

ABSTRACT
Type II toxin-antitoxin (TA) systems play a critical role in the establishment and maintenance of bacterial dormancy. They are composed of a protein toxin and its cognate protein antitoxin. They function to regulate growth under conditions of stress, such as starvation or antibiotic treatment. As cellular proteases degrade the antitoxin, which normally binds and neutralizes the toxin, this frees the toxin to act on its cellular targets and arrest bacterial growth. TA systems are of particular concern in regard to pathogenic organisms, such as nontypeable Haemophilus influenzae (NTHi), as dormancy may lead to chronic infections and failure of antibiotic treatment. Many targets of VapC toxins have not been identified, to date, and this knowledge is crucial to understanding how toxins control the establishment and maintenance of bacterial dormancy. Accordingly, we characterized the target specificity of the VapC toxins from the two paralogous NTHi vapBC TA systems. RNA sequencing and Northern blot analysis revealed that VapC1 and VapC2 cleave tRNAfMet in the anticodon loop. Overexpression of tRNAfMet suppresses VapC toxicity, suggesting that translation inhibition results from the depletion of tRNAfMet. These experiments also identified base pairs in the tRNAfMet anticodon stem that play a key role in VapC-specific cleavage of the tRNA. Together these findings suggest the potential for NTHi VapC1 and VapC2 to induce dormancy by sequence-specific cleavage of tRNAfMet.
IMPORTANCE Bacterial persistence is a significant concern in regard to pathogenic organisms, such as nontypeable Haemophilus influenzae, as it can result in recurrent and chronic infections. Toxin-antitoxin systems can lead to persistence by causing bacteria to enter a slow-growing state that renders them antibiotic tolerant. Type II toxin components affect a wide variety of bacterial targets in order to elicit dormancy, and for many toxin-antitoxin systems, these mechanisms are not well understood. Thus, in order to understand how vapBC toxin-antitoxin systems cause dormancy, it is crucial to investigate the substrate specificity of VapC toxins. This study identifies the target of the VapC1 and VapC2 toxins from NTHi and takes important steps toward understanding the specificity of these toxins for their tRNA target.
KEYWORDS: NTHi, RNA, endonuclease, persisters, tRNA, toxin-antitoxin
INTRODUCTION
Toxin-antitoxin (TA) systems are ubiquitous in bacteria and archaea, where they play an important role in the establishment and maintenance of dormancy. These systems are composed of a protein toxin and its cognate antitoxin, which can function as a protein (types II, IV, and V) or an RNA (types I and III). Under ideal growth conditions, the toxin is neutralized by its cognate antitoxin, allowing bacteria to grow exponentially. However, under conditions of stress, such as starvation or antibiotic treatment, the antitoxin is inactivated, leaving the toxin free to inhibit growth (1–3). Growth inhibition due to TA systems has been implicated in bacterial persistence (4–6). Persisters occur when a small fraction of a population of cells stochastically enter a slow-growing state, allowing them to survive under stressful conditions. This is of particular concern in regard to pathogenic organisms, as these dormant bacteria are antibiotic tolerant, which leads to recurrent and chronic infections. Indeed, gradual deletion of 10 of the 12 type II TA systems in Escherichia coli led to a decrease in persistence during antibiotic treatment, suggesting that these systems play an important role in persistence and multidrug tolerance (7).
There are five types of TA systems, which are classified based on the antitoxin's mechanism of action. Among these five types, type II systems are the most abundant and the best characterized. For example, there are 88 type II systems in Mycobacterium tuberculosis (8). In type II systems, the antitoxin is a protein that directly binds the toxin, preventing its activity. Generally, type II TA systems are cotranscribed from a single operon, with the antitoxin gene typically found upstream of the toxin gene (3). The operon is autoregulated by the antitoxin or the toxin-antitoxin complex, which binds to operators near the promoter to repress transcription (1, 2).
Type II TA systems are classified into families based on sequence homology (9). Their mechanisms of toxicity have been characterized, but not all have been well studied. The MazF family of toxins inhibits translation by cleaving mRNA, 16S rRNA, 23S rRNA, and some tRNAs in a sequence-specific manner (10–14). The HipA family acts through phosphorylation of glutamyl-tRNA synthetase (4). RelE toxins cleave mRNA in the ribosomal A site (15), while the HicA and Kid families cleave mRNA in a manner that is independent of the ribosome (16, 17). Doc toxins phosphorylate elongation factor Tu to inhibit translation elongation (18, 19). ParE and CcdB toxins inactivate DNA gyrase and thus inhibit DNA replication (20, 21). The most common, though less studied, family of type II TA systems is the VapBC family.
The VapC toxin is a PIN domain endoribonuclease that inhibits translation through the hydrolytic cleavage of RNAs (22, 23). The PIN domain coordinates Mg2+ ions in the active site to facilitate RNA cleavage. PIN domain proteins are found throughout all domains of life. In eukaryotes, they are known to function in RNA processing and RNA degradation (24–27). While the cleavage targets of most VapC toxins have not been identified, those characterized thus far suggest that VapC toxins can be grouped into two categories: those that cleave tRNAs and those that cleave rRNAs (28). The VapC toxins from Salmonella enterica, Shigella flexneri, and Leptospira interrogans all cleave tRNAfMet (29, 30). VapC4 from Mycobacterium tuberculosis cleaves tRNACys-GCA, while VapC32, VapC11, and VapC15 cleave tRNALeu-CAG (28). Additionally, VapC28 and VapC30 from M. tuberculosis cleave tRNASer-TGA,CGA, and five other M. tuberculosis VapCs (VapC25, VapC33, VapC37, VapC29, and VapC39) cleave tRNATrp-CCA (28). On the other hand, VapC20 and VapC26 from M. tuberculosis cleave 23S rRNA at the sarcin-ricin loop (SRL) (28, 31).
The pathogenic organism nontypeable Haemophilus influenzae (NTHi) contains two vapBC systems: vapB1C1 and vapB2C2. These two systems are homologous; however, there is no cross talk between them, as the antitoxins are highly specific for their cognate toxins (32). Studies have shown that these systems are upregulated during NTHi infection and facilitate enhanced survival and growth regulation (33, 34). The present study aimed to identify and characterize the RNA targets of VapC1 and VapC2 from NTHi. In particular, we were interested in whether these homologous toxins employ different or redundant mechanisms to induce dormancy. Metabolic labeling experiments showed that both toxins slow cell growth by inhibiting protein synthesis. RNA sequencing (RNA-seq) and Northern blot analysis demonstrated that both VapC toxins cleave tRNAfMet. Overexpression of this tRNA suppresses the toxicity of NTHi VapC1, suggesting that the depletion of tRNAfMet causes toxin-induced inhibition of translation. Finally, mutation of a G-C pair at the tRNAfMet anticodon stem-loop (ASL) junction reduced VapC cleavage, suggesting that the endoribonucleases recognize features of the anticodon stem. These findings reveal that NTHi VapC toxins inhibit bacterial cell growth by sequence-specific cleavage of tRNAfMet.
RESULTS
VapC1NTHi and VapC2NTHi slow cell growth by inhibiting protein synthesis.
Previous work with VapC1NTHi revealed that its conditional expression from the arabinose promoter on a plasmid (pBAD) results in inhibition of growth of E. coli and that this effect requires conserved amino acid residues necessary for the function of VapC1NTHi proteins (35). We compared the timing and degree of growth inhibition by VapC1NTHi to those for VapC2NTHi and found that VapC1NTHi acted more rapidly and slowed cell growth to a greater degree than that with VapC2NTHi expressed under the same conditions (Fig. 1). In both cases, IPTG (isopropyl-β-d-thiogalactopyranoside)-induced expression of the cognate antitoxins (VapB1NTHi and VapB2NTHi) from the tac promoter on a compatible plasmid suppressed the toxicity. We concluded that this expression system reflects the known actions of these TA systems, and the results suggest that under these conditions, VapC1NTHi acts more quickly than VapC2NTHi to inhibit protein synthesis and cell growth.
FIG 1.

VapC1NTHi and VapC2NTHi slow cell growth by inhibiting protein synthesis. (A) The growth rates of E. coli Top10 cells carrying pBAD-VapC1 and pJSB31-VapB1-sfGFP under different induction conditions were compared using the bacterial growth curve assay as described in Materials and Methods. Cultures were grown in M9 medium containing 0.2% glucose and appropriate antibiotics. pBAD-VapC1 was induced with 0.2% l-arabinose (ara), and pJSB31-VapB1-sfGFP was induced with 0.5 mM IPTG. (B) The growth rates of E. coli Top10 cells carrying pBAD-VapC2 and pJSB31-VapB2-sfGFP under different induction conditions were compared as described for panel A. (C and D) Rates of translation (C) and transcription (D) were compared between E. coli Top10 cells carrying the pBAD empty vector, pBAD-VapC1, or pBAD-VapC2 by pulse labeling with [35S]methionine (C) or [3H]uracil (D) according to the metabolic labeling protocol described in Materials and Methods. Induction occurred by addition of 0.2% l-arabinose at 0 min. Counts per minute were normalized by use of the A600 value for each time point and plotted relative to the measurement at −10 min. Data are averages for two biological replicates, and error bars represent standard deviations.
To determine how NTHi VapC toxins inhibit cell growth, we measured the rates of [35S]methionine and [3H]uracil incorporation into protein and RNA, respectively, by pulse labeling cells at specific times before (−10 min) and after (0 min) induction of protein expression in E. coli (Fig. 1C and D). The results revealed 70% and 50% inhibition of [35S]methionine incorporation due to VapC1NTHi and VapC2NTHi expression, respectively, compared to the incorporation with the empty vector control (Fig. 1C). However, the rates of [3H]uracil incorporation changed relatively little compared to the vector control rate for each toxin (Fig. 1D). These results support our conclusion that NTHi VapCs primarily inhibit protein synthesis.
VapC1NTHi and VapC2NTHi cleave the tRNAfMet anticodon loop.
To identify the RNA targets of the VapC1NTHi and VapC2NTHi toxins, we modified a method designed to capture, amplify, and sequence small RNA products produced by endoribonucleolytic hydrolysis (36) (Fig. 2A). Since this method relies on the chemical nature of the 5′ end of the 3′ product of endoribonucleolytic cleavage of the RNA target, and because metallo-endoribonucleases typically produce a 3′ cleavage product with a 5′ phosphate, we carried out an experiment to determine if the known 3′ product of tRNAfMet cleavage by the related VapCLT2 toxin from Salmonella enterica serovar Typhimurium carries a 5′ phosphate or 5′ hydroxyl group. We expressed S. Typhimurium VapCLT2 in Escherichia coli to produce its previously identified tRNAfMet cleavage products (29; also see below). A 5′ monophosphate-specific terminator exoribonuclease treatment of total RNA isolated after expression of VapCLT2 destroyed the 5′ but not 3′ tRNAfMet product of VapCLT2 cleavage, indicating that the resistant 3′ product likely carried a 5′-OH (Fig. 2B, compare lanes 4 and 8). Next, all RNAs in the sample received (i) a preadenylated 3′ DNA adapter by ligation and (ii) a 5′ phosphate to mark the position of VapCLT2 cleavage on the 3′ product of tRNAfMet. After ligation of a 5′ RNA adapter to this position, reverse transcription-PCR (RT-PCR) verified the production of the VapCLT2-tRNAfMet cDNA containing both flanking adapters (data not shown). These steps indicated that this procedure captures the 3′ tRNAfMet cleavage product of VapCLT2 as a cDNA flanked by the adapter sequences.
FIG 2.
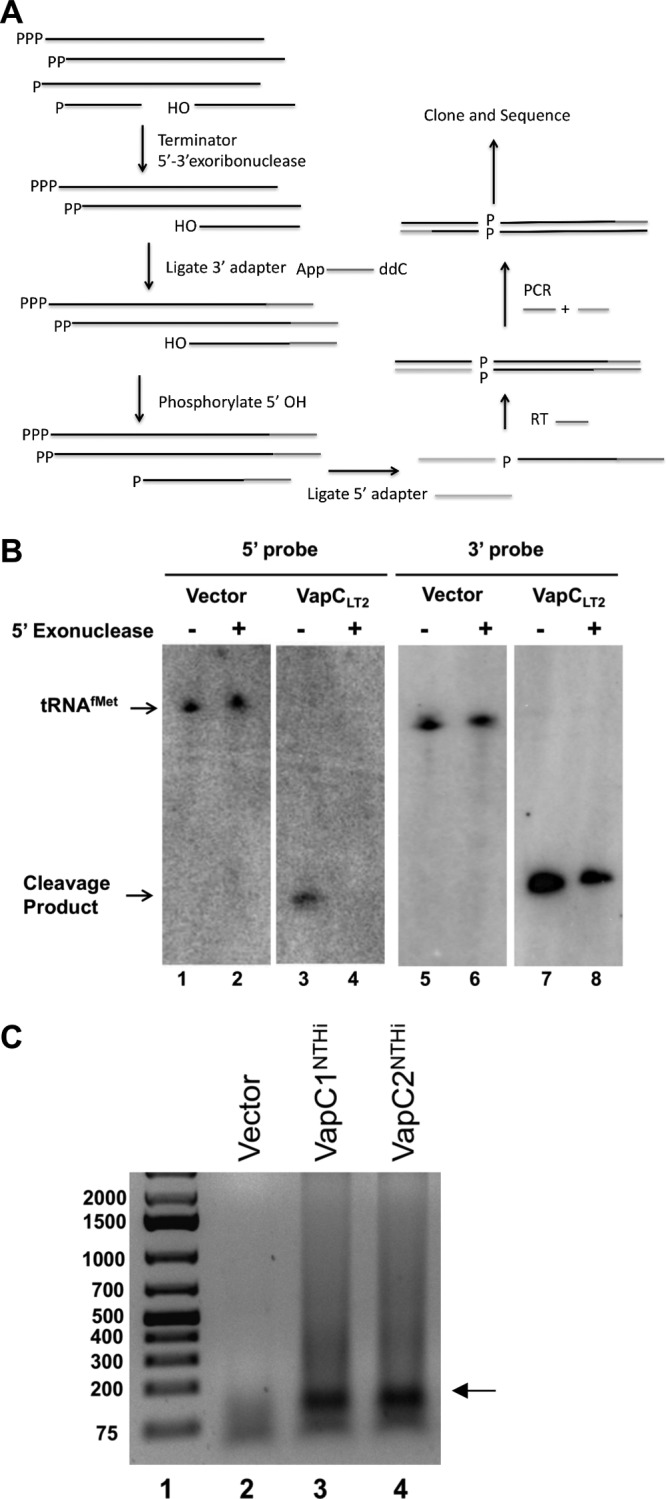
RNA sequencing protocol enriches for VapC cleavage fragments. (A) RNA sequencing protocol to specifically amplify VapC cleavage products as described in Materials and Methods. (B) Northern blot of total RNA harvested after induction of the pBAD empty vector or pBAD-VapCLT2 by use of 0.2% l-arabinose. RNA was either untreated or treated with XrnI, a 5′ monophosphate-dependent exonuclease. The first two panels were probed with an oligonucleotide that hybridizes to the 3′ cleavage fragment of tRNAfMet, while the third and fourth panels were probed with an oligonucleotide that hybridizes to the 5′ cleavage fragment. (C) cDNA was amplified following the RNA sequencing protocol shown in panel A and as described in Materials and Methods, and the fragments were separated by agarose gel electrophoresis. The arrow indicates the potential VapC cleavage products.
Next, we applied this procedure to total RNA samples from cells expressing or not expressing VapC1NTHi or VapC2NTHi. Analysis of the products revealed strong production of cDNAs with lengths in the 150- to 200-bp range for each toxin, but not from cells carrying the empty vector (Fig. 2C). Cloning and DNA sequence analysis of the cDNAs from VapC1NTHi-expressing cells identified plasmids with fragments of tRNAfMet, tRNALeu-CAG, and tRNAVal-GAC flanked by the ligated adapters (Fig. 3A). Each of these potential targets was identified once, and these were the only targets found by sequencing. We tested the effect of NTHi VapC expression on each of these tRNAs by Northern blotting of total RNA preparations from cells expressing each toxin as well as those carrying empty vector or expressing VapCLT2, as a negative or positive control, respectively (Fig. 3B). The results revealed that the NTHi and S. Typhimurium VapCs produced fragments of tRNAfMet and tRNALeu-CAG but not tRNAVal-GAC or the elongator tRNAMet-CAT. Notably, expression of the VapCs resulted in a nearly complete loss of tRNAfMet but had relatively little effect on the amount of tRNALeu-CAG and no observable effect on tRNAVal-GAC and tRNAMet-CAT. The antitoxins VapB1NTHi and VapB2NTHi bind to their cognate toxins and block growth arrest, presumably by interfering with the endonuclease function of the enzymes. This predicts that coexpression of the antitoxins should inhibit the activity of the toxins and spare tRNAfMet from cleavage. Indeed, VapB1NTHi and VapB2NTHi blocked cleavage of tRNAfMet by their cognate toxins (Fig. 3C). These collective findings support our conclusion that VapC1NTHi and VapC2NTHi preferentially cleave tRNAfMet in vivo.
FIG 3.

VapC1NTHi and VapC2NTHi cleave the tRNAfMet anticodon loop. (A) Sequences and structure diagrams of tRNAs identified by RNA sequencing as potential VapC cleavage products. Italic and underlined sequences show the 5′ and 3′ adaptors, respectively, and those in bold show the identified cleavage fragments. On each tRNA diagram, an arrow indicates the site of cleavage based on sequencing results. (B) Northern blot analysis of total RNA from E. coli BW25113Δ6 carrying pBAD, pBAD-VapC1, pBAD-VapC2, or pBAD-VapCLT2 (induced with 0.2% l-arabinose), as described in Materials and Methods. Blots were probed with radiolabeled oligonucleotides specific to the tRNAs listed above the panels. Full-length or cleaved tRNAs are indicated with arrows. (C) Northern blot analysis of total RNA from E. coli BW25113Δ6 carrying pBAD, pBAD-VapC1, or pBAD-VapC2 or expressing each toxin coexpressed with its cognate antitoxin (pJSB31-VapB1-sfGFP or pJSB31-VapB2-sfGFP), as described in Materials and Methods. Blots were probed with a radiolabeled oligonucleotide specific for tRNAfMet. Full-length or cleaved tRNAfMet is indicated with an arrow.
Conditional expression of tRNAfMet suppresses the growth defect caused by VapC1NTHi.
If hydrolysis of tRNAfMet by NTHi VapCs inhibits cell growth by depletion of the tRNA, then increased expression of tRNAfMet should suppress the growth defect, but tRNAMet-CAT or tRNALeu-CAG should not. We tested this by expressing these tRNAs from an IPTG-inducible promoter in the presence or absence of VapC1NTHi (Fig. 4A). Induction of expression of tRNAfMet in cells not expressing VapC1NTHi had no effect on the growth of cells in the presence or absence of arabinose (Fig. 4B). However, cells carrying a plasmid encoding VapC1NTHi grew more slowly than those containing empty vector, even in the absence of induction by arabinose, suggesting a leaky toxicity of VapC1NTHi (Fig. 4B and C). Induction of tRNAfMet expression by use of IPTG suppressed the leaky toxicity of VapC1NTHi (Fig. 4C). Likewise, expression of tRNAfMet suppressed the growth defect caused by induction of VapC1NTHi by arabinose addition (Fig. 4D). In contrast, expression of tRNALeu-CAG or tRNAMet-CAT did not suppress toxicity caused by induction of VapC1NTHi (Fig. 4E and F). These findings support the conclusion that the growth defect caused by VapC1NTHi likely results from depletion of intact tRNAfMet caused by endoribonucleolytic cleavage.
FIG 4.

Conditional expression of tRNAfMet suppresses the growth defect caused by VapC1NTHi. (A) Diagram illustrating the expression of tRNAfMet from the T7 promoter of plasmid pJSB31 in trans to the VapC gene expressed from the ara promoter on pBAD/Myc-His B, which results in cleavage of the tRNA in the anticodon loop. (B to F) Growth curve analysis of E. coli BL21(DE3) strains carrying pBAD or pBAD-VapC1 and pJSB31-T7-tRNAfMet, pJSB31-T7-tRNAMet-CAT, or pJSB31-T7-tRNALeu-CAG, as indicated. Cultures were grown in M9 glycerol medium, with induction of pBAD or pJSB31 by use of 0.01% l-arabinose or 0.5 mM IPTG, respectively, as indicated. The A600 values were normalized to the A600 at 0 min. Data are averages for two biological replicates, and error bars indicate standard deviations.
VapC1NTHi and VapC2NTHi cleave NTHi tRNAfMets.
While the experiments described above clearly indicated that NTHi VapCs cleave tRNAfMet in E. coli, it remained unclear if they effectively target tRNAfMet from NTHi. Accordingly, we tested the ability of NTHi VapCs to cleave NTHi tRNAfMet. Two tRNAfMet genes in NTHi produce initiator tRNAs (tRNAfMet-14 and tRNAfMet-23) that differ in sequence, at several positions, from each other and from the single E. coli tRNAfMet (Fig. 5A). Thus, differential hybridization of oligonucleotide probes specific to each of the NTHi tRNAs allows their detection in total RNA preparations from cells expressing combinations of E. coli and NTHi tRNAs. Oligonucleotide probes with a single mismatch to E. coli tRNAfMet but full complementarity to either NTHi tRNAfMet allowed detection of both tRNAs at 42°C but only the NTHi tRNA at 68°C (Fig. 5B). At the higher hybridization temperature, the probe complementary to NTHi tRNAfMet-14 distinguished it from tRNAfMet-23 and E. coli tRNAfMet, and the probe complementary to NTHi tRNAfMet-23 distinguished it from tRNAfMet-14 and E. coli tRNAfMet (Fig. 5B). The results indicate that VapC1NTHi and VapC2NTHi cleave tRNAfMets from NTHi and E. coli with similar efficiencies under these conditions.
FIG 5.

VapC1NTHi and VapC2NTHi cleave NTHi tRNAfMets. (A) Sequence alignment of E. coli tRNAfMet and NTHi tRNAfMet-14 and tRNAfMet-23. Sequence differences are highlighted in bold, and the anticodon sequence is underlined. (B) Differential hybridization Northern blot analysis of total RNAs isolated from E. coli BL21(DE3) cells expressing VapC toxins (induced with 0.01% l-arabinose) and tRNAs (induced with 0.5 mM IPTG) as indicated, performed as described in Materials and Methods. The blots were hybridized with an oligonucleotide specific to both E. coli tRNAfMet and the NTHi tRNAfMet of interest, but with one nucleotide mismatch for the E. coli tRNAfMet. After hybridization of the probe, blots were washed at 42°C and then again at 68°C, as indicated above each panel.
Efficient cleavage of tRNAfMet by VapC1NTHi and VapC2NTHi requires key base pairs in the anticodon stem structure.
A complete understanding of the mechanism of VapC-induced bacterial dormancy requires determination of the sequence and structural features that govern recognition of the VapC RNA targets. VapC toxins characterized to date act as endonucleases that cleave tRNAs or the sarcin-ricin loop (SRL) of 23S rRNA (28, 30, 31, 37). Winther et al. proposed that the enzymes may recognize similar RNA structures, since the tRNA anticodon stem-loop (ASL) and the 23S rRNA SRL share some structural similarities (31). Indeed, studies of M. tuberculosis VapC endonucleases indicate that they recognize specific structures in the SRL of 23S rRNA and the ASLs of specific tRNAs (31, 37). In the case of VapC1NTHi and VapC2NTHi, consideration of the ASLs of several tRNAs that are not cleaved suggests the identities of specificity determinants in the stem of the ASL of tRNAfMet (Fig. 6A). The identical loop sequences in tRNAfMet and the nonsubstrate elongator tRNAMet-CAT indicate that the loop does not specify cleavage by itself (Fig. 6A). The three consecutive G-C pairs (numbered 29-41, 30-40, and 31-39 in Fig. 6A) in the tRNAfMet stem are required for efficient function of the tRNA in protein synthesis and facilitate its entry directly into the ribosomal P site by virtue of interactions with the 16S rRNA (38, 39). Notably, the noninitiator, nonsubstrate tRNAs tRNAMet-CAT, tRNAPhe-GAA, and tRNALeu-TAA each contain similar G-C paired stems, with the exception of the loop-closing A-U pair (Fig. 6A). Accordingly, we changed the loop-closing G-C pair (31–39) in NTHi tRNAfMet-14 to A-U and measured the extent of its cleavage by VapC1NTHi and VapC2NTHi in vivo. The result revealed that the G-C-to-A-U mutation decreased cleavage of NTHi tRNAfMet-14 by 30 to 40%, suggesting that this G-C pair plays a key role in recognition of the substrate by VapCs (Fig. 6B and C).
FIG 6.
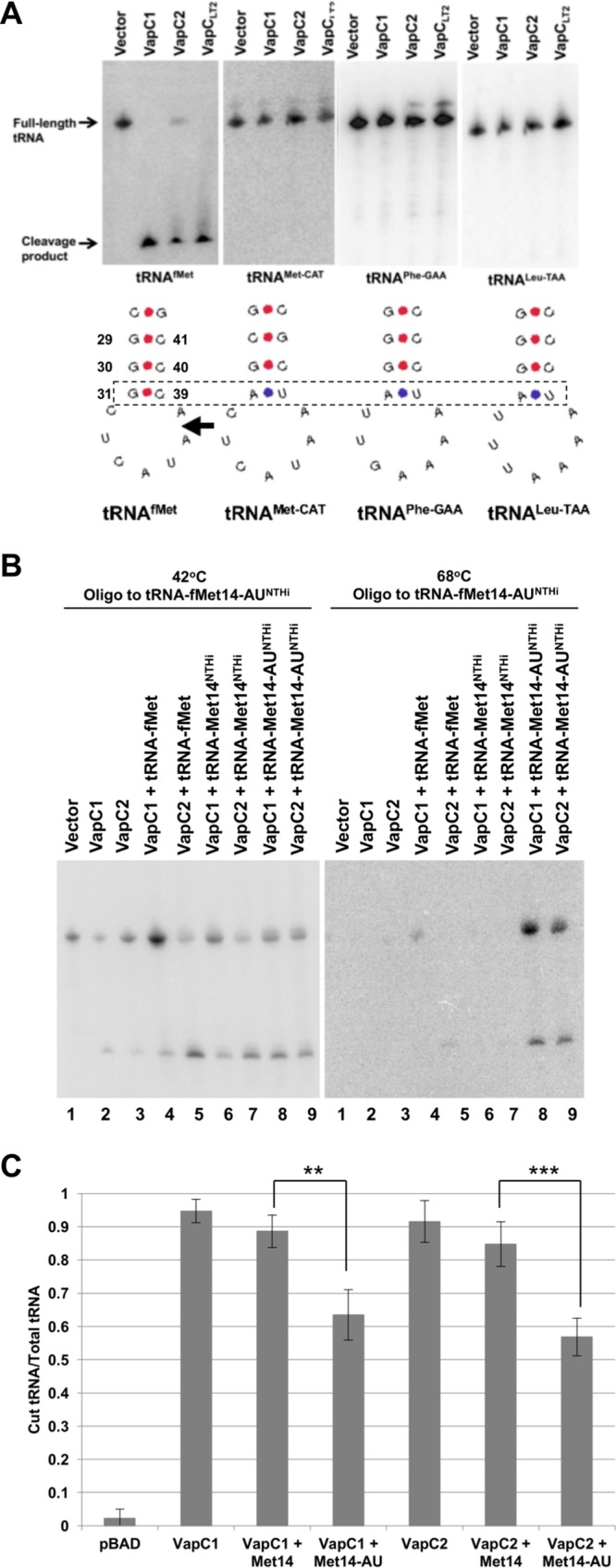
Efficient cleavage of tRNAfMet by VapC1NTHi and VapC2NTHi requires a key base pair in the anticodon stem structure. (A) Diagram showing anticodon stem-loop sequences and structures for various tRNAs. Above each structure is a Northern blot of total RNA from E. coli carrying pBAD, pBAD-VapC1, or pBAD-VapC2 (induced with 0.2% l-arabinose), probed for each tRNA. The box denotes the base pairs of interest that differ between tRNAs that are cleaved and those that are not. The arrow indicates the site of cleavage in tRNAfMet. (B) Differential hybridization Northern blot analysis of total RNAs isolated from E. coli BL21(DE3) cells expressing VapC toxins (induced with 0.01% l-arabinose) and tRNAs (induced with 0.5 mM IPTG) as indicated, performed as described in Materials and Methods. The blots were hybridized with an oligonucleotide specific to both E. coli tRNAfMet and NTHi tRNA14-AUfMet, but with one nucleotide mismatch for the E. coli tRNAfMet. After hybridization of the probe, blots were washed at 42°C and then again at 68°C, as indicated above each panel. (C) Quantification of cleaved tRNA/total tRNA from Northern blots, measured using ImageQuant. Quantification of the histogram bar representing VapC1 + Met14 was carried out by probing the Northern blots from panel B with a probe specific to tRNAfMet14 (not shown). The data are averages for four biological replicates, and error bars represent standard deviations. **, P < 0.01; ***, P < 0.001.
The effect of the G-C-to-A-U mutation on cleavage of tRNAfMet by VapCs suggested that conversion of A-U to G-C at this position in the elongator tRNAMet-CAT might convert this nonsubstrate into a target for the VapCs. To test this, we changed the relevant portion of the stem of tRNAMet-CAT to match that of tRNAfMet within the three base pairs critical for the function of the stem (Fig. 7A). This mutant, tRNAMet-CAT-M13, became a relatively weak but clear substrate for both VapCs (Fig. 7B and C). Finally, we switched the C-G base pair at positions 28 and 42 in tRNAfMet to produce tRNAfMet-M4, whose sequence matches the sequence of tRNAMet-CAT-M13. This mutation caused a slight increase in cleavage by VapC1 but no significant change for VapC2 (Fig. 7B and C). Interestingly, cleavage of the two mutant tRNAs resulted in multiple cleavage products, suggesting that these base pair changes altered the site specificity of VapC1 and VapC2 (Fig. 7B). Collectively, these experiments indicate that the VapC enzymes recognize structural features of the anticodon stem which influence the efficiency and site of tRNA cleavage.
FIG 7.
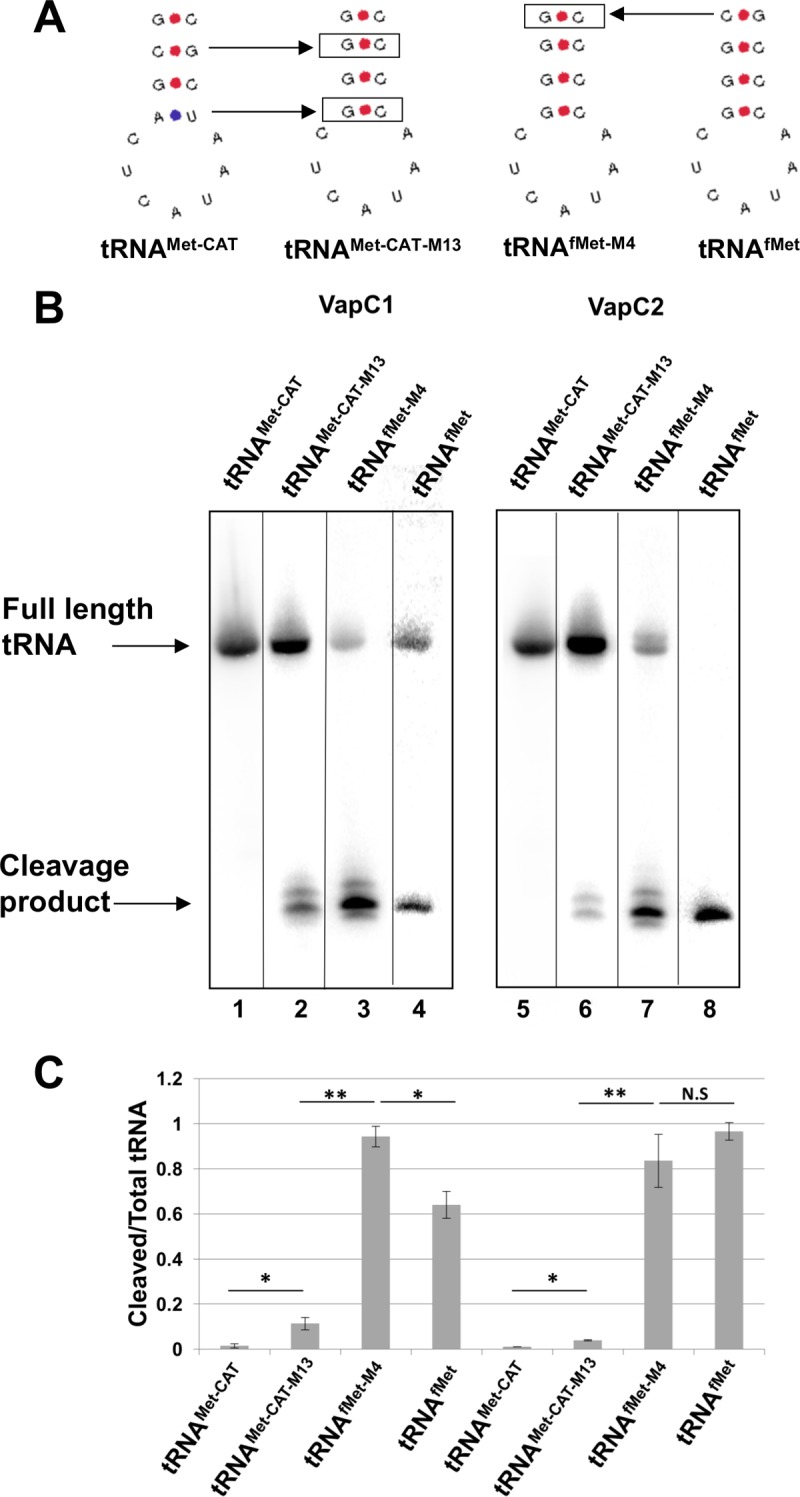
Efficient cleavage of tRNAfMet by VapC1NTHi and VapC2NTHi requires key base pairs in the anticodon stem structure. (A) Diagram showing anticodon stem-loop sequences and structures of tRNAs and mutants. Boxed base pairs indicate mutations made from either tRNAMet-CAT or tRNAfMet. (B) Differential hybridization Northern blot analysis of total RNAs isolated from E. coli BL21(DE3) cells expressing the VapC1 (left) or VapC2 (right) toxin (induced with 0.01% l-arabinose) and tRNAs (induced with 0.5 mM IPTG) as indicated, performed as described in Materials and Methods. The blots were hybridized with an oligonucleotide specific to both E. coli tRNAfMet or tRNAMet-CAT and the mutant tRNA, but with one nucleotide mismatch for the wild-type tRNA. After hybridization of the probe, blots were washed at 42°C and then again at 68°C. The image is a composite, as each tRNA required hybridization of a different oligonucleotide. Complete images may be seen in Fig. S1 in the supplemental material. (C) Quantification of cleaved tRNA/total tRNA from Northern blots, measured using ImageQuant. The data are averages for two biological replicates, and error bars represent standard deviations. *, P < 0.05; **, P < 0.01; N.S, not significant.
DISCUSSION
Bacterial persistence is a significant problem in pathogenic organisms because it results in antibiotic tolerance, leading to chronic and recurrent infections. Toxin-antitoxin systems are known to cause bacterial dormancy and formation of persisters. Therefore, to address the issue of bacterial persistence, it is crucial to understand the mechanisms behind toxin-induced dormancy. We approached the question of substrate specificity for the two VapC toxins from NTHi by identifying their targets in vivo. Using RNA sequencing and Northern blot analysis, we identified the initiator tRNAfMet as the target of VapC1NTHi and VapC2NTHi. Overexpression of tRNAfMet specifically suppresses VapC1 toxicity, suggesting that depletion of tRNAfMet rather than accumulation of its degradation products causes the observed inhibition of protein synthesis and cell growth caused by NTHi VapC expression. These findings are consistent with observations in other organisms, which revealed that VapCs from S. enterica, S. flexneri, and L. interrogans inhibit translation through cleavage of tRNAfMet, while a number of other VapC toxins from M. tuberculosis cleave specific tRNAs and rRNAs (28–31, 37). In the case of NTHi and these other organisms, the results suggest that VapCs may give rise to persister cells by tRNA or rRNA cleavage resulting in the inhibition of protein synthesis.
We identified the targets of NTHi VapC toxins by using E. coli as a surrogate. Nevertheless, we found that the NTHi toxins cleave tRNAfMet from NTHi with a high efficiency similar to that observed for E. coli tRNAfMet, indicating that the few sequence differences between these substrates likely have minimal effects on recognition by the endonucleases and that it is unlikely that the toxins require any factors specific to NTHi for tRNA cleavage. These considerations justify the use of E. coli for these experiments, but further work in NTHi is necessary to determine if the VapC toxins are capable of causing the formation of dormant persisters.
Several observations indicate that cleavage of tRNAfMet by VapC1NTHi and VapC2NTHi is relatively specific. First, although RNA-seq identified tRNALeu-CAG and tRNAVal-GAC as potential substrates in addition to tRNAfMet, further Northern blot analysis of cellular RNA revealed that tRNALeu-CAG is cleaved very inefficiently, and tRNAVal-GAC not at all. These findings suggest that the sensitivity of the RNA-seq method probably allows the identification of VapC-independent tRNA degradation intermediates (i.e., tRNAVal-GAC) and, possibly, off-target cleavage (i.e., tRNALeu-CAG). The common use of high-copy-number plasmids and strong inducible promoters to express VapCs in cells may contribute to the latter effect. In the case of the NTHi VapC toxins, we found that expression of tRNAfMet, but not that of tRNALeu-CAG or tRNAMet-CAT, could suppress the growth defect caused by expression of VapC1, suggesting that tRNAfMet is the primary target whose cleavage inhibits cell growth.
The specificity of VapC toxins is of considerable interest for structural and functional reasons. Accordingly, we analyzed the structural requirements for tRNAfMet cleavage. The analysis focused on three G-C base pairs that close the anticodon loop of the tRNA, since these provide the critical information for the unique function of P-site binding to the initiator tRNA. The results revealed that the G-C pair at the junction of the anticodon stem and loop plays an important role in efficient cleavage of the tRNA. Moreover, switching this pair from A-U to G-C in the elongator tRNAMet-CAT converted this nonsubstrate tRNA into a substrate for the endonucleases. However, this G-C pair is clearly not the only specificity determinant for VapC cleavage of tRNAfMet. Two mutant tRNAs created for our experiments, the initiator tRNAfMet-M4 and the elongator tRNAMet-CAT-M13, have identical loop sequences as well as the G-C base pairs necessary for tRNAfMet interaction with the ribosomal P site. Nevertheless, the initiator tRNAfMet-M4 is a far better substrate than the elongator tRNAMet-CAT-M13. This indicates that the endonucleases likely recognize structural aspects of tRNAfMet in addition to those found in the ASL. Other work has shown that VapC toxins require both the stem-loop structure and conservation of the cleavage site sequence in order to recognize and cleave their RNA targets. A study from Winther et al. showed that VapC20 could no longer cleave the sarcin-ricin loop of 23S rRNA when either the stem-loop structure was altered or the cleavage site sequence was mutated (31). Although our studies did not address the importance of the loop sequence, its presence in the nonsubstrate tRNA tRNAMet-CAT indicates that it is not sufficient for cleavage. However, our findings suggest that the position of the cleavage site is likely influenced by the structure of the stem, as the base pair changes within the stem alter the cleavage site. Clearly, a thorough understanding of the requirements for the specificity of the NTHi VapCs will require experiments in a defined system in vitro.
In summary, this study identified that growth inhibition by VapC1NTHi and VapC2NTHi results from translation arrest due to cleavage of the initiator tRNAfMet. The enzymes cleave both E. coli and NTHi initiator tRNAs in vivo in E. coli, suggesting that no NTHi factors are necessary for recognition of the substrates. Additionally, we found that the efficiency of tRNA cleavage depends in part on the presence of G-C base pairs at the junction of the anticodon stem and loop. Overall, our work demonstrates the highly specific function of VapC toxins.
MATERIALS AND METHODS
Bacterial strains and growth media.
Escherichia coli BL21(DE3) [fhuA2 (lon) ompT gal dcm (λ DE3) ΔhsdS], E. coli BW25113Δ6 [F− DE(araD-araB)567 lacZ4787(del)::rrnB-3 LAM− rph-1 DE(rhaD-rhaB)568 hsdR514 Δ6] (TA Systems), and E. coli Top10 [F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 deoR recA1 araD139 Δ(araA-leu)7697 galU galK rpsL endA1 nupG] carrying the indicated plasmids were grown at 37°C in M9 medium supplemented with 0.2% glucose, 0.2% Casamino Acids, 1 mM thiamine, and appropriate antibiotics or in LB medium supplemented with 0.2% glucose and appropriate antibiotics.
Plasmid construction. (i) pJSB31-sfGFP.
The superfolder green fluorescent protein (sfGFP)-encoding DNA was amplified from the SuperFolder GFP expression plasmid (Sandia Biotech) by PCR with primers OSB1233 and OSB1234. The PCR product was digested with BglII and KpnI and cloned into the same sites of the pJSB31 plasmid (40). The pJSB31-sfGFP plasmid was constructed to encode a 6×-glycine linker at the N terminus of the superfolder GFP. This plasmid contains a p15A origin of replication, which is compatible with the pBR322 origin of replication in the pBAD/Myc-His B plasmid.
(ii) pJSB31-VapB1-sfGFP and pJSB31-VapB2-sfGFP.
The VapB1 (HI0321)- and VapB2 (HI0946)-encoding DNAs were PCR amplified using primers OSB1385 and OSB1386 (VapB1) or OSB1289 and OSB1352 (VapB2). The PCR products were digested with BglII and NcoI and ligated into the corresponding sites of pJSB31-sfGFP. The resulting plasmids express C-terminally sfGFP-tagged VapBs in the presence of IPTG.
(iii) pBAD-VapC1, pBAD-VapC2, and pBAD-VapCLT2.
VapC1 (HI0322)-, VapC2 (HI0947)-, and VapCLT2 (STM3033)-encoding DNAs were PCR amplified using primers OSB829 and OSB830 (VapC1), OSB834 and OSB835 (VapC2), or OSB1103 and OSB1104 (VapCLT2). The PCR products were digested with NcoI and XbaI and ligated into the corresponding sites of pBAD/Myc-His B (Invitrogen). The resulting plasmids express C-terminally Myc/His-tagged VapCs in the presence of l-arabinose.
(iv) pJSB31-T7-tRNA plasmids.
The tRNA-MetV-, tRNA-Met68-, tRNA-LeuP-, and tRNA-Val16-encoding DNAs were PCR amplified from colonies of E. coli Top10. The tRNA-Met14NTHi (HI0116.1)- and tRNA-Met23NTHi (HI1281.1)-encoding DNAs were PCR amplified from colonies of NTHi 86-028NP. The PCR primers included the T7 promoter at the 5′ end of each tRNA and an additional 42 nucleotides downstream of each tRNA for 3′ processing. The PCR products were digested with PvuI and BglII and ligated into the corresponding sites of pJSB31. The resulting plasmids express each tRNA in the presence of IPTG by induction of the T7 polymerase in E. coli BL21(DE3).
All plasmid sequences were verified by DNA sequencing.
RNA isolation.
E. coli cells carrying the indicated plasmids were grown at 37°C to an A600 of ∼0.5 and induced by the addition of appropriate inducers for 1 h. Fifty milliliters of each cell culture was removed, and the cells were collected by centrifugation at 8,000 × g for 10 min at 4°C. Cells were resuspended in 0.4 ml RNA isolation buffer (RIB) (0.2 M Tris-HCl, 0.5 M NaCl, 0.01 M EDTA, 1% SDS). The cell suspension was then combined with 0.4 ml phenol-chloroform-isoamyl alcohol (50:49:1) saturated with RIB containing 0.1% 2-hydroxyquinoline (PCI-RIB), 2 μl 2-mercaptoethanol, 2 μl diethyl pyrocarbonate (DEPC), and 0.4 ml sterile beads. The cells were then lysed by bead beating at 5.0 m/s for 20 s followed by 4.5 m/s for 20 s. Debris was removed by centrifugation for 2 min at 13,000 rpm, and the supernatant was washed twice with 0.4 ml PCI-RIB. RNA was precipitated on dry ice with 0.8 ml DEPC-treated 95% ethanol. The RNA pellet was collected by centrifugation for 10 min at 13,000 rpm and washed with 70% ethanol before it was dried and resuspended in 100 μl RNase-free water.
MORE-RNA-seq.
The protocol for mapping by overexpression of an RNase in Escherichia coli and RNA-seq (MORE-RNA-seq) was modified from the work of Schifano et al. (36). First, total RNA was harvested from E. coli BW25113Δ6 as described above, after induction of either the pBAD empty vector or VapC toxins for 1 h. The RNA was then treated with 10 U XrnI (NEB) and incubated for 1 h at 37°C, followed by ligation of a preadenylated 3′ adaptor by use of T4 RNA ligase 2, truncated K227Q (NEB), at 16°C overnight. Next, the RNA was incubated with OptiKinase (Affymetrix) for 30 min at 37°C to phosphorylate the 5′ hydroxyl group and allow for ligation of the 5′ adaptor by T4 RNA ligase (NEB) at 25°C overnight. Finally, RT-PCR was performed using Superscript III reverse transcriptase (200 U; ThermoFisher) and primers specific to the 5′ and 3′ adaptors. Reverse transcription was performed by heating the ligation mixture and primer for 3 min at 85°C and then 5 min at 5°C, followed by addition of 1× first-strand buffer, a 0.5 mM concentration of each deoxynucleoside triphosphate (dNTP), 0.5 mM dithiothreitol (DTT), 40 U RiboLock RNase inhibitor (ThermoFisher), and 200 U Superscript III reverse transcriptase. The mixture was incubated for 5 min at 25°C and 60 min at 55°C and finally inactivated for 15 min at 70°C. PCR conditions consisted of 1 cycle of 98°C for 3 min, 30 cycles of 98°C for 30 s, 65°C for 30 s, and 72°C for 2 min, and a final extension step at 72°C for 5 min.
Site-directed mutagenesis.
Oligonucleotide-directed site-specific mutagenesis was carried out by a modification of the method of Fisher and Pei (41). The template plasmid was amplified in 50 μl of reaction mixture containing 10 ng DNA template, a 0.2 mM concentration of each dNTP, a 0.2 μM concentration of each primer with the appropriate base changes, 9% dimethyl sulfoxide (DMSO), and 1 U of iProof polymerase (Bio-Rad) in the supplied reaction buffer. PCR conditions consisted of 1 cycle of 98°C for 3 min, 30 cycles of 98°C for 30 s, 65°C for 30 s, and 72°C for 2 min, and a final extension step at 72°C for 5 min. The PCR product was digested with 5 U of DpnI at 37°C for 1 h. E. coli BL21(DE3) cells were transformed with 20 μl of the DpnI-treated PCR product. All constructs were verified by DNA sequencing analysis.
Bacterial growth curves.
E. coli strains were grown at 37°C overnight in M9 glucose (0.2%) medium supplemented with 100 μg/ml ampicillin and 30 μg/ml chloramphenicol. The saturated cultures were diluted to an optical density at 600 nm (OD600) of 0.025 in a 96-well plate, using M9 glucose (0.2%) medium supplemented with antibiotics and inducers as indicated. Growth in the plates was monitored in a BioTek PowerWave XS plate reader for 8 h at 37°C, with measurement of the OD600 after shaking every 15 min. Data were normalized to the starting OD600.
Northern blot analysis.
Fifteen micrograms of total RNA isolated as described above was combined with 15 μl FBX (formamide, bromophenol blue, xylene cyanol [8:1:1]) loading dye and boiled for 3 min. Samples were separated by electrophoresis on an 8% polyacrylamide-urea gel. RNA was then transferred to a GeneScreen Plus hybridization membrane by electroblotting at 8 V overnight at 4°C. The RNA was cross-linked to the membrane by use of a Stratagene UV Stratalinker 1800. Blots were incubated with rotation in a glass hybridization cylinder for 4 h at 37°C with hybridization buffer (50 mM KH2PO4, 10× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 0.001 mM Ficoll, 0.01 mM polyvinyl pyrrolidone, 6 μM bovine serum albumin [BSA], 1% SDS, 100 μl salmon sperm DNA). The temperature was then raised to 75°C before adding 5 × 105 cpm/ml of 32P-radiolabeled DNA probe. The temperature was then gradually returned to 37°C, and the probe was hybridized to the blot overnight. The membrane was then washed with wash buffer (0.1% SDS, 1× SSC) for 30 min at 42°C. For differential hybridization Northern blotting, blots were first washed for 30 min at 42°C and then for 15 min at 68°C. Northern blots were then exposed to a storage phosphor screen for 4 h, imaged with a Typhoon 9410 or Typhoon FLA 9500 imager (GE Biosciences), and quantitated with ImageQuant.
Metabolic labeling.
Saturated cultures of E. coli Top10 were diluted 1:100 in 50 ml of M9 medium supplemented with 0.2% glucose and appropriate antibiotics. Cultures were grown with shaking at 37°C to an OD600 of ∼0.2. At 0 min, toxin expression was induced with 0.2% l-arabinose. At each time point, 1 ml of culture was removed for measurement of the OD600, and 1 ml of culture was removed and incubated with 10 μCi [3H]uracil (40 Ci/mmol) or 2 μCi [35S]methionine (1,175 Ci/mmol) at 37°C for 1 min. After labeling, 1 ml cold 10% trichloroacetic acid (TCA) was added to each sample, followed by incubation on ice for 30 min. Pellets were collected by centrifugation at 4,000 rpm for 10 min and washed three times with 1 ml cold 5% TCA. The final pellet was resuspended in 1 ml 0.1 M NaOH. This was added to 9 ml Scintiverse, and radioactivity was measured using a Beckman LS 6000SC liquid scintillation counter. The amount of radioisotope incorporated was normalized to the OD600 at each time point.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Public Health Service grants GM099731 (J.S.B.), T32-GM068411 (L.R.W.), and T32AI118689 (L.R.W.) from the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00582-17.
REFERENCES
- 1.Dienemann C, Boggild A, Winther KS, Gerdes K, Brodersen DE. 2011. Crystal structure of the VapBC toxin-antitoxin complex from Shigella flexneri reveals a hetero-octameric DNA-binding assembly. J Mol Biol 414:713–722. doi: 10.1016/j.jmb.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Winther KS, Gerdes K. 2012. Regulation of enteric vapBC transcription: induction by VapC toxin dimer-breaking. Nucleic Acids Res 40:4347–4357. doi: 10.1093/nar/gks029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerdes K, Christensen SK, Lobner-Olesen A. 2005. Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microbiol 3:371–382. doi: 10.1038/nrmicro1147. [DOI] [PubMed] [Google Scholar]
- 4.Germain E, Castro-Roa D, Zenkin N, Gerdes K. 2013. Molecular mechanism of bacterial persistence by HipA. Mol Cell 52:248–254. doi: 10.1016/j.molcel.2013.08.045. [DOI] [PubMed] [Google Scholar]
- 5.Maisonneuve E, Castro-Camargo M, Gerdes K. 2013. (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 154:1140–1150. doi: 10.1016/j.cell.2013.07.048. [DOI] [PubMed] [Google Scholar]
- 6.Maisonneuve E, Gerdes K. 2014. Molecular mechanisms underlying bacterial persisters. Cell 157:539–548. doi: 10.1016/j.cell.2014.02.050. [DOI] [PubMed] [Google Scholar]
- 7.Maisonneuve E, Shakespeare LJ, Jorgensen MG, Gerdes K. 2011. Bacterial persistence by RNA endonucleases. Proc Natl Acad Sci U S A 108:13206–13211. doi: 10.1073/pnas.1100186108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Ramage HR, Connolly LE, Cox JS. 2009. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet 5:e1000767. doi: 10.1371/journal.pgen.1000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leplae R, Geeraerts D, Hallez R, Guglielmini J, Dreze P, Van Melderen L. 2011. Diversity of bacterial type II toxin-antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res 39:5513–5525. doi: 10.1093/nar/gkr131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schifano JM, Cruz JW, Vvedenskaya IO, Edifor R, Ouyang M, Husson RN, Nickels BE, Woychik NA. 2016. tRNA is a new target for cleavage by a MazF toxin. Nucleic Acids Res 44:1256–1270. doi: 10.1093/nar/gkv1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schifano JM, Edifor R, Sharp JD, Ouyang M, Konkimalla A, Husson RN, Woychik NA. 2013. Mycobacterial toxin MazF-mt6 inhibits translation through cleavage of 23S rRNA at the ribosomal A site. Proc Natl Acad Sci U S A 110:8501–8506. doi: 10.1073/pnas.1222031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vesper O, Amitai S, Belitsky M, Byrgazov K, Kaberdina AC, Engelberg-Kulka H, Moll I. 2011. Selective translation of leaderless mRNAs by specialized ribosomes generated by MazF in Escherichia coli. Cell 147:147–157. doi: 10.1016/j.cell.2011.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Zhang J, Hoeflich KP, Ikura M, Qing G, Inouye M. 2003. MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol Cell 12:913–923. doi: 10.1016/S1097-2765(03)00402-7. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Zhu L, Zhang J, Inouye M. 2005. Characterization of ChpBK, an mRNA interferase from Escherichia coli. J Biol Chem 280:26080–26088. doi: 10.1074/jbc.M502050200. [DOI] [PubMed] [Google Scholar]
- 15.Pedersen K, Zavialov AV, Pavlov MY, Elf J, Gerdes K, Ehrenberg M. 2003. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell 112:131–140. doi: 10.1016/S0092-8674(02)01248-5. [DOI] [PubMed] [Google Scholar]
- 16.Jorgensen MG, Pandey DP, Jaskolska M, Gerdes K. 2009. HicA of Escherichia coli defines a novel family of translation-independent mRNA interferases in bacteria and archaea. J Bacteriol 191:1191–1199. doi: 10.1128/JB.01013-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munoz-Gomez AJ, Lemonnier M, Santos-Sierra S, Berzal-Herranz A, Diaz-Orejas R. 2005. RNase/anti-RNase activities of the bacterial parD toxin-antitoxin system. J Bacteriol 187:3151–3157. doi: 10.1128/JB.187.9.3151-3157.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castro-Roa D, Garcia-Pino A, De Gieter S, van Nuland NA, Loris R, Zenkin N. 2013. The Fic protein Doc uses an inverted substrate to phosphorylate and inactivate EF-Tu. Nat Chem Biol 9:811–817. doi: 10.1038/nchembio.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cruz JW, Rothenbacher FP, Maehigashi T, Lane WS, Dunham CM, Woychik NA. 2014. Doc toxin is a kinase that inactivates elongation factor Tu. J Biol Chem 289:7788–7798. doi: 10.1074/jbc.M113.544429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang Y, Pogliano J, Helinski DR, Konieczny I. 2002. ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol Microbiol 44:971–979. doi: 10.1046/j.1365-2958.2002.02921.x. [DOI] [PubMed] [Google Scholar]
- 21.Miki T, Park JA, Nagao K, Murayama N, Horiuchi T. 1992. Control of segregation of chromosomal DNA by sex factor F in Escherichia coli. Mutants of DNA gyrase subunit A suppress letD (ccdB) product growth inhibition. J Mol Biol 225:39–52. [DOI] [PubMed] [Google Scholar]
- 22.Arcus VL, McKenzie JL, Robson J, Cook GM. 2011. The PIN-domain ribonucleases and the prokaryotic VapBC toxin-antitoxin array. Protein Eng Des Sel 24:33–40. doi: 10.1093/protein/gzq081. [DOI] [PubMed] [Google Scholar]
- 23.Arcus VL, Rainey PB, Turner SJ. 2005. The PIN-domain toxin-antitoxin array in mycobacteria. Trends Microbiol 13:360–365. doi: 10.1016/j.tim.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 24.Bleichert F, Granneman S, Osheim YN, Beyer AL, Baserga SJ. 2006. The PINc domain protein Utp24, a putative nuclease, is required for the early cleavage steps in 18S rRNA maturation. Proc Natl Acad Sci U S A 103:9464–9469. doi: 10.1073/pnas.0603673103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huntzinger E, Kashima I, Fauser M, Sauliere J, Izaurralde E. 2008. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 14:2609–2617. doi: 10.1261/rna.1386208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamanna AC, Karbstein K. 2009. Nob1 binds the single-stranded cleavage site D at the 3′-end of 18S rRNA with its PIN domain. Proc Natl Acad Sci U S A 106:14259–14264. doi: 10.1073/pnas.0905403106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneider C, Leung E, Brown J, Tollervey D. 2009. The N-terminal PIN domain of the exosome subunit Rrp44 harbors endonuclease activity and tethers Rrp44 to the yeast core exosome. Nucleic Acids Res 37:1127–1140. doi: 10.1093/nar/gkn1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Winther K, Tree JJ, Tollervey D, Gerdes K. 2016. VapCs of Mycobacterium tuberculosis cleave RNAs essential for translation. Nucleic Acids Res 44:9860–9871. doi: 10.1093/nar/gkw781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winther KS, Gerdes K. 2011. Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc Natl Acad Sci U S A 108:7403–7407. doi: 10.1073/pnas.1019587108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopes AP, Lopes LM, Fraga TR, Chura-Chambi RM, Sanson AL, Cheng E, Nakajima E, Morganti L, Martins EA. 2014. VapC from the leptospiral VapBC toxin-antitoxin module displays ribonuclease activity on the initiator tRNA. PLoS One 9:e101678. doi: 10.1371/journal.pone.0101678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winther KS, Brodersen DE, Brown AK, Gerdes K. 2013. VapC20 of Mycobacterium tuberculosis cleaves the sarcin-ricin loop of 23S rRNA. Nat Commun 4:2796. doi: 10.1038/ncomms3796. [DOI] [PubMed] [Google Scholar]
- 32.Walling LR, Butler JS. 2016. Structural determinants for antitoxin identity and insulation of cross talk between homologous toxin-antitoxin systems. J Bacteriol 198:3287–3295. doi: 10.1128/JB.00529-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baddal B, Muzzi A, Censini S, Calogero RA, Torricelli G, Guidotti S, Taddei AR, Covacci A, Pizza M, Rappuoli R, Soriani M, Pezzicoli A. 2015. Dual RNA-seq of nontypeable Haemophilus influenzae and host cell transcriptomes reveals novel insights into host-pathogen cross talk. mBio 6:e01765-. doi: 10.1128/mBio.01765-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren D, Walker AN, Daines DA. 2012. Toxin-antitoxin loci vapBC-1 and vapXD contribute to survival and virulence in nontypeable Haemophilus influenzae. BMC Microbiol 12:263. doi: 10.1186/1471-2180-12-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamilton B, Manzella A, Schmidt K, DiMarco V, Butler JS. 2014. Analysis of non-typeable Haemophilus influenzae VapC1 mutations reveals structural features required for toxicity and flexibility in the active site. PLoS One 9:e112921. doi: 10.1371/journal.pone.0112921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schifano JM, Vvedenskaya IO, Knoblauch JG, Ouyang M, Nickels BE, Woychik NA. 2014. An RNA-seq method for defining endoribonuclease cleavage specificity identifies dual rRNA substrates for toxin MazF-mt3. Nat Commun 5:3538. doi: 10.1038/ncomms4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cruz JW, Sharp JD, Hoffer ED, Maehigashi T, Vvedenskaya IO, Konkimalla A, Husson RN, Nickels BE, Dunham CM, Woychik NA. 2015. Growth-regulating Mycobacterium tuberculosis VapC-mt4 toxin is an isoacceptor-specific tRNase. Nat Commun 6:7480. doi: 10.1038/ncomms8480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lancaster L, Noller HF. 2005. Involvement of 16S rRNA nucleotides G1338 and A1339 in discrimination of initiator tRNA. Mol Cell 20:623–632. doi: 10.1016/j.molcel.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 39.Seong BL, RajBhandary UL. 1987. Escherichia coli formylmethionine tRNA: mutations in GGGCCC sequence conserved in anticodon stem of initiator tRNAs affect initiation of protein synthesis and conformation of anticodon loop. Proc Natl Acad Sci U S A 84:334–338. doi: 10.1073/pnas.84.2.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin G, Pavelka MS Jr, Butler JS. 2015. Structure-function analysis of VapB4 antitoxin identifies critical features of a minimal VapC4 toxin-binding module. J Bacteriol 197:1197–1207. doi: 10.1128/JB.02508-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fisher CL, Pei GK. 1997. Modification of a PCR-based site-directed mutagenesis method. Biotechniques 23:570–571, 574. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.