by FACS. Proc Natl Acad Sci USA. 2007b;104:175–180. doi: 10.1073/pnas.0609490104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan BA, Hollister-Lock J, Bonner-Weir S, Weir GC. Reduced Ki67 Staining in the Postmortem State Calls Into Question Past Conclusions About the Lack of Turnover of Adult Human beta-Cells. Diabetes. 2015;64:1698–1702. doi: 10.2337/db14-1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell. 2012;150:1223–1234. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellez N, Montanya E. Gastrin induces ductal cell dedifferentiation and beta-cell neogenesis after 90% pancreatectomy. J Endocrinol. 2014;223:67–78. doi: 10.1530/JOE-14-0222. [DOI] [PubMed] [Google Scholar]
- Tellez N, Vilaseca M, Marti Y, Pla A, Montanya E. beta-Cell dedifferentiation, reduced duct cell plasticity, and impaired beta-cell mass regeneration in middle-aged rats. Am J Physiol Endocrinol Metab. 2016;311:E554–563. doi: 10.1152/ajpendo.00502.2015. [DOI] [PubMed] [Google Scholar]
- Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell. 2007;12:817–826. doi: 10.1016/j.devcel.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Thorel F, Nepote V, Avril I, Kohno K, Desgraz R, Chera S, Herrera PL. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature. 2010;464:1149–1154. doi: 10.1038/nature08894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornovsky-Babeay S, Dadon D, Ziv O, Tzipilevich E, Kadosh T, Schyr-Ben Haroush R, Hija A, Stolovich-Rain M, Furth-Lavi J, Granot Z, et al. Type 2 diabetes and congenital hyperinsulinism cause DNA double-strand breaks and p53 activity in beta cells. Cell metabolism. 2014;19:109–121. doi: 10.1016/j.cmet.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Toselli C, Hyslop CM, Hughes M, Natale DR, Santamaria P, Huang CT. Contribution of a non-beta-cell source to beta-cell mass during pregnancy. PLoS One. 2014;9:e100398. doi: 10.1371/journal.pone.0100398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrberg B, Ustinov J, Otonkoski T, Andersson A. Stimulated endocrine cell proliferation and differentiation in transplanted human pancreatic islets: effects of the ob gene and compensatory growth of the implantation organ. Diabetes. 2001;50:301–307. doi: 10.2337/diabetes.50.2.301. [DOI] [PubMed] [Google Scholar]
- Valdez IA, Dirice E, Gupta MK, Shirakawa J, Teo AK, Kulkarni RN. Proinflammatory Cytokines Induce Endocrine Differentiation in Pancreatic Ductal Cells via STAT3-Dependent NGN3 Activation. Cell Rep. 2016;15:460–470. doi: 10.1016/j.celrep.2016.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Casteele M, Leuckx G, Cai Y, Yuchi Y, Coppens V, De Groef S, Van Gassen N, Baeyens L, Heremans Y, Wright CV, et al. Partial duct ligation: beta-cell proliferation and beyond. Diabetes. 2014;63:2567–2577. doi: 10.2337/db13-0831. [DOI] [PubMed] [Google Scholar]
- van der Meulen T, Mawla AM, DiGruccio MR, Adams MW, Nies V, Dolleman S, Liu S, Ackermann AM, Caceres E, Hunter AE, et al. Virgin Beta Cells Persist throughout Life at a Neogenic Niche within Pancreatic Islets. Cell metabolism. 2017;25:911–926 e916. doi: 10.1016/j.cmet.2017.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Racine JJ, Song X, Li X, Nair I, Liu H, Avakian-Mansoorian A, Johnston HF, Liu C, Shen C, et al. Mixed chimerism and growth factors augment beta cell regeneration and reverse late-stage type 1 diabetes. Sci Transl Med. 2012;4:133ra159. doi: 10.1126/scitranslmed.3003835. [DOI] [PubMed] [Google Scholar]
- Wang P, Alvarez-Perez JC, Felsenfeld DP, Liu H, Sivendran S, Bender A, Kumar A, Sanchez R, Scott DK, Garcia-Ocana A, et al. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat Med. 2015;21:383–388. doi: 10.1038/nm.3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RN, Kloppel G, Bouwens L. Duct- to islet-cell differentiation and islet growth in the pancreas of duct-ligated adult rats. Diabetologia. 1995;38:1405–1411. doi: 10.1007/BF00400600. [DOI] [PubMed] [Google Scholar]
- Wang TC, Bonner-Weir S, Oates PS, Chulak M, Simon B, Merlino GT, Schmidt EV, Brand SJ. Pancreatic gastrin stimulates islet differentiation of transforming growth factor alpha-induced ductular precursor cells. J Clin Invest. 1993;92:1349–1356. doi: 10.1172/JCI116708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Golson ML, Schug J, Traum D, Liu C, Vivek K, Dorrell C, Naji A, Powers AC, Chang KM, et al. Single-Cell Mass Cytometry Analysis of the Human Endocrine Pancreas. Cell metabolism. 2016;24:616–626. doi: 10.1016/j.cmet.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, York Nathaniel W, Nichols Colin G, Remedi Maria S. Pancreatic beta Cell Dedifferentiation in Diabetes and Redifferentiation following Insulin Therapy. Cell metabolism. 2014;19:872–882. doi: 10.1016/j.cmet.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir GC. Islet encapsulation: advances and obstacles. Diabetologia. 2013;56:1458–1461. doi: 10.1007/s00125-013-2921-1. [DOI] [PubMed] [Google Scholar]
- Weir GC, Aguayo-Mazzucato C, Bonner-Weir S. Beta-cell dedifferentiation in diabetes is important, but what is it? Islets. 2013;5:233–237. doi: 10.4161/isl.27494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir GC, Bonner-Weir S, Leahy JL. Islet mass and function in diabetes and transplantation. Diabetes. 1990;39:401–405. doi: 10.2337/diab.39.4.401. [DOI] [PubMed] [Google Scholar]
- Westphalen CB, Takemoto Y, Tanaka T, Macchini M, Jiang Z, Renz BW, Chen X, Ormanns S, Nagar K, Tailor Y, et al. Dclk1 Defines Quiescent Pancreatic Progenitors that Promote Injury-Induced Regeneration and Tumorigenesis. Cell Stem Cell. 2016;18:441–455. doi: 10.1016/j.stem.2016.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Guo L, Jakubowski A, Su L, Li WC, Bonner-Weir S, Burkly LC. TNF-like weak inducer of apoptosis (TWEAK) promotes beta cell neogenesis from pancreatic ductal epithelium in adult mice. PLoS One. 2013;8:e72132. doi: 10.1371/journal.pone.0072132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Y, Kim J, Okamoto H, Ni M, Wei Y, Adler C, Murphy AJ, Yancopoulos GD, Lin C, Gromada J. RNA Sequencing of Single Human Islet Cells Reveals Type 2 Diabetes Genes. Cell metabolism. 2016;24:608–615. doi: 10.1016/j.cmet.2016.08.018. [DOI] [PubMed] [Google Scholar]
- Xu X, D’Hoker J, Stange G, Bonne S, De Leu N, Xiao X, Van De Casteele M, Mellitzer G, Ling Z, Pipeleers D, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132:197–207. doi: 10.1016/j.cell.2007.12.015. [DOI] [PubMed] [Google Scholar]
- Yatoh S, Dodge R, Akashi T, Omer A, Sharma A, Weir GC, Bonner-Weir S. Differentiation of affinity-purified human pancreatic duct cells to beta-cells. Diabetes. 2007;56:1802–1809. doi: 10.2337/db06-1670. [DOI] [PubMed] [Google Scholar]
- Yoneda S, Uno S, Iwahashi H, Fujita Y, Yoshikawa A, Kozawa J, Okita K, Takiuchi D, Eguchi H, Nagano H, et al. Predominance of beta-cell neogenesis rather than replication in humans with an impaired glucose tolerance and newly diagnosed diabetes. The Journal of clinical endocrinology and metabolism. 2013;98:2053–2061. doi: 10.1210/jc.2012-3832. [DOI] [PubMed] [Google Scholar]
- Yoon KH, Ko SH, Cho JH, Lee JM, Ahn YB, Song KH, Yoo SJ, Kang MI, Cha BY, Lee KW, et al. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. The Journal of clinical endocrinology and metabolism. 2003;88:2300–2308. doi: 10.1210/jc.2002-020735. [DOI] [PubMed] [Google Scholar]
- Zhang H, Ackermann AM, Gusarova GA, Lowe D, Feng X, Kopsombut UG, Costa RH, Gannon M. The FoxM1 transcription factor is required to maintain pancreatic beta-cell mass. Mol Endocrinol. 2006;20:1853–1866. doi: 10.1210/me.2006-0056. [DOI] [PubMed] [Google Scholar]
- Zhang M, Lin Q, Qi T, Wang T, Chen CC, Riggs AD, Zeng D. Growth factors and medium hyperglycemia induce Sox9+ ductal cell differentiation into beta cells in mice with reversal of diabetes. Proc Natl Acad Sci USA. 2016;113:650–655. doi: 10.1073/pnas.1524200113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JX, Dhawan S, Fu H, Snyder E, Bottino R, Kundu S, Kim SK, Bhushan A. Combined modulation of polycomb and trithorax genes rejuvenates beta cell replication. J Clin Invest. 2013;123:4849–4858. doi: 10.1172/JCI69468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]