

ABSTRACT
In 1680, Antonie van Leeuwenhoek noted compositional differences in his oral and fecal microbiota, pioneering the study of the diversity of the human microbiome. From Leeuwenhoek's time to successful modern attempts at changing the gut microbial landscape to cure disease, there has been an exponential increase in the recognition of our resident microbes as part of ourselves. Thus, the human host and microbiome have evolved in parallel to configure a balanced system in which microbes survive in homeostasis with our innate and acquired immune systems, unless disease occurs. A growing number of studies have demonstrated a correlation between the presence/absence of microbial taxa and some of their functional molecules (i.e., genes, proteins, and metabolites) with health and disease states. Nevertheless, misleading experimental design on human subjects and the cost and lack of standardized animal models pose challenges to answering the question of whether changes in microbiome composition are cause or consequence of a certain biological state. In this review, we evaluate the state of the art of methodologies that enable the study of the gut microbiome, encouraging a change in broadly used analytic strategies by choosing effector molecules (proteins and metabolites) in combination with coding nucleic acids. We further explore microbial and effector microbial product imbalances that relate to disease and health.
KEYWORDS: gut microbiota, health, microbiome
INTRODUCTION
We are populated by microbes, which comprise what is commonly known as the microbiota. The gastrointestinal tract harbors the most diverse fraction, constituting an ecosystem with many millions of microbes from several thousand genera, which collectively weigh between 1.5 and 2 kg in a reference 70-kg human (1). From our births onward, we establish an intimate symbiotic relationship with our gut microbiota and their genes, known as the gut microbiome (2), which is inherited from one generation to the next throughout the evolution of primates (3). It is speculated that the gut microbiome has an active role in the absorption/synthesis/degradation of around 40% of molecules inside the human body (4). This exemplifies its quantitative importance in our daily life. To date, at least 105 diseases or disorders (e.g., systemic lupus erythematosus [5–7], Clostridium difficile infection [8–10], human immunodeficiency virus [HIV] infection [11–13], and colorectal cancer [14–16]), 22 different types of major covariates (e.g., aging [17–19], delivery mode [20, 21], sexual preference [22], and residence zone [23–25]), and 68 antibiotic treatments (alone [26–28] or in cocktails [29, 30]) have been associated with changes in our microbes. As our gut microbiome plays an essential role in our immune response by, for example, outcompeting and occasionally actively engaged in direct antimicrobial activity, these changes may influence our life and health condition. During development, the human microbiome is continuously responding to environmental stimuli. In a recent study in preterm infants, it was suggested that the more microbial species we shelter and the more balanced our microbial community is, the better we are able to compensate for environmentally induced alterations, such as those induced by antibiotic uptake (30). Still, this association needs to be demonstrated in a broad sense in controls and case individuals; while healthier individuals tend to have more diverse microbiomes than those with disease, this is not a characteristic that is that robust. The effects of having altered populations and different numbers and abundances of bacterial species may have still-unpredictable consequences yet to be demonstrated, e.g., at the level of our predisposition to suffer diseases. However, the concept of what constitutes a balanced (or healthy) microbiome remains to be defined. This is difficult, given the inter- and intrapersonal variation among individuals and the resistance and redundancy level of different microbial communities when confronted with alterations (31). Indeed, although in constant evolution (32), the microbiome's composition is highly resilient to changes (33, 34). Moreover, there is another factor complicating efforts to unambiguously find what a balanced microbiota is and how our health status is affected, depending on how far or close from this status we are. This factor is that health status is most often discussed at the clinical level but not often discussed in the context of the microbiome, or for that matter in the context of disease.
As has recently been reported, a correlation exists between taxonomic imbalances in the microbiome and metabolic alterations associated with them, thus raising the question of how this influences our health status (32, 35). In the present report, we briefly discuss general information about complementary technologies for gut microbiome analysis and the main findings that revealed their utility to find close associations between gut microbiome components (taxa, genes, proteins, and metabolites) and human health status.
TECHNIQUES FOR GUT MICROBIOTA ANALYSIS
During the last few decades, we have experienced great development in high-throughput techniques to unveil the impact that the gut microbes have on our health and vice versa (36). The focus of these studies has been the analysis of stool samples. Researchers and clinicians consider stool as the most convenient sample to study gut microbiota, although they have also recognized that monitoring mucosal microorganisms is crucial to understand disease progression and complications. Endoscopy is a current standard to access this material, but it is an expensive and invasive procedure with a risk of complications; this is why stool samples are commonly considered for investigations of gut microbiota in large cohort studies or clinical trials.
Scientists have mostly focused on decoding the identities of microbial taxa by sequencing the gene encoding the small subunit of the ribosome, which is considered to be an evolutionary chronometer. Using this technology, we know that there are over 5,000 species of microbes in the human body and that not all microbes inhabiting our organism are active (37). Nevertheless, taxonomic diversity does not allow us to infer gene content and function of the microbiome per se (38). Function can be identified by directly sequencing the microbial nucleic acids, then quantifying the expression level of genes and their encoded proteins (39–44), and finally by measuring the metabolites (45) produced by the proteins being expressed. A graphical summary of all technologies that can be used for assessing a complete understanding of our microbiota can be found in Fig. 1.
FIG 1.

The strong impact of today's techniques for the analysis of our microbiota. The figure illustrates the different components of the microbes living inside, on, or around our body, whose identity can be achieved by using multiple complementary techniques. As shown, the first step is high-throughput DNA and cDNA/mRNA sequencing, which enables us to quantify alteration at the level of genes which can be directly related with the total (genomics) or with the expressed functionality (transcriptomics). The next logical step is assessing the proteome and the metabolome, the truly active components in the functional hierarchy. In more detail, the microbial genes encode proteins whose expression level can be studied by (proteomic) MS analysis. Finally, active proteins participate in biochemical reactions in which a number of substrates are converted into products. All of these metabolites can be studied by metabolomics.
A selected series of methods for phenotyping (46–48), genomics, transcriptomics (49, 50), proteomics (51), and more recently, metabolomics (52) of fecal material are available. We would like to highlight that depending on each laboratory and based on the type of sample and the period for which it will be preserved until processing, it will be necessary to choose the most suitable sample collection method according to different criteria. These include the price, availability, difficulty of use, time required for management, and whether the collection method is compatible with other methods that would be applied to the same sample. In any case, before proceeding to the nucleic acid, RNA, protein, and metabolite isolation, it is recommended to collect a minimum amount of 0.4 to 1.0 g of feces and to freeze it immediately at −80°C, although higher temperatures are also acceptable (up to −20°C). Freezing is likely to prevent changes in microbial communities until microbial product extraction(s) can be performed. Quick-freezing is especially important if RNA will be extracted, because RNA is easily degraded at room temperature. Frozen samples can undergo up to four freeze-thaw cycles without significantly affecting the composition (5).
Each of the techniques mentioned above has its own pros and cons (5). For example, sequencing the gene encoding the 16S subunit of the bacterial ribosome (the 16S rRNA gene) offers a fast and cheap method to analyze the community structure; however, the short reads obtained after sequencing are not ideal for a deep phylogenetic assignation or to identify microbial lineages present in low abundance. On the other hand, genomics and transcriptomics are convenient tools for uncovering microbial diversity and to find new genes and presumptive functions associated with them, but the cost of shotgun sequencing is still higher than that of targeted amplicon sequencing, and subsequent bioinformatics analyses remain a challenge for small research institutions lacking well-established computation facilities. Moreover, the instability of mRNA and its difficult purification complicate transcriptomic analysis. On the other hand, high-throughput proteomics requires complex and time-consuming wet lab procedures, as well as specialized and difficult bioinformatics analyses. Moreover, the number of proteins commonly identified as being synthesized is in the order of a few thousand, which is much lower than the millions of genes that can routinely be identified by DNA sequencing. Finally, metabolomics has a relevant advantage compared to previous techniques, namely, that the metabolome produced by microbes is the best representative of the host-microbiota interaction (5). However, the technical challenges are more evident in metabolomics (52, 53) than in any other technique previously mentioned. For example, genes and proteins can be identified by a single platform, namely, shotgun sequencing and mass spectrometry (MS) with a coupled separation technique, respectively. However, metabolites need to be analyzed using multiple extraction methods (water, single solvents, or mixtures), and analytical platforms (MS or nuclear magnetic resonance [NMR]) coupled with multiple separation techniques liquid chromatography (LC), capillary electrophoresis (CE), or gas chromatography (GC), which are required because each analytical technique is suited to a certain fraction of the metabolome (54–57). For example, NMR is most recommended for the analysis of short-chain fatty acids, amino acids, and amines; LC-MS for bile acids, lipids, and aromatic compounds, CE-MS for amino acids, amines and carboxylic acids; and GC-MS for short-chain fatty acids, amino acids, alcohols, aldehydes, ketones, and esters. In addition, not everyone is following the Metabolomics Standards Initiatives (58), and the availability of large MS equipment remains a challenge for small research institutions. Important aspects in the metabolomics workflow, such as experimental design, sample storage, analytical quality assurance, and data treatment have been recently reviewed (5).
THE IMPACT OF DISEASES AS SEEN BY 16S RIBOSOMAL GENE SEQUENCING
Different factors that produce alterations in the microbes residing inside and on our bodies have recently been listed. Each of these disturbances differs in nature, strength, and duration, and can be categorized into three main groups: diseases, antibiotics, and others. Focusing on diseases, literature records exists for at least 105 diseases; they have been associated to changes in the total composition of our gut microbiota as revealed by sequencing the gene encoding the 16S subunit of the total bacterial ribosome (the 16S rRNA gene) (5). A review of the effect of each of these illnesses has revealed significant changes in only 231 out of the 5,000 bacterial species inhabiting our gut (5). This suggests that not all microbes residing in our body are affected by diseases, but a limited number of them are. While there are microbial lineages that are affected by only one disease, there are others that are affected by multiple diseases; some of those are affected by at least 50% of the illnesses, and therefore they are the most susceptible to disease. This can be seen in Fig. 2, which shows the major archaeal and bacterial lineages representing the human microbiome (5). Families assigned to archaea and bacteria that are commonly affected by disease are indicated in boxes, with the most susceptible ones being indicated in bold letters. These include bacteria of the orders Lactobacillales (genus Lactobacillus), Clostridiales (genus Faecalibacterium), and Bifidobacteriales (genus Bifidobacterium) that are essential for our bodies to function properly (59–63).
FIG 2.
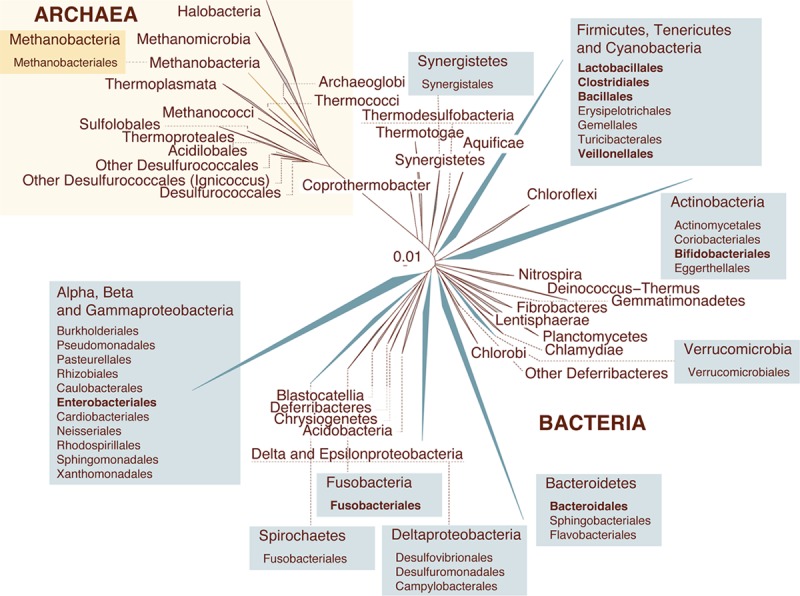
The gut microbiota is affected by diseases. The figure illustrates the all-species living tree highlighting all clades for which gut microbial isolates have been obtained (archaea are presented in yellow, and bacteria are in blue). Collapsed clades illustrate phyla, and colored boxes include the archaeal/bacterial orders. Order-level taxa that have been reported to be influenced mainly by disease are indicated in boldface. Database obtained from “The All-Species Living Tree” Project (https://www.arb-silva.de/projects/living-tree/). Information about the microbiome isolates was obtained from both the Human Microbiome Project (http://www.hmpdacc.org/; 103) and the MetaHIT (http://www.metahit.eu/; 43, 104, 105) initiatives.
We now also know that not all of the microbes inhabiting a healthy person's colon are active (5). Their identities can be established by sequencing the gene encoding the 16S subunit of the bacterial mRNA (16S rRNA). By using this technique, it has been found that bacteria of the phylum Bacteroidetes are the most dormant, dead, or quiescent microbiome fraction in a healthy state, whereas those of the phylum Firmicutes are much more metabolically active (12, 13, 40). When confronted with disease, microorganisms from taxa other than Firmicutes become activated (8, 11, 13, 64–74). Thus, bacteria of the phyla Actinobacteria, Proteobacteria, Fusobacteria, Verrucomicrobia, archaea of the phylum Euryarchaeota, and fungi of the division Basidiomycota also become the most transcriptionally active. Bacteria of the phylum Bacteroidetes were also found to become activated when confronted with disease. This clearly indicates that some microbes only become activated under certain conditions, including the presence of a disease.
It is also interesting to point out the existence of a common core microbiome, which is crucial in the maintenance of ecosystem balance (34). The core of the human microbiome has been reported to include the following species: Faecalibacterium prausnitzii, Bifidobacterium spp., Akkermansia spp., Prevotella melaninogenica, Ruminococcus bromii, and Bacteroides fragilis, all of them with a clear association with host health (34). In the future, it will be important not only to decipher the microbial taxa that are altered in the health/disease context but also to assess the conditions under which they become activated. This is relevant in order to gain insight into the diagnosis of disease through the knowledge of its effects on the microbiota. It is increasingly becoming clearer that medical treatments do not influence only the human host; the microbiome is also affected and vice versa (35).
THE IMPACT OF DISEASES AS SEEN BY GENE AND PROTEIN ANALYSIS
Although taxonomic studies are useful, the actual function of our microbiome is mainly related to gene expression and protein activity (33). As mentioned above, minor or major illnesses can have huge impacts on our microbes and, furthermore, on in our predisposition to illness. Diseases can likely influence the microbial gene and protein contents and their expression levels. By using techniques based on direct sequencing of microbial DNA or cDNA, we know which of the microbial gene functions are altered in the frame of different diseases (5). For example, gut microbial genes and proteins involved in pathogenicity, cell wall component biosynthesis, transport, bacterial translocation, and in amino acid, energy and short-chain fatty acid metabolism have been found to be altered when humans are confronted with HIV infection (13), colorectal cancer (14, 75), nonalcoholic fatty liver disease (76), type 1 diabetes (64), inflammatory bowel disease (65), and Crohn's disease (77, 78). Systemic lupus erythematosus (6), C. difficile infection (79), and obesity (41, 66, 80–82) are also associated with an overrepresentation of genes implicated in oxidative phosphorylation and phosphotransferase transport systems. Furthermore, abnormalities in genes implicated in the metabolism of carbohydrates and sugar alcohols have also been observed in the contexts of C. difficile infection (79), colorectal cancer (14), nonalcoholic fatty liver disease (76), inflammatory bowel disease (65, 83), systemic lupus erythematosus (6), and Crohn's disease (78). Genes implicated in aromatic amino acid biosynthesis are significantly underrepresented during C. difficile infection (79). This information suggests that a certain number of core functions are the most sensitive to change during disease, although more conclusive evidence is needed (5, 34), as frequently the same diseases are quoted in different context.
To the best of our knowledge, there are just a few investigations in which protein analysis by MS was undertaken in the context of different diseases (5). For example, a recent proteomic analysis has revealed that a different cocktail of glycoside hydrolases is expressed in obese than in lean individuals (81, 84). At a different scope, it was shown that a group of transport proteins from the bacterial family Succinivibrionaceae are actively synthesized during HIV infection; these proteins actively transport molecules that help reduce inflammation and immune recovery during infection (12).
THE IMPACT OF DISEASES AS SEEN BY METABOLITE FINGERPRINT
The metabolites are the final outputs of the functional hierarchy of the “omics” cascade (Fig. 1). Since they represent the phenotype, they provide the most reliable snapshot of the human-microbial exchange and health condition (5), are considered the active agents in health (5, 35), and some of them are beneficial. Some metabolites produced by microbiota are neurotransmitters that regulate the gut-brain axis (85); others help to maintain the large intestine, to control intestinal inflammation (86) or cancer cell proliferation (87). Nevertheless, their influence may be also negative, as in the expansion of enteric pathogens due to the host sugars released (88), the pathogenicity of C. difficile (67, 89), carcinogenesis in different regions of the intestine (68), or atherosclerosis (69).
To date, the metabolite content of stool samples, which is somewhat representative of the microbes colonizing a person's colon, from groups of individual confronted with any one of 37 diseases has been investigated through fingerprint metabolomics (5). Through extensive analysis of the metabolome, we now know that diseases which mostly affect the colon produce major disturbances in the gut microbial metabolism, that some microbes become activated only under certain health conditions but not under others, and that the same microbial taxa can undergo different metabolic alterations when faced with two different situations. For example, lactobacilli are less able to metabolize sugars or synthesize vitamins when faced with dietary change, whereas their ability to produce amino acids is hindered in inflammatory bowel diseases (5). This evidence, which previously was known by microbiologists from culture techniques, is now being confirmed at the genomic and metabolic levels.
Analogous to the alterations in microbial genes and protein content, the first question to answer is which metabolites are most altered in the context of the above diseases, and possibly in others yet to be investigated. To answer this, we have recently provided a panoramic view of a large set of metabolites identified by multiple analytic platforms (5). A comparative analysis revealed that not all metabolites from microbes residing in our gut are affected in disease, but a limited number of them (<0.1%) are some of the most influenced. Short-chain fatty acids are among these, and their alterations have been associated to intestinal inflammatory diseases such as Hirschsprung's-associated enterocolitis (70), irritable bowel syndrome (71–74) or ulcerative colitis (73), autoimmune diseases (7), celiac disease (90), chronic kidney disease (91), colorectal cancer (16), nonalcoholic fatty liver disease (76), or mental disorders such as autism and pervasive developmental disorder (92), to mention a few. Analogous examples can be found for other compounds, such as bile acids and amino acids (5, 35).
ANALYZING THE IMPACTS OF DISEASES: CRITICAL COMMENTS
As mentioned above, the bacterial constituents of a microbial population are identified by high-throughput analytical approaches and the changes associated to diseases. However, in many cases there is a lack of agreement between the studies, leaving the search for biomarkers (bacterial species, genes, proteins, and metabolites) in the gut sadly lacking. Maybe it reflects individual differences because of different environmental contexts, perhaps it reflects the complexity of some diseases, or perhaps it reflects biases because of technical considerations (see below). Hopefully, as follow-up research into the field of gut microbiota continues, a consensus will begin to appear. It should also be highlighted that most of the studies are based on single fecal samples collected from limited cohorts at just one time point. A better experimental design would be to collect multiple samples over time, allowing for variability within and between individuals and study of patients in relation to disease progress and clinical variables. In addition, it is recognized that environment can alter disease risk and susceptibility and thus our microbes. Analyzing differences between case individuals in multiple contexts is crucial to understand the gut components (bacteria, genes, proteins, and metabolites) implicated in disease status.
EXPLORING THE MICROBIOME REQUIRES HIGH-THROUGHPUT PROTOCOLS FOR SAMPLING AND STORAGE
With the recent progress in multiomics and bioinformatics, we can now track the total microbiota and also the active microbiota with unprecedented resolution using multiple and complementary high-throughput analytical approaches by measuring the expressed genes, proteins and metabolites. Nevertheless, there is an urgent need to standardize these methods (5) with regard to RNA processing and turnover, protein synthesis rate, and synthesis, processing, and transport of metabolites. Does sample processing induce any potential bias in the results? Few studies really take this into consideration with sufficient detail. For example, storage of a stool sample in different preservation reagents (i.e., PowerMicrobiome kit, RNA Later, or RNA Protect) introduces a bias into the mRNA profile which may also vary from person to person depending, among other factors, on the relative abundance of hard-to-lyse species versus the relative abundance of species that are easier to lyse (50). The preservation reagents have also been found to influence the transcripts for, e.g., the cluster of orthologous groups (COG) category of “carbohydrate metabolism and transport” (50). Other examples, which consider the effect of sample storage and shipping, have been mentioned above. Some studies recommend immediate freezing to prevent changes in microbial communities (5). Others recommend adding RNA Later as preservation reagent when stool samples need to be shipped to the laboratory at ambient temperature, since the RNA will be stable for up to 6 days. When shipping can be realized within 24 h or samples can be shipped on ice, RNA Protect may represent a better alternative as preservation reagent, since the introduced bias is smaller (50). The take-home message for cohort studies or clinical trials is that investigations directed to unambiguously defining the best protocols for sampling and stabilization of samples may be an area where more research is needed. This should be of practical importance for both patients and hospital staff in order to guarantee sample quality. Focusing on the metabolites (56), it is also crucial to maximize their stability, which is mainly affected by storage duration, temperature, and freeze-thaw cycles. In that sense, it can be pointed out that a processed sample is more stable compared to crude feces. Ideally, the metabolome should be extracted from a homogenized sample within 24 h after collection and then stored at −80°C; however, it can be impossible in the case of large cohorts where samples cannot be simultaneously collected from the donors and processed. In any case it is better to process all the samples at the same time in order to minimize interbatch variability. Finally, in a multiplatform analysis, it is recommended to aliquot the material, avoiding more than one freeze-thaw cycle.
BIGGEST CHALLENGE IN MICROBIOME RESEARCH: DISTINGUISHING ASSOCIATION FROM CAUSATION
As described above, a clear link exists between the cooccurrence of dysbiosis and multiple diseases; however, in the majority of the cases there is insufficient evidence of a cause-effect relationship (5). In the last decade, a multitude of studies have been conducted to determine the existing deviations in diversity, community structure, and its metabolic landscape in case and control individuals. Among them, we can cite ulcerative colitis, intestinal inflammatory diseases, Crohn's disease, enterocolitis, gastrointestinal cancer, etc. (64, 65, 68–70). These relationships have also been explored in disorders that affect organs or functions whose relation with any microbial community is only indirect, such as autism, depression, Parkinson's disease, and diabetes, to cite just a few (5, 67).
Once the taxonomic dysbiosis has been deciphered and the differences in genes, proteins, and metabolites are determined, the opportunity arises to correct those alterations affecting the microbiome. This correction to reestablish the normal functional abundances requires determining with higher precision the taxonomic groups that are needed to promote or to inhibit a function. While this may seem obvious attending to the taxonomic differences between case and controls, it is not so much to the extent that the contribution of each taxon to the abundance of a specific function may not be indicative of its contribution to a specific functional deviation. In other words, a taxon may be highly abundant in case and controls and contribute to a specific function but may not be responsible for the deviations related to a disease. In general, this means that whereas the abundance of a gene can be seen as the sum of all contributions at taxonomic level, an analysis is needed that defines strictly how to spread among taxa the differential abundances of each gene and the activity supported by the proteins encoded by each gene. Today, integration of both types of data, taxonomic and functional, is still lacking in the study of microbiota to unambiguously identify cause-effect relationships and to define the contribution of specific microbes and functions supported by specific genes, proteins, and metabolites to disease.
In this line, Manor and Borenstein (93) have recently developed the FishTaco software, which allows tracking of the functional deviation of a microbiome in the context of a disease and conversion of these deviations into deviation in the taxonomic composition. That allows clarification of which taxa positively or negatively influence the observed functional deviation. And, what is more, for each of the altered functions, different taxa may exist in case or control individuals that attenuate the alterations in a positive or negative way. The quantification of the contribution of each altered taxon will allow, from one side, unveiling which of these alterations are the cause or consequence of a disease and, from the other side, the design of therapies directed to correct these alterations in diseases for which a clear evidence of causality exists. This will require the exploration of imbalances in microbiome composition and effector microbial products to understand the strong influence of microbiome-dependent metabolic pathways on critical aspects of pathogenesis.
CONCLUDING REMARKS AND FUTURE PERSPECTIVES
Microbiome profiling by targeting of the hypervariable regions of the 16S subunit of the microbial ribosome (generated from DNA or RNA) is a widespread analytic tool to begin decoding the identities of microbes residing inside our bodies. In spite of the existence of differences with direct shotgun sequencing of microbial nucleic acids, 16S ribosomal gene sequencing can provide a comprehensive comparative tool to understand how our relationship with them is altered. The presence of curated repositories for this phylogenetic marker (Greengenes, http://greengenes.lbl.gov/; Ribosomal Database Project, https://rdp.cme.msu.edu/; EZBioCloud, http://www.ezbiocloud.net/; or the SILVA database, https://www.arb-silva.de/) constitutes one of the main advantages for its use. The lack of resolution using short sequences has been stated previously, but new approaches using a combination of 16S ribosomal gene hypervariable regions have been proposed to enhance the accuracy of the technique (http://acai.igb.uiuc.edu/bio/tutorial.html). Nevertheless, amplification of a DNA-encoded gene, such as the 16S ribosomal gene, or of any other of the at least eight million microbial genes that coexist in our organism alongside the 23,000 human genes, does not ensure that they are being expressed. Studies based on selection of RNA followed by amplification of the target 16S ribosomal gene region from the corresponding cDNA are scarce. A recently published protocol explores the possibility of performing direct 16S rRNA-seq from bacterial communities using a PCR-independent approach (94). Most studies reporting microbiome nucleic acid profiling are conducted without previous selection of microbial cells, that is, by using raw fecal or tissue material (5). In contrast, when a negative selection of microbial cells in subsequent filtering procedures is avoided, the host's DNA is normally extracted together with microbial DNA. The use of cell preenrichment methods by multiple low-speed centrifugation steps (5) or the use of a gradient media such as Nycodenz (95) has been proved to yield 1010 viable bacteria per 2 g of feces without affecting the original microbial composition, as assessed by shotgun metagenomics (96). One of the challenges to be addressed in the future is the creation of a comprehensive and reproducible bioinformatics workflow (Fig. 3) to efficiently integrate such data sets and those generated by the analysis of effector molecules (proteins and metabolites) by MS analysis. Through the reconstruction of the so-called genome-scale metabolic models (GEMs), genome sequences and similarity-based annotations are correlated with biochemical reactions based on gene-protein associations of the target organism. The stoichiometric coefficients of a given biochemical reaction are then used to construct a stoichiometric matrix, in which rows and columns consist of all metabolites involved in the network, and in which the values correspond to conversion of substrates and generation of products. This formulation of the different microbial, genetic, protein, and metabolite networks constitutes a key feature for computational tool development, which will pave the path toward integrated medicine and microbiota tinkering in the near future (97).
FIG 3.

A focus on metabolomics as the methodology of choice for direct pathway reconstruction. Microbiome alterations in health and disease through sequencing of the 16S rRNA gene might not represent real changes in the active fraction (the 16S rRNA gene that is being transcribed) of microbes that change under a certain condition. Nevertheless, integration of 16S rRNA gene (rDNA) sequencing with shotgun metagenome sequencing and metaproteomics provides a more precise understanding of the function of the system. As an alternative solution, metabolomics alone could provide the pieces missing in pathway reconstruction and allow direct interference of function. Further integration with shotgun metagenomics, metaproteomics, or target gene amplicon sequencing would incorporate all layers of complexity in the biological system, allowing deep understanding of the mechanisms within.
We must identify all microbial members of the resident microbiome and discover how they react to factors influencing well-being by integrating multiple data sets. This should be crucial for future microbiota transplantation (35), an efficient therapeutic tool in vivo to restore disease-related alterations that cannot be restored by common therapeutic methods. Fecal microbiota transplantation (FMT) has consistently demonstrated high success rates against recurrent C. difficile infection in hospitals since the early 1950s. Contemporary research is trying to decipher the dynamics of FMT. For example, a recent publication tracked the composition of the gut microbiome through analysis using shotgun metagenomics of compositions of donor and recipient feces after the transplantation event. About 22% of the metagenome-assembled genomes (MAGs) with high level of completeness from the donor successfully colonized and prevailed in the recipients for at least 8 weeks (98). In order to avoid undesirable outcomes, such as weight gain after fecal microbiota transplantation (99), controlled transplants of the microbiota by using defined microbial cocktails have been suggested in the literature as a synthetic stool substitute (microbiota ecosystem therapeutics, MET) (99–101). Research in this area is promising, since the advantages of this approach are numerous, including standardization of the microbial composition and enhanced stability of the inoculum to be transferred, pathogen exclusion, or inclusion of antibiotic-sensitive organisms only. This is just one example of how the integration of multiple data sets may help treat recurrent infections.
In summary, as recently mentioned in the literature, the human microbiome should be viewed as a complex landscape, and its modulation with therapeutic purposes should account for all of this complexity (34). Future efforts should be directed toward the unification and optimization of metabolomics strategies in microbiome research, as this methodology provides an accurate snapshot of the system's function. The phenotyping era is mature enough to open new research avenues that will enable a better understanding of the mechanisms from a systems microbiology perspective (102), which will greatly impact modern medicine.
ACKNOWLEDGMENTS
This work was supported by the Spanish Ministry of Economy, Industry and Competitiveness (CTQ2014-55279-R and BIO2014-54494-R), from which C.B., D.R., and M.F. acknowledge funding.
We express our gratitude to Renae R. Geier for her support in the review of English grammatical correctness.
REFERENCES
- 1.Falony G, Joossens M, Vieira-Silva S, Wang J, Darzi Y, Faust K, Kurilshikov A, Bonder MJ, Valles-Colomer M, Vandeputte D, Tito RY, Chaffron S, Rymenans L, Verspecht C, De Sutter L, Lima-Mendez G, D'Hoe K, Jonckheere K, Homola D, Garcia R, Tigchelaar EF, Eeckhaudt L, Fu J, Henckaerts L, Zhernakova A, Wijmenga C, Raes J. 2016. Population-level analysis of gut microbiome variation. Science 352:560–564. doi: 10.1126/science.aad3503. [DOI] [PubMed] [Google Scholar]
- 2.Sender R, Fuchs S, Milo R. 2016. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol 14:e1002533. doi: 10.1371/journal.pbio.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moeller AH, Caro-Quintero A, Mjungu D, Georgiev AV, Lonsdorf EV, Muller MN, Pusey AE, Peeters M, Hahn BH, Ochman H. 2016. Cospeciation of gut microbiota with hominids. Science 353:380–382. doi: 10.1126/science.aaf3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vernocchi P, Del Chierico F, Putignani L. 2016. Gut microbiota profiling: metabolomics based approach to unravel compounds affecting human health. Front Microbiol 7:1144. doi: 10.3389/fmicb.2016.01144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rojo D, Mendez-Garcia C, Raczkowska BA, Bargiela R, Moya A, Ferrer M, Barbas C. 2017. Exploring the human microbiome from multiple perspectives: factors altering its composition and function. FEMS Microbiol Rev 41:453–478. doi: 10.1093/femsre/fuw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hevia A, Milani C, Lopez P, Cuervo A, Arboleya S, Duranti S, Turroni F, Gonzalez S, Suarez A, Gueimonde M, Ventura M, Sanchez B, Margolles A. 2014. Intestinal dysbiosis associated with systemic lupus erythematosus. mBio 5:e01548-. doi: 10.1128/mBio.01548-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rojo D, Hevia A, Bargiela R, Lopez P, Cuervo A, Gonzalez S, Suarez A, Sanchez B, Martinez-Martinez M, Milani C, Ventura M, Barbas C, Moya A, Suarez A, Margolles A, Ferrer M. 2015. Ranking the impact of human health disorders on gut metabolism: systemic lupus erythematosus and obesity as study cases. Sci Rep 5:8310. doi: 10.1038/srep08310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rojo D, Gosalbes MJ, Ferrari R, Perez-Cobas AE, Hernandez E, Oltra R, Buesa J, Latorre A, Barbas C, Ferrer M, Moya A. 2015. Clostridium difficile heterogeneously impacts intestinal community architecture but drives stable metabolome responses. ISME J 9:2206–2220. doi: 10.1038/ismej.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dzunkova M, D'Auria G, Moya A. 2015. Direct sequencing of human gut virome fractions obtained by flow cytometry. Front Microbiol 6:955. doi: 10.3389/fmicb.2015.00955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dzunkova M, Moya A, Vazquez-Castellanos JF, Artacho A, Chen X, Kelly C, D'Auria G. 2016. Active and secretory IgA-coated bacterial fractions elucidate dysbiosis in Clostridium difficile infection. mSphere 1:e00101-. doi: 10.1128/mSphere.00101-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mutlu EA, Keshavarzian A, Losurdo J, Swanson G, Siewe B, Forsyth C, French A, Demarais P, Sun Y, Koenig L, Cox S, Engen P, Chakradeo P, Abbasi R, Gorenz A, Burns C, Landay A. 2014. A compositional look at the human gastrointestinal microbiome and immune activation parameters in HIV infected subjects. PLoS Pathog 10:e1003829. doi: 10.1371/journal.ppat.1003829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serrano-Villar S, Rojo D, Martinez-Martinez M, Deusch S, Vazquez-Castellanos JF, Bargiela R, Sainz T, Vera M, Moreno S, Estrada V, Gosalbes MJ, Latorre A, Seifert J, Barbas C, Moya A, Ferrer M. 2016. Gut bacteria metabolism impacts immune recovery in HIV-infected individuals. EBioMedicine 8:203–216. doi: 10.1016/j.ebiom.2016.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vazquez-Castellanos JF, Serrano-Villar S, Latorre A, Artacho A, Ferrus ML, Madrid N, Vallejo A, Sainz T, Martinez-Botas J, Ferrando-Martinez S, Vera M, Dronda F, Leal M, Del Romero J, Moreno S, Estrada V, Gosalbes MJ, Moya A. 2015. Altered metabolism of gut microbiota contributes to chronic immune activation in HIV-infected individuals. Mucosal Immunol 8:760–772. doi: 10.1038/mi.2014.107. [DOI] [PubMed] [Google Scholar]
- 14.Zeller G, Tap J, Voigt AY, Sunagawa S, Kultima JR, Costea PI, Amiot A, Bohm J, Brunetti F, Habermann N, Hercog R, Koch M, Luciani A, Mende DR, Schneider MA, Schrotz-King P, Tournigand C, Tran Van Nhieu J, Yamada T, Zimmermann J, Benes V, Kloor M, Ulrich CM, von Knebel Doeberitz M, Sobhani I, Bork P. 2014. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol Syst Biol 10:766. doi: 10.15252/msb.20145645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weir TL, Manter DK, Sheflin AM, Barnett BA, Heuberger AL, Ryan EP. 2013. Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS One 8:e70803. doi: 10.1371/journal.pone.0070803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goedert JJ, Sampson JN, Moore SC, Xiao Q, Xiong X, Hayes RB, Ahn J, Shi J, Sinha R. 2014. Fecal metabolomics: assay performance and association with colorectal cancer. Carcinogenesis 35:2089–2096. doi: 10.1093/carcin/bgu131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, Heath AC, Warner B, Reeder J, Kuczynski J, Caporaso JG, Lozupone CA, Lauber C, Clemente JC, Knights D, Knight R, Gordon JI. 2012. Human gut microbiome viewed across age and geography. Nature 486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Festi D, Schiumerini R, Eusebi LH, Marasco G, Taddia M, Colecchia A. 2014. Gut microbiota and metabolic syndrome. World J Gastroenterol 20:16079–16094. doi: 10.3748/wjg.v20.i43.16079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Claesson MJ, Cusack S, O'Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, Marchesi JR, Falush D, Dinan T, Fitzgerald G, Stanton C, van Sinderen D, O'Connor M, Harnedy N, O'Connor K, Henry C, O'Mahony D, Fitzgerald AP, Shanahan F, Twomey C, Hill C, Ross RP, O'Toole PW. 2011. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci U S A 108(Suppl 1):S4586–S91. doi: 10.1073/pnas.1000097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu CY, Ni YH. 2015. Gut microbiota and the development of pediatric diseases. J Gastroenterol 50:720–726. doi: 10.1007/s00535-015-1082-z. [DOI] [PubMed] [Google Scholar]
- 21.Goedert JJ, Hua X, Yu G, Shi J. 2014. Diversity and composition of the adult fecal microbiome associated with history of cesarean birth or appendectomy: analysis of the American Gut Project. EBioMedicine 1:167–172. doi: 10.1016/j.ebiom.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noguera-Julian M, Rocafort M, Guillen Y, Rivera J, Casadella M, Nowak P, Hildebrand F, Zeller G, Parera M, Bellido R, Rodriguez C, Carrillo J, Mothe B, Coll J, Bravo I, Estany C, Herrero C, Saz J, Sirera G, Torrela A, Navarro J, Crespo M, Brander C, Negredo E, Blanco J, Guarner F, Calle ML, Bork P, Sonnerborg A, Clotet B, Paredes R. 2016. Gut microbiota linked to sexual preference and HIV infection. EBioMedicine 5:135–146. doi: 10.1016/j.ebiom.2016.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yap GC, Chee KK, Hong PY, Lay C, Satria CD, Sumadiono Soenarto Y, Haksari EL, Aw M, Shek LP, Chua KY, Zhao Y, Leow D, Lee BW. 2011. Evaluation of stool microbiota signatures in two cohorts of Asian (Singapore and Indonesia) newborns at risk of atopy. BMC Microbiol 11:193. doi: 10.1186/1471-2180-11-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kemppainen KM, Ardissone AN, Davis-Richardson AG, Fagen JR, Gano KA, Leon-Novelo LG, Vehik K, Casella G, Simell O, Ziegler AG, Rewers MJ, Lernmark A, Hagopian W, She JX, Krischer JP, Akolkar B, Schatz DA, Atkinson MA, Triplett EW; TEDDY Study Group. 2015. Early childhood gut microbiomes show strong geographic differences among subjects at high risk for type 1 diabetes. Diabetes Care 38:329–332. doi: 10.2337/dc14-0850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Del Chierico F, Vernocchi P, Dallapiccola B, Putignani L. 2014. Mediterranean diet and health: food effects on gut microbiota and disease control. Int J Mol Sci 15:11678–11699. doi: 10.3390/ijms150711678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iapichino G, Callegari ML, Marzorati S, Cigada M, Corbella D, Ferrari S, Morelli L. 2008. Impact of antibiotics on the gut microbiota of critically ill patients. J Med Microbiol 57:1007–1014. doi: 10.1099/jmm.0.47387-0. [DOI] [PubMed] [Google Scholar]
- 27.Soares GM, Teles F, Starr JR, Feres M, Patel M, Martin L, Teles R. 2015. Effects of azithromycin, metronidazole, amoxicillin, and metronidazole plus amoxicillin on an in vitro polymicrobial subgingival biofilm model. Antimicrob Agents Chemother 59:2791–2798. doi: 10.1128/AAC.04974-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perez-Cobas AE, Artacho A, Knecht H, Ferrus ML, Friedrichs A, Ott SJ, Moya A, Latorre A, Gosalbes MJ. 2013. Differential effects of antibiotic therapy on the structure and function of human gut microbiota. PLoS One 8:e80201. doi: 10.1371/journal.pone.0080201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perez-Cobas AE, Gosalbes MJ, Friedrichs A, Knecht H, Artacho A, Eismann K, Otto W, Rojo D, Bargiela R, von Bergen M, Neulinger SC, Daumer C, Heinsen FA, Latorre A, Barbas C, Seifert J, dos Santos VM, Ott SJ, Ferrer M, Moya A. 2013. Gut microbiota disturbance during antibiotic therapy: a multi-omic approach. Gut 62:1591–1601. doi: 10.1136/gutjnl-2012-303184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greenwood C, Morrow AL, Lagomarcino AJ, Altaye M, Taft DH, Yu Z, Newburg DS, Ward DV, Schibler KR. 2014. Early empiric antibiotic use in preterm infants is associated with lower bacterial diversity and higher relative abundance of Enterobacter. J Pediatr 165:23–29. doi: 10.1016/j.jpeds.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ding T, Schloss PD. 2014. Dynamics and associations of microbial community types across the human body. Nature 509:357–360. doi: 10.1038/nature13178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goodrich JK, Davenport ER, Waters JL, Clark AG, Ley RE. 2016. Cross-species comparisons of host genetic associations with the microbiome. Science 352:532–535. doi: 10.1126/science.aad9379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moya A, Ferrer M. 2016. Functional redundancy-induced stability of gut microbiota subjected to disturbance. Trends Microbiol 24:402–413. doi: 10.1016/j.tim.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 34.Shetty SA, Hugenholtz F, Lahti L, Smidt H, de Vos WM. 2017. Intestinal microbiome landscaping: insight in community assemblage and implications for microbial modulation strategies. FEMS Microbiol Rev 41:182–199. doi: 10.1093/femsre/fuw045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suez J, Elinav E. 2017. The path towards microbiome-based metabolite treatment. Nat Microbiol 2:17075. doi: 10.1038/nmicrobiol.2017.75. [DOI] [PubMed] [Google Scholar]
- 36.Tuddenham S, Sears CL. 2015. The intestinal microbiome and health. Curr Opin Infect Dis 28:464–470. doi: 10.1097/QCO.0000000000000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shin NR, Whon TW, Bae JW. 2015. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol 33:496–503. doi: 10.1016/j.tibtech.2015.06.011. [DOI] [PubMed] [Google Scholar]
- 38.Bikel S, Valdez-Lara A, Cornejo-Granados F, Rico K, Canizales-Quinteros S, Soberon X, Del Pozo-Yauner L, Ochoa-Leyva A. 2015. Combining metagenomics, metatranscriptomics and viromics to explore novel microbial interactions: towards a systems-level understanding of human microbiome. Comput Struct Biotechnol J 13:390–401. doi: 10.1016/j.csbj.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aguiar-Pulido V, Huang W, Suarez-Ulloa V, Cickovski T, Mathee K, Narasimhan G. 2016. Metagenomics, metatranscriptomics, and metabolomics approaches for microbiome analysis. Evol Bioinform Online 12(Suppl 1):5–16. doi: 10.4137/EBO.S36436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gosalbes MJ, Durban A, Pignatelli M, Abellan JJ, Jimenez-Hernandez N, Perez-Cobas AE, Latorre A, Moya A. 2011. Metatranscriptomic approach to analyze the functional human gut microbiota. PLoS One 6:e17447. doi: 10.1371/journal.pone.0017447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greenblum S, Turnbaugh PJ, Borenstein E. 2012. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc Natl Acad Sci U S A 109:594–599. doi: 10.1073/pnas.1116053109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lepage P, Leclerc MC, Joossens M, Mondot S, Blottiere HM, Raes J, Ehrlich D, Dore J. 2013. A metagenomic insight into our gut's microbiome. Gut 62:146–158. doi: 10.1136/gutjnl-2011-301805. [DOI] [PubMed] [Google Scholar]
- 43.Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, Arumugam M, Kultima JR, Prifti E, Nielsen T, Juncker AS, Manichanh C, Chen B, Zhang W, Levenez F, Wang J, Xu X, Xiao L, Liang S, Zhang D, Zhang Z, Chen W, Zhao H, Al-Aama JY, Edris S, Yang H, Wang J, Hansen T, Nielsen HB, Brunak S, Kristiansen K, Guarner F, Pedersen O, Dore J, Ehrlich SD, Meta HITC, Bork P, Wang J, Meta HITC. 2014. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol 32:834–841. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 44.Bashiardes S, Zilberman-Schapira G, Elinav E. 2016. Use of metatranscriptomics in microbiome research. Bioinform Biol Insights 10:19–25. doi: 10.4137/BBI.S34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beger RD, Dunn W, Schmidt MA, Gross SS, Kirwan JA, Cascante M, Brennan L, Wishart DS, Oresic M, Hankemeier T, Broadhurst DI, Lane AN, Suhre K, Kastenmuller G, Sumner SJ, Thiele I, Fiehn O, Kaddurah-Daouk R. 2016. Metabolomics enables precision medicine: “A White Paper, Community Perspective.” Metabolomics 12:149. doi: 10.1007/s11306-016-1094-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Logares R, Sunagawa S, Salazar G, Cornejo-Castillo FM, Ferrera I, Sarmento H, Hingamp P, Ogata H, de Vargas C, Lima-Mendez G, Raes J, Poulain J, Jaillon O, Wincker P, Kandels-Lewis S, Karsenti E, Bork P, Acinas SG. 2014. Metagenomic 16S rDNA Illumina tags are a powerful alternative to amplicon sequencing to explore diversity and structure of microbial communities. Environ Microbiol 16:2659–2671. doi: 10.1111/1462-2920.12250. [DOI] [PubMed] [Google Scholar]
- 47.Takahashi S, Tomita J, Nishioka K, Hisada T, Nishijima M. 2014. Development of a prokaryotic universal primer for simultaneous analysis of bacteria and archaea using next-generation sequencing. PLoS One 9:e105592. doi: 10.1371/journal.pone.0105592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jovel J, Patterson J, Wang W, Hotte N, O'Keefe S, Mitchel T, Perry T, Kao D, Mason AL, Madsen KL. 2016. Characterization of the gut microbiome using 16S or shotgun metagenomics. Front Microbiol 7:459. doi: 10.3389/fmicb.2016.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hampton-Marcell JT, Moormann SM, Owens SM, Gilbert JA. 2013. Preparation and metatranscriptomic analyses of host-microbe systems. Methods Enzymol 531:169–185. doi: 10.1016/B978-0-12-407863-5.00009-5. [DOI] [PubMed] [Google Scholar]
- 50.Reck M, Tomasch J, Deng Z, Jarek M, Husemann P, Wagner-Dobler I, Consortium C. 2015. Stool metatranscriptomics: a technical guideline for mRNA stabilisation and isolation. BMC Genomics 16:494. doi: 10.1186/s12864-015-1694-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tanca A, Palomba A, Pisanu S, Addis MF, Uzzau S. 2015. Enrichment or depletion? The impact of stool pretreatment on metaproteomic characterization of the human gut microbiota. Proteomics 15:3474–3485. doi: 10.1002/pmic.201400573. [DOI] [PubMed] [Google Scholar]
- 52.Smirnov KS, Maier TV, Walker A, Heinzmann SS, Forcisi S, Martinez I, Walter J, Schmitt-Kopplin P. 2016. Challenges of metabolomics in human gut microbiota research. Int J Med Microbiol 306:266–279. doi: 10.1016/j.ijmm.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 53.Stasulli NM, Shank EA. 2016. Profiling the metabolic signals involved in chemical communication between microbes using imaging mass spectrometry. FEMS Microbiol Rev 40:807–813. doi: 10.1093/femsre/fuw032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bargiela R, Mapelli F, Rojo D, Chouaia B, Tornes J, Borin S, Richter M, Del Pozo MV, Cappello S, Gertler C, Genovese M, Denaro R, Martinez-Martinez M, Fodelianakis S, Amer RA, Bigazzi D, Han X, Chen J, Chernikova TN, Golyshina OV, Mahjoubi M, Jaouanil A, Benzha F, Magagnini M, Hussein E, Al-Horani F, Cherif A, Blaghen M, Abdel-Fattah YR, Kalogerakis N, Barbas C, Malkawi HI, Golyshin PN, Yakimov MM, Daffonchio D, Ferrer M. 2015. Bacterial population and biodegradation potential in chronically crude oil-contaminated marine sediments are strongly linked to temperature. Sci Rep 5:11651. doi: 10.1038/srep11651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xie G, Zhang S, Zheng X, Jia W. 2013. Metabolomics approaches for characterizing metabolic interactions between host and its commensal microbes. Electrophoresis 34:2787–2798. [DOI] [PubMed] [Google Scholar]
- 56.Gratton J, Phetcharaburanin J, Mullish BH, Williams HR, Thursz M, Nicholson JK, Holmes E, Marchesi JR, Li JV. 2016. Optimized sample handling strategy for metabolic profiling of human feces. Anal Chem 88:4661–4668. doi: 10.1021/acs.analchem.5b04159. [DOI] [PubMed] [Google Scholar]
- 57.Godzien J, Alonso-Herranz V, Barbas C, Armitage EG. 2015. Controlling the quality of metabolomics data: new strategies to get the best out of the QC sample. Metabolomics 11:518–528. doi: 10.1007/s11306-014-0712-4. [DOI] [Google Scholar]
- 58.Salek RM, Steinbeck C, Viant MR, Goodacre R, Dunn WB. 2013. The role of reporting standards for metabolite annotation and identification in metabolomic studies. Gigascience 2:13. doi: 10.1186/2047-217X-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, Grangette C, Vasquez N, Pochart P, Trugnan G, Thomas G, Blottiere HM, Dore J, Marteau P, Seksik P, Langella P. 2008. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A 105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sarrabayrouse G, Bossard C, Chauvin JM, Jarry A, Meurette G, Quevrain E, Bridonneau C, Preisser L, Asehnoune K, Labarriere N, Altare F, Sokol H, Jotereau F. 2014. CD4CD8αα lymphocytes, a novel human regulatory T cell subset induced by colonic bacteria and deficient in patients with inflammatory bowel disease. PLoS Biol 12:e1001833. doi: 10.1371/journal.pbio.1001833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laval L, Martin R, Natividad JN, Chain F, Miquel S, Desclee de Maredsous C, Capronnier S, Sokol H, Verdu EF, van Hylckama Vlieg JE, Bermudez-Humaran LG, Smokvina T, Langella P. 2015. Lactobacillus rhamnosus CNCM I-3690 and the commensal bacterium Faecalibacterium prausnitzii A2-165 exhibit similar protective effects to induced barrier hyper-permeability in mice. Gut Microbes 6:1–9. doi: 10.4161/19490976.2014.990784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fujimura KE, Slusher NA, Cabana MD, Lynch SV. 2010. Role of the gut microbiota in defining human health. Expert Rev Anti Infect Ther 8:435–454. doi: 10.1586/eri.10.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O'Callaghan A, van Sinderen D. 2016. Bifidobacteria and their role as members of the human gut microbiota. Front Microbiol 7:925. doi: 10.3389/fmicb.2016.00925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Consortium HMP. 2012. Structure, function and diversity of the healthy human microbiome. Nature 486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, Bousvaros A, Korzenik J, Sands BE, Xavier RJ, Huttenhower C. 2012. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 13:R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Papathanasopoulos A, Camilleri M. 2010. Dietary fiber supplements: effects in obesity and metabolic syndrome and relationship to gastrointestinal functions. Gastroenterology 138:65–72 e1–2. doi: 10.1053/j.gastro.2009.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shen A. 2015. A Gut Odyssey: The Impact of the Microbiota on Clostridium difficile spore formation and germination. PLoS Pathog 11:e1005157. doi: 10.1371/journal.ppat.1005157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kibe R, Kurihara S, Sakai Y, Suzuki H, Ooga T, Sawaki E, Muramatsu K, Nakamura A, Yamashita A, Kitada Y, Kakeyama M, Benno Y, Matsumoto M. 2014. Upregulation of colonic luminal polyamines produced by intestinal microbiota delays senescence in mice. Sci Rep 4:4548. doi: 10.1038/srep04548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tang WH, Hazen SL. 2014. The contributory role of gut microbiota in cardiovascular disease. J Clin Invest 124:4204–4211. doi: 10.1172/JCI72331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Demehri FR, Frykman PK, Cheng Z, Ruan C, Wester T, Nordenskjold A, Kawaguchi A, Hui TT, Granstrom AL, Funari V, Teitelbaum DH; HAEC Collaborative Research Group. 2016. Altered fecal short chain fatty acid composition in children with a history of Hirschsprung-associated enterocolitis. J Pediatr Surg 51:81–86. doi: 10.1016/j.jpedsurg.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dior M, Delagreverie H, Duboc H, Jouet P, Coffin B, Brot L, Humbert L, Trugnan G, Seksik P, Sokol H, Rainteau D, Sabate JM. 2016. Interplay between bile acid metabolism and microbiota in irritable bowel syndrome. Neurogastroenterol Motil 28:1330–1340. doi: 10.1111/nmo.12829. [DOI] [PubMed] [Google Scholar]
- 72.Duboc H, Rainteau D, Rajca S, Humbert L, Farabos D, Maubert M, Grondin V, Jouet P, Bouhassira D, Seksik P, Sokol H, Coffin B, Sabate JM. 2012. Increase in fecal primary bile acids and dysbiosis in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol Motil 24:513–520. doi: 10.1111/j.1365-2982.2012.01893.x. [DOI] [PubMed] [Google Scholar]
- 73.Le Gall G, Noor SO, Ridgway K, Scovell L, Jamieson C, Johnson IT, Colquhoun IJ, Kemsley EK, Narbad A. 2011. Metabolomics of fecal extracts detects altered metabolic activity of gut microbiota in ulcerative colitis and irritable bowel syndrome. J Proteome Res 10:4208–4218. doi: 10.1021/pr2003598. [DOI] [PubMed] [Google Scholar]
- 74.Ponnusamy K, Choi JN, Kim J, Lee SY, Lee CH. 2011. Microbial community and metabolomic comparison of irritable bowel syndrome faeces. J Med Microbiol 60:817–827. doi: 10.1099/jmm.0.028126-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vogtmann E, Hua X, Zeller G, Sunagawa S, Voigt AY, Hercog R, Goedert JJ, Shi J, Bork P, Sinha R. 2016. colorectal cancer and the human gut microbiome: reproducibility with whole-genome shotgun sequencing. PLoS One 11:e0155362. doi: 10.1371/journal.pone.0155362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Michail S, Lin M, Frey MR, Fanter R, Paliy O, Hilbush B, Reo NV. 2015. Altered gut microbial energy and metabolism in children with non-alcoholic fatty liver disease. FEMS Microbiol Ecol 91:1–9. doi: 10.1093/femsec/fiu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Erickson AR, Cantarel BL, Lamendella R, Darzi Y, Mongodin EF, Pan C, Shah M, Halfvarson J, Tysk C, Henrissat B, Raes J, Verberkmoes NC, Fraser CM, Hettich RL, Jansson JK. 2012. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn's disease. PLoS One 7:e49138. doi: 10.1371/journal.pone.0049138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Quince C, Ijaz UZ, Loman N, Eren AM, Saulnier D, Russell J, Haig SJ, Calus ST, Quick J, Barclay A, Bertz M, Blaut M, Hansen R, McGrogan P, Russell RK, Edwards CA, Gerasimidis K. 2015. Extensive modulation of the fecal metagenome in children with Crohn's disease during exclusive enteral nutrition. Am J Gastroenterol 110:1718–1729; quiz 1730. doi: 10.1038/ajg.2015.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Perez-Cobas AE, Artacho A, Ott SJ, Moya A, Gosalbes MJ, Latorre A. 2014. Structural and functional changes in the gut microbiota associated to Clostridium difficile infection. Front Microbiol 5:335. doi: 10.3389/fmicb.2014.00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cox LM, Blaser MJ. 2013. Pathways in microbe-induced obesity. Cell Metab 17:883–894. doi: 10.1016/j.cmet.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ferrer M, Ruiz A, Lanza F, Haange SB, Oberbach A, Till H, Bargiela R, Campoy C, Segura MT, Richter M, von Bergen M, Seifert J, Suarez A. 2013. Microbiota from the distal guts of lean and obese adolescents exhibit partial functional redundancy besides clear differences in community structure. Environ Microbiol 15:211–226. doi: 10.1111/j.1462-2920.2012.02845.x. [DOI] [PubMed] [Google Scholar]
- 82.Subramanian S, Huq S, Yatsunenko T, Haque R, Mahfuz M, Alam MA, Benezra A, DeStefano J, Meier MF, Muegge BD, Barratt MJ, VanArendonk LG, Zhang Q, Province MA, Petri WA Jr, Ahmed T, Gordon JI. 2014. Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature 510:417–421. doi: 10.1038/nature13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rooks MG, Veiga P, Wardwell-Scott LH, Tickle T, Segata N, Michaud M, Gallini CA, Beal C, van Hylckama-Vlieg JE, Ballal SA, Morgan XC, Glickman JN, Gevers D, Huttenhower C, Garrett WS. 2014. Gut microbiome composition and function in experimental colitis during active disease and treatment-induced remission. ISME J 8:1403–1417. doi: 10.1038/ismej.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hernandez E, Bargiela R, Diez MS, Friedrichs A, Perez-Cobas AE, Gosalbes MJ, Knecht H, Martinez-Martinez M, Seifert J, von Bergen M, Artacho A, Ruiz A, Campoy C, Latorre A, Ott SJ, Moya A, Suarez A, Martins dos Santos VA, Ferrer M. 2013. Functional consequences of microbial shifts in the human gastrointestinal tract linked to antibiotic treatment and obesity. Gut Microbes 4:306–315. doi: 10.4161/gmic.25321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Matsumoto M, Kibe R, Ooga T, Aiba Y, Sawaki E, Koga Y, Benno Y. 2013. Cerebral low-molecular metabolites influenced by intestinal microbiota: a pilot study. Front Syst Neurosci 7:9. doi: 10.3389/fnsys.2013.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Louis P, Hold GL, Flint HJ. 2014. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 12:661–672. doi: 10.1038/nrmicro3344. [DOI] [PubMed] [Google Scholar]
- 87.Bindels LB, Porporato P, Dewulf EM, Verrax J, Neyrinck AM, Martin JC, Scott KP, Buc Calderon P, Feron O, Muccioli GG, Sonveaux P, Cani PD, Delzenne NM. 2012. Gut microbiota-derived propionate reduces cancer cell proliferation in the liver. Br J Cancer 107:1337–1344. doi: 10.1038/bjc.2012.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, Sonnenburg JL. 2013. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502:96–99. doi: 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bender KO, Garland M, Ferreyra JA, Hryckowian AJ, Child MA, Puri AW, Solow-Cordero DE, Higginbottom SK, Segal E, Banaei N, Shen A, Sonnenburg JL, Bogyo M. 2015. A small-molecule antivirulence agent for treating Clostridium difficile infection. Sci Transl Med 7:306ra148. doi: 10.1126/scitranslmed.aac9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sellitto M, Bai G, Serena G, Fricke WF, Sturgeon C, Gajer P, White JR, Koenig SS, Sakamoto J, Boothe D, Gicquelais R, Kryszak D, Puppa E, Catassi C, Ravel J, Fasano A. 2012. Proof of concept of microbiome-metabolome analysis and delayed gluten exposure on celiac disease autoimmunity in genetically at-risk infants. PLoS One 7:e33387. doi: 10.1371/journal.pone.0033387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Poesen R, Windey K, Neven E, Kuypers D, De Preter V, Augustijns P, D'Haese P, Evenepoel P, Verbeke K, Meijers B. 2016. The influence of CKD on colonic microbial metabolism. J Am Soc Nephrol 27:1389–1399. doi: 10.1681/ASN.2015030279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.De Angelis M, Piccolo M, Vannini L, Siragusa S, De Giacomo A, Serrazzanetti DI, Cristofori F, Guerzoni ME, Gobbetti M, Francavilla R. 2013. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS One 8:e76993. doi: 10.1371/journal.pone.0076993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Manor O, Borenstein E. 2017. Systematic characterization and analysis of the taxonomic drivers of functional shifts in the human microbiome. Cell Host Microbe 21:254–267. doi: 10.1016/j.chom.2016.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rosselli R, Romoli O, Vitulo N, Vezzi A, Campanaro S, de Pascale F, Schiavon R, Tiarca M, Poletto F, Concheri G, Valle G, Squartini A. 2016. Direct 16S rRNA-seq from bacterial communities: a PCR-independent approach to simultaneously assess microbial diversity and functional activity potential of each taxon. Sci Rep 6:32165. doi: 10.1038/srep32165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rickwood D, Ford T, Graham J. 1982. Nycodenz: a new nonionic iodinated gradient medium. Anal Biochem 123:23–31. doi: 10.1016/0003-2697(82)90618-2. [DOI] [PubMed] [Google Scholar]
- 96.Hevia A, Delgado S, Margolles A, Sanchez B. 2015. Application of density gradient for the isolation of the fecal microbial stool component and the potential use thereof. Sci Rep 5:16807. doi: 10.1038/srep16807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Witherden EA, Moyes DL, Bruce KD, Ehrlich SD, Shoaie S. 2017. Using systems biology approaches to elucidate cause and effect in host–microbiome interactions. Curr Opin Syst Biol 3:141–146. doi: 10.1016/j.coisb.2017.05.003. [DOI] [Google Scholar]
- 98.Lee STM, Kahn SA, Delmont TO, Shaiber A, Esen OC, Hubert NA, Morrison HG, Antonopoulos DA, Rubin DT, Eren AM. 2017. Tracking microbial colonization in fecal microbiota transplantation experiments via genome-resolved metagenomics. Microbiome 5:50. doi: 10.1186/s40168-017-0270-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Alang N, Kelly CR. 2015. Weight gain after fecal microbiota transplantation. Open Forum Infect Dis 2:ofv004. doi: 10.1093/ofid/ofv004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Petrof EO, Gloor GB, Vanner SJ, Weese SJ, Carter D, Daigneault MC, Brown EM, Schroeter K, Allen-Vercoe E. 2013. Stool substitute transplant therapy for the eradication of Clostridium difficile infection: ‘RePOOPulating’ the gut. Microbiome 1:3. doi: 10.1186/2049-2618-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Petrof EO, Khoruts A. 2014. From stool transplants to next-generation microbiota therapeutics. Gastroenterology 146:1573–1582. doi: 10.1053/j.gastro.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Blaser MJ. 2014. The microbiome revolution. J Clin Invest 124:4162–4165. doi: 10.1172/JCI78366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Group NHW, Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, Baker CC, Di Francesco V, Howcroft TK, Karp RW, Lunsford RD, Wellington CR, Belachew T, Wright M, Giblin C, David H, Mills M, Salomon R, Mullins C, Akolkar B, Begg L, Davis C, Grandison L, Humble M, Khalsa J, Little AR, Peavy H, Pontzer C, Portnoy M, Sayre MH, Starke-Reed P, Zakhari S, Read J, Watson B, Guyer M. 2009. The NIH Human Microbiome Project. Genome Res 19:2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Dore J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, et al. . 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, Bertalan M, Borruel N, Casellas F, Fernandez L, Gautier L, Hansen T, Hattori M, Hayashi T, Kleerebezem M, Kurokawa K, Leclerc M, Levenez F, Manichanh C, Nielsen HB, Nielsen T, Pons N, Poulain J, Qin J, Sicheritz-Ponten T, Tims S, Torrents D, Ugarte E, Zoetendal EG, Wang J, Guarner F, Pedersen O, de Vos WM, Brunak S, Dore J, Meta HITC, Antolin M, Artiguenave F, Blottiere HM, Almeida M, Brechot C, Cara C, Chervaux C, Cultrone A, Delorme C, Denariaz G, et al. . 2011. Enterotypes of the human gut microbiome. Nature 473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]