

Summary
Caloric restriction rapidly reverses type 2 diabetes (T2D) but the mechanism(s) of this reversal are poorly understood. Here we show that three days of a very low-calorie diet (VLCD, 1/4 their typical intake) lowered plasma glucose and insulin concentrations in a rat model of T2D without altering body weight. The lower plasma glucose was associated with a 30% reduction in hepatic glucose production resulting from suppression of both gluconeogenesis from pyruvate carboxylase (VPC), explained by a reduction in hepatic acetyl-CoA content, and net hepatic glycogenolysis. In addition, VLCD resulted in reductions in hepatic triglyceride and diacylglycerol content and PKCε translocation, associated with improved hepatic insulin sensitivity. Taken together these data show that there are pleotropic mechanisms by which VLCD reverses hyperglycemia in a rat model of T2D, including reduced DAG-PKCε-induced hepatic insulin resistance, reduced hepatic glycogenolysis, and reduced hepatic acetyl-CoA content, PC flux and gluconeogenesis.
eToc Blurb
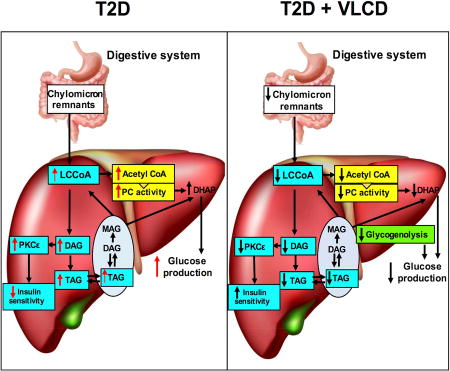
XXX show that short-term (3 days) low calorie diet improves glucose metabolism before weight loss in a rat model of T2 diabetes and trace the beneficial metabolic effects to improved liver metabolism due to reductions in hepatic glycogenolysis, acetyl-CoA driven pyruvate carboxylase flux, and TAG-DAG-PKC mediated insulin resistance.
Introduction
With type 2 diabetes (T2D) projected to affect one-third of Americans by the year 2050 (Boyle et al., 2010), interventions to reverse diabetes are of particular clinical interest. Recent meta-analyses report remission of T2D with discontinuation of all diabetes medications in 70–80% of cases soon after bariatric surgery (Baskota et al., 2015; Buchwald et al., 2009). Of note, T2D often resolves prior to significant weight loss (Buchwald et al., 2004; Buchwald et al., 2009; Clements et al., 2004; Schauer et al., 2003), calling into question the mechanism by which bariatric surgery rapidly reduces hyperglycemia. In this regard short-term consumption of a very low calorie diet has been shown to be as effective as bariatric surgery in reversing T2D in humans (Campos et al., 2010; Henry et al., 1985; Isbell et al., 2010; Jackness et al., 2013; Laferrere et al., 2008; Lim et al., 2011; Plum et al., 2011), suggesting that part or all of the metabolic improvements of bariatric surgery may have little to do with the surgery itself, but rather with the forced reduction in caloric intake caused by bariatric procedures. The key question, then, becomes “How does short-term VLCD reverse T2D prior to clinically significant weight loss?”
Given the key roles of ectopic lipid and increased rates of hepatic glucose production in promoting hepatic insulin resistance and fasting hyperglycemia in T2D (Perry et al., 2014; Samuel and Shulman, 2016), we undertook a comprehensive study to examine the impact of short-term VLCD on rates of hepatic glucose production and hepatic triacylglycerol (TAG), diacylglycerol (DAG), ceramide content and PKCepsilon (PKCε) activity in a well-established rodent model of type 2 diabetes (Reed et al., 2000) in which rats exhibit hyperglycemia, hyperinsulinemia, obesity, and NAFLD – the hallmarks of T2D in humans. In order to determine which intra-hepatocellular and extra-hepatocellular processes might contribute to VLCD-induced reductions in plasma glucose concentrations, we also examined the impact of VLCD on rates of hepatic gluconeogenesis, white adipose tissue (WAT) lipolysis and the respective contribution of pyruvate carboxylase flux (VPC) and glycerol (Vglycerol) to hepatic gluconeogenesis as well as net rates of hepatic glycogenolysis and rates of hepatic mitochondrial oxidation using a novel Positional Isotopomer NMR Tracer Analysis method (Perry et al., in press).
Results
Similar to patients with type 2 diabetes, plasma glucose and insulin concentrations as well as liver triglyceride concentrations were increased in our T2D rats as compared to non-diabetic, chow fed control rats (122±5 mg/dL, 24±5 µU/mL, and 13.5±1.9 mg/g, respectively). Three days of VLCD lowered plasma glucose concentrations by ~75 mg/dL, and plasma insulin concentrations by ~50%, despite a lack of differences in body weight (Fig. 1A–B; non-diabetic, chow fed controls’ body weight: 328±9 g). VLCD also led to reductions in plasma non-esterified fatty acids and plasma triglyceride concentrations (Fig. 1C–D) as well as marked reductions in hepatic triglyceride and hepatic DAG content but no change in hepatic ceramide content (Fig. 1E–H, Fig. S1A, Table S1). The VLCD-induced reduction in hepatic DAG content was associated with a ~40% reduction in the PKCε membrane/cytosol ratio, consistent with reduced PKCε translocation. In contrast, VLCD had no effect on plasma concentrations of lactate, alanine, glycine, serine, leucine, isoleucine, aspartate+asparagine, phenylalanine, glutamate+glutamine, glucagon, corticosterone, FGF-21, liver enzymes, or on hepatic inflammatory cytokine concentrations or markers of endoplasmic reticulum stress (Fig. S1B–L).
Fig. 1. Caloric restriction ameliorates fasting hyperglycemia and reduces ectopic hepatic lipid accumulation.

(A) Body weight after 3 days of caloric restriction (or ad lib caloric intake in the controls). (B)–(D) Fasting plasma glucose, insulin, and non-esterified fatty acid concentrations. (E)–(F) Plasma and liver triglyceride concentrations. (G) Liver membrane diacylglycerol. (H) PKCε membrane/cytosol ratio. In all panels, data are the mean±S.E.M. of n=6 per group, with comparisons by t-test. *P<0.05, **P<0.01, ***P<0.001. See also Fig. S1 and Table S1.
Next to examine the cause of the lower plasma glucose concentrations exhibited by VLCD fed rats, we applied a novel PINTA method (Perry et al., in press) to evaluate hepatic flux rates that contribute to hepatic glucose production. We found that caloric restriction reduced rates of both net hepatic glycogenolysis, which accounted for ~1/3 of total glucose production in our 8 hr fasted rats, and gluconeogenesis from oxaloacetate (VPC), but did not alter gluconeogenesis from glycerol, which comprised ~10–15% of total glucose turnover in both T2D control and VLCD rats (Fig. 2A, Fig. S2A). There were no differences in pyruvate kinase flux (VPK) relative to the sum of VPC flux and pyruvate dehydrogenase flux (VPDH) in VLCD fed rats, with this ratio minimal (<5%) in both groups (Fig. S2B). In addition, the observed alterations in gluconeogenic flux occurred in the absence of any differences in the protein expression of key gluconeogenic enzymes (PC, cytosolic phosphoenolpyruvate carboxykinase, glucose-6-phosphatase) in liver (Fig. 2B). Given the reductions in hepatic TAG and DAG concentrations observed in VLCD-fed rats, we next hypothesized that hepatic insulin sensitivity would improve with VLCD feeding in T2D rats. Consistent with this hypothesis, we found that insulin-mediated suppression of hepatic glucose production was increased 2.5-fold in VLCD rats, associated with increased Akt2 phosphorylation under clamp conditions in the T2D + VLCD rats but not T2D controls (Fig. 2C–E, Fig. S2C). In contrast, there was no difference in the glucose infusion rate required to maintain euglycemia during the hyperinsulinemic-euglycemic clamp, in the rate of insulin-stimulated whole-body glucose disposal, or in insulin-mediated suppression of plasma NEFA or glycerol concentrations (Fig. S2D–I), demonstrating that the insulin-sensitizing effect of short-term VLCD is confined to the liver.
Fig. 2. Caloric restriction lowers the rate of hepatic glucose production due to contributions from both net hepatic glycogenolysis and pyruvate carboxylase (VPC) flux and improves hepatic insulin sensitivity.

(A) Glucose production from glycogenolysis, glycerol, and oxaloacetate. The asterisk in the T2D + VLCD bar indicates P<0.05 comparing gluconeogenesis from OAA between T2D and T2D + VLCD rats. (B) Gluconeogenic enzyme protein expression, normalized to GAPDH. (C) Endogenous glucose turnover under basal and hyperinsulinemic-euglycemic clamp conditions. Comparisons between basal and clamp were performed using the 2-tailed paired Student’s t-test. (D) Suppression of hepatic glucose production in the clamp. (E) Ratio of phosphorylated Akt2 to total Akt2. In all panels, *P<0.05, **P<0.01, ***P<0.001. Data are the mean±S.E.M. of n=6 per group, with comparisons by the 2-tailed unpaired Student’s t-test unless otherwise specified. See also Fig. S2.
We next hypothesized that the reductions in VPC could be explained by suppression of hepatic acetyl-CoA content, a well-known allosteric activator of pyruvate carboxylase (Perry et al., 2015; Utter and Keech, 1963). Consistent with this hypothesis, VLCD rats exhibited reductions in both hepatic long-chain acyl-CoA and acetyl-CoA, as well as whole-body βOHB turnover, which is an excellent surrogate of hepatic acetyl-CoA concentrations (Perry et al., 2017b); however there was no difference in plasma βOHB concentrations, suggesting that there may be a reduction in βOHB clearance with caloric restriction (Fig. 3A–C, Fig. S3A). In contrast there were no differences in hepatic malonyl-CoA, in WAT lipolysis as assessed by rates of [U-13C16]palmitate and [2H5]glycerol turnover, in mitochondrial oxidation rates (citrate synthase flux, VCS) measured by PINTA, or in liver PGC1α or PPARγ protein expression (Fig. 3D–F, Fig. S3B–C). Consistent with their reduced hepatic lipid content, VLCD rats exhibited 30% lower rates of hepatic triglyceride export (Fig. 3G). Finally, to demonstrate whether the reduction in hepatic lipid content could primarily be attributed directly to their lower caloric intake, we measured [13C] labeled fatty acid, derived from ingested [1-13C]triolein into hepatic DAG by administering a meal (half the rats’ daily caloric intake in a bolus of Ensure containing [1-13C]triolein, or one-quarter that quantity) to T2D and T2D-VLCD rats. Using this approach we found that there was no difference in the two groups’ ability to synthesize fatty acids into DAG when given the same size meal, but that more 13C label from ingested [1-13C]triolein was incorporated into hepatic DAG when rats were fed a large meal as opposed to a small meal (Fig. 3H).
Fig. 3. Caloric restriction lowers hepatic acetyl-CoA content but does not alter substrate oxidation or lipolysis.

(A)–(B) Hepatic long-chain and acetyl-CoA. (C) Whole-body βOHB turnover. (D)–(E) Whole-body palmitate and glycerol turnover. (F) Liver VCS flux. (G) Hepatic triglyceride export. (H) Hepatic membrane C18:1 C18:0 DAG enrichment following a mixed-meal tolerance with [1-13C]triolein. n=7 T2D-small meal, 8 T2D + VLCD-small meal, 7 T2D-large meal, and 7 T2D + VLCD-large meal. **P<0.01, ***P<0.001, referring to the comparison of rats within the same group (control or VLCD) fed a small vs. large meal. In all panels, groups were compared by t-test, and *P<0.05. Unless otherwise specified, the data are the mean±S.E.M. of n=6 per group. See also Fig. S3.
We next sought to demonstrate whether the reductions in VPC flux and in hepatic glycogenolysis measured in caloric restricted rats were directly responsible for these animals’ lower plasma glucose concentrations. To test the role of hepatic acetyl-CoA in driving hepatic glucose production, we infused acetate [60 µmol/(kg-min)] to increase hepatic acetyl-CoA content in VLCD-T2D rats to the concentrations measured in T2D controls (Fig. 4A). This intervention resulted in a ~10 µmol/(kg-min) increase in hepatic glucose production (Fig. 4B), very similar to the decrement in VPC measured between T2D and T2D-VLCD rats (Fig. 2A). Finally we treated a group of T2D rats with a small molecule inhibitor of glycogen phosphorylase (Nagy et al., 2013). The treated rats exhibited a doubling in their liver glycogen content which was associated with a 40% reduction in hepatic glucose production (Fig. 4C–D), consistent with the ~30–40% contribution of net hepatic glycogenolysis to hepatic glucose production which we previously measured in T2D and T2D-VLCD rats (Fig. 2A).
Fig. 4. Increased hepatic acetyl-CoA content and increased rates of net hepatic glycogenolysis contribute to increased rates hepatic glucose production in T2D rats.

(A) Acetyl-CoA content in T2D + VLCD rats (copied from Fig. 3B for comparison) and rats treated with an infusion of acetate. In panels (A) and (C), comparisons were performed using the 2-tailed unpaired Student’s t-test. (B) EGP before and after acetate infusion. Data in panels (B) and (D) were compared by the 2-tailed paired Student’s t-test. (C) Liver glycogen in T2D rats (copied from Fig. S2) and T2D rats treated with a glycogen phosphorylase inhibitor. (D) EGP before and after treatment with the phosphorylase inhibitor. In all panels, *P<0.05, ***P<0.001. Data are the mean±S.E.M. of n=6 per group. See also Fig. S4.
Finally to determine whether the effects of VLCD to ameliorate hyperglycemia in high fat fed T2D rats depends on the diet administered, we fed rats a “fast food” Western diet: a safflower oil-based high fat diet supplemented with 5% sucrose water, resulting in ~51% caloric intake from carbohydrate, ~39% from fat, and ~10% from protein based on their typical ad lib daily food and water intake. Three days of caloric restriction did not change body weight in these rats, but lowered 8 hr fasted plasma glucose and insulin concentrations, liver glycogen, TAG, and DAG concentrations, as well as the membrane/cytosol ratio of PKCε (Fig. S4A–G).
Discussion
Multiple studies have demonstrated that caloric restriction is as effective as bariatric surgery at reversing fasting hyperglycemia in patients with T2D within a few days, independent of significant reductions in body weight (Campos et al., 2010; Henry et al., 1985; Isbell et al., 2010; Jackness et al., 2013; Laferrere et al., 2008; Lim et al., 2011; Plum et al., 2011). These results raise the question of how VLCD rapidly reverses T2D and whether these mechanisms could be harnessed as a potential therapeutic approach for T2D. Our group has previously shown that a hypocaloric (1200 Kcal) diet resulting in an ~10% weight loss over the course of ~2 months was sufficient to reverse fasting hyperglycemia, reduce rates of hepatic glucose production and improve hepatic insulin sensitivity in patients with type 2 diabetes (Petersen et al., 2005). These marked improvements in hepatic glucose metabolism were associated with resolution of NAFLD in these individuals. Previous studies have also shown that lipid-induced hepatic insulin resistance is mediated in part through DAG activation of PKCε resulting in phosphorylation of the insulin receptor tyrosine kinase (IRTK) on threonine 1160 leading to decreased IRTK activity (Petersen et al., 2016; Samuel et al., 2004; Samuel et al., 2007). We therefore hypothesized that reductions in hepatic TAG-DAG-PKCε activation leading to improved hepatic insulin sensitivity may be associated with reduced plasma glucose concentrations in a calorically restricted rat model of T2D. To test this hypothesis, we utilized a well-established rat model of T2D (Reed et al., 2000) and subjected them to three days of a VLCD, feeding them ¼ their typical daily caloric intake. Importantly this intervention did not significantly alter body weight between the T2D control and VLCD animals within the three-day caloric restriction period, allowing us to identify weight-independent effects of caloric restriction on metabolism. Despite unchanged body weight, the VLCD was associated with reductions in ectopic lipid content, with liver TAG, DAG, and PKCε membrane/cytosol ratio all lowered in VLCD rats. Importantly, there was no difference in the incorporation of dietary lipids into DAG between control T2D and T2D-VLCD rats when they were fed the same size meal, but administration of a smaller meal led to a reduction in lipid incorporation into DAG (Fig. 3H), identifying the smaller meal size as the proximal cause of the reductions in ectopic lipid in liver. We found that VLCD lowered plasma glucose concentrations by ~75 mg/dL and plasma insulin concentrations by ~50%, demonstrating a striking effect of only three days of caloric restriction to ameliorate fasting hyperglycemia and fasting hyperinsulinemia, similar to what is observed in T2D patients undergoing caloric restriction. This was associated with improved hepatic insulin sensitivity demonstrated by increased suppression of hepatic glucose production during a hyperinsulinemic-euglycemic clamp, as well as increased insulin stimulation of Akt2 phosphorylation (Fig. 2C–E). However the VLCD-induced reversal of hyperglycemia, hyperinsulinemia and hepatic insulin resistance was independent of any changes in other putative mediators of hepatic insulin resistance, including hepatic ceramide content, hepatic inflammatory cytokine concentrations, or changes in plasma branched-chain amino acids, glucagon, corticosterone, or FGF-21 concentrations.
To identify the mechanism(s) responsible for the VLCD-induced reversal of fasting hyperglycemia in these diabetic rats, we applied a novel PINTA method (Perry et al., in press) to measure the relative contributions of net hepatic glycogenolysis and gluconeogenesis from both oxaloacetate and glycerol to total hepatic glucose production. This PINTA approach allowed us to measure the impact of a VCLD on all three key contributors to hepatic glucose production for the first time in awake rats in vivo, and demonstrated that both rates of net hepatic glycogenolysis and VPC flux were reduced with VLCD. The 40% reduction in rates of hepatic glucose production measured in T2D rats treated with a pharmacologic inhibitor of glycogen phosphorylase (Nagy et al., 2013) confirmed a key role for net hepatic glycogenolysis in driving hepatic glucose production and fasting hyperglycemia in these 8 hr fasted T2D rats.
In addition, we observed a ~25% reduction in rates of hepatic gluconeogenesis from oxaloacetate (VPC flux), which could be attributed to a ~20% suppression of hepatic acetyl-CoA content. This VLCD-induced reduction in VPC flux was associated with a ~20% suppression in rates of βOHB turnover and no changes in rates of hepatic mitochondrial oxidation (VCS), VPK/(VPC+VPDH) flux, or whole-body WAT lipolysis assessed by [U-13C16]palmitate and [2H5]glycerol turnover. Taken together these data suggest that VLCD-induced reductions in hepatic acetyl-CoA content are driven mostly by reductions in intra-hepatic lipolysis and decreased β-oxidation as reflected by decreased hepatic acyl-CoA content and reduced rates of βOHB turnover. To confirm the impact of acetyl-CoA on rates of hepatic glucose production we infused acetate in VLCD rats to increase liver acetyl-CoA content to match hepatic acetyl-CoA concentrations measured in untreated diabetic control animals. This intervention resulted in a ~25% increase in hepatic glucose turnover (Fig. 4B), demonstrating that matching hepatic acetyl-CoA concentrations promotes similar increases in VPC flux as observed in the T2D rats.
In summary these data demonstrate that there are multiple mechanisms by which a VLCD acutely reduces fasting hyperglycemia in a rat model of poorly controlled type 2 diabetes involving: 1) reductions of hepatic DAG content resulting in decreased PKCε activation and improved hepatic insulin sensitivity, 2) reductions in rates of net hepatic glycogenolysis, and 3) reductions in hepatic acetyl-CoA content leading to decreased hepatic gluconeogenesis due to decreased hepatic pyruvate carboxylase (VPC) flux. Furthermore VLCD-induced reversal of fasting hyperglycemia in these diabetic rodents occurred independently of any changes in hepatic ceramide content, hepatic inflammatory cytokine concentrations, or changes in plasma branched-chain amino acids, glucagon, corticosterone, or FGF-21 concentrations. Taken together these data provide new insights into the mechanisms by which a VLCD leads to reversal of fasting hyperglycemia and identifies key pathways that could confer therapeutic benefit to ameliorate fasting hyperglycemia in individuals with poorly-controlled T2D.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Dr. Gerald I. Shulman, gerald.shulman@yale.edu.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal studies were approved by the Yale University Institutional Animal Care and Use Committee and were performed in accordance with all regulatory standards. 250 g male Sprague-Dawley rats were obtained from Charles River Laboratories (Wilmington, MA) and were group housed (3 per cage) while they were fed either a high fat diet (Dyets #112245, Bethlehem, PA; 59% calories from fat, 26% from carbohydrate, 15% from protein) or a chow diet (Harlan Teklad #2018, Madison, WI; 18% calories from fat, 58% from carbohydrate, 24% from protein), ad lib for 3 weeks. Rats given a Western diet were given ad lib access to the same high fat diet as well as to 5% sucrose water for the same period of time. All rats then underwent surgery under general isoflurane anesthesia for placement of polyethylene catheters in the common carotid artery (PE50 tubing, Instech Solomon, Plymouth Meeting, PA) and the jugular vein (PE90 tubing, Instech), after which they were singly housed until sacrifice. A subset of rats underwent surgery to place a catheter in the antrum of the stomach (PE50 tubing, Instech) in preparation for mixed-meal tolerance tests. The rats continued on ad lib high fat diet for an additional four days, after which T2D was induced by an injection of nicotinamide (85 mg/kg; Sigma, St. Louis, MO) and, 15 min later, streptozotocin (40 mg/kg; Sigma). Starting 24 hr after injection of streptozotocin and nicotinamide, blood glucose concentrations were checked by tail prick and those with random glucose >250 mg/dL were randomly divided into control or caloric restricted groups. Control rats were allowed to eat ad lib for 3 days, while VLCD rats were given 5.0 g of high fat diet per day (2.5 g of the safflower at 8 AM and 2.5 g at 6 PM). VLCD rats on the Western diet were given the same quantity of high fat diet as well as ad lib access to 1.25% sucrose water during the caloric restriction period. Unless otherwise specified, all in vivo studies in both VLCD and control rats were performed following an 8 hr fast. At the conclusion of each study, rats were euthanized by IV pentobarbital.
METHOD DETAILS
Flux Analysis
Hepatic glycogenolysis rates were determined by measuring hepatic glycogen content in 6 hr and 8 hr fasted rats. We calculated the difference in glycogen content between the two time points and assumed a linear rate of net hepatic glycogenolysis over the 120 min between the two time points. To measure gluconeogenic flux, rats were infused with [1,2,3,4,5,6,6-2H7]glucose (prime 0.3 mg/[kg-min] for 5 min, continuous infusion rate 0.1 mg/[kg-min]) and [3-13C]sodium lactate (prime 120 µmol/[kg-min] for 5 min, continuous infusion rate 40 µmol/[kg-min]) for a total of 120 min, after which blood (600 µl whole blood) was obtained from the venous catheter and immediately centrifuged and the plasma obtained, and rats were sacrificed and their livers snap-frozen in situ using metal tongs pre-chilled in liquid N2. We then applied PINTA analysis to measure whole-body glucose production, VPC flux, and VCS flux as we have described (Perry et al., in press). To measure the contribution of glycerol to gluconeogenesis, we subtracted the sum of the rates of glycogenolysis and VPC from the total EGP, assuming that the three sources of glucose production are glycogenolysis, VPC, and gluconeogenesis from glycerol. To measure hepatic triglyceride export (i.e. VLDL export), we injected Poloxamer 407 and measured plasma triglyceride concentrations as we have previously reported (Lee et al., 2011).
To measure whole-body rates of lipolysis and ketone turnover, rats were infused with [1,1,2,2,3-2H5]glycerol (0.1 mmol/[kg-min]), [U-13C]palmitate (0.2 mmol/[kg-min]), and [U-13C]βOHB (0.1 mg/[kg-min]) (Sigma, Cambridge Isotopes, Tewksbury, MA, and Cambridge Isotopes, respectively). GC/MS was used to measure plasma [2H5]glycerol and [U-13C]palmitate enrichment, from which we calculated the rates of lipolysis, correcting for the percent fatty acids that palmitate comprises, as we have described (Perry et al., 2015; Vatner et al., 2015). GC/MS was also used to assess βOHB turnover as previously reported (Perry et al., 2017a). The same extraction method as was utilized to measure βOHB turnover was also used to measure plasma lactate [m+1] enrichment in rats infused with [3-13C]lactate, with turnover calculated using the same formula as for all other turnover measurements: .
To assess triglyceride incorporation into DAG, we fed rats a mixed-meal tolerance test consisting of 100 kcal/kg Ensure Plus (“large meal”, ~half the ad lib fed rats’ daily caloric intake) supplemented with 10 mg/ml [1-13C]glyceryl-trioleate (Sigma), whereas the rats receiving a small meal were given 25 kcal/kg of the same mixture. The mixed-meal was administered through a gastric catheter.
Perturbations to Modulate Hepatic Glycogen and Acetyl-CoA
To increase hepatic acetyl-CoA in caloric restricted rats to concentrations measured in the T2D group, we infused sodium acetate (60 µmol/[kg-min]) throughout the duration of a [2H7]glucose infusion as described above. Samples were processed for measurement of glucose turnover rates as described (Perry et al., in press). Rats treated with a glycogen phosphorylase inhibitor (Sigma #361515) were injected with a 5 mg/kg dose of the drug IV and the same [2H7]glucose infusion was immediately begun. Both groups of rats were sacrificed by IV pentobarbital after 120 min of tracer infusion, and hepatic glycogen and acetyl-CoA and whole-body glucose turnover were measured as described in the following sections.
Hyperinsulinemic-Euglycemic Clamps
Beginning 6.5 hours after food was withdrawn, T2D and T2D-VLCD rats underwent a basal intra-arterial infusion of [1,2,3,4,5,6,6-2H]glucose (prime 1.5 mg/[kg-min] for 5 min, followed by a continuous infusion at a rate of 0.5 mg/[kg-min]) for 85 min. Blood samples were taken from a catheter in the jugular vein at 70, 80, and 90 min of the basal infusion for measurement of [2H7]glucose enrichment and calculation of basal glucose turnover. A hyperinsulinemic-euglycemic clamp was then performed: rats were given a 40 mU/kg bolus of Regular insulin followed by a 2.5 hr insulin infusion at a rate of 4.0 mU/(kg-min). Blood samples were drawn from the venous catheter at 0, 15, 30, 40, 50, 60, 75, 90, 105, 120, 130, 140, and 150 min of the clamp, with the samples from the 130, 140, and 150 min time points used to measure clamp glucose turnover. Plasma insulin was measured by radioimmunoassay by the Yale Diabetes Research Center at the 90 and 150 min time points of the basal infusion and clamp, respectively. Glucose turnover in the clamp was calculated as above ( ), and the rate of whole-body glucose disposal was calculated as the sum of the glucose infusion rate plus the glucose turnover rate measured in the clamp.
Biochemical Analysis
Plasma glucose concentrations were measured enzymatically using the YSI Glucose Analyzer (Yellow Springs, OH). Plasma insulin (other than in clamp samples, in which insulin was measured by radioimmunoassay), corticosterone, and FGF-21 were measured by ELISA (Mercodia, Winston-Salem, NC; Abcam, Cambridge, MA; and R&D Systems, Minneapolis, MN, respectively). Plasma lactate, βOHB, and transaminase concentrations were measured by COBAS (Roche Diagnostics, Indianapolis, IN). Plasma NEFA and triglyceride concentrations were measured spectrophotometrically using a Wako reagent (Wako Diagnostics, Mountain View, CA). To measure glycerol concentrations, plasma samples were spiked with an equal volume of [2-13C]glycerol (89 µM) and derivitized as described for glucose above. Glycerol was measured by GC/MS (EI mode), comparing the ratio of [m0] to [m+1] glycerol in the samples to a standard curve. To measure plasma amino acid concentrations by GC/MS, plasma samples (100 µl) were spiked with an internal standard containing 1 mM of each analyte of interest with an [m+1] 13C isotopic label, then dried under N2 gas and derivatized with 75 µl N-butanol 4N HCl (Sigma). Next the samples were heated for 60 minutes at 60°C and dried under N2 gas. They were then reacted with 100 µl of 1:7 trifluoroacetic acid (Thermo Fisher Scientific, Waltham, MA):methylene chloride (Sigma). GC/MS (CI mode) was used to measure concentrations of alanine, glycine, serine, leucine, isoleucine, aspartate+asparagine, phenylalanine, glutamate+glutamine by comparing the ratio of [m+1] 13C to natural abundance peak areas in the plasma samples to a standard curve.
Tissue Analysis
Liver glycogen was assessed as previously reported (Jurczak et al., 2011). Liver DAG and ceramide concentrations (Yu et al., 2002), long-chain acyl-CoA (Yu et al., 2002), acetyl- and malonyl-CoA (Perry et al., 2015; Perry et al., 2017a) were measured as we have described. Liver TAG content was measured using the method of Bligh and Dyer (Bligh and Dyer, 1959). Hepatic PKCε membrane/cytosol ratio (Samuel et al., 2004), gluconeogenic enzyme protein expression (Perry et al., 2015), ER stress markers (Jurczak et al., 2012) and PPARγ (Marx et al., 1998) and PGC1α (Erion et al., 2009) expression were assessed by Western blot as previously reported. Hepatic inflammatory cytokine concentrations were measured by ELISA. For measurement of inflammatory cytokine concentrations, liver samples (~200 mg) were homogenized in 5× volume of PBS, and the Qiagen Rat Inflammatory Cytokine Multi-Analyte ELISA was used to measure relative cytokine concentrations. Akt2 phosphorylation relative to total Akt2 was measured by Western blot using antibodies from Cell Signaling.
QUANTIFICATION AND STATISTICAL ANALYSIS
Comparisons were performed using the 2-tailed Student’s t-test, unpaired unless otherwise specified in the figure legends, with significance defined as a p-value <0.05. GraphPad Prism 7.0 (San Diego, CA) was used for all statistical analysis. In most cases, n=6 rats per group, unless otherwise indicated in the figure legends. Data are presented as the mean±S.E.M.
Supplementary Material
Highlights.
A 3 day very low calorie diet (VLCD) reverses T2D in rats by multiple mechanisms.
A VLCD lowers hepatic acetyl-CoA, glycogenolysis, and TAG-DAG-PKCε activation.
A VLCD markedly improves hepatic, not peripheral, insulin sensitivity in T2D rats.
Acknowledgments
The authors thank Jianying Dong, Xian-Man Zhang, Mario Kahn, Gina Butrico, John Stack, and Irina Smolgovsky for their excellent technical assistance. This study was supported by grants from the United States Public Health Service (R01 DK40936, P30 DK059635, R01 AG23686, T32 DK101019, K99 CA215315, R01 NS087568).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
The study was designed by R.J.P., K.F.P., and G.I.S. Experiments were performed and data analyzed by R.J.P., L.P., G.W.C., Y.W., A.R.-C., J.D.S., D.Z., Y.N., and S.D. The manuscript was written by R.J.P. and G.I.S., with input from all authors.
The authors declare that no conflicts of interest exist.
References
- Baskota A, Li S, Dhakal N, Liu G, Tian H. Bariatric Surgery for Type 2 Diabetes Mellitus in Patients with BMI <30 kg/m2: A Systematic Review and Meta-Analysis. PloS one. 2015;10:e0132335. doi: 10.1371/journal.pone.0132335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Canadian journal of biochemistry and physiology. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Boyle JP, Thompson TJ, Gregg EW, Barker LE, Williamson DF. Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Population health metrics. 2010;8:29. doi: 10.1186/1478-7954-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwald H, Avidor Y, Braunwald E, Jensen MD, Pories W, Fahrbach K, Schoelles K. Bariatric surgery: a systematic review and meta-analysis. JAMA. 2004;292:1724–1737. doi: 10.1001/jama.292.14.1724. [DOI] [PubMed] [Google Scholar]
- Buchwald H, Estok R, Fahrbach K, Banel D, Jensen MD, Pories WJ, Bantle JP, Sledge I. Weight and type 2 diabetes after bariatric surgery: systematic review and meta-analysis. The American journal of medicine. 2009;122:248–256. e245. doi: 10.1016/j.amjmed.2008.09.041. [DOI] [PubMed] [Google Scholar]
- Campos GM, Rabl C, Peeva S, Ciovica R, Rao M, Schwarz JM, Havel P, Schambelan M, Mulligan K. Improvement in peripheral glucose uptake after gastric bypass surgery is observed only after substantial weight loss has occurred and correlates with the magnitude of weight lost. J Gastrointest Surg. 2010;14:15–23. doi: 10.1007/s11605-009-1060-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements RH, Gonzalez QH, Long CI, Wittert G, Laws HL. Hormonal changes after Roux-en Y gastric bypass for morbid obesity and the control of type-II diabetes mellitus. Am Surg. 2004;70:1–4. discussion 4–5. [PubMed] [Google Scholar]
- Erion DM, Yonemitsu S, Nie Y, Nagai Y, Gillum MP, Hsiao JJ, Iwasaki T, Stark R, Weismann D, Yu XX, et al. SirT1 knockdown in liver decreases basal hepatic glucose production and increases hepatic insulin responsiveness in diabetic rats. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:11288–11293. doi: 10.1073/pnas.0812931106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry RR, Scheaffer L, Olefsky JM. Glycemic effects of intensive caloric restriction and isocaloric refeeding in noninsulin-dependent diabetes mellitus. The Journal of clinical endocrinology and metabolism. 1985;61:917–925. doi: 10.1210/jcem-61-5-917. [DOI] [PubMed] [Google Scholar]
- Isbell JM, Tamboli RA, Hansen EN, Saliba J, Dunn JP, Phillips SE, Marks-Shulman PA, Abumrad NN. The importance of caloric restriction in the early improvements in insulin sensitivity after Roux-en-Y gastric bypass surgery. Diabetes care. 2010;33:1438–1442. doi: 10.2337/dc09-2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackness C, Karmally W, Febres G, Conwell IM, Ahmed L, Bessler M, McMahon DJ, Korner J. Very low-calorie diet mimics the early beneficial effect of Roux-en-Y gastric bypass on insulin sensitivity and beta-cell Function in type 2 diabetic patients. Diabetes. 2013;62:3027–3032. doi: 10.2337/db12-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurczak MJ, Lee AH, Jornayvaz FR, Lee HY, Birkenfeld AL, Guigni BA, Kahn M, Samuel VT, Glimcher LH, Shulman GI. Dissociation of inositol-requiring enzyme (IRE1alpha)-mediated c-Jun N-terminal kinase activation from hepatic insulin resistance in conditional X-box-binding protein-1 (XBP1) knock-out mice. The Journal of biological chemistry. 2012;287:2558–2567. doi: 10.1074/jbc.M111.316760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurczak MJ, Lee HY, Birkenfeld AL, Jornayvaz FR, Frederick DW, Pongratz RL, Zhao X, Moeckel GW, Samuel VT, Whaley JM, et al. SGLT2 deletion improves glucose homeostasis and preserves pancreatic beta-cell function. Diabetes. 2011;60:890–898. doi: 10.2337/db10-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laferrere B, Teixeira J, McGinty J, Tran H, Egger JR, Colarusso A, Kovack B, Bawa B, Koshy N, Lee H, et al. Effect of weight loss by gastric bypass surgery versus hypocaloric diet on glucose and incretin levels in patients with type 2 diabetes. The Journal of clinical endocrinology and metabolism. 2008;93:2479–2485. doi: 10.1210/jc.2007-2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Birkenfeld AL, Jornayvaz FR, Jurczak MJ, Kanda S, Popov V, Frederick DW, Zhang D, Guigni B, Bharadwaj KG, et al. Apolipoprotein CIII overexpressing mice are predisposed to diet-induced hepatic steatosis and hepatic insulin resistance. Hepatology. 2011;54:1650–1660. doi: 10.1002/hep.24571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, Taylor R. Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia. 2011;54:2506–2514. doi: 10.1007/s00125-011-2204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx N, Schonbeck U, Lazar MA, Libby P, Plutzky J. Peroxisome proliferator-activated receptor gamma activators inhibit gene expression and migration in human vascular smooth muscle cells. Circulation research. 1998;83:1097–1103. doi: 10.1161/01.res.83.11.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy L, Docsa T, Szanto M, Brunyanszki A, Hegedus C, Marton J, Konya B, Virag L, Somsak L, Gergely P, et al. Glycogen phosphorylase inhibitor N-(3,5-dimethyl-Benzoyl)-N'-(beta-D-glucopyranosyl)urea improves glucose tolerance under normoglycemic and diabetic conditions and rearranges hepatic metabolism. PloS one. 2013;8:e69420. doi: 10.1371/journal.pone.0069420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM, et al. Hepatic Acetyl CoA Links Adipose Tissue Inflammation to Hepatic Insulin Resistance and Type 2 Diabetes. Cell. 2015;160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Peng L, Abulizi A, Kennedy L, Cline GW, Shulman GI. Mechanism for leptin's acute insulin-independent effect to reverse diabetic ketoacidosis. The Journal of clinical investigation. 2017a;127:657–669. doi: 10.1172/JCI88477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Peng L, Cline GW, Butrico GM, Wang Y, Zhang XM, Rothman DL, Petersen KF, Shulman GI. Non-Invasive Assessment of Hepatic Mitochondrial Metabolism by Positional Isotopomer NMR Tracer Analysis (PINTA) Nature communications. doi: 10.1038/s41467-017-01143-w. (in press) Please see Supplement. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Peng L, Cline GW, Petersen KF, Shulman GI. A Non-invasive Method to Assess Hepatic Acetyl-CoA In Vivo. Cell metabolism. 2017b;25:749–756. doi: 10.1016/j.cmet.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Samuel VT, Petersen KF, Shulman GI. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature. 2014;510:84–91. doi: 10.1038/nature13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes. 2005;54:603–608. doi: 10.2337/diabetes.54.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MC, Madiraju AK, Gassaway BM, Marcel M, Nasiri AR, Butrico G, Marcucci MJ, Zhang D, Abulizi A, Zhang XM, et al. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. The Journal of clinical investigation. 2016;126:4361–4371. doi: 10.1172/JCI86013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plum L, Ahmed L, Febres G, Bessler M, Inabnet W, Kunreuther E, McMahon DJ, Korner J. Comparison of glucostatic parameters after hypocaloric diet or bariatric surgery and equivalent weight loss. Obesity. 2011;19:2149–2157. doi: 10.1038/oby.2011.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed MJ, Meszaros K, Entes LJ, Claypool MD, Pinkett JG, Gadbois TM, Reaven GM. A new rat model of type 2 diabetes: the fat-fed, streptozotocin-treated rat. Metabolism: clinical and experimental. 2000;49:1390–1394. doi: 10.1053/meta.2000.17721. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. The Journal of biological chemistry. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Liu ZX, Wang A, Beddow SA, Geisler JG, Kahn M, Zhang XM, Monia BP, Bhanot S, Shulman GI. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. The Journal of clinical investigation. 2007;117:739–745. doi: 10.1172/JCI30400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. The Journal of clinical investigation. 2016;126:12–22. doi: 10.1172/JCI77812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer PR, Burguera B, Ikramuddin S, Cottam D, Gourash W, Hamad G, Eid GM, Mattar S, Ramanathan R, Barinas-Mitchel E, et al. Effect of laparoscopic Roux-en Y gastric bypass on type 2 diabetes mellitus. Annals of surgery. 2003;238:467–484. doi: 10.1097/01.sla.0000089851.41115.1b. discussion 484-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrutton MC, Keech DB, Utter MF. Pyruvate Carboxylase. Iv. Partial Reactions and the Locus of Activation by Acetyl Coenzyme A. The Journal of biological chemistry. 1965;240:574–581. [PubMed] [Google Scholar]
- Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. The New England journal of medicine. 2014;371:1131–1141. doi: 10.1056/NEJMra1011035. [DOI] [PubMed] [Google Scholar]
- Utter MF, Keech DB. Pyruvate Carboxylase. I. Nature of the Reaction. The Journal of biological chemistry. 1963;238:2603–2608. [PubMed] [Google Scholar]
- Utter MF, Keech DB, Scrutton MC. A possible role for acetyl CoA in the control of gluconeogenesis. Advances in enzyme regulation. 1964;2:49–68. doi: 10.1016/s0065-2571(64)80005-4. [DOI] [PubMed] [Google Scholar]
- Vatner DF, Majumdar SK, Kumashiro N, Petersen MC, Rahimi Y, Gattu AK, Bears M, Camporez JP, Cline GW, Jurczak MJ, et al. Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:1143–1148. doi: 10.1073/pnas.1423952112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. The Journal of biological chemistry. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.