

Abstract
A better understanding of natural variation in neutralization resistance and fitness of diverse hepatitis C virus (HCV) envelope (E1E2) variants will be critical to guide rational development of an HCV vaccine. This work has been hindered by inadequate genetic diversity in viral panels and by a lack of standardization of HCV entry assays. Neutralization assays generally use lentiviral pseudoparticles expressing HCV envelope proteins (HCVpp) or chimeric full-length viruses that are replication competent in cell culture (HCVcc). There have been few systematic comparisons of specific infectivities of E1E2-matched HCVcc and HCVpp, and to our knowledge, neutralization of E1E2-matched HCVpp and HCVcc has never been compared using a diverse panel of human broadly neutralizing monoclonal antibodies (bNAbs) targeting distinct epitopes. Here, we describe an efficient method for introduction of naturally occurring E1E2 genes into a full-length HCV genome, producing replication-competent chimeric HCVcc. We generated diverse panels of E1E2-matched HCVcc and HCVpp and measured the entry-mediating fitness of E1E2 variants using the two systems. We also compared neutralization of E1E2-matched HCVcc and HCVpp by a diverse panel of human bNAbs targeting epitopes across E1E2. We found no correlation between specific infectivities of E1E2-matched HCVcc versus HCVpp, but found a very strong positive correlation between relative neutralization resistance of these same E1E2-matched HCVcc and HCVpp variants. These results suggest that quantitative comparisons of neutralization resistance of E1E2 variants can be made with confidence using either HCVcc or HCVpp, allowing the use of either or both systems to maximize diversity of neutralization panels.
Keywords: neutralizing antibody, broadly neutralizing antibody, hepatitis C virus, pseudoparticle, HCVcc
Introduction
Hepatitis C virus (HCV) infects over 170 million people worldwide (Thomas et al., 2000), and kills more people in the USA annually than human immunodeficiency virus (HIV) (Holmberg et al., 2013). While direct-acting antiviral therapy has revolutionized care for some patients with HCV, control of the HCV pandemic remains challenging due to poor access to care, frequent nosocomial transmission in developing countries (Averhoff et al., 2012), reinfection in high-risk individuals and the high proportion of infected individuals who are unaware of asymptomatic carriers (Armstrong et al., 2006). Since direct-acting antiviral therapy is very unlikely to control the HCV pandemic, development of a vaccine against HCV remains essential.
Understanding of virus neutralization by antibodies is important for vaccine development, since prior studies have shown that antibodies that neutralize HCV play an important role in immune-mediated control of the virus, and that neutralizing antibody resistance mutations may reduce viral replicative fitness (Gottwein et al., 2009; Lavie et al., 2014; Osburn et al., 2014; Pestka et al., 2007). However, study of HCV neutralizing antibodies has been hindered by lack of standardized methods to measure virus entry and neutralization, and by inadequate genetic diversity in viral panels. Entry and neutralization assays generally rely on either lentiviral particles with HCV envelope proteins (E1and E2) on their surface [HCV pseudoparticles (HCVpp)], or full-length hepatitis viruses that are replication competent in cell culture (HCVcc).
HCVpp have greatly advanced the understanding of HCV entry and neutralization by antibodies (Giang et al., 2012; Law et al., 2008; Morin et al., 2012; Osburn et al., 2014; Pestka et al., 2007). This model system has been used extensively in identifying HCV cell surface receptors required for viral entry (Bartosch et al., 2003; Cormier et al., 2004; McKeating et al., 2004; Zhang et al., 2004), and studies using HCVpp have established that neutralizing antibodies can drive evolution in vivo of HCV E1E2, leading to neutralization escape (Dowd et al., 2009; Pestka et al., 2007). HCVpp panels expressing diverse E1E2 variants have also been used to define neutralizing breadth of antibodies and to show that development of broadly neutralizing antibodies is associated with spontaneous clearance of HCV infection (Osburn et al., 2014; Pestka et al., 2007), and reduced liver fibrosis in chronic infection (Swann et al., 2016).
More recently, chimeric full-length viruses that are HCVcc have been produced which express diverse E1E2 variants and reproduce the full replication cycle of HCV in vitro and in animal models (Gottwein et al., 2007, 2009; Scheel et al., 2011). These HCVcc have been used to further define viral entry pathways (Brimacombe et al., 2011; Carlsen et al., 2013; Mathiesen et al., 2015; Timpe et al., 2008) and to show that broadly neutralizing monoclonal antibodies (bNAbs) can prevent HCV infection in animal models (Forns et al., 2000; Morin et al., 2012; Youn et al., 2005). Panels of HCVcc expressing E1E2 from multiple genotypes have also been used to measure neutralizing breadth of bNAbs (Carlsen et al., 2014; Keck et al., 2008, 2013; Law et al., 2008).
Overall, HCVpp and HCVcc both have advantages for investigation of HCV entry and antibody neutralization. HCVpp can be used to quantitate single round entry and neutralization in a high-throughput format. Recently, large panels of HCVpp have been produced that encompass more of the diversity of circulating HCV variants, which is critical for accurate measurement of antibody neutralizing breadth (Osburn et al., 2014; Urbanowicz et al., 2015). While significantly fewer replication-competent HCVcc variants are available, progress has been made in expanding the number of variants (Mathiesen et al., 2014; McClure et al., 2016; Urbanowicz et al., 2015). E1E2 on surface of HCVcc may resemble more closely the envelopes of viruses circulating in vivo, given incorporation of human apolipoproteins into HCVcc virions (Hishiki et al., 2010; Jiang et al., 2012; Meunier et al., 2008).
As viral entry, neutralization and vaccine development studies proceed using HCVpp, HCVcc or both, it is critical to understand whether specific infectivity and neutralization results measured in one assay system are reproducible in the other. Neutralization results obtained using HCVcc and HCVpp have generally been similar (Fofana et al., 2012; Meunier et al., 2005; Scheel et al., 2008; Swann et al., 2016; Urbanowicz et al., 2015), but comparisons of specific infectivity and neutralization of HCVpp and HCVcc expressing genetically identical E1E2 variants have previously been performed only on a fairly limited basis, and to our knowledge neutralization of E1E2-matched HCVpp and HCVcc has never been compared using a diverse panel of bNAbs targeting distinct epitopes.
Here, we describe an efficient method for insertion of naturally occurring E1E2 genes into a full-length HCV genome, producing a panel of replication-competent HCVcc chimeras expressing naturally occurring E1E2 proteins. Identical E1E2 genes were also used to produce HCVpp, allowing a systematic comparison of specific infectivities of E1E2-matched HCVcc and HCVpp, as well as comparison of neutralization of E1E2-matched HCVcc and HCVpp by a panel of eight well-characterized human bNAbs targeting distinct epitopes.
Results
Efficient cloning of naturally occurring E1E2 genes to generate E1E2-matched HCVcc and HCVpp
We previously generated a modified H77/JFH-1 expression plasmid with the E1E2 gene sequence replaced by an AfeI restriction site, allowing rapid in-frame introduction of natural E1E2 genes by In-Fusion cloning and expression of chimeric virus (Wasilewski et al., 2016) (Fig. 1). For this study, we selected 19 natural E1E2 variants previously shown to produce functional HCVpp, and used this HCVcc production method to produce a panel of HCVcc chimeras expressing these same naturally occurring E1E2 protein variants. Of these 19 HCVcc chimeras, differing at an average of 14 % of their E1E2 amino acids (0.3–22 %), 13 produced replication-competent virus and 6 were non-functional in multiple experiments (Fig. 2). Subsets of this panel of replication-competent HCVcc, along with two previously described HCVcc chimeras, H77/JFH-1 (genotype 1a E1E2) (Scheel et al., 2008) and S52/JFH-1 (genotype 3a E1E2) (Gottwein et al., 2007), were used to compare specific infectivity of E1E2-matched HCVcc and HCVpp, and also to compare neutralization of E1E2-matched HCVpp and HCVcc by a panel of human bNAbs targeting distinct epitopes across E1 and E2.
Fig. 1.

Cloning strategy to generate HCVcc and HCVpp with matched E1E2 proteins. Using PCR and In-Fusion cloning, the H77 E1E2 gene sequence was deleted from the H77/JFH-1 chimeric full-length replication-competent HCV (HCVcc) genome, and an AfeI restriction site was introduced between the Core and P7 genes. This plasmid was then linearized by AfeI digestion and various E1E2 genes from naturally circulating HCVs inserted, generating chimeric H77/natural isolate E1E2/JFH-1 HCVcc genomes. RNA was produced via in vitro transcription and transfected into Huh7.5.1 cells to generate replication-competent virus. Matching natural isolate E1E2 gene sequences were also cloned into the expression vector pcDNA3.2, and this plasmid was co-transfected with the lentiviral luciferase reporter plasmid pNL4-3.Luc.R−E− to produce HCVpp. Colours in the HCVcc genome indicate the HCV variant from which each segment is derived (red, JFH-1; blue, H77; orange, naturally circulating HCV sequence).
Fig. 2.
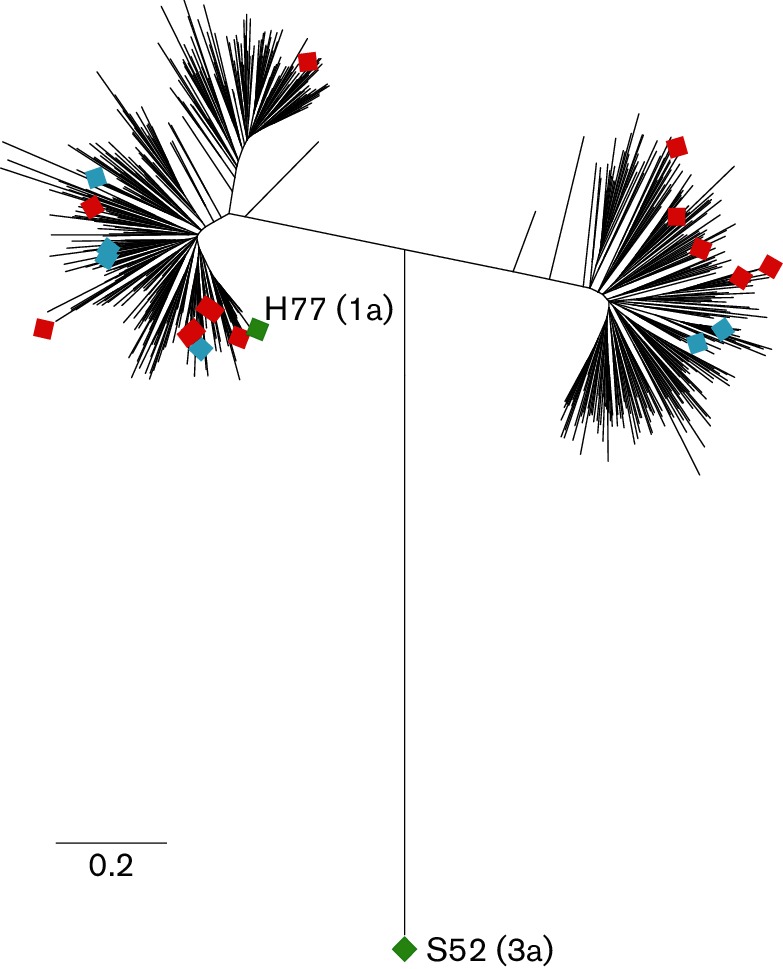
E1E2 genes used to generate E1E2-matched HCVcc and HCVpp are genetically diverse. Maximum-likelihood tree of E1E2 amino acid sequences of envelope-matched HCVcc and HCVpp (diamonds) with 634 genotype 1 reference sequences from GenBank (lines without symbols). Nineteen primary isolate E1E2 genes that were functional in HCVpp (12 genotype 1a and 7 genotype 1b) were used to generate HCVcc chimeras: 13 HCVcc chimeras were replication competent (red symbols) and 6 were replication incompetent (blue symbols). These novel replication-competent HCVcc as well as two previously described HCVcc chimeras, H77/JFH-1 (genotype 1a E1E2) and S52/JFH-1 (genotype 3a E1E2) (green symbols), were used in infectivity and neutralization experiments.
There is no correlation between specific infectivities of E1E2-matched HCVcc and HCVpp
To determine whether in vitro function of E1E2 quantified using HCVcc or HCVpp is related, specific infectivities [spot forming units (SFU) per HCV IU for HCVcc and relative light units (RLU) per HIV IU for HCVpp] of 12 functional E1E2-matched HCVcc and HCVpp were compared. To limit HCVcc as much as possible to single round infection, virus was removed from cell supernatant after 12 h, and infected cells were fixed and stained after a 72 h incubation period. For both HCVpp and HCVcc, dilution of supernatant and level of infection showed a linear relationship (data not shown). Specific infectivities of HCVcc [range: 0.03–0.67 SFU (HCV IU)−1] and HCVpp [range: 0.002–0.079 RLU (HIV IU)−1] expressing different E1E2 variants varied considerably (Fig. 3a and Table S1, available in the online Supplementary Material). Interestingly, no correlation in relative specific infectivities of E1E2-matched HCVcc and HCVpp was observed. These results suggest that relative specific infectivity of different E1E2 variants measured with either HCVpp or HCVcc may not predict results that would be obtained using the other model system.
Fig. 3.
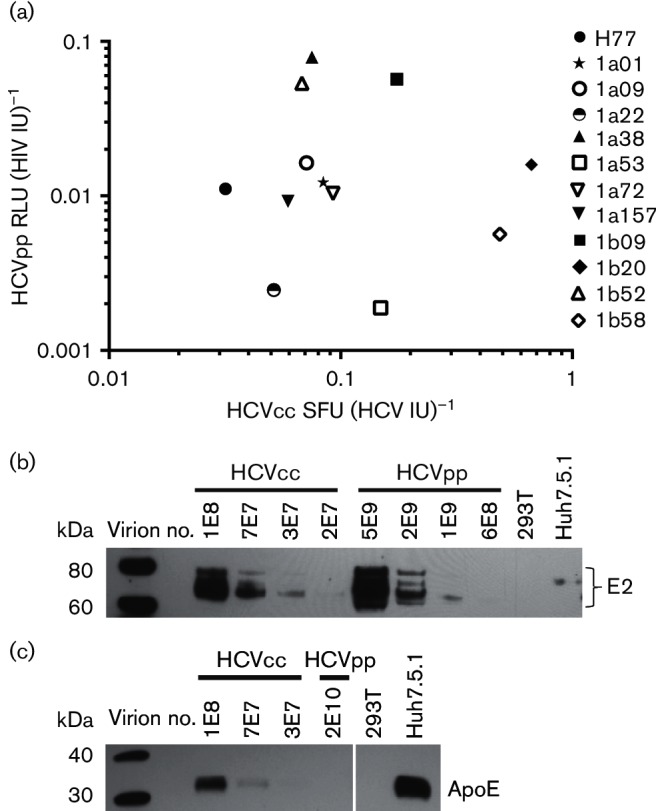
No correlation between relative specific infectivities of E1E2-matched HCVcc and HCVpp. (a) Serial dilutions of 12 different E1E2-matched HCVcc and HCVpp were used to infect Huh7.5.1 cells. Using a data point in the linear range of each infectivity assay, entry was quantified for HCVpp (RLU per millilitre of supernatant) and HCVcc (SFU per millilitre of supernatant). Viral RNA was extracted from these supernatants and RNA viral load (IU per millilitre) quantitated using real-time PCR and an IU viral load standard. Specific infectivity for HCVpp was calculated as RLU per HIV IU and for HCVcc as SFU per HCV IU. Each point represents specific infectivity of HCVcc on the x-axis and HCVpp on the y-axis of E1E2-matched HCVpp and HCVcc. Values represent the average of two independent experiments performed in duplicate or triplicate. (b) E1E2-matched HCVcc and HCVpp (1a38 variant) were purified by ultracentrifugation through a 20 % sucrose cushion. Viral RNA was extracted from the purified supernatants and viral copy number (IU) measured. A dilution series of the purified virus was analysed by Western blotting. Mock-transfected 293T supernatants (293T) and uninfected Huh7.5.1 cell supernatants (Huh7.5.1) were also tested. Blots were probed with human anti-E2 (HC33.1.53). (c) Purified E1E2-matched HCVcc and HCVpp (1a38 variant), mock-transfected 293T supernatants (293T) and uninfected Huh7.5.1 supernatants (Huh7.5.1) were analysed by Western blotting probed with anti-ApoE.
Given these differences in specific infectivity between E1E2-matched HCVcc and HCVpp, we investigated biochemical differences between HCVcc and HCVpp. To determine whether the amount of E2 incorporated per virion differs between E1E2-matched HCVcc and HCVpp, we performed a Western blot analysis on HCVcc and HCVpp which had been purified by ultracentrifugation through a 20 % sucrose cushion (Fig. 3b). HCVcc incorporated significantly more E2 per virion, with HCVcc E2 becoming undetectable by Western blot below a dilution of 3E7 virions (virions quantitated by HCV IU), whereas HCVpp E2 became undetectable below 1E9 virions (virions quantitated by HIV IU). The complex E2 banding pattern observed has been reported elsewhere and has been attributed to the presence of FBS protein (Vieyres et al., 2010) or the high mannose content of E2 glycans (Falkowska et al., 2007). Together, these results suggest that HCVcc incorporate a larger number of E2 molecules per virion than HCVpp.
Multiple prior studies have demonstrated that human apolipoproteins are present on the surface of HCVcc virions. To investigate the presence of apolipoproteins in our model systems, we performed a Western blot analysis on purified E1E2-matched HCVcc and HCVpp. We found that we can detect ApoE protein in purified HCVcc preparations and in uninfected Huh7.5.1 supernatants (Fig. 3c). It has been previously demonstrated that some of this ApoE is associated with HCVcc virions (Boyer et al., 2014; Catanese et al., 2013; Jiang & Luo, 2009). Despite testing a much higher number of purified HCVpp virions as well as mock-transfected 293T supernatants, we could not detect any ApoE protein. These data show that ApoE protein is present in HCVcc supernatants, but is not present in HCVpp supernatants or the 293T cells used to produce them.
E1E2-matched HCVcc and HCVpp show the same hierarchy of resistance to a diverse panel of human broadly neutralizing antibodies
To compare antibody neutralization measured using HCVcc and HCVpp model systems, five E1E2-matched HCVpp and HCVcc (H77, 1a38, 1a53, 1b09 and 1b52) were tested for neutralization by serial dilutions of up to four bNAbs each to quantitate neutralization sensitivity, with a total of 12 E1E2/bNAb combinations tested in this manner. Despite the differences in HCVcc- and HCVpp-specific infectivity, E2 incorporation and ApoE incorporation, relative neutralization of E1E2-matched HCVcc and HCVpp by bNAbs was strikingly similar. Representative neutralization curves are shown in Fig. 4, with all other curves in Fig. S1. IC50 values for all E1E2-matched HCVcc and HCVpp tested with full bNAb dilution curves are summarized in Table 1.
Fig. 4.

E1E2-matched HCVcc and HCVpp show the same hierarchy of neutralization resistance to a panel of broadly neutralizing antibodies targeting distinct epitopes. Representative neutralization curves for H77 and 1b09 (natural isolate) HCVcc and HCVpp. E1E2-matched HCVcc and HCVpp were tested for sensitivity to neutralization by serial dilutions of anti-HCV bNAbs or non-specific IgG. Colours indicate bNAb tested (dark blue, CBH-5; red, HC84.26; green, HC84.22; purple, HC33.4; orange, AR3A). Error bars indicate sd between duplicate wells. IC50 results for all E1E2/bNAb combinations tested with full mAb dilution curves for both HCVcc and HCVpp are summarized in Table 1, and all neutralization curves are shown in Fig. S1.
Table 1. E1E2-matched HCVpp and HCVcc IC50 values.
| E1E2 | mAb | HCVpp IC50 (µg ml−1)* | HCVcc IC50 (µg ml−1)* |
|---|---|---|---|
| H77 | AR3A | 0.10 | 9.56 |
| CBH-5 | >50 | >50 | |
| HC33.4 | 0.12 | 1.16 | |
| HC84.22 | 0.38 | ~50 | |
| 1a38 | AR4A | >50 | >50 |
| 1a53 | AR3C | 0.14 | 0.76 |
| AR4A | 0.36 | 0.68 | |
| 1b09 | AR3A | 0.05 | 0.51 |
| CBH-5 | 0.30 | 1.84 | |
| HC84.22 | 0.37 | 3.20 | |
| HC84.26 | 0.03 | 5.4e−03 | |
| 1b52 | AR5A | ~50 | 34.67 |
*For neutralization curves with only the highest antibody concentration producing more than 50 % neutralization, IC50 is reported as ~50 µg ml−1 and for curves with maximum neutralization less than 50 %, IC50 is reported as >50 µg ml−1.
Using Spearman correlation, rank order of IC50s of 12 different bNAb/HCVpp combinations were compared to the rank order of IC50s of the same bNAbs tested against E1E2-matched HCVcc (Fig. 5). A highly significant positive correlation between IC50s determined using HCVpp or HCVcc was observed (r=0.8, P=0.002), suggesting that comparison of relative neutralization resistance of different E1E2 variants expressed on either HCVcc or HCVpp reliably yields the same result. Interestingly, while relative neutralization of E1E2-matched HCVpp and HCVcc was very tightly correlated, there are examples of E1E2 that were neutralized to similar levels in HCVpp, but neutralized differently in HCVcc (i.e. H77 and 1b09 neutralized by mAb HC84.22, Fig. 5). Also, the absolute IC50 values for each bNAb/HCVpp were generally lower than the IC50s measured for the same bNAbs with E1E2-matched HCVcc (Fig. 5 and Table 1), suggesting that HCVpp may be generally more neutralization sensitive than HCVcc. Notable exceptions are combinations like mAb CBH-5/E1E2 variant H77 and mAb AR5A/E1E2 variant 1b52, where both HCVcc and HCVpp were fully or almost fully resistant. Also, bNAb HC84.26 had a lower IC50 measured against 1b09 HCVcc than against 1b09 HCVpp (IC50 5.4e−03 vs 0.03 µg ml−1).
Fig. 5.
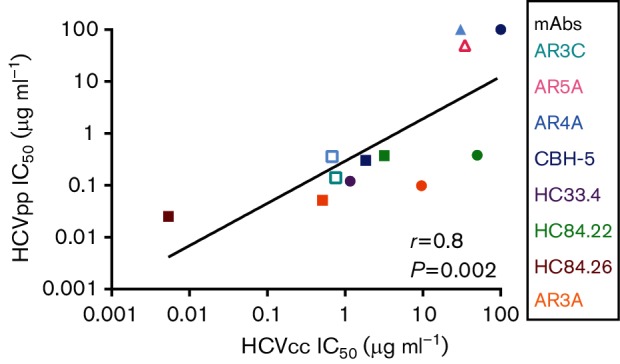
Positive correlation between the rank orders of IC50 values measured using E1E2-matched HCVpp and HCVcc. E1E2-matched HCVcc and HCVpp with five different E1E2 proteins (H77, 1a38, 1a53, 1b09 and 1b52) were tested in duplicate in neutralization assays with serial dilutions of up to four different bNAbs targeting distinct epitopes or control non-specific IgG (12 total bNAb/E1E2 combinations). HCVpp and HCVcc IC50 values were calculated from these curves. Each point indicates the HCVcc IC50 on the x-axis and HCVpp IC50 on the y-axis of E1E2-matched HCVcc and HCVpp. For neutralization curves with only the highest antibody concentration (50 µg ml−1) producing more than 50 % neutralization, IC50 is graphed as 50 µg ml−1 and for curves with maximum neutralization less than 50 %, IC50 is graphed as 100 µg ml−1. Colours indicate mAb tested (cyan, AR3C; pink, AR5A; light blue, AR4A; dark blue, CBH-5; purple, HC33.4; green, HC84.22; red, HC84.26; orange, AR3A) and shapes indicate E1E2 sequence (▲, 1a38; □, 1a53; ⚫, H77; △, 1b52; ■, 1b09). Correlation (r) and P value were computed using the Spearman method.
To improve throughput of HCVpp neutralization assays, neutralization of HCVpp is sometimes measured at a single concentration of antibody or a single dilution of serum rather than with full antibody dilution series. This quantitative neutralization value can be expressed as a fraction unaffected (Fu), which is infection in the presence of a given concentration of antibody relative to infection in the presence of an equivalent concentration of non-specific IgG. Seven HCVpp were tested for neutralization by 10 µg ml−1 of up to seven bNAbs each to quantitate neutralization sensitivity, with a total of 34 bNAb/HCVpp combinations tested in this manner (Table 2). E1E2 variants tested included four different genotype 1a isolates (H77, 1a09, 1a38 and 1a53), two different genotype 1b isolates (1b09, 1b52) and one genotype 3a isolate (S52). HCVcc expressing these same E1E2 variants were tested for neutralization by full dilution series of the same bNAbs, allowing calculation of IC50 (Table 2 and Fig. S1). Of note, IC50s measured for H77 HCVcc and S52 HCVcc with bNAbs CBH-5, HC33.4, HC84.26 and AR3A were all consistent with values measured in a previously published study (Carlsen et al., 2014). As has been shown in prior publications (Bailey et al., 2015a; Keck et al., 2009; Wasilewski et al., 2016), relative resistance of HCVcc variants to most bNAbs did not show any relationship to polymorphisms present at known bNAb binding residues (Fig. S2). One exception was the presence of F442L in E1E2 variants with resistance to HC84.22 and HC84.26. This polymorphism was previously shown to confer resistance to these mAbs (Bailey et al., 2015a).
Table 2. HCVpp Fu and HCVcc IC50 values.
| E1E2 | mAb | HCVpp Fu (mAb at 10 µg ml−1) | HCVcc IC50 (µg ml−1)* |
|---|---|---|---|
| H77 | AR3A | 0.23 | 9.56† |
| CBH-5 | 0.67 | >50† | |
| HC33.4 | 3.9e−03 | 1.16† | |
| HC84.22 | 0.17 | ~50† | |
| HC84.26 | 0.003 | 0.1 | |
| 1a09 | AR3A | 0.51 | >50 |
| CBH-2 | 0.67 | >50 | |
| CBH-5 | 0.56 | >50 | |
| HC33.4 | 0.23 | >50 | |
| HC84.22 | 0.66 | >50 | |
| HC84.26 | 0.49 | >50 | |
| 1a53 | AR3A | 0.09 | 8.0 |
| AR3C | 0.01 | 0.76† | |
| AR4A | 0.02 | 0.68† | |
| CBH-5 | 0.17 | ~50 | |
| HC33.4 | 0.04 | 5.71 | |
| HC84.22 | 0.16 | >50 | |
| HC84.26 | 8.7e−03 | 0.08 | |
| 1b09 | AR3A | 0.12 | 0.51† |
| CBH-5 | 0.06 | 1.84† | |
| HC33.4 | 0.14 | 2.17 | |
| HC84.22 | 0.09 | 3.2† | |
| HC84.26 | 2.9e−04 | 5.4e−03† | |
| 1b52 | AR3A | 0.59 | >50 |
| AR5A | 0.88 | 34.67† | |
| CBH-5 | 0.39 | >50 | |
| HC33.4 | 0.38 | ~50 | |
| HC84.22 | 0.37 | >50 | |
| HC84.26 | 1.6e−03 | 1.12 | |
| S52 | AR3A | 0.68 | >50 |
| CBH-5 | 0.79 | >50 | |
| HC33.4 | 0.35 | ~50 | |
| HC84.22 | 0.84 | >50 | |
| HC84.26 | 0.53 | 0.53 |
*For neutralization curves with only the highest antibody concentration producing more than 50 % neutralization, IC50 is reported as ~50 µg ml−1 and for curves with maximum neutralization less than 50 %, IC50 is reported as >50 µg ml−1.
†Values also reported in Table 1.
To determine whether HCVpp Fu also has a positive correlation with E1E2-matched HCVcc IC50, we compared the rank order of 10 µg ml−1 Fu of 34 different bNAb/HCVpp combinations to the IC50s of the same bNAbs measured using E1E2-matched HCVcc (Fig. 6). These values also showed a highly significant positive correlation (r=0.7, P<0.0001), suggesting that even if neutralization of HCVpp is tested at a single mAb concentration, Fu values varying more than 3000-fold can be quantitated, and comparison of relative neutralization resistance of different E1E2 variants expressed on either HCVcc or HCVpp reliably yields the same result.
Fig. 6.
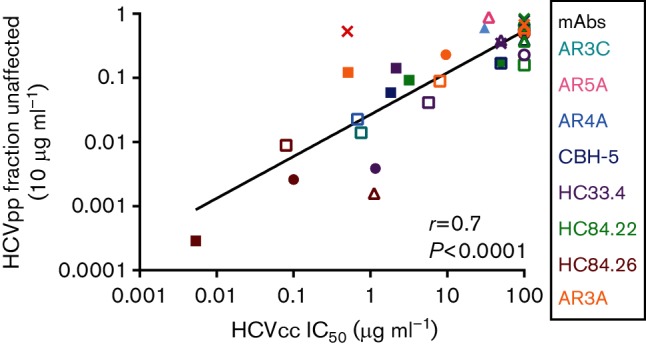
Positive correlation between the rank orders of Fu values measured using HCVpp and IC50 values measured using E1E2-matched HCVcc. E1E2-matched HCVcc and HCVpp with six different E1E2 proteins (H77, 1a38, 1a53, 1b09, 1b52 and S52) were tested in duplicate in neutralization assays with up to seven different bNabs or control non-specific IgG (34 total bNAb/E1E2 combinations). For HCVpp, neutralization was tested at a single concentration of mAb (10 µg ml−1), and Fu was calculated (infection in the presence of 10 µg ml−1 of mAb/infection in the presence of non-specific IgG). HCVcc were tested with serial dilutions of these bNabs and IC50 values were calculated from these curves. Values are summarized in Table 2. Relative HCVpp Fu values were compared to IC50 values of E1E2-matched HCVcc neutralized by the same bNAbs. Each point indicates the HCVcc IC50 on the x-axis and HCVpp Fu on the y-axis of E1E2-matched HCVcc and HCVpp. Colours indicate mAb tested (cyan, AR3C; pink, AR5A; light blue, AR4A; dark blue, CBH-5; purple, HC33.4; green, HC84.22; red, HC84.26; orange, AR3A) and shapes indicate E1E2 (○, 1a09; ▲, 1a38; □, 1a53; ⚫, H77; △, 1b52; ■, 1b09; ×, S52). Correlation (r) and P value were computed using the Spearman method.
Discussion
For this study, we produced infectious HCVcc chimeras expressing naturally occurring E1E2 genes. We used paired HCVcc and HCVpp with genetically identical E1E2 to compare the two model systems with respect to specific infectivity and bNAb neutralization resistance. We found that specific infectivities of E1E2-matched HCVcc and HCVpp showed no correlation, perhaps due to different amounts of E2 on HCVcc virions relative to HCVpp, and presence of ApoE on HCVcc virions and not HCVpp. Despite these differences, there was a very strong correlation between relative bNAb neutralization resistance of E1E2-matched HCVcc and HCVpp variants.
Our work supports prior more limited studies showing that HCVcc and HCVpp with identical E1E2 have similar relative neutralization resistance (Bailey et al., 2015a; Fofana et al., 2012; Swann et al., 2016; Urbanowicz et al., 2015). Importantly, to our knowledge, this is the first study to show this correlation using a diverse panel of human bNAbs targeting distinct epitopes across E1E2 and a large panel of diverse natural E1E2 variants, suggesting that these results are broadly applicable to other E1E2 variants and other antibodies. This work also increases the impact of prior studies showing in both HCVcc and HCVpp model systems that different E1E2 variants show very different sensitivities to antibody neutralization (Keck et al., 2012; Urbanowicz et al., 2015; Wasilewski et al., 2016).
These results have important implications for HCV fitness and antibody neutralization testing. First, this study suggests that specific infectivities measured with HCVpp or HCVcc may not predict results that would be obtained using the same E1E2 in the other model system. Prior studies have shown that small variations in amino acid sequence of E1E2 proteins can render HCVpp non-functional, which may be an artefact of the assay itself as these changes are sometimes tolerated differently in HCVcc models (Keck et al., 2009; Russell et al., 2009). In addition, a recent study by Urbanowicz et al. (2016) highlights multiple technical factors that can influence production of functional HCVpp, including plasmid transfection ratios and use of either murine leukaemia virus or HIV reporter constructs. In this study, we demonstrate that HCVcc incorporate more strain 1a38 E1E2 per virion than HCVpp. Together, these studies suggest that variable incorporation of different E1E2 variants into HCVpp may complicate measurement of E1E2 fitness using HCVpp, which may explain the lack of correlation between HCVcc- and HCVpp-specific infectivities. We were only able to quantitate incorporation of one E1E2 variant (1a38) into HCVpp and HCVcc, so further studies are needed to determine whether these findings are applicable to multiple E1E2 variants.
Further studies are also needed to determine which model system more accurately reflects E1E2 fitness in vivo. Measurement of HCVpp entry may be more quantitative, since only a single round of infection can occur, but the incorporation of E2 and apolipoproteins into HCVcc virions may be more similar to natural HCV virions. It is important to note, however, that some studies have shown that HCVcc particles produced in Huh-7 cells also differ from HCV particles produced in vivo or in primary human hepatocytes in their biophysical properties (Lindenbach et al., 2006; Podevin et al., 2010), and that mutations that enhance HCVcc replication in vitro can reduce efficient replication in animal models (Bukh et al., 2002). Measurement of specific infectivity in both systems is also potentially complicated by the presence of viral RNA that is not associated with authentic virions. Ultimately, understanding of E1E2 fitness will likely require correlation of in vitro testing with persistence of particular variants in vivo.
Importantly, we also showed for the first time that as an alternative to serial mAb dilution testing with IC50 measurement, neutralization of HCVpp can also be measured as an Fu at a single antibody concentration, with results that correlate with those that would be obtained using HCVcc and full antibody dilution curves. Testing of single antibody concentrations for neutralization of HCVpp could allow extremely high throughput neutralizing breadth measurements against large numbers of diverse variants.
These studies also confirm that natural E1E2 isolates show a wide range of neutralization resistance whether they are expressed on HCVpp or HCVcc, suggesting that large, representative panels of HCV variants are needed to accurately define neutralizing antibody breadth. It is noteworthy that in this study, we identified a natural E1E2 variant, 1a09, which is resistant to all human bNAbs tested. High-level resistance of this variant despite the conservation of known binding residues for most bNAbs (shown in Fig. S2) highlights the influence of polymorphisms outside of binding epitopes on neutralization resistance, as has been described in other studies (Bailey et al., 2015a; Keck et al., 2009; Wasilewski et al., 2016). The lack of correlation in specific infectivities between HCVcc and HCVpp indicates that some E1E2 variants are more robustly functional and therefore, may be more useful as reagents in either HCVcc or HCVpp systems. Therefore, neutralization panels may need to incorporate both HCVpp and HCVcc to maximize the diversity of E1E2 variants that can be tested.
This study also confirms prior work showing that IC50 values measured using HCVpp are generally lower than IC50 values of the same mAb measured using HCVcc, which could lead to higher estimates of neutralizing antibody breadth using panels of HCVpp and lower estimates using HCVcc expressing the same E1E2 variants (Urbanowicz et al., 2015). This quantitative difference in HCVpp and HCVcc neutralization may be explained by the recent demonstration by Fauvelle et al. (2016) that ApoE increases HCVcc neutralization resistance.
In conclusion, we have developed an efficient method to produce HCVcc bearing natural E1E2 protein variants. With this tool, we produced HCVcc and HCVpp bearing matched E1E2 proteins and conducted a systematic comparison of specific infectivities, also comparing for the first time resistance of E1E2-matched HCVcc and HCVpp to a diverse panel of human bNAbs. We found no correlation between specific infectivities of E1E2 variants quantitated using HCVcc or HCVpp, whereas we found a very strong positive correlation between relative neutralization resistance of these same E1E2-matched HCVcc and HCVpp variants. These results suggest that quantitative comparisons of relative neutralization resistance of E1E2 variants can be made with confidence using either HCVcc or HCVpp, allowing the use of either or both systems to maximize diversity of neutralization panels.
Methods
Source of HCVcc.
H77/JFH-1 (Scheel et al., 2008) and S52/JFH-1 (Gottwein et al., 2007) were a gift of Jens Bukh (Copenhagen University Hospital, Copenhagen, Denmark).
Source of E1E2.
Plasma samples were obtained from HCV-infected subjects as previously described (Osburn et al., 2014). E1E2 clones used are found in GenBank (accession numbers FJ828970.1, KF589884.1, KJ187972.1–KJ187975.1, KJ187977.1, KJ187983.1, KJ187984.1, KJ187986.1, KJ187989.1 and KJ187990.1).
Source of mAbs.
CBH-5 (Hadlock et al., 2000), HC84.22, HC84.26 (Keck et al., 2012) and HC33.4 (Keck et al., 2013) were a gift of Steven Foung (Stanford University School of Medicine, Stanford, CA, USA). AR3A, AR3C (Law et al., 2008), AR4A and AR5A (Giang et al., 2012) were a gift from Mansun Law (The Scripps Research Institute, La Jolla, CA, USA).
HCVcc neutralization assays.
HCVcc neutralization assays were performed as described elsewhere (Wasilewski et al., 2016). Briefly, human hepatoma Huh7.5.1 cells (a gift of Jake Liang, NIH, Bethesda, MD, USA) were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10 % FBS, 1 % sodium pyruvate and 1 % l-glutamate. Ten thousand Huh7.5.1 cells per well were plated in flat bottom 96-well tissue culture plates and incubated overnight at 37 °C. The following day, HCVcc were mixed with mAb (2.5-fold dilutions started at 50 µg ml−1) then incubated at 37 °C for 1 h. Medium was removed from the cells and replaced with 50 µl of HCVcc/antibody mixture. The plates were placed in a CO2 incubator at 37 °C overnight, after which the HCVcc were removed and replaced with 100 µl of Huh7.5.1 medium and incubated for 48 h at 37 °C. After 48 h, medium was removed and cells were fixed and stained. Images were acquired and SFU were counted in the presence of mAb (HCVccSFUtest) or non-specific IgG (HCVccSFUcontrol) using an AID iSpot Reader Spectrum operating AID ELISpot Reader version 7.0. Percentage neutralization was calculated as 100 %×[1−(HCVccSFUtest/HCVccSFUcontrol)].
HCV NS5A immunostaining.
HCV NS5A immunostaining was conducted as described elsewhere (Wasilewski et al., 2016). Briefly, cells were fixed with 4 % formaldehyde then stained for HCV NS5A using primary anti-NS5A antibody 9E10 (a gift of Charles Rice, The Rockefeller University, New York City, NY, USA) at 1 : 10 000 dilution for 1 h at room temperature. Cells were washed twice with PBS and stained using secondary antibody Alexa Daylight 488 conjugated goat anti-mouse IgG (Life Technologies) at 1 : 500 dilution for 1 h at room temperature. Cells were washed twice in PBS and then stored covered in 100 µl PBS at 4 °C.
HCVpp neutralization assays.
HCVpp were generated by cotransfection of pNL4-3.Luc.R−E− plasmid and an expression plasmid containing HCV E1E2 as described elsewhere (Bailey et al., 2015b; Hsu et al., 2003; Logvinoff et al., 2004). Virus-containing medium was collected at 48 and 72 h, pooled and stored at −4 °C. For infectivity and neutralization testing of HCVpp 8000 Hep3B cells (American Type Culture Collection) or 10 000 Huh7.5.1 cells per well were plated in flat bottom 96-well tissue culture plates and incubated overnight at 37 °C. The following day, HCVpp were mixed with mAb (2.5-fold dilutions started at 50 µg ml−1 or a single concentration of 10 µg ml−1) then incubated at 37 °C for 1 h. Medium was removed from the cells and replaced with 50 µl of HCVpp/antibody mixture. The plates were placed in a CO2 incubator at 37 °C for 5 h, after which the HCVpp were removed and replaced with 100 µl of fresh medium and incubated for 72 h at 37 °C. Medium was removed from the cells and 50 µl of 1× Cell Culture Lysis Reagent (Promega) added and left to incubate for >5 min, then 45 µl from each well was then transferred to a white, low-luminescence 96-well plate (Berthold Technologies) and luciferase activity measured in RLU in a Berthold luminometer (Centro LB 960; Berthold Technologies). Pseudoparticle infection was measured in the presence of mAb (HCVppRLUtest) or non-specific IgG/HCV-negative normal human plasma (HCVppRLUcontrol) at the same dilution. The percentage of neutralization was calculated as 100 %×[1−(HCVppRLUtest/HCVppRLUcontrol)]. Fu was measured as described above using mAb and IgG at a concentration of 10 µg ml−1. This was then calculated as (HCVppRLUtest/HCVppRLUcontrol). Each sample was tested in duplicate. A mock pseudoparticle (no envelope) was used as a negative control. Neutralization was tested only for HCVpp with absolute infectivity (RLU) at least 10× greater than typical RLU values of mock pseudoparticles with no envelope proteins.
HCVcc-specific infectivity quantitation.
HCVcc RNA levels in infection supernatants (IU per millilitre) were quantified using a process of RNA extraction and utilization of commercial real-time reagents (Abbot HCV RealTime assay) migrated onto a research-based real-time PCR platform (LightCycler 480; Roche). HCVcc were titred and SFU per millilitre were calculated in the linear range. Specific infectivity was calculated as: SFU per millilitre/HCV IU per millilitre.
HCVpp-specific infectivity quantitation.
HCVpp RNA viral load (IU per millilitre) was quantitated using real-time PCR and an IU viral load standard (Shan et al., 2013). The amount of infection produced in Huh7.5.1 cells by a measured volume of HCVpp supernatant was measured in RLU (HCVppRLU) as described in HCVpp neutralization assays. To determine specific infectivity, the amount of infection per millilitre was divided by viral load per millilitre and calculated as: HCVppRLU per millilitre/HIV IU per millilitre.
Generation of HCVcc chimeras.
HCVcc chimeras were generated as described previously (Wasilewski et al., 2016). Briefly, HCVcc chimera H77/JFH1 (Scheel et al., 2008) was amplified in three sections, omitting nucleotides 916–2579 (E1E2). Amplified sections were re-assembled using In-Fusion cloning (Clontech), and this reassembly generated an AfeI restriction site at the location of the omitted nucleotides. After digestion of this HCVcc backbone with enzyme AfeI (New England Biolabs), E1E2 inserts amplified from library plasmids were inserted in frame using In-Fusion cloning.
To make HCVcc RNA, 2 µg plasmid DNA was linearized using XbaI (New England Biolabs) then used for in vitro RNA transcription using the T7 MEGAscript kit (Ambion). RNA clean-up was performed using RNeasy mini kit (Qiagen), quantified using a NanoDrop 1000 spectrophotometer (Thermo Scientific) and stored at −80 °C. Five micrograms of RNA was transfected into 1.8e6 Huh7.5.1 cells using Nucleofector kit T (Amaxa) and plated in a 6 cm plate. Transfection supernatants were collected 4–6 days later and stored at −80 °C for titring by HCV NS5A immunostaining. High-titre transfection supernatants were used to infect Huh7.5.1 cells and infection supernatants collected, titred and stored at −80 °C for use in infectivity and neutralization experiments.
Determination of IC50 values.
Neutralization data were plotted and analysed in GraphPad Prism version 6.00. Curves were plotted by non-linear regression and IC50 values calculated. Since IC50 could not be accurately quantitated for curves that did not exceed 50 % neutralization at the highest antibody concentration tested, or for curves that exceeded 50 % neutralization at only the highest antibody concentration (50 µg ml−1), these curves were assigned an IC50 of >50 µg ml−1 or ~50 µg ml−1, respectively. Any curve with a calculated IC50 value greater than 50 µg ml−1 was also assigned an IC50 >50 µg ml−1. IC50 values >50 µg ml−1 and ~50 µg ml−1 were plotted for correlation analysis as 100 µg ml−1 or 50 µg ml−1, respectively. Curves generating ambiguous IC50 values were discarded.
Generation of phylogenetic tree.
A maximum-likelihood tree was generated from E1E2 amino acid sequences using the Jones–Taylor–Thornton (JTT) model with gamma distribution and invariant sites, deletion of columns with gaps, with maximum-likelihood tree inferred by the nearest-neighbour interchange method, with analyses carried out in mega v6 (Tamura & Nei, 1993; Tamura et al., 2013). Six hundred and thirty-four genotype 1a and 1b sequences from GenBank were included for reference.
Western blotting.
HCVpp and HCVcc supernatants were concentrated and purified by ultracentrifugation through a 20 % sucrose cushion (123 000 g, 2 h, 4 °C) with pellets resuspended in PBS. Viral RNA was extracted from purified supernatants and RNA viral concentration quantitated using real-time PCR. Equal copy numbers of each viral supernatant were denatured and run on a 4–12 % Bis-Tris gel. After transfer, blots were probed with human anti-E2 HC33.1.53 (Keck et al., 2013) (a gift of Dr Steven Foung, Stanford University School of Medicine, Stanford, CA, USA) and goat anti-ApoE (50A-G1b, Academy Bio-Medical). Binding was detected with HRP-conjugated anti-human IgG secondary antibody (no. 555788, BD Pharmingen) and HRP-conjugated anti-goat IgG (sc-2020; Santa Cruz Biotechnology).
Acknowledgements
The authors would like to thank Anna E. Snider, Jillian K. Brady and Jeffrey Quinn (the Johns Hopkins University Viral Hepatitis Center) for excellent technical assistance, and Ashwin Balagopal, David Thomas, Andrea Cox and Choe Thio (the Johns Hopkins University Viral Hepatitis Center) for useful discussions. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. This study was supported by NIH grants K08 AI102761 (J. B.), U19 AI088791 (S. R.) and R37 DA013806 (S. R.) and the Johns Hopkins University Center for AIDS Research grant P30 AI094189 (J. B.).
Supplementary Data
References
- Armstrong G. L., Wasley A., Simard E. P., McQuillan G. M., Kuhnert W. L., Alter M. J.(2006). The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 144705–714. 10.7326/0003-4819-144-10-200605160-00004 [DOI] [PubMed] [Google Scholar]
- Averhoff F. M., Glass N., Holtzman D.(2012). Global burden of hepatitis C: considerations for healthcare providers in the United States. Clin Infect Dis 55S10–S15. 10.1093/cid/cis361 [DOI] [PubMed] [Google Scholar]
- Bailey J. R., Wasilewski L. N., Snider A. E., El-Diwany R., Osburn W. O., Keck Z., Foung S. K. H., Ray S. C.(2015a). Naturally selected hepatitis C virus polymorphisms confer broad neutralizing antibody resistance. J Clin Invest 125437–447. 10.1172/JCI78794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey J. R., Dowd K. A., Snider A. E., Osburn W. O., Mehta S. H., Kirk G. D., Thomas D. L., Ray S. C.(2015b). CD4 T-cell-dependent reduction in hepatitis C virus-specific neutralizing antibody responses after coinfection with human immunodeficiency virus. J Infect Dis 212914–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartosch B., Vitelli A., Granier C., Goujon C., Dubuisson J., Pascale S., Scarselli E., Cortese R., Nicosia A., Cosset F. L.(2003). Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J Biol Chem 27841624–41630. 10.1074/jbc.M305289200 [DOI] [PubMed] [Google Scholar]
- Boyer A., Dumans A., Beaumont E., Etienne L., Roingeard P., Meunier J. C.(2014). The association of hepatitis C virus glycoproteins with apolipoproteins E and B early in assembly is conserved in lipoviral particles. J Biol Chem 28918904–18913. 10.1074/jbc.M113.538256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimacombe C. L., Grove J., Meredith L. W., Hu K., Syder A. J., Flores M. V., Timpe J. M., Krieger S. E., Baumert T. F., et al. (2011). Neutralizing antibody-resistant hepatitis C virus cell-to-cell transmission. J Virol 85596–605. 10.1128/JVI.01592-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukh J., Pietschmann T., Lohmann V., Krieger N., Faulk K., Engle R. E., Govindarajan S., Shapiro M., St Claire M., Bartenschlager R.(2002). Mutations that permit efficient replication of hepatitis C virus RNA in Huh-7 cells prevent productive replication in chimpanzees. Proc Natl Acad Sci U S A 9914416–14421. 10.1073/pnas.212532699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsen T. H., Scheel T. K., Ramirez S., Foung S. K., Bukh J.(2013). Characterization of hepatitis C virus recombinants with chimeric E1/E2 envelope proteins and identification of single amino acids in the E2 stem region important for entry. J Virol 871385–1399. 10.1128/JVI.00684-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsen T. H., Pedersen J., Prentoe J. C., Giang E., Keck Z. Y., Mikkelsen L. S., Law M., Foung S. K., Bukh J.(2014). Breadth of neutralization and synergy of clinically relevant human monoclonal antibodies against HCV genotypes 1a, 1b, 2a, 2b, 2c, and 3a. Hepatology 601551–1562. 10.1002/hep.27298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catanese M. T., Uryu K., Kopp M., Edwards T. J., Andrus L., Rice W. J., Silvestry M., Kuhn R. J., Rice C. M.(2013). Ultrastructural analysis of hepatitis C virus particles. Proc Natl Acad Sci U S A 1109505–9510. 10.1073/pnas.1307527110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormier E. G., Tsamis F., Kajumo F., Durso R. J., Gardner J. P., Dragic T.(2004). CD81 is an entry coreceptor for hepatitis C virus. Proc Natl Acad Sci U S A 1017270–7274. 10.1073/pnas.0402253101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowd K. A., Netski D. M., Wang X. H., Cox A. L., Ray S. C.(2009). Selection pressure from neutralizing antibodies drives sequence evolution during acute infection with hepatitis C virus. Gastroenterology 1362377–2386. 10.1053/j.gastro.2009.02.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkowska E., Kajumo F., Garcia E., Reinus J., Dragic T.(2007). Hepatitis C virus envelope glycoprotein E2 glycans modulate entry, CD81 binding, and neutralization. J Virol 818072–8079. 10.1128/JVI.00459-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauvelle C., Felmlee D. J., Crouchet E., Lee J., Heydmann L., Lefèvre M., Magri A., Hiet M. S., Fofana I., et al. (2016). Apolipoprotein E mediates evasion from hepatitis C virus neutralizing antibodies. Gastroenterology 150206–217. 10.1053/j.gastro.2015.09.014 [DOI] [PubMed] [Google Scholar]
- Fofana I., Fafi-Kremer S., Carolla P., Fauvelle C., Zahid M. N., Turek M., Heydmann L., Cury K., Hayer J., et al. (2012). Mutations that alter use of hepatitis C virus cell entry factors mediate escape from neutralizing antibodies. Gastroenterology 143223–233. 10.1053/j.gastro.2012.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forns X., Payette P. J., Ma X., Satterfield W., Eder G., Mushahwar I. K., Govindarajan S., Davis H. L., Emerson S. U., et al. (2000). Vaccination of chimpanzees with plasmid DNA encoding the hepatitis C virus (HCV) envelope E2 protein modified the infection after challenge with homologous monoclonal HCV. Hepatology 32618–625. 10.1053/jhep.2000.9877 [DOI] [PubMed] [Google Scholar]
- Giang E., Dorner M., Prentoe J. C., Dreux M., Evans M. J., Bukh J., Rice C. M., Ploss A., Burton D. R., Law M.(2012). Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc Natl Acad Sci U S A 1096205–6210. 10.1073/pnas.1114927109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottwein J. M., Scheel T. K., Hoegh A. M., Lademann J. B., Eugen-Olsen J., Lisby G., Bukh J.(2007). Robust hepatitis C genotype 3a cell culture releasing adapted intergenotypic 3a/2a (S52/JFH1) viruses. Gastroenterology 1331614–1626. 10.1053/j.gastro.2007.08.005 [DOI] [PubMed] [Google Scholar]
- Gottwein J. M., Scheel T. K., Jensen T. B., Lademann J. B., Prentoe J. C., Knudsen M. L., Hoegh A. M., Bukh J.(2009). Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 49364–377. 10.1002/hep.22673 [DOI] [PubMed] [Google Scholar]
- Hadlock K. G., Lanford R. E., Perkins S., Rowe J., Yang Q., Levy S., Pileri P., Abrignani S., Foung S. K.(2000). Human monoclonal antibodies that inhibit binding of hepatitis C virus E2 protein to CD81 and recognize conserved conformational epitopes. J Virol 7410407–10416. 10.1128/JVI.74.22.10407-10416.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishiki T., Shimizu Y., Tobita R., Sugiyama K., Ogawa K., Funami K., Ohsaki Y., Fujimoto T., Takaku H., et al. (2010). Infectivity of hepatitis C virus is influenced by association with apolipoprotein E isoforms. J Virol 8412048–12057. 10.1128/JVI.01063-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg S. D., Spradling P. R., Moorman A. C., Denniston M. M.(2013). Hepatitis C in the United States. N Engl J Med 3681859–1861. 10.1056/NEJMp1302973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu M., Zhang J., Flint M., Logvinoff C., Cheng-Mayer C., Rice C. M., McKeating J. A.(2003). Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci U S A 1007271–7276. 10.1073/pnas.0832180100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J., Luo G.(2009). Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J Virol 8312680–12691. 10.1128/JVI.01476-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J., Cun W., Wu X., Shi Q., Tang H., Luo G.(2012). Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J Virol 867256–7267. 10.1128/JVI.07222-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck Z. Y., Olson O., Gal-Tanamy M., Xia J., Patel A. H., Dreux M., Cosset F. L., Lemon S. M., Foung S. K.(2008). A point mutation leading to hepatitis C virus escape from neutralization by a monoclonal antibody to a conserved conformational epitope. J Virol 826067–6072. 10.1128/JVI.00252-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck Z. Y., Li S. H., Xia J., von Hahn T., Balfe P., McKeating J. A., Witteveldt J., Patel A. H., Alter H., et al. (2009). Mutations in hepatitis C virus E2 located outside the CD81 binding sites lead to escape from broadly neutralizing antibodies but compromise virus infectivity. J Virol 836149–6160. 10.1128/JVI.00248-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck Z. Y., Xia J., Wang Y., Wang W., Krey T., Prentoe J., Carlsen T., Li A. Y., Patel A. H., et al. (2012). Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog 8e1002653. 10.1371/journal.ppat.1002653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck Z., Wang W., Wang Y., Lau P., Carlsen T. H., Prentoe J., Xia J., Patel A. H., Bukh J., Foung S. K.(2013). Cooperativity in virus neutralization by human monoclonal antibodies to two adjacent regions located at the amino terminus of hepatitis C virus E2 glycoprotein. J Virol 8737–51. 10.1128/JVI.01941-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavie M., Sarrazin S., Montserret R., Descamps V., Baumert T. F., Duverlie G., Séron K., Penin F., Dubuisson J.(2014). Identification of conserved residues in hepatitis C virus envelope glycoprotein E2 that modulate virus dependence on CD81 and SRB1 entry factors. J Virol 8810584–10597. 10.1128/JVI.01402-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law M., Maruyama T., Lewis J., Giang E., Tarr A. W., Stamataki Z., Gastaminza P., Chisari F. V., Jones I. M., et al. (2008). Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med 1425–27. 10.1038/nm1698 [DOI] [PubMed] [Google Scholar]
- Lindenbach B. D., Meuleman P., Ploss A., Vanwolleghem T., Syder A. J., McKeating J. A., Lanford R. E., Feinstone S. M., Major M. E., et al. (2006). Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc Natl Acad Sci U S A 1033805–3809. 10.1073/pnas.0511218103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logvinoff C., Major M. E., Oldach D., Heyward S., Talal A., Balfe P., Feinstone S. M., Alter H., Rice C. M., McKeating J. A.(2004). Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc Natl Acad Sci U S A 10110149–10154. 10.1073/pnas.0403519101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathiesen C. K., Jensen T. B., Prentoe J., Krarup H., Nicosia A., Law M., Bukh J., Gottwein J. M.(2014). Production and characterization of high-titer serum-free cell culture grown hepatitis C virus particles of genotype 1-6. Virology 458–459190–208. 10.1016/j.virol.2014.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathiesen C. K., Prentoe J., Meredith L. W., Jensen T. B., Krarup H., McKeating J. A., Gottwein J. M., Bukh J.(2015). Adaptive mutations enhance assembly and cell-to-cell transmission of a high-titer hepatitis C virus genotype 5a core-NS2 JFH1-based recombinant. J Virol 897758–7775. 10.1128/JVI.00039-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure C. P., Urbanowicz R. A., King B. J., Cano-Crespo S., Tarr A. W., Ball J. K.(2016). Flexible and rapid construction of viral chimeras applied to hepatitis C virus. J Gen Virol 972187–2193. 10.1099/jgv.0.000530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeating J. A., Zhang L. Q., Logvinoff C., Flint M., Zhang J., Yu J., Butera D., Ho D. D., Dustin L. B., et al. (2004). Diverse hepatitis C virus glycoproteins mediate viral infection in a CD81-dependent manner. J Virol 788496–8505. 10.1128/JVI.78.16.8496-8505.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J. C., Engle R. E., Faulk K., Zhao M., Bartosch B., Alter H., Emerson S. U., Cosset F. L., Purcell R. H., Bukh J.(2005). Evidence for cross-genotype neutralization of hepatitis C virus pseudo-particles and enhancement of infectivity by apolipoprotein C1. Proc Natl Acad Sci U S A 1024560–4565. 10.1073/pnas.0501275102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J. C., Russell R. S., Engle R. E., Faulk K. N., Purcell R. H., Emerson S. U.(2008). Apolipoprotein c1 association with hepatitis C virus. J Virol 829647–9656. 10.1128/JVI.00914-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin T. J., Broering T. J., Leav B. A., Blair B. M., Rowley K. J., Boucher E. N., Wang Y., Cheslock P. S., Knauber M., et al. (2012). Human monoclonal antibody HCV1 effectively prevents and treats HCV infection in chimpanzees. PLoS Pathog 8e1002895. 10.1371/journal.ppat.1002895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osburn W. O., Snider A. E., Wells B. L., Latanich R., Bailey J. R., Thomas D. L., Cox A. L., Ray S. C.(2014). Clearance of hepatitis C infection is associated with the early appearance of broad neutralizing antibody responses. Hepatology 592140–2151. 10.1002/hep.27013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka J. M., Zeisel M. B., Bläser E., Schürmann P., Bartosch B., Cosset F. L., Patel A. H., Meisel H., Baumert J., et al. (2007). Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc Natl Acad Sci U S A 1046025–6030. 10.1073/pnas.0607026104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podevin P., Carpentier A., Pène V., Aoudjehane L., Carrière M., Zaïdi S., Hernandez C., Calle V., Méritet J. F., et al. (2010). Production of infectious hepatitis C virus in primary cultures of human adult hepatocytes. Gastroenterology 1391355–1364. 10.1053/j.gastro.2010.06.058 [DOI] [PubMed] [Google Scholar]
- Russell R. S., Kawaguchi K., Meunier J. C., Takikawa S., Faulk K., Bukh J., Purcell R. H., Emerson S. U.(2009). Mutational analysis of the hepatitis C virus E1 glycoprotein in retroviral pseudoparticles and cell-culture-derived H77/JFH1 chimeric infectious virus particles. J Viral Hepat 16621–632. 10.1111/j.1365-2893.2009.01111.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel T. K., Gottwein J. M., Jensen T. B., Prentoe J. C., Hoegh A. M., Alter H. J., Eugen-Olsen J., Bukh J.(2008). Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc Natl Acad Sci U S A 105997–1002. 10.1073/pnas.0711044105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel T. K., Gottwein J. M., Carlsen T. H., Li Y. P., Jensen T. B., Spengler U., Weis N., Bukh J.(2011). Efficient culture adaptation of hepatitis C virus recombinants with genotype-specific core-NS2 by using previously identified mutations. J Virol 852891–2906. 10.1128/JVI.01605-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan L., Rabi S. A., Laird G. M., Eisele E. E., Zhang H., Margolick J. B., Siliciano R. F.(2013). A novel PCR assay for quantification of HIV-1 RNA. J Virol 876521–6525. 10.1128/JVI.00006-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann R. E., Cowton V. M., Robinson M. W., Cole S. J., Barclay S. T., Mills P. R., Thomson E. C., McLauchlan J., Patel A. H.(2016). Broad anti-HCV antibody responses are associated with improved clinical disease parameters in chronic HCV infection. J Virol 904530–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Nei M.(1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10512–526. [DOI] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A., Kumar S.(2013). mega6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 302725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D. L., Astemborski J., Rai R. M., Anania F. A., Schaeffer M., Galai N., Nolt K., Nelson K. E., Strathdee S. A., et al. (2000). The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA 284450–456. [DOI] [PubMed] [Google Scholar]
- Timpe J. M., Stamataki Z., Jennings A., Hu K., Farquhar M. J., Harris H. J., Schwarz A., Desombere I., Roels G. L., et al. (2008). Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 4717–24. 10.1002/hep.21959 [DOI] [PubMed] [Google Scholar]
- Urbanowicz R. A., McClure C. P., Brown R. J., Tsoleridis T., Persson M. A., Krey T., Irving W. L., Ball J. K., Tarr A. W.(2015). A diverse panel of hepatitis C virus glycoproteins for use in vaccine research reveals extremes of monoclonal antibody neutralization resistance. J Virol 903288–3301. 10.1128/JVI.02700-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanowicz R. A., McClure C. P., King B., Mason C. P., Ball J. K., Tarr A. W.(2016). Novel functional hepatitis C virus glycoprotein isolates identified using an optimized viral pseudotype entry assay. J Gen Virol 972265–2279. 10.1099/jgv.0.000537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieyres G., Thomas X., Descamps V., Duverlie G., Patel A. H., Dubuisson J.(2010). Characterization of the envelope glycoproteins associated with infectious hepatitis C virus. J Virol 8410159–10168. 10.1128/JVI.01180-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasilewski L. N., El-Diwany R., Munshaw S., Snider A. E., Brady J. K., Osburn W. O., Ray S. C., Bailey J. R.(2016). A hepatitis C virus envelope polymorphism confers resistance to neutralization by polyclonal sera and broadly neutralizing monoclonal antibodies. J Virol 903773–3782. 10.1128/JVI.02837-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn J. W., Park S. H., Lavillette D., Cosset F. L., Yang S. H., Lee C. G., Jin H. T., Kim C. M., Shata M. T., et al. (2005). Sustained E2 antibody response correlates with reduced peak viremia after hepatitis C virus infection in the chimpanzee. Hepatology 421429–1436. 10.1002/hep.20934 [DOI] [PubMed] [Google Scholar]
- Zhang J., Randall G., Higginbottom A., Monk P., Rice C. M., McKeating J. A.(2004). CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J Virol 781448–1455. 10.1128/JVI.78.3.1448-1455.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.