

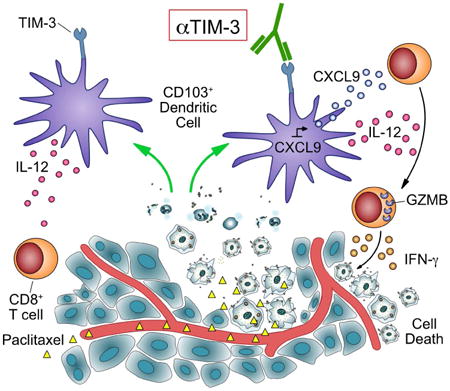
Summary
Intratumoral CD103+ dendritic cells (DCs) are necessary for anti-tumor immunity. Here we evaluated expression of immune regulators by CD103+ DCs in a murine model of breast cancer and identify expression of TIM-3 as a target for therapy. Anti-TIM-3 antibody improved response to paclitaxel chemotherapy in models of triple-negative and luminal B disease, with no evidence of toxicity. Combined efficacy was CD8+ T cell-dependent and associated with increased granzyme B expression; however, TIM-3 expression was predominantly localized to myeloid cells in both human and murine tumors. Gene expression analysis identified upregulation of Cxcl9 within intratumoral DCs during combination therapy, and therapeutic efficacy was ablated by CXCR3 blockade, Batf3-, or Irf8-deficiency.
Significance
Immunotherapeutic approaches are particularly lacking in breast cancer, and thus we sought to identify potential therapeutic targets in a murine model. Herein we report that TIM-3 expression by intratumoral CD103+ DCs regulates chemokine expression during paclitaxel treatment, with anti-TIM-3 antibody administration leading to enhanced granzyme B expression by CD8+ T cells and an immune-mediated response to chemotherapy. These findings expand upon the potential targets of TIM-3 antibodies currently in clinical trials, and offer a rationale for combinatorial studies with chemotherapy in breast cancer and other solid malignancies.
Keywords: TIM-3, galectin-9, dendritic cells, chemotherapy, paclitaxel, breast cancer, immunotherapy
Graphical abstract
Introduction
The presence of cytotoxic CD8+ T cells within tumors has a strong association with positive outcomes across a range of malignancies (Fridman et al., 2012), and they are important mediators of response to therapeutic interventions, including chemotherapy, radiotherapy, and targeted agents (Coffelt and de Visser, 2015). This is also true in breast cancer, and tumor-infiltrating lymphocytes are increasingly recognized as an emerging prognostic and predictive biomarker (DeNardo et al., 2011; Salgado et al., 2015). Enhancing the presence or functional activity of CD8+ T cells is thus the central goal of most immunotherapies, but despite recent successes, clinical response rates remain low, and it has become increasingly clear that response correlates with mutational burden (Le et al., 2015; Rizvi et al., 2015; Van Allen et al., 2015) and the extent of an anti-tumor immune response prior to therapy initiation (Herbst et al., 2014; Tumeh et al., 2014). This a problem not only for individual patients, but also for tumor types that have a comparatively low mutational frequency and/or cytotoxic T cell response.
Conventional dendritic cells (cDCs) are well established as the central inducers of a T cell response through their ability to present antigenic peptides on major histocompatibility complex (MHC) I and II following activation/maturation. It is generally thought that migratory tumor DCs are required to prime a de novo T cell response within the draining lymph nodes (Chen and Mellman, 2013). These can be divided into two lineages in mice: the predominant CD11b+ cDC2 population depends upon the transcription factor interferon regulatory factor 4 (IRF4); while the minor CD8α/CD103+ cDC1 population depends upon the transcription factors IRF8 and basic leucine zipper transcription factor ATF-like 3 (BATF3) (Broz et al., 2014). Anti-tumor immunity is absent in Batf3-deficient mice (Hildner et al., 2008; Sanchez-Paulete et al., 2015), and the recruitment of CD103+ cDCs into tumors is necessary for a CD8+ T cell response to develop (Spranger et al., 2015), implicating migratory CD103+ cDCs as the inducers of a systemic CD8+ T cell response under non-therapeutic conditions. This is consistent with their superior ability to transport and cross-present tumor antigens in the draining lymph nodes (Desch et al., 2011; Headley et al., 2016), including from spontaneously developing tumors (Roberts et al., 2016; Salmon et al., 2016).
In addition to their role in inducing de novo T cell responses, cDCs may be important in maintaining an effective T cell response within peripheral tissues (Iijima and Iwasaki, 2014; Natsuaki et al., 2014). Within tumors, CD103+ cDC1 have also been shown to restimulate CD8+ T cells and to mediate the efficacy of adoptive cell transfer therapy (Broz et al., 2014). Similarly, we have found that expression of interleukin (IL)-12 by CD103+ cDC1 promotes a CD8+ T cell response to chemotherapy following blockade of select immunosuppressive pathways (Ruffell et al., 2014). Based upon this data we therefore sought to determine whether tumor cDC1s could be therapeutically targeted through their expression of immune checkpoint molecules.
Results
TIM-3 is highly expressed by cDCs in breast cancer
To identify potential targets expressed by cDCs within tumors, single cell suspensions from transgenic MMTV-PyMT mammary carcinomas were screened by flow cytometry for surface expression of proteins associated with immune regulation (Figure 1A). Programmed death ligand-1 (PD-L1) was broadly expressed by leukocytes, and PD-1 was expressed by T lymphocytes; however, αPD-1 treatment had no effect on tumor growth alone or in combination with paclitaxel (PTX) (Figure S1A), consistent with previous findings in combination with radiotherapy (Bos et al., 2013).
Figure 1. TIM-3 is expressed by tumor-associated cDCs and macrophages.
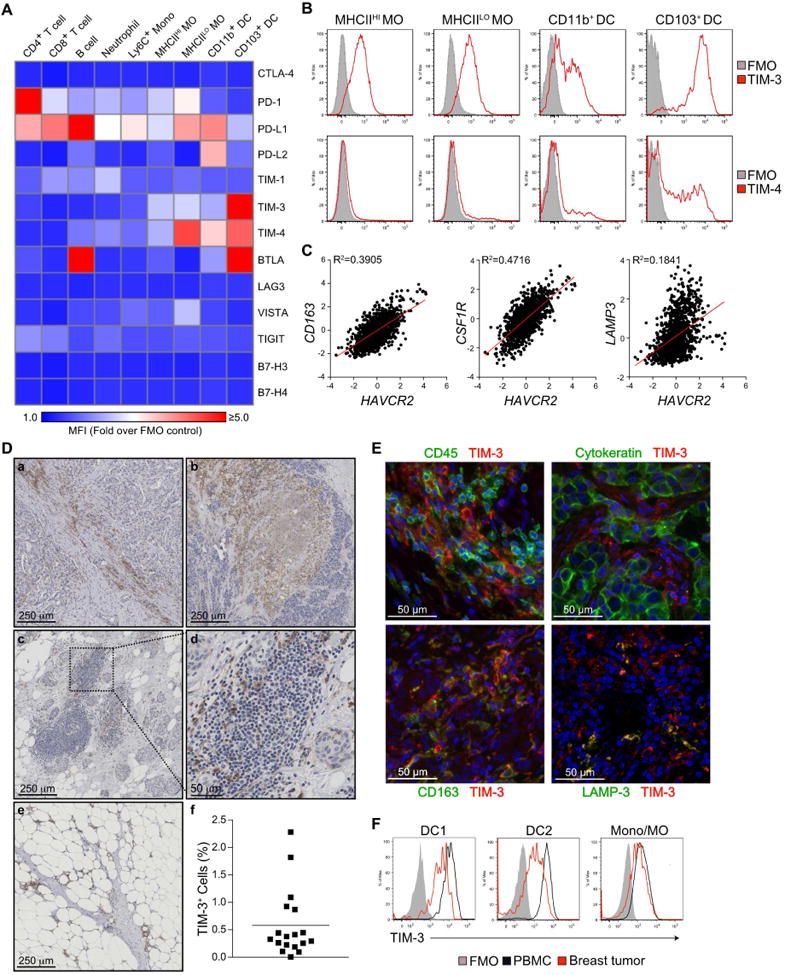
(A) Surface expression of immune checkpoint markers on leukocyte populations within late stage MMTV-PyMT tumors, as determined by flow cytometry. Results are shown as a heat map of the mean fluorescence intensity (MFI) divided by the background of fluorescence minus one (FMO) controls. n=3, one of three representative experiments shown. Mono, monocyte; MO, macrophage. (B) Representative histograms from A displaying surface expression of TIM-3 and TIM-4 on macrophages and cDCs within MMTV-PyMT tumors. (C) Correlation between HAVCR2 expression and myeloid genes (CD163, CSF1R, LAMP3) in human breast cancer samples from the TCGA dataset (n=1161, R2 values by linear regression). (D) TIM-3 immunohistochemistry in human breast cancer tissue samples. Representative images from 18 patients display positive staining in stromal regions (a), necrotic areas (b), tertiary lymphoid structures (c, d) and adjacent normal tissue (e). Cellular positivity for TIM-3 is shown at (f), with the horizontal bar representing the mean. (E) Immunofluorescent staining for TIM-3 (red) and CD45, cytokeratin, CD163, or LAMP-3 (green) in human breast cancer. DNA was visualized with Hoechst 33342 (blue). 3 patient samples were analyzed for each combination. (F) Representative histograms of TIM-3 expression by CD141+ cDC1, CD1c+ cDC2 or CD14+ monocytes/macrophages in the peripheral blood of healthy volunteers (n=5) or breast tumors (n=9). See also Figure S1.
In addition to PD-1/PD-L1 expression, we observed clustered expression of TIM-3 and TIM-4 on myeloid cells within tumors, particularly CD103+ cDCs (Figure 1B). The gene for TIM-3, HAVCR2, also strongly correlated with macrophage-associated genes CD163 and CSF1R, along with the DC-associated gene LAMP3 in the TCGA dataset (Figure 1C). In contrast, TIMD4 expression displayed poor correlation with these same genes (Figure S1B). Based upon this data, we focused on analyzing the TIM-3 expression pattern in breast cancer using samples from 18 patients that had not received neoadjuvant therapy prior to surgical resection. TIM-3 cellular positivity by immunohistochemistry was found to be variable between individual tumors, ranging from over 2% to less than 0.1% (Figure 1D). Positive cells predominantly included those with a myeloid morphology in areas with high extracellular matrix deposition, cell death/necrosis, and invasive fronts. Based upon the apparent staining of myeloid cells, we performed immunofluorescent staining in conjunction with pan-cytokeratin, CD45, CD163, or lysosomal associated membrane protein 3 (LAMP-3, DC-LAMP, CD208). TIM-3 was not observed on cytokeratin-expressing tumor cells (Figure 1E), and instead was largely observed on cells expressing lower levels of CD45, consistent with a myeloid localization. Indeed, TIM-3 showed a high degree of overlap with CD163+ macrophages, with high TIM-3 expression also noted on LAMP-3HI DCs. Expression by both CD141+ cDC1 and CD1c+ cDC2 populations within peripheral blood and breast tumors was confirmed using flow cytometry (Figure 1F, S1C-D). These data demonstrate that TIM-3 is predominantly expressed by myeloid cells in breast and mammary carcinomas, and suggest that high expression of TIM-3 by cDCs could be a viable therapeutic target.
αTIM-3 antibody improves response to chemotherapy
As TIM-3 and TIM-4 were both expressed in the murine model, and combinatorial efficacy has been observed (Baghdadi et al., 2013), we first evaluated the effect of dual αTIM-3 and αTIM-4 antibodies in MMTV-PyMT transgenic mice. Although αTIM-3/αTIM-4 treatment alone did not alter tumor growth, in combination with PTX there was a significant reduction in growth for the duration of the experiment, as compared to treatment with PTX and an isotype control antibody (Figure 2A). These findings were extended to the C3(1)-TAg model of triple negative breast cancer, where similar efficacy was observed in combination with PTX (Figure 2B). To determine which antibody was required, they were individually combined with PTX. αTIM-4 did not affect tumor growth, whereas αTIM-3 improved response to PTX equivalent to the combination of αTIM-3/αTIM-4 (Figure 2C). αTIM-3 also led to an increase in cell death within tumors compared to PTX alone, as seen by increased staining for cleaved caspase 3 (Figure S2A), and could improve response to the chemotherapeutic agent carboplatin, albeit not to the degree observed with PTX (Figure 2D). Notwithstanding the effects of αTIM-3 on the primary tumor, there was no difference in the number or the size of the pulmonary metastatic foci in MMTV-PyMT animals across any of the treatment groups (Figure S2B). This failure to impact metastasis may relate to the late stage of intervention and/or the relative inability of CD8+ T cells to suppress metastasis in the transgenic PyMT model (Bos et al., 2013; DeNardo et al., 2011). Importantly however, αTIM-3 efficacy was not associated with clinical measures of toxicity as revealed by liver or kidney function tests (Figure S2C, D), thus demonstrating safety and efficacy against the primary tumor with the combination of αTIM-3 and PTX.
Figure 2. αTIM-3 improves response to chemotherapy.
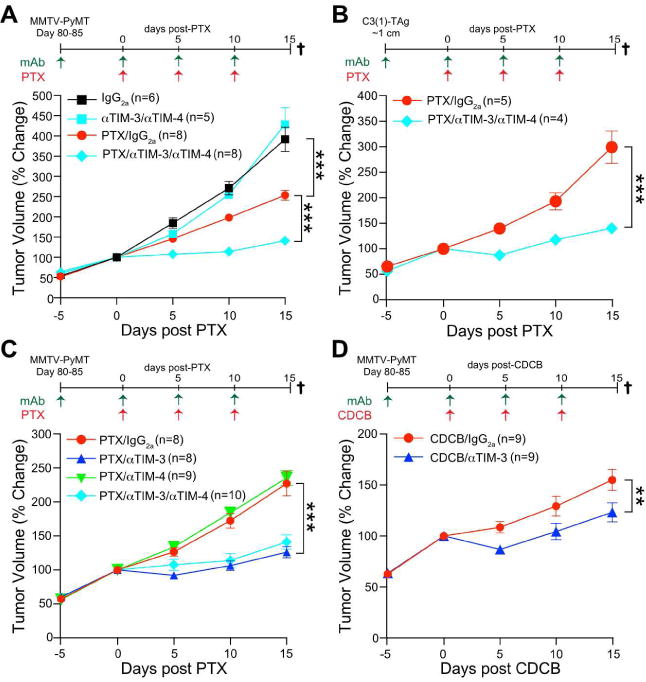
(A) Tumor volume shown as a relative change from the initiation of chemotherapy (day 0) in MMTV-PyMT animals. Mice were treated with an IgG2a isotype control or the combination of αTIM-3 and αTIM-4 antibodies, alone or together with 10 mg/kg PTX as indicated. n=5-8 mice per group, pooled over 4 cohorts. (B) Same as A, except C(3)1-TAg animals were treated when a single tumor reached ∼1 cm in diameter. n=4-5 mice per group, pooled over 4 cohorts. (C) Same as A, except MMTV-PyMT animals were treated individually with αTIM-3 or αTIM-4 antibodies. n=8-10 mice per group, pooled over 4 cohorts. Mice in the αTIM-3/αTIM-4/PTX group overlap with those in A and are shown for comparison. (D) Same as A, except MMTV-PyMT animals were treated with αTIM-3 in combination with 20 mg/kg carboplatin (CDCB). n=9 mice per group, pooled over 3 cohorts. Data in A-D are mean ± SEM; **p<0.01, *** p<0.001, with statistical significance determined by two-way ANOVA. See also Figure S2.
cDC1 are necessary for response to αTIM-3
Although CD103+ cDC1 expressed the highest levels of TIM-3 within tumors, it was possible that other TIM-3-expressing myeloid subsets were the actual targets of therapy. To confirm that cDC1 were functionally important, we acquired Batf3 knockout animals and backcrossed them onto the FVB/NJ background. Only Batf3+/- MMTV-PyMT animals responded to αTIM-3, with no difference in tumor volume observed between Batf3-/- mice treated with IgG2a/PTX compared to αTIM-3/PTX (Figure 3A). Surprisingly, this phenotype was not due to the absence of CD103/CD8α+ cDC1 (Figure S3A), perhaps related to the secondary role of BATF3 in maintaining Irf8-depdendent cDC development (Grajales-Reyes et al., 2015), and was instead most likely due to a defect in cross-presentation in Batf3-deficient cDCs (Jackson et al., 2011; Seillet et al., 2013). To clarify the importance of the cDC1 subset we generated Itgax-cre.Irf8fl/fl bone marrow chimeric animals on the C57BL/6J background, and confirmed the absence of cDC1s (Figure S3B). Following orthotopic implantation of syngeneic PyMT tumor cells, mice were treated with a combination of αTIM-3 and PTX, and as expected, Itgax-cre.Irf8fl/fl animals failed to respond to the combination therapy (Figure 3B).
Figure 3. cDC1 are necessary for response to αTIM-3.
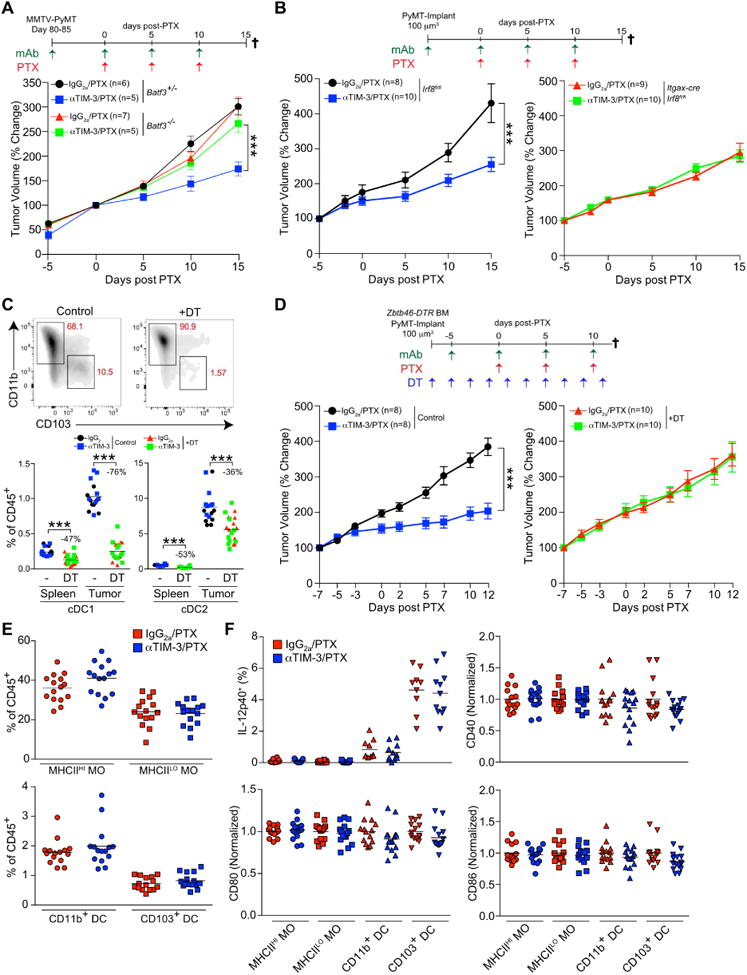
(A) Tumor volume shown as a relative change from the initiation of chemotherapy (day 0 to 15) in MMTV-PyMT animals either Batf3-proficient (+/-) or Batf3-deficient (-/-). Mice were treated with an IgG2a isotype control or αTIM-3 antibody in combination with 10 mg/kg PTX as indicated. n=5-7 per group, data pooled over 5 cohorts. (B) Orthotopic PyMT tumor volume in chimeric C57BL/6J animals reconstituted with either Itgax-cre;Irf8fl/fl or Irf8fl/fl bone marrow. n=8-10 per group, with one of two representative experiments shown. (C) Top, cDC subsets (gated on CD45+CD11c+MHCII+F4/80-) within PyMT tumors of chimeric C57BL/6J animals reconstituted with Zbtb46-DTR bone marrow and treated with diphtheria toxin (DT) as indicated. Bottom, the percentage of each cDC subset within the spleens or tumors of mice treated as in D. The percent reduction in the population resulting from DT administration is shown. n=8-10 per group, with one of two representative experiments shown. (D) Tumor volume in chimeric C57BL/6J animals reconstituted with Zbtb46-DTR bone marrow and treated with DT as indicated. n=8-10 per group, with one of two representative experiments shown. (E) Frequency of macrophage (MO) and cDC subsets as a percent of total CD45+ cells within tumors of MMTV-PyMT animals treated with PTX and αTIM-3 antibody, determined by flow cytometry (day 7). n=15 per group, data pooled over 3 cohorts. (F) Intracellular flow cytometric analysis of IL-12p40, along with surface expression of CD40, CD80, and CD86 on macrophages and cDCs from MMTV-PyMT animals treated with PTX in conjunction with IgG2a or αTIM-3 (day 7). n=13-15 per group, data pooled over 3 experiments by normalizing to 1 as indicated. Data in A, B, D are shown as mean ± SEM; ***p<0.001, with significance determined by two-way ANOVA. Horizontal bars in C, E, F represent the mean; ***p<0.001, with significance determined by unpaired t-test. See also Figure S3.
A caveat to these findings is that mice lacking cDC1s prior to exposure to tumors would also be expected to lack an endogenous anti-tumor CD8+ T cell response, making it unclear whether CD103+ cDCs were the functional target of αTIM-3. We therefore created chimeric animals using donor Zbtb46-DTR bone marrow to allow depletion of cDCs after tumor implantation (Broz et al., 2014). Administration of diphtheria toxin preferentially depleted the CD103+ cDC1 subset within tumors (Figure 3C) and prevented response to αTIM-3/PTX (Figure 3D). These results demonstrate that CD103+ cDC1s are functionally necessary for response to therapy, and further support cDC1s being an important therapeutic target of TIM-3 blocking antibodies.
Despite the importance of cDC1 in mediating response to αTIM-3, neither infiltration by cDCs nor macrophages was altered by αTIM-3 administration, whether measured 2 or 5 days after PTX administration (Figure 3E, S3C). As TIM-3 has previously been reported to suppress intracellular TLR-induced activation of CD11c+ myeloid cells (Chiba et al., 2012), we also examined whether the activation status of cDCs was altered within tumors from mice treated with αTIM-3/PTX. However, there was no difference in the surface expression of the activation/maturation markers CD80, CD86, CD40, MHCI and MHCII, nor in expression of IL-12p40 by CD103+ tumor cDC1s as measured by ex vivo intracellular staining (Figure 3F, S3D).
αTIM-3 indirectly promotes an intratumoral CD8+ T cell response
To determine whether αTIM-3 was enhancing a T cell response during PTX treatment, we began by evaluating whether CD4+ or CD8+ T cells were necessary for response to αTIM-3/PTX via depletion studies. CD4-depletion had no effect on response to therapy, while CD8-depletion prior to the initiation of chemotherapy prevented the improved response (Figure 4A). These studies were extended to determine the importance of cytokines implicated in promoting CD8/TH1 responses to chemotherapy (Kroemer et al., 2013; Ruffell et al., 2014; Sistigu et al., 2014): obstructing type I interferon (IFN) signaling by blocking IFN alpha receptor 1 (IFNAR1) abrogated the therapeutic effect of αTIM-3, a phenotype that was also observed when neutralizing antibodies against either IL-12p70 or IFN-γ were administered (Figure 4A). We next measured TIM-3 expression in tumor-bearing MMTV-PyMT animals and found that TIM-3 was not expressed on T lymphocytes in the blood, lymph node, or spleen (Figure 4B, 4C). TIM-3 was also barely detectable on CD8+ T cells within most tumors, but was consistently expressed by about 20% of CD4+ T cells (19.1±1.3%). Minimal surface expression of TIM-3 by T cells was matched by lower expression of Havcr2 compared to macrophages or cDCs (Figure S4A). Similarly, TIM-3 was not detectable on CD4+ or CD8+ T cells in breast cancer samples by immunofluorescence (Figure S4B). To better quantify expression on human T cells we also analyzed TIM-3 expression by flow cytometry, and similar to murine mammary tumors, found only low expression by a small population of CD4+ (8.1±2.1%) and CD8+ T cells (6.6±3.1%) within breast tumors (Figure 4B, 4D). Therefore, while αTIM-3 enhances a CD8+ T cell-dependent response to chemotherapy, the data indicate that this occurs via an indirect mechanism of action, consistent with high TIM-3 expression (Figure 1B) and the functional importance (Figure 3A-D) of the cDC1 subset.
Figure 4. αTIM-3 indirectly enhances a CD8+ T cell response.
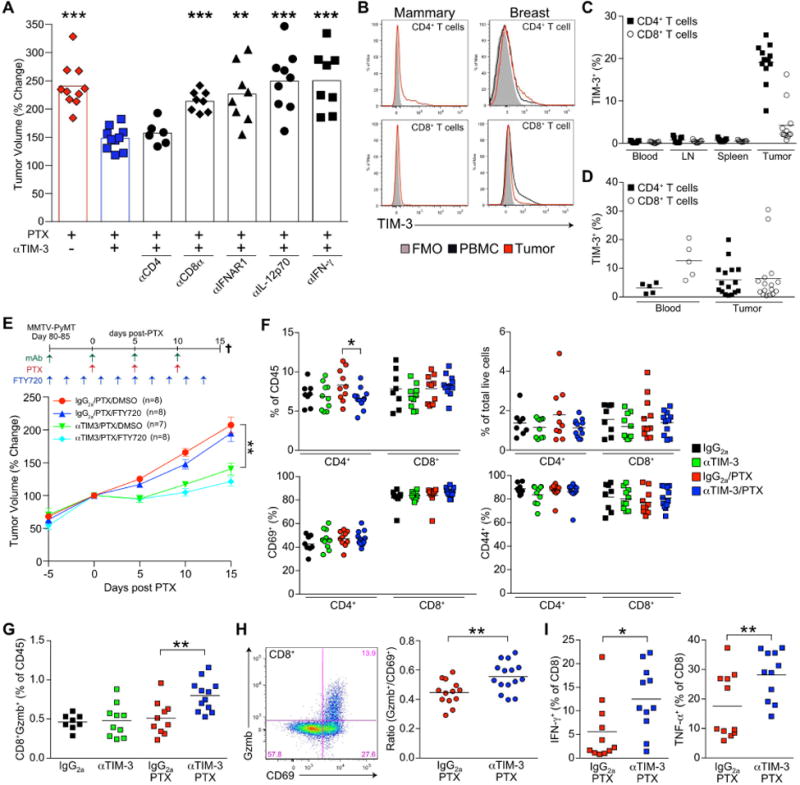
(A) Relative tumor volume after 3 rounds of PTX in MMTV-PyMT transgenic mice (day 15). Blocking (αIFNAR1), neutralizing (αIL-12p70, αIFN-γ) and depleting (αCD8α, αCD4) antibodies were administered concurrently with αTIM-3, 5 days prior to and then in conjunction with PTX every 5 days. n=8-12 per group, data pooled over 7 cohorts. Bar graphs represent the mean; **p<0.01, ***p<0.001, with significance determined by an unpaired t-test with Welch's correction compared to αTIM-3. (B) Representative histograms of TIM-3 surface expression on T cells from MMTV-PyMT animals (left) or human breast tumors (right). (C) TIM-3 expression on murine T cells from MMTV-PyMT animals shown as a percentage of the total positive cells. n=9-12 per tissue, data merged from 3 experiments. (D) Percentage of TIM-3+ human T cells in the peripheral blood of healthy volunteers (n=5) or within breast tumors (n=16). (E) Relative tumor volume in MMTV-PyMT animals treated with IgG2a/PTX or αTIM-3/PTX in combination with twice daily doses of FTY720 or DMSO as indicated. n=7-8 mice per group, pooled over 4 cohorts. Data shown as mean ± SEM; **p<0.01, with significance determined by two-way ANOVA. (F) Frequency of CD8+ and CD4+ T cells within tumors as a percent of total CD45+ (top left) or live cells (top right), and the percent of T cells expressing CD69 (bottom left) or CD44 (bottom right) in tumors. (G) CD8+Gzmb+ T cells shown as percentage of total leukocyte infiltration. (H) Ratio of CD8+Gzmb+ to CD8+CD69+ T cells in tumors from MMTV-PyMT animals treated with IgG2a/PTX or αTIM-3/PTX (day 7). n=13-15 per group, data pooled over 3 cohorts. Representative staining for CD69 and Gzmb is shown to the left. (I) Percentage of IFN-γ or TNF-α expressing CD8+ T cells. Data for F, G, and I are from mice bearing PyMT implantable tumors treated with IgG2a, αTIM-3, or PTX (day 7). n=8-12 per group, data pooled from 2 experiments. Horizontal bars in C-D, F-I represent the mean; *p<0.05, **p<0.01, with significance determined by unpaired t-test. See also Figure S4.
In order to differentiate between the induction of a systemic de novo immune response and enhancement of local effector function, we first evaluated the clonality of T cells within the peripheral blood, but observed no effect of αTIM-3 (Figure S4C). We then administered FTY720 to animals during the course of treatment to determine whether retention of T cells within the secondary lymphoid organs would prevent response to therapy. While, as expected, this retained T cells within the spleen and reduced the circulating population, it did not decrease infiltration by intratumoral T cells (Figure S4D), and no impact on tumor growth was observed in either the IgG2a/PTX or αTIM-3/PTX treatment groups (Figure 4E). We therefore focused on identifying changes in T cells within tumors. There were no quantitative changes in tumor infiltration as a result of αTIM-3, regardless of whether tumors were examined 2 days or 5 days following PTX in the MMTV-PyMT transgenic model (Figure S4E-F), or in an orthotopic PyMT implantable model (Figure 4F). Expression of the T cell activation markers CD44 and CD69 by intratumoral T cells was highly variable between transgenic tumors but was similarly unchanged (Figure S4G). Given the high variability in T cell activation in the transgenic model we evaluated T cell activation status in the PyMT implantable model, but again found no differences between treatment groups (Figure 4F). We next measured granzyme B within tumor T cells as a surrogate for cytotoxic potential using the PyMT implantable model and found that αTIM-3 in combination with PTX significantly increased the frequency of CD8+ T cells expressing granzyme B, measured as either a percentage of total leukocyte infiltration or the total population of CD8+ T cells (Figure 4G, S4H). A comparable increase in granzyme B expression was observed with αTIM-3/PTX treatment in MMTV-PyMT animals after controlling for the highly variable level of T cell activation (Figure 4H). Ex vivo activation and intracellular staining for IFN-γ and TNF-α also revealed a significant increase in the percentage of cells expressing these cytokines (Figure 4I).
αTIM-3 increases CXCR3 chemokine ligand expression by tumor cDCs
As the data indicated that αTIM-3 improved the ability of CD103+ cDC1 to enhance the effector function of CD8 T cells within tumors, we sought to identify a potential mechanism of action. Binding of phosphatidylserine (PS) to TIM-3 has been shown to promote uptake of antigens and cross-presentation by CD8α+ cDC1 (Nakayama et al., 2009). Although in this case blockade of TIM-3 would be expected to suppress the induction of anti-tumor immunity, we examined the uptake of tumor antigens by flow cytometry using transgenic FVB/NJ animals expressing PyMT, mCherry, and ovalbumin under the control of the MMTV promoter (i.e. PyMTchOVA). As previously reported, macrophages were the dominant antigen-presenting population within tumors, with both CD11b and CD103+ cDC1 subsets displaying lower levels of mCherry uptake (Broz et al., 2014). However, no difference was observed between mice treated with αTIM-3 versus an isotype control in terms of the percentage of cells or their overall fluorescence (Figure 5A).
Figure 5. αTIM-3 increases CXCR3 ligand expression by cDCs.
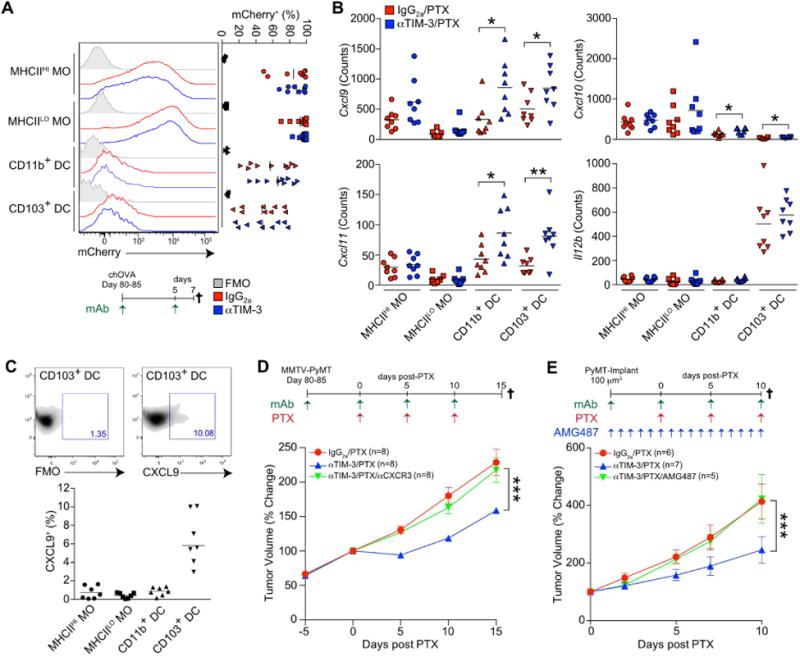
(A) Detection of mCherry fluorescence in macrophages and cDCs from PyMTchOVA tumors. Mice were treated with isotype control or αTIM-3 for 7 days prior to analysis. n=9 per group, data pooled over 5 experiments. (B) mRNA expression levels in tumor macrophages and cDCs isolated from mice bearing orthotopically implanted PyMT tumors 2 days following second dose of PTX (day 7). Expression of Cxcl9, Cxcl10, Cxcl11, and Il12b were determined by NanoString, with normalized counts displayed. n=8 per group, data pooled from 2 experiments. (C) Intracellular flow cytometric analysis of CXCL9 in macrophages and cDCs from MMTV-PyMT animals following i.v. injection of brefeldin A for 4-6 hr. Representative staining as well as a fluorescence minus one (FMO) control is shown above. n=7, data pooled over 2 experiments. (D) Tumor volume shown as a relative change from the initiation of chemotherapy (day 0) in MMTV-PyMT animals. Mice were treated with an IgG2a isotype control, αTIM-3 and/or αCXCR3 antibodies, together with 10 mg/kg PTX as indicated. n=8 mice per group, pooled over 4 cohorts. (E) Orthotopic PyMT tumor volume in C57BL/6J animals treated with an IgG2a isotype control, αTIM-3 and/or (±)-AMG 487, together with 10 mg/kg PTX as indicated. n=5-7 per group, with one of two representative experiments shown. Horizontal bars in A-C represent the mean; *p<0.05, **p<0.01, with significance determined by unpaired t-test. Data in D-E are shown as mean ± SEM; ***p<0.001, with significance determined by two-way ANOVA. See also Figure S5.
To take a more unbiased approach in determining how cDC1s were altered by αTIM-3, we isolated macrophages and cDCs from orthotopically implanted PyMT tumors (to minimize the impact of variation inherent in the transgenic model), and performed gene expression analysis using the NanoString nCounter Mouse Immunology Panel. Significant changes (p<0.05) that exceeded 1.5-fold between animals treated with αTIM-3/PTX and IgG2a/PTX are shown in Figure S5A. CD103+ cDC1s displayed increased expression of only 5 genes (Cxcl11, Cxcl10, Cxcl9, Tagap, and Cd40), with a corresponding increase in chemokine expression also observed in CD11b+ cDCs (Figure 5B). As Cxcl9 was expressed at much higher levels in cDCs than Cxcl10 or Cxcl11 (more than 10-fold in CD103+ cDC1), we next examined whether CXCL9 could be detected at the protein level within tumor leukocytes. In transgenic MMTV-PyMT tumors CXCL9 was only detectable in CD103+ cDCs isolated directly from mice following in vivo administration of brefeldin A (Figure 5C), consistent with preferential expression of Cxcl9 mRNA by this subset (Figure S5B). Only following ex vivo stimulation with IFN-γ could CXCL9 be detected in all of the myeloid subsets examined (Figure S5C). Based upon the increase in Cxcl9 expression and specific expression of CXCL9 by CD103 cDC1 in vivo, a blocking antibody against CXCR3 (receptor for CXCL9, CXCL10, and CXCL11) was administered during combination therapy, and as shown in Figure 5D, blocking CXCR3 abrogated the effect of αTIM-3. Similar results were obtained with a specific inhibitor of CXCR3 (Figure 5E) that blocked migration of activated splenic CD8 T cells towards either CXCL9 or CXCL10 in vitro (Figure S5D). Cumulatively, these data point to a potential role for CXCL9-expressing CD103+ cDC1 in promoting a cytotoxic T cell response following TIM-3 blockade.
αTIM-3 and αGalectin-9 antibodies promote CXCL9 expression
To determine if αTIM-3 could directly regulate CXCL9 expression, bone marrow DCs (BMDCs) were first generated using Fms-Related Tyrosine Kinase 3 Ligand (FLT-3L). However, these cells only expressed low levels of TIM-3 (Figure S6A), and despite previous reports (Chiba et al., 2012) we did not find that IL-10 or vascular endothelial growth factor (VEGF)-A increased TIM-3 expression in vitro (Figure S6B, S6C). In addition, blockade of the IL-10 receptor had no effect on TIM-3 expression on macrophages or cDCs within MMTV-PyMT tumors (Figure S6D), indicating that this pathway was not a major driver of TIM-3 expression by tumor myeloid cells. We therefore enriched for splenic cDCs, as CD8α+ cDC1 displayed expression levels of TIM-3 comparable to tumor CD103+ cDC1 (Figure S6E), and stimulated the cells with poly(I:C) or CpG in the presence or absence of αTIM-3. However, neither TLR ligand increased CXCL9 expression, and there was no impact of αTIM-3 on either CXCL9 or IL-12 expression in either cDC subset (Figure 6A). As these agonists do not reflect the ligands that would be present with tumors, we next utilized the supernatant of PyMT tumor cells killed by irradiation or heat shock in vitro. Tumor cell debris alone had little to no impact on CXCL9 expression; however, CXCL9 expression was consistently enhanced in CD8α+ cDC1s by the addition of the αTIM-3, with a small increase also observed for CD11b+ cDC2s (Figure 6B). This was consistent with the increase in Cxcl9 mRNA expression we observed within tumor CD103+ cDC1s (Figure 5B). We also found no increase in expression of surface activation markers in response to tumor cell debris in vitro (Figure S6F), similar to our in vivo observations (Figure 3F, S3D). Together these results suggest that TIM-3 can directly regulate CXCL9 expression by CD103+ cDC1 within tumors.
Figure 6. αTIM-3 and αGalectin-9 antibodies promote CXCL9 expression.
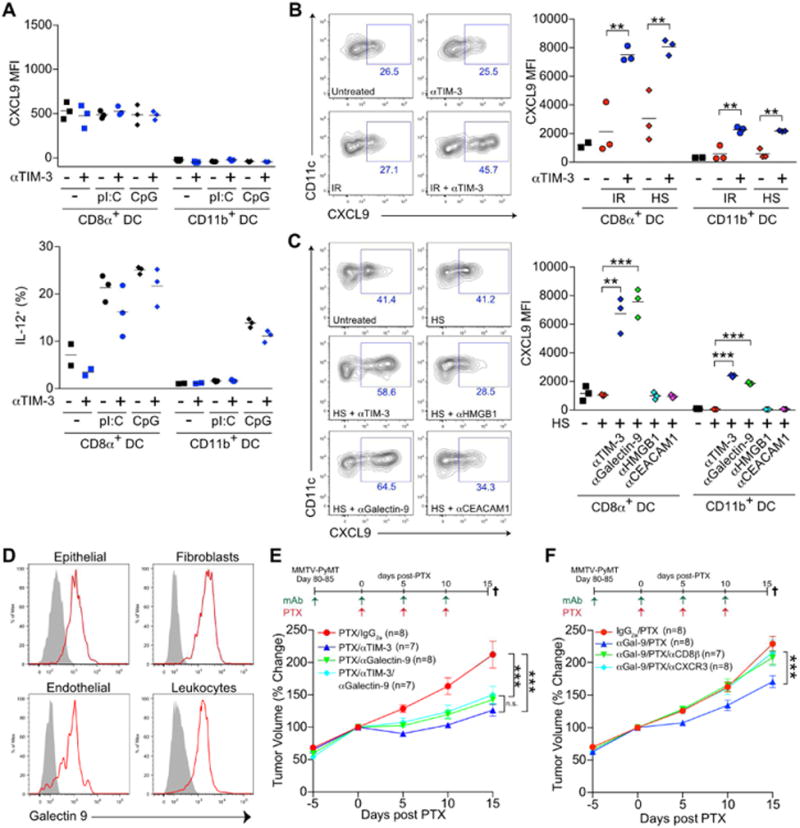
(A) CXCL9 (top) or IL-12 (bottom) expression by splenic cDCs following stimulation with Poly(I:C) [pI:C] or CpG (1 μg/ml). One of two representative experiments is shown. (B) CXCL9 expression by splenic cDCs following incubation with αTIM-3 and/or tumor cell debris generated by irradiation (IR) or heat shock (HS). One of three representative experiments is shown. (C) Same as B, but with αTIM-3, αGalectin-9, αHMGB1, or αCEACAM-1 antibodies added in combination with tumor cell debris generated by HS. One of two representative experiments is shown. (D) Detection of galectin-9 on the surface of EpCAM+ epithelial cells, PDGFRα+ fibroblasts, CD31+ endothelial cells, or CD45+ leukocytes from MMTV-PyMT tumors, as determined by flow cytometry. Grey histograms represent FMO control. n=3, one of three representative experiments shown. (E) Tumor volume shown as a relative change from the initiation of chemotherapy (day 0) in MMTV-PyMT animals. Mice were treated with an IgG2a isotype control or the combination of αTIM-3 and αGalectin-9 antibodies, alone or together with 10 mg/kg PTX as indicated. (F) Same as E, but mice were treated with αGalectin-9/PTX alone or with αCXCR3 or αCD8β antibodies. Horizontal bars in A-C represent the mean of technical replicates; **p<0.01, ***p<0.001, with significance determined by an unpaired t-test. Data in E-F represent 7-8 mice per group, pooled over 4 cohorts, and are shown as mean ± SEM; ***p<0.001, with significance determined by two-way ANOVA. See also Figure S6.
We next sought to evaluate whether neutralizing antibodies against identified TIM-3 ligands (Anderson et al., 2016) could recapitulate the increase in CXCL9 expression observed with αTIM-3. As shown in Figure 6C, neither antibodies against high mobility group box 1 (αHMGB1) nor αCEACAM1 impacted CXCL9 expression, while αGalectin-9 led to an increase comparable to αTIM-3. Galectin-9 was found on the surface of all cells examined within MMTV-PyMT tumors, including epithelial cells, fibroblasts, and leukocytes (Figure 6D). Similarly, galectin-9 was found throughout human breast tumors, with strong staining within the stromal regions by immunohistochemistry, and variable levels of staining observed in carcinoma cells (Figure S6G, H). We therefore examined whether galectin-9 neutralization could improve response to PTX, and found that αGalectin-9 was equivalent to αTIM-3 in suppressing tumor growth during PTX treatment (Figure 6E). The efficacy of αGalectin-9/PTX was also CD8+ T cell- and CXCR3-dependent (Figure 6F). While both TIM-3 and galectin-9 have multiple potential binding partners, the data suggests that an interaction between these molecules may be involved in regulated the function of cDCs within tumors.
DC infiltration correlates with CXCL9 expression and response to chemotherapy
Murine cDC1s expressed TIM-3 (Figure 1B) and were an important source of CXCL9 within mammary tumors (Figure 5C, S5B). As human cDC1s constitutively expressed TIM-3 (Figure 1F, S1D), we next sought to determine whether they might also be an important source of CXCL9 within breast tumors. Indeed, CXCL9 gene expression correlated with expression of both LAMP3 and IRF8 (R2=0.5884; R2=0.4834), consistent with expression by the human cDC1 equivalent, while displaying minimal correlation with CSF1R or IRF4 (R2=0.1202; R2=0.2084) (Figure 7A, S7A). Similarly, CXCL9 expression was detected by immunofluorescence in LAMP3+ DCs but not CD163+ macrophages (Figure 7B). Thus, both human and murine cDC1s express TIM-3 and CXCL9, suggesting that αTIM-3 antibodies may be a viable method to enhance the function of cDCs in breast cancer.
Figure 7. DC infiltration correlates with CXCL9 expression and response to chemotherapy.
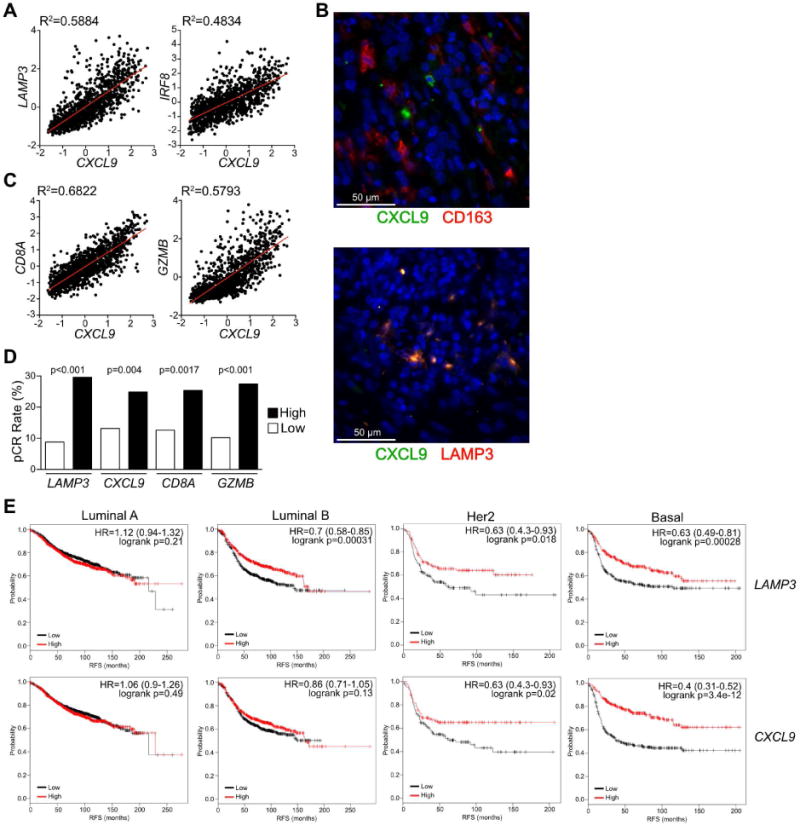
(A) Linear regression analysis between CXCL9 expression and DC-associated genes (LAMP3, IRF8) in human breast cancer samples from the TCGA dataset (n=1161). (B) Immunofluorescent staining for CXCL9 (green) and CD163 or LAMP3 (red) in human breast cancer. DNA was visualized with Hoechst 33342 (blue). 3 patient samples were analyzed for each combination. (C) Linear regression analysis between CXCL9 expression and various cytotoxic lymphocyte-associated genes (CD8A, GZMB) in human breast cancer samples from the TCGA dataset. (D) Frequency of pathologic complete response (pCR) in patients separated by median expression for the indicated genes. Data reflects a cohort of 379 patients constructed from 2 independent datasets, with significance determined by chi-square. (E) Recurrence free survival (RFS) based upon median expression of LAMP3 or CXCL9 in breast tumor tissue. Data is shown for intrinsic luminal A (n=1933), luminal B (n=1149), Her2 (n=251) and basal (n=618) molecular subtypes. Hazard ratio (HR) and logrank p values are shown in the upper right of each Kaplan-Meier Plot. See also Figure S7.
HAVCR2 gene expression in tumors was largely due to expression by macrophages (Figure 1C-E), and therefore was not useful as a marker of cDCs or the importance of TIM-3 expression by this population. We thus examined whether CXCL9 expression correlated with the presence of cytotoxic T cells, and observed an association with both CD8A and GZMB (Figure 7C). These associations were largely consistent across molecular subtypes (Figure S7B, C). We have previously described that expression of CD8A or GZMB is associated with a higher rate of pathological complete response to chemotherapy in breast cancer patients (Ruffell et al., 2014) using published datasets (Hess et al., 2006; Tabchy et al., 2010). We therefore evaluated whether the same was true for LAMP3 or CXCL9 and found a comparable ∼2-fold segregation in response rates for each gene (Figure 7D). Similarly, CD8A, LAMP3 and CXCL9 expression all correlated with recurrence free survival in patients with basal or Her2 disease subtypes (Figure 7E, S7D). Collectively these data hint at an important role for cDC1 in promoting a cytotoxic T cell response through CXCL9 production, and suggest that αTIM-3 antibodies could enhance this expression, promote response to neoadjuvant chemotherapy, and improve survival.
Discussion
TIM-3 was originally identified based upon its preferential expression by TH1-polarized CD4+ T cells, and αTIM-3 antibodies can promote TH1-responses by reducing cell death and exhaustion in autoimmunity and infection models (Kane, 2010; Monney et al., 2002). TIM-3 expression is also associated with an exhausted phenotype in CD8+ T cells during chronic infection, graft-versus-host disease, and cancer, and TIM-3 blockade increases IFN-γ expression in vivo and ex vivo (Jin et al., 2010; Jones et al., 2008; Oikawa et al., 2006; Sakuishi et al., 2010). Cumulatively these studies have led to interest in developing therapeutic antibodies against TIM-3 to enhance T cell immunity. Within MMTV-PyMT tumors, TIM-3 was expressed by a fraction of CD4+ T cells, consistent with previous reports using subcutaneously implanted cell lines (Ngiow et al., 2011; Sakuishi et al., 2010). However, CD4 depletion had no effect on response to combination therapy with αTIM-3 and PTX, indicating that any effects on this population were irrelevant in our model. We were unable to detect TIM-3 on CD8+ T cells within most MMTV-PyMT tumors, despite expression on a large proportion of CD8+ T cells in the subcutaneous tumor models mentioned above (Ngiow et al., 2011; Sakuishi et al., 2010). These differences may reflect the intensity of an antigen-specific response, as TIM-3 expression by CD8+ T cells is associated with antigen-specificity in melanoma patients (Baitsch et al., 2011; Fourcade et al., 2010), and transgenic tumor models show a lower frequency of neo-epitopes (Yadav et al., 2014). Breast tumors also display a lower average frequency of somatic mutations (Alexandrov et al., 2013), and our findings in mice were consistent with our observations in human breast tumors. Despite the paucity of TIM-3 on the surface of CD8+ T cells, they were required for response to αTIM-3 combination therapy. Thus, αTIM-3 antibodies can promote T cell immunity without directly targeting T cells, a finding that has implications for clinical trial design and patient selection criteria, for example, by not restricting treatment to only patients who display TIM-3+ lymphocytes.
Instead of T cells, we found that TIM-3 expression was localized to macrophages and cDCs in tumors and normal tissues, with the highest levels consistently found on the cDC1 subset. We are far from the first to describe TIM-3 expression by myeloid cells under homeostatic conditions, as TIM-3 is found on mast cells, monocytes, microglia, splenic cDCs, and human circulating cDCs (Anderson et al., 2007; Nakayama et al., 2009; Phong et al., 2015). Inflammation-induced TIM-3 expression by peritoneal macrophages and microglia has also been observed, though not in human glioblastoma multiforme (Anderson et al., 2007; Nakayama et al., 2009). Finally, CD11c+ cells within subcutaneous murine tumors express TIM-3, although whether these represent macrophages or cDCs is unclear, and TIM-3 expression by CD11c+ cells was not apparent in the spleens or lymph nodes from the same study (Chiba et al., 2012), despite previous reports and our own observations (Nakayama et al., 2009).
Little is understood regarding the molecular mechanisms by which TIM-3 regulates immune responses. In T cells, galectin-9 binding induces tyrosine phosphorylation and prevents an association with nuclear factor HLA-B-associated transcript 3 (BAT3) (Rangachari et al., 2012; van de Weyer et al., 2006; Zhu et al., 2005). However, TIM-3 expression has also been shown to enhance early T cell receptor signaling and promote acute CD8+ T cell responses (Gorman et al., 2014; Lee et al., 2011). Similarly, antibodies against TIM-3 can both suppress and induce NF-κB activation in BMDCs and DC cell lines, respectively (Anderson et al., 2007; Maurya et al., 2014), and have been shown to promote Fc receptor signaling in mast cells (Phong et al., 2015). Some of these differences might relate to the fact that TIM-3 is not the only receptor for galectin-9 (Katoh et al., 2007; Su et al., 2011), or that as galectin-9 binds to carbohydrate moieties on TIM-3, protein expression alone does not ensure binding will occur (Leitner et al., 2013). Furthermore, as TIM-3 is also a receptor for PS, HMGB1 and CEACAM-1, there may be important interplay between these ligands under pathological conditions such as those found within tumors. Finally, while the RMT3-23 antibody clone used in this study has been shown to block TIM-3 binding to galectin-9, PS and HMGB1 (Chiba et al., 2012; Kanzaki et al., 2012; Nakayama et al., 2009), it remains possible that RMT3-23 could directly induce tyrosine phosphorylation, as has been described for polyclonal antibodies and other αTIM-3 antibody clones (Maurya et al., 2014; Phong et al., 2015). The mechanism by which αTIM-3 antibody promotes CXCL9 expression by cDCs is currently under investigation.
It has recently been described that CD103+ cDC1s are necessary to promote antigen-specific T cell recruitment into immunogenic melanoma tumors through their ability to express CXCL10 (Spranger et al., 2017). In contrast, in mammary tumors we found that CD103+ cDC1s expressed minimal levels of Cxcl10, and instead expressed the highest levels of Cxcl9 when compared to other leukocyte subsets within tumors. Despite the increase in Cxcl9 expression observed following αTIM-3/PTX treatment however, we did not detect an increase in T cell infiltration. This may be due to expression of multiple T cell-attracting chemokines within tumors (e.g. CCL5, CXCL9, and CXCL10), or selective recruitment of antigen-specific cells that is not detected when measuring bulk T cell infiltration. Alternatively, as FTY720 administration did not impact response to αTIM-3/PTX, it may be that local promotion of a CD8+ T cell effector response explains our observations. Interestingly, CXCL9 expression by cDCs has previously been described to mediate DC-T cell clustering within lymph nodes (Kastenmuller et al., 2013), and as DC-T cell interactions are infrequent within tumors (Broz et al., 2014), it is possible that increased expression of CXCL9 could facilitate these interactions to promote T cell effector function. However, whether CXCL9 expression by CD103+ cDC1s is functionally important remains to be shown. αTIM-3 antibodies have been successfully combined with αPD-1 or αPD-L1 blockade to suppress tumor growth (Ngiow et al., 2011; Sakuishi et al., 2010), and this combination is likely to be the first evaluated clinically. Surprisingly, single agent efficacy with αTIM-3 has been shown to occur largely independent of CD11c+ cells (Ngiow et al., 2011), whereas our data indicate that cDC1s are necessary mediators of response to combination cytotoxic therapy. This may relate to the restricted expression of IL-12 in CD103+ cDCs and its critical role in promoting cytotoxic T cell responses within MMTV-PyMT tumors during PTX chemotherapy (Ruffell et al., 2014). As PTX is one of the preferred chemotherapies for use in breast cancer (Rugo et al., 2015), our findings indicate that αTIM-3 antibodies currently in clinical development should also be considered in this setting as a means to improve upon the immune-dependent response to chemotherapy.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Brian Ruffell (Brian.Ruffell@moffitt.org).
Experimental Model and Subject Details
Human Studies: Human biospecimens were consented and collected through Moffitt Cancer Center's Total Cancer Care general banking protocol (MCC#14690/Chesapeake IRB approval #Pro00014441). De-identified formalin-fixed paraffin embedded breast tissues were released in support of this study with an SRC and IRB approved protocol (MCC#50168/Chesapeake IRB Pro00019964). Breast tumors for flow cytometry were obtained from adult female patients under Chesapeake IRB approval #Pro00050168. De-identified peripheral blood mononuclear cells of unknown gender were purchased from OneBlood. Patient consent forms for all samples were obtained at the time of tissue acquisition.
Animal Studies: Animals were maintained in either the Oregon Health & Science University or University of South Florida Department of Comparative Medicine barrier facility, and the respective Institutional Animal Care and Use Committee approved all experiments. Female FVB/NJ strain background mice harboring the polyoma middle T (PyMT) transgene under the control of the mouse mammary tumor virus (MMTV) promoter (Guy et al., 1992), and the simian virus 40 large tumor antigen (SV40 TAg) under control of the rat prostatic steroid binding protein gene [C3(1)] (Maroulakou et al., 1994) have been previously described. PyMTchOVA mice expressing PyMT, mCherry, and ovalbumin under control of the MMTV promoter (Engelhardt et al., 2012) were backcrossed onto FVB/NJ mice 10 generations. Batf3-deficient mice (Hildner, 2008) were a kind gift of Kenneth Murphy (Washington University School of Medicine, St. Louis) and were backcrossed onto FVB/NJ mice 5 generations. Itgax-cre, Irf8fl/fl, Zbtb46-DTR, and MMTV-PyMT mice on the C57BL/6J background mice were acquired from The Jackson Laboratory. Bone marrow chimeric mice were generated by irradiating recipient mice with 2 doses of 500 rads, followed by a bone marrow transfer from donor animals, with tumors implanted after an additional 6 weeks. Implantation of orthotopic mammary tumors was performed in female mice (approximately 2-4 months of age) by using single-cell suspensions isolated from mammary tumors of MMTV-PyMT transgenic mice combined 1:1 with matrigel (Corning), and injecting 106 cells/100 μl into the right 2/3 mammary gland. For MMTV-PyMT animals treatment schedules were initiated in non-blinded fashion with age-matched littermates (day 80-85) randomized to treatment groups as indicated in the respective figures. C3(1)-TAg animals were treated when tumors reached 1 cm in diameter (approximately 5-8 months of age). Monoclonal antibodies (IgG1/HRPN, IgG2a/2A3, αTIM-3/RMT3-23, αTIM-4/RMT4-53, αCD8a/2.43, αCD8p/53-5.8, αIL-12p75/R2-9A5, αIFN-γ/XMG1.2, αIFNAR1/MAR1-5A3, αGalectin-9/RG9-1, αPD-1/RMP1-14, αCXCR3/CXCR3-173) were obtained from BioXCell and were administered by intraperitoneal (i.p.) injection at 1.0 mg/mouse, with follow-up doses of 0.5 mg every 5 days. FTY720 from Sigma-Aldrich was administered i.p. every 2 days at 20 μg per animal. DT (Sigma-Aldrich) was injected i.p. at 20 ng/g to start, and then at 4 ng/g every 2nd day. (±)-AMG 487 from R&D Systems was dissolved in 20% (2-hydroxypropyl)-β-cyclodextrin (Sigma-Aldrich) and was administered i.p twice daily at 5 μg/g as described (Walser et al., 2006). Clinical grade PTX (Hospira or Mylan) or carboplatin (Teva Pharmaceutical) was administered intravenously every 5 days at 10 mg/kg or 20 mg/kg, respectively.
Method Detail
Quantitation of metastatic burden
Following resection, lungs from transgenic MMTV-PyMT animals were injected with neutral buffered formalin via the trachea and incubated overnight in formalin prior to ethanol dehydration and paraffin embedding. Five lungs sections, each 100 μm apart, were haematoxylin and eosin stained and digitally scanned with an Aperio ScanScope CS Slide Scanner. Frequency and size of the metastatic foci were determined by manual circling in a blinded fashion using Imagescope software (Aperio).
Flow cytometry
Mice were cardiac-perfused with PBS containing 10 U/ml heparin to clear peripheral blood, and single cell suspensions were prepared by incubating minced tissue in 1 mg/ml collagenase (Roche) and 50 U/ml DNase I (Roche) at 35°C with agitation. Cells were used immediately or stored in 10% DMSO at -80°C. Immune populations were identified with a previously described gating strategy (Ruffell et al., 2014) using antibodies described in the Key Resource Table. Ex vivo intracellular staining for IL-12p40 (clone C15.6), granyzme B (clone GB11), or CXCL9 (clone MIG-2F5.5,) was performed on isolated cells 4-6 hr following an intravenous injection of 0.25 mg brefeldin A (Sigma-Aldrich). Alternatively, a single cell suspension was stimulated for 4 hr in vitro with Cell Activation Cocktail with Brefeldin A (BioLegend) or IFN-γ (40 ng/ml) in the presence of 5 μg/ml brefeldin A (BioLegend), and then stained for intracellular CXCL9, IFN-γ (clone XMG1.2), or TNF-α (clone MP6-XT22). Data was collected with either a LSRII or Fortessa flow cytometer (BD Bioscience). Human breast tumors were prepared as described above using antibodies listed in the Key Resource Table and the gating strategy shown in Supplemental Figure 1 (Ruffell et al., 2012), with data collected using a BD FACSymphony. All analysis was performed using FlowJo version 9 or 10 (FlowJo LLC).
Gene Expression
Fluorescent-activated cell sorting (FACS) was conducted on a FACSAriaII (BD Biosciences), with 2,000 to 50,000 sorted cells flash frozen in liquid nitrogen as a cell pellet. For realtime PCR analysis RNA was prepared using RNeasy Micro kit guidelines (Qiagen). Contaminating DNA was removed with DNAse I (Life Technologies), and then SuperScript III (Life Technologies) was used to reverse transcribe purified RNA into cDNA according to manufacturer's directions. PCR was performed using individual TaqMan Assays following a preamplification step (Life Technologies). The comparative threshold cycle method was used to calculate fold change in gene expression, which was normalized to a single (Tbp) reference gene. For gene expression analysis by Nanostring nCounter, cell lysates were hybridized to the 561 gene Mouse Immunology Panel according to the manufacturer's protocol (NanoString Technologies). Briefly, 10 μl of Ambion Cells-to-Ct buffer (Thermo Fisher Scientific) was added to a cell pellet and a 5.0 μl volume of lysate was hybridized to the NanoString reporter and capture probes in a thermal cycler for 16 hr at 65°C. Washing and cartridge immobilization were performed on the NanoString nCounter PrepStation, and the cartridge was scanned at 555 fields of view on the nCounter Digital Analyzer. The resulting RCC files containing raw counts were reviewed for quality and normalized in the NanoString nSolver Analysis Software v2.5, followed by exportation and analysis.
Immunohistochemistry and Immunofluoresence
5 μm sections of formalin fixed, paraffin embedded tissue were deparaffinized with xylene, rehydrated, and subjected to antigen retrieval with heated antigen unmasking solution (1.0 mM EDTA, 0.05% Tween 20, pH 8.0). After 1 hr in horse serum blocking buffer, primary antibodies were applied for 3 hr at room temperature or overnight at 4°C. Anti-human antibodies included TIM-3 (1:100 for IF, 1:400 for IHC, Clone D5D5R, Cell Signaling), Galectin-9 (1:100, Clone 1D12, Novus Biologicals), DC-LAMP (1:100, #AF4087, R&D Systems), pancytokeratin Alexa 488 (1:100, Clone C1 1, Cell Signaling); and the following from Thermo Scientific: CD45 (1:100, Clone PD7/26/16+2B11), CD3ε (1:100, Clone PS1), CD8 (1:50, Clone C8/144B), CD4 (1:50, Clone 4B12), CD163 (1:50, Clone 10D6), CXCL9 (1:100, Rabbit Polyclonal). Anti-mouse antibodies included cleaved caspase 3 (1:200, Cell Signaling #9661) and Ki67 (1:400, Clone D3B5, Cell Signaling). For immunohistochemistry, the ImmPRESS detection system was used with DAB chromogen, followed by counterstaining with hematoxylin QS (all from Vector Labs). Slides were digitally scanned using the Aperio ScanScope CS Slide Scanner with a 40× objective, and automated quantitative image analysis was performed using Imagescope and the nuclear detection algorithm (Leica Biosystems). For immunofluorescence, secondary antibodies were used at 1:500 for 1 hr at room temperature, followed by incubation with 1.0 μg/ml Hoechst 33342 for 15 min (all from Invitrogen). Slides were then washed and mounted with ProLong Gold anti-fade mounting medium (Invitrogen), and images were acquired with a Zeiss Axio Imager Z1.
In vitro DC stimulation
Bone marrow was harvested from FVB/NJ female mice and red blood cells lysed with 150 mM NH4Cl/10 mM NaHCO3/1 mM EDTA. Remaining cells were plated at 2×106 per ml in RPMI 1640 containing 2.0 mM L-glutamine and 25 mM HEPES, supplemented with 10 mM Sodium Pyruvate, nonessential amino acids, 100 U/ml penicillin/streptomycin, 55 μM β-ME, and 10% fetal calf serum (Life Technologies). Recombinant human Flt-3 Ligand Immunoglobulin (Flt-3L-Ig; BioXCell) was added at 100 ng/ml and cells were incubated untouched for 7 days. Cells in suspension were removed by pipetting (>90% CD11c+), resuspended at 106 per ml in RPMI1640 with 100 ng/ml Flt-3L-Ig, and incubated for 24 hr with the following reagents: IFN-γ (40 ng/ml; Peprotech), αCD40 (10 (μg/ml; FGK4.5; BioXCell), Poly(I:C)-LMW, LPS-EB Ultrapure, Imiquimod, or CpG ODN2395 (all at 1 μg/ml; InvivoGen), IL-10 (1-100 U/ml; Peprotech), and/or VEGFA (1-100 U/ml; Peprotech).
Splenic cDC were enriched (∼50% purity) by negative selection using biotinylated antibodies against CD3, B220, Ly6G, CD49b and Ter119 in combination with MojoSort magnetic beads (BioLegend). Cells were plated at 1×106 per ml in serum free RPMI 1640 and stimulated for 6 hr with the agents described above in the presence of 5 μg/ml brefeldin A (BioLegend), or suspended in supernatant containing tumor cell debris created by irradiation (15000 Rads, harvest after 48 hr) or heat shock (55°C for 1 hr) of PyMT cells at 70-80% confluence. Blocking antibodies against TIM-3 (clone RMT3-23, BioLegend), Galectin-9 (clone RG9-1, BioXCell), HMGB1 (clone 3E8, BioLegend, dialyzed to remove sodium azide), or CEACAM-1 (clone MAb-CC1, BioLegend) were added to the supernatant at 10 μg/ml.
Transwell assay
Splenic CD8+ T cells were isolated by negative selection using the MojoSort Mouse CD8+ T Cell Isolation Kit (Biolegend) following the manufacturer's instructions. Isolated cells were plated at 2×106 per mL in RPMI 1640 containing 2.0 mM L-glutamine and 25 mM HEPES, supplemented with 10 mM Sodium Pyruvate, nonessential amino acids, 100 U/ml penicillin/streptomycin, 55 μM β-ME, and 10% fetal calf serum (complete media). Cells were stimulated with 1 μg/mL ionomycin (Invivogen) and 1 ng/mL phorbol myristate acetate (PMA, Invivogen) for 7 days, with complete media supplemented with 200 U/mL recombinant human IL-2 added on days 3 and 6. Following stimulation, cells were resuspended at 5×105 per mL in RPMI 1640 supplemented with 0.1% bovine serum albumin, and were incubated at 4°C for 60 min with or without 100 nM (±)-AMG 487 (R&D Systems). Cells were then plated in the top well of 96 well transwell plate (3 μm polycarbonate membrane pore, Corning). The bottom well of the plate contained RPMI 1640 supplemented with 0.1% BSA, either with or without recombinant mouse CXCL9 or CXCL10 (Biolegend). Cells were allowed to migrate for 1 hr at 37°C, prior to data collection with a MACSQuant VYB flow cytometer (Miltenyi Biotech) and analysis using FlowJo version 10.
Quantification and statistical analysis
Statistical analyses were performed using Prism 6-7 (GraphPad). Data points represent biological replicates and are shown as the mean ± SEM unless otherwise indicated. Statistical significance was determined as indicated in the figure legends. For growth curves significance was determined via 2-way ANOVA with Tukey's multiple comparisons test, with significance shown for the final data point. A 2-way unpaired t-test or 2-way unpaired t-test with Welch's correction was used for comparison between groups with equal or unequal variance, respectively. Mann-Whitney and Kruskal-Wallis was used for data failing the D’Agostino & Pearson omnibus normality test. Significance is shown as *p<0.05, **p <0.01, ***p<0.001 as described in each figure legend. Heat maps were generated with GENE-E software (http://www.broadinstitute.org/cancer/software/GENE-E/), with hierarchical clustering performed with a one minus Pearson correlation. Linear regression analysis in breast cancer was performed in Prism using the dataset from The Cancer Genome Atlas Network (TCGA, 2012). Gene expression data from fine needle aspirate obtained prior to neoadjuvant chemotherapy in breast cancer patients was obtained from 2 datasets (GSE20194, GSE20271) annotated for pathologic complete response (Hess et al., 2006; Tabchy et al., 2010). Survival analysis was performed using Kaplan-Meier Plotter (kmplot.com) (Gyorffy et al., 2010).
Supplementary Material
Highlights.
TIM-3 is highly expressed by intratumoral CD103+ dendritic cells
TIM-3 antibody indirectly enhances a CD8+ T cell response during chemotherapy
TIM-3 antibody increases CXCL9 expression by dendritic cells
CXCL9 expression correlates with response to chemotherapy in breast cancer
de Mingo Pulido et al. show that intratumoral CD103+ dendritic cells (DC) highly express TIM-3. Anti-TIM-3 antibody promotes these DC to express CXCL9, which enhances the function of CD8+ T cells. In breast cancer models, anti-TIM-3 antibody improves therapeutic activity, which requires CD8+ T cells.
Acknowledgments
This work was supported by the Moffitt Cancer Center Flow Cytometry, Molecular Genomics, Analytic Microscopy, and Tissue Core Facilities, all comprehensive cancer center facilities designated by the National Cancer Institute (P30-CA076292). The authors would like to thank Vivian Lee, Asmaa E. El-Kenawi, Jodi Kroeger, Sean Yoder and Daniel Abate-Daga for technical assistance. Research reported herein was supported by a Breast Cancer Research Foundation grant to H.S.R, Komen Promise Award to H.S.R and L.M.C, DoD Era of Hope Expansion and NCI/NIH grants to L.M.C (R01CA15531-06, U54CA163123-05), and the Moffitt Cancer Center's Shula Breast Cancer Award and NCI/NIH grants (K99CA185325-01A1 and R00CA185325-02) to B.R.
Footnotes
Author Contributions: Conceptualization, L.M.C. and B.R.; Investigation, A.M.P., A.G., S.H. and B.R.; Materials, M.F.K.; Writing – Original Draft, B.R.; Writing – Review & Editing, A.M.P., A.G., H.S., M.F.K, L.M.C. and B.R.; Supervision, B.R.; Funding Acquisition, H.S.R., L.M.C. and B.R.
Declaration of Interests: This work was supported in part by a grant from Tesaro, Inc. H.S. has received payments from Novartis International AG for consulting and advisory boards. B.R. and H.S. have courtesy faculty appointments at the University of South Florida, Tampa, FL 33620.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, Chandwaskar R, Karman J, Su EW, Hirashima M, et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science. 2007;318:1141–1143. doi: 10.1126/science.1148536. [DOI] [PubMed] [Google Scholar]
- Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity. 2016;44:989–1004. doi: 10.1016/j.immuni.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baghdadi M, Nagao H, Yoshiyama H, Akiba H, Yagita H, Dosaka-Akita H, Jinushi M. Combined blockade of TIM-3 and TIM-4 augments cancer vaccine efficacy against established melanomas. Cancer Immunol Immunother. 2013;62:629–637. doi: 10.1007/s00262-012-1371-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, Wieckowski S, Bouzourene H, Deplancke B, Romero P, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. 2011;121:2350–2360. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. The Journal of experimental medicine. 2013;210:2435–2466. doi: 10.1084/jem.20130762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer cell. 2014;26:638–652. doi: 10.1016/j.ccell.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nature immunology. 2012;13:832–842. doi: 10.1038/ni.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffelt SB, de Visser KE. Immune-mediated mechanisms influencing the efficacy of anticancer therapies. Trends in immunology. 2015;36:198–216. doi: 10.1016/j.it.2015.02.006. [DOI] [PubMed] [Google Scholar]
- DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer discovery. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desch AN, Randolph GJ, Murphy K, Gautier EL, Kedl RM, Lahoud MH, Caminschi I, Shortman K, Henson PM, Jakubzick CV. CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. The Journal of experimental medicine. 2011;208:1789–1797. doi: 10.1084/jem.20110538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, Egeblad M, Krummel MF. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer cell. 2012;21:402–417. doi: 10.1016/j.ccr.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Kuchroo V, Zarour HM. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. The Journal of experimental medicine. 2010;207:2175–2186. doi: 10.1084/jem.20100637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nature reviews Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- Gorman JV, Starbeck-Miller G, Pham NL, Traver GL, Rothman PB, Harty JT, Colgan JD. Tim-3 directly enhances CD8 T cell responses to acute Listeria monocytogenes infection. Journal of immunology. 2014;192:3133–3142. doi: 10.4049/jimmunol.1302290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grajales-Reyes GE, Iwata A, Albring J, Wu X, Tussiwand R, Kc W, Kretzer NM, Briseno CG, Durai V, Bagadia P, et al. Batf3 maintains autoactivation of Irf8 for commitment of a CD8alpha(+) conventional DC clonogenic progenitor. Nature immunology. 2015;16:708–717. doi: 10.1038/ni.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123:725–731. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- Headley MB, Bins A, Nip A, Roberts EW, Looney MR, Gerard A, Krummel MF. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature. 2016 doi: 10.1038/nature16985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess KR, Anderson K, Symmans WF, Valero V, Ibrahim N, Mejia JA, Booser D, Theriault RL, Buzdar AU, Dempsey PJ, et al. Pharmacogenomic predictor of sensitivity to preoperative chemotherapy with paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide in breast cancer. J Clin Oncol. 2006;24:4236–4244. doi: 10.1200/JCO.2006.05.6861. [DOI] [PubMed] [Google Scholar]
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildner KEBT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM. Batf3 Deficiency Reveals a Critical Role for CD8a+ Dendritic Cells in Cytotoxic T Cell Immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima N, Iwasaki A. T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science. 2014;346:93–98. doi: 10.1126/science.1257530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson JT, Hu Y, Liu R, Masson F, D'Amico A, Carotta S, Xin A, Camilleri MJ, Mount AM, Kallies A, et al. Id2 expression delineates differential checkpoints in the genetic program of CD8alpha+ and CD103+ dendritic cell lineages. EMBO J. 2011;30:2690–2704. doi: 10.1038/emboj.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, Freeman GJ, Kuchroo VK, Ahmed R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14733–14738. doi: 10.1073/pnas.1009731107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, Wong JC, Satkunarajah M, Schweneker M, Chapman JM, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. The Journal of experimental medicine. 2008;205:2763–2779. doi: 10.1084/jem.20081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LP. T cell Ig and mucin domain proteins and immunity. Journal of immunology. 2010;184:2743–2749. doi: 10.4049/jimmunol.0902937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzaki M, Wada J, Sugiyama K, Nakatsuka A, Teshigawara S, Murakami K, Inoue K, Terami T, Katayama A, Eguchi J, et al. Galectin-9 and T cell immunoglobulin mucin-3 pathway is a therapeutic target for type 1 diabetes. Endocrinology. 2012;153:612–620. doi: 10.1210/en.2011-1579. [DOI] [PubMed] [Google Scholar]
- Kastenmuller W, Brandes M, Wang Z, Herz J, Egen JG, Germain RN. Peripheral prepositioning and local CXCL9 chemokine-mediated guidance orchestrate rapid memory CD8+ T cell responses in the lymph node. Immunity. 2013;38:502–513. doi: 10.1016/j.immuni.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh S, Ishii N, Nobumoto A, Takeshita K, Dai SY, Shinonaga R, Niki T, Nishi N, Tominaga A, Yamauchi A, Hirashima M. Galectin-9 inhibits CD44-hyaluronan interaction and suppresses a murine model of allergic asthma. Am J Respir Crit Care Med. 2007;176:27–35. doi: 10.1164/rccm.200608-1243OC. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annual review of immunology. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. The New England journal of medicine. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Su EW, Zhu C, Hainline S, Phuah J, Moroco JA, Smithgall TE, Kuchroo VK, Kane LP. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol Cell Biol. 2011;31:3963–3974. doi: 10.1128/MCB.05297-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner J, Rieger A, Pickl WF, Zlabinger G, Grabmeier-Pfistershammer K, Steinberger P. TIM-3 does not act as a receptor for galectin-9. PLoS Pathog. 2013;9:e1003253. doi: 10.1371/journal.ppat.1003253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroulakou IG, Anver M, Garrett L, Green JE. Prostate and mammary adenocarcinoma in transgenic mice carrying a rat C3(1) simian virus 40 large tumor antigen fusion gene. Proc Natl Acad Sci USA. 1994;91:11236–11240. doi: 10.1073/pnas.91.23.11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurya N, Gujar R, Gupta M, Yadav V, Verma S, Sen P. Immunoregulation of dendritic cells by the receptor T cell Ig and mucin protein-3 via Bruton's tyrosine kinase and c-Src. Journal of immunology. 2014;193:3417–3425. doi: 10.4049/jimmunol.1400395. [DOI] [PubMed] [Google Scholar]
- Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, Manning S, Greenfield EA, Coyle AJ, Sobel RA, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536–541. doi: 10.1038/415536a. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, Yagita H, Okumura K. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. 2009;113:3821–3830. doi: 10.1182/blood-2008-10-185884. [DOI] [PubMed] [Google Scholar]
- Natsuaki Y, Egawa G, Nakamizo S, Ono S, Hanakawa S, Okada T, Kusuba N, Otsuka A, Kitoh A, Honda T, et al. Perivascular leukocyte clusters are essential for efficient activation of effector T cells in the skin. Nature immunology. 2014;15:1064–1069. doi: 10.1038/ni.2992. [DOI] [PubMed] [Google Scholar]
- Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer research. 2011;71:3540–3551. doi: 10.1158/0008-5472.CAN-11-0096. [DOI] [PubMed] [Google Scholar]
- Oikawa T, Kamimura Y, Akiba H, Yagita H, Okumura K, Takahashi H, Zeniya M, Tajiri H, Azuma M. Preferential involvement of Tim-3 in the regulation of hepatic CD8+ T cells in murine acute graft-versus-host disease. Journal of immunology. 2006;177:4281–4287. doi: 10.4049/jimmunol.177.7.4281. [DOI] [PubMed] [Google Scholar]
- Phong BL, Avery L, Sumpter TL, Gorman JV, Watkins SC, Colgan JD, Kane LP. Tim-3 enhances FcepsilonRI-proximal signaling to modulate mast cell activation. The Journal of experimental medicine. 2015;212:2289–2304. doi: 10.1084/jem.20150388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangachari M, Zhu C, Sakuishi K, Xiao S, Karman J, Chen A, Angin M, Wakeham A, Greenfield EA, Sobel RA, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3-mediated cell death and exhaustion. Nature medicine. 2012;18:1394–1400. doi: 10.1038/nm.2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, Krummel MF. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer cell. 2016;30:324–336. doi: 10.1016/j.ccell.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell B, Au A, Rugo HS, Esserman LJ, Hwang ES, Coussens LM. Leukocyte composition of human breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2796–2801. doi: 10.1073/pnas.1104303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, Daniel D, Hwang ES, Rugo HS, Coussens LM. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer cell. 2014;26:623–637. doi: 10.1016/j.ccell.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugo HS, Barry WT, Moreno-Aspitia A, Lyss AP, Cirrincione C, Leung E, Mayer EL, Naughton M, Toppmeyer D, Carey LA, et al. Randomized Phase III Trial of Paclitaxel Once Per Week Compared With Nanoparticle Albumin-Bound Nab-Paclitaxel Once Per Week or Ixabepilone With Bevacizumab As First-Line Chemotherapy for Locally Recurrent or Metastatic Breast Cancer: CALGB 40502/NCCTG N063H (Alliance) Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2015;33:2361–2369. doi: 10.1200/JCO.2014.59.5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. The Journal of experimental medicine. 2010;207:2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado R, Denkert C, Demaria S, Sirtaine N, Klauschen F, Pruneri G, Wienert S, Van den Eynden G, Baehner FL, Penault-Llorca F, et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an International TILs Working Group 2014. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2015;26:259–271. doi: 10.1093/annonc/mdu450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity. 2016;44:924–938. doi: 10.1016/j.immuni.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Paulete AR, Cueto FJ, Martinez-Lopez M, Labiano S, Morales-Kastresana A, Rodriguez-Ruiz ME, Jure-Kunkel M, Azpilikueta A, Aznar MA, Quetglas JI, et al. Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer discovery. 2015 doi: 10.1158/2159-8290.CD-15-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seillet C, Jackson JT, Markey KA, Brady HJ, Hill GR, Macdonald KP, Nutt SL, Belz GT. CD8alpha+ DCs can be induced in the absence of transcription factors Id2, Nfil3, and Batf3. Blood. 2013;121:1574–1583. doi: 10.1182/blood-2012-07-445650. [DOI] [PubMed] [Google Scholar]
- Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remedios C, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nature medicine. 2014;20:1301–1309. doi: 10.1038/nm.3708. [DOI] [PubMed] [Google Scholar]
- Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015 doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer cell. 2017;31:711–723. doi: 10.1016/j.ccell.2017.04.003. e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su EW, Bi S, Kane LP. Galectin-9 regulates T helper cell function independently of Tim-3. Glycobiology. 2011;21:1258–1265. doi: 10.1093/glycob/cwq214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabchy A, Valero V, Vidaurre T, Lluch A, Gomez H, Martin M, Qi Y, Barajas-Figueroa LJ, Souchon E, Coutant C, et al. Evaluation of a 30-gene Paclitaxel, Fluorouracil, Doxorubicin, and cyclophosphamide chemotherapy response predictor in a multicenter randomized trial in breast cancer. Clin Cancer Res. 2010;16:5351–5361. doi: 10.1158/1078-0432.CCR-10-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MH, Goldinger SM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Weyer PS, Muehlfeit M, Klose C, Bonventre JV, Walz G, Kuehn EW. A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem Biophys Res Commun. 2006;351:571–576. doi: 10.1016/j.bbrc.2006.10.079. [DOI] [PubMed] [Google Scholar]
- Walser TC, Rifat S, Ma X, Kundu N, Ward C, Goloubeva O, Johnson MG, Medina JC, Collins TL, Fulton AM. Antagonism of CXCR3 inhibits lung metastasis in a murine model of metastatic breast cancer. Cancer research. 2006;66:7701–7707. doi: 10.1158/0008-5472.CAN-06-0709. [DOI] [PubMed] [Google Scholar]
- Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515:572–576. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, Zheng XX, Strom TB, Kuchroo VK. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nature immunology. 2005;6:1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.