

Abstract
Mitochondrial (MT) dysfunction is known to occur in chondrocytes isolated from end-stage osteoarthritis (OA) patients, but the role of MT dysfunction in the initiation and early pathogenesis of post-traumatic OA (PTOA) remains unclear. The objective of this study was to investigate chondrocyte MT function immediately following mechanical injury in cartilage, and to determine if the response to injury differed between a weight bearing region (medial femoral condyle; MFC) and a non-weight bearing region (distal patellofemoral groove; PFG) of the same joint. Cartilage was harvested from the MFC and PFG of 10 neonatal bovids, and subjected to injurious compression at varying magnitudes (5–17 MPa, 5–34 GPa/s) using a rapid single-impact model. Chondrocyte MT respiratory function, MT membrane polarity, chondrocyte viability, and cell membrane damage were assessed in situ. Cartilage impact resulted in MT depolarization and impaired MT respiratory function within 2 h of injury. Cartilage from a non-weight bearing region of the joint (PFG) was more sensitive to impact-induced MT dysfunction and chondrocyte death than cartilage from a weight-bearing surface (MFC). Our findings suggest that MT dysfunction is an acute response of chondrocytes to cartilage injury, and that MT may play a key mechanobiological role in the initiation and early pathogenesis of PTOA. Clinical significance: Direct therapeutic targeting of MT function in the early post-injury time frame may provide a strategy to block perpetuation of tissue damage and prevent the development of PTOA.
Keywords: cartilage, mechanobiology, osteoarthritis, posttraumatic, mitochondria
Despite decades of evidence establishing a link between mechanical injury to cartilage and post-traumatic osteoarthritis (PTOA),1,2 no effective therapies exist to prevent or slow progression of the disease.3 A sizeable body of research has focused on the molecular, cellular, and structural changes occurring in the days to weeks following cartilage injury. However, mounting evidence suggests that interventions must target the earliest pathologic events in order to modify the disease course.4,5 Therefore, an improved understanding of the acute (within hours) events after cartilage injury is necessary to develop novel preventative therapies.
Mitochondria (MT) are subcellular organelles that drive tissue development, repair, and aging.6 In addition to their role in cellular ATP production by oxidative phosphorylation, MT act as intracellular mechanotransducers via strain-mediated release of reactive oxygen species (ROS), which function in normal cell signaling.7,8 When MT dysfunction, cellular energy production declines and ROS are produced in excess, leading to degradation of lipid membranes, accumulation of DNA damage, initiation of catabolic signaling cascades, and cell death.9 Apoptosis is triggered when cytochrome C dissociates from the inner MT membrane and activates the caspase cascade in the cytosol.10
Bioenergetic failure, cumulative oxidative stress, and MT-mediated apoptosis are implicated in the pathogenesis of many complex degenerative diseases,11 and substantial evidence supports a role for MT dysfunction in the chronic stages of osteoarthritis (OA).12–14 In chondrocytes isolated from patients with end-stage OA, MT dysfunction is associated with later-stage pathologic events including upregulation of matrix metalloproteinase-1, –3, and –13, decreased synthesis of collagen and proteoglycans, and pathologic calcification of cartilage.15,16 Additionally, MT dysfunction and oxidative stress were recently linked to chronic overloading of chondrocytes.17 Several studies provide indirect evidence that MT play a central mechanobiological role in impact-induced chondrocyte death. For example, MT-derived ROS contribute to chondrocyte death after cartilage injury,18 and antioxidants prevent impact-induced cell death.19,20 Furthermore, inhibition of calcium signaling and activation of the caspase cascade after cartilage injury can prevent impact-induced chondrocyte death.21 These findings suggest a causal link between MT-dysfunction and PTOA pathogenesis, however, a significant knowledge gap remains in identifying the relationship between mechanical injury, in situ chondrocyte MT respiratory function, and cell death immediately after impact injury.
The objective of this study was to investigate chondrocyte MT function in situ immediately following cartilage impact injury using a real-time microscale respirometry assay modified for explanted cartilage. Our hypothesis was that MT dysfunction is an acute response of chondrocytes to mechanical injury. Furthermore, we hypothesized that a joint surface adapted to withstand predominantly compressive loads during weight-bearing (the MFC) would be less sensitive to impact-induced cell death and MT dysfunction than a non-weight-bearing region within the same joint.
METHODS
Cartilage Harvest and Mechanical Injury
Healthy bovids (n = 10; 1–3 days of age) were obtained from a livestock auction and humanely euthanized in accordance with AVMA guidelines.22 Within 12 h of sacrifice, full thickness cartilage explants were harvested from both the left and right knee joints, using an 8 mm biopsy punch (Figure 1a). Explants were rinsed in phosphate-buffered saline (PBS), trimmed to a uniform thickness of 3 mm from the articular surface using a custom jig, and placed in cartilage explant media (phenol-free DMEM containing FBS 10%, HEPES 0.025 ml/ml, penicillin 100 U/ml, and streptomycin 100 U/ml). All animal experiments were approved by the Institutional Animal Care and Use Committee.
Figure 1.

Experimental design and methods. (A) Cartilage explants were harvested from the medial femoral condyle (MFC) for the first set of experiments, and from two sites for the second set of experiments; the MFC and the distal patellofemoral groove (PFG). One explant from each region was impacted at a higher impact magnitude, one was impacted at a lower magnitude, and one served as an un-impacted control. Explants were then divided for use in several assays; chondrocyte viability was quantified using live/dead staining, mitochondrial (MT) membrane polarity was determined as red to green fluorescent intensity (R:G) ratio on confocal imaging and MT respiratory function was assessed via microrespirometry. Cell membrane damage was assessed by measuring lactate dehydrogenase (LDH) activity in cartilage conditioned media. (B) MT respiratory function was quantified by measuring oxygen consumption rate (OCR) over time. After basal OCR was determined, a MT stress test was performed by sequential addition of (i) oligomycin, an ATP synthase inhibitor; (ii) FCCP, a proton circuit uncoupler; and (iii) rotenone (Rot) and antimycin A (Ant A), inhibitors of MT complexes I and III. Parameters of respiratory control were calculated as depicted by the shaded areas under the curve.
Explants were subjected to a single, rapid impact injury using a validated model, as previously described, or served as un-injured controls.23 Briefly, explants were positioned in a well containing media under the plane-ended tip of a spring-loaded impacting device.23,24 Impact magnitude was adjusted by setting the deflection of the impactor’s internal spring. During impact, force was measured at 50 kHz by an in-line load cell (PCB Piezotronics, Depew, NY). Voltages from the load cell were recorded simultaneously with a custom Lab-VIEW program (NI, Austin TX) and mechanical parameters for each impact were calculated as previously described.23
Characterization of In Situ Chondrocyte Mitochondrial Respiratory Function Immediately Following Cartilage Injury
The goal of the first set of experiments was to determine the effect of acute mechanical injury on chondrocyte MT function. Microrespirometry has previously been used to investigate respiratory function of cultured chondrocytes,17,25 but has not been applied to chondrocytes in situ. Studying chondrocytes within their native extracellular matrix minimizes alterations in the mechanobiological environment and shortens the time frame between injury and observation. Therefore, based on previously described modifications of this technique for use in whole tissues26,27 and after assay optimization (Supplement 1, Detailed Methods), real-time microscale respirometry was used to measure chondrocyte MT respiratory function in explanted cartilage, as follows. Explants (n = 65 total) from the medial femoral condyle (MFC) were harvested and impacted over a broad range of injury magnitudes (M1–M4; 5–17 MPa, 5–34 GPa/s; Table 1), as described above. This range was selected based on preliminary trials (160 explants, 8 trials), and previous work23 to determine the stress and stress rate thresholds associated with cell death, and extracellular matrix damage in this system. The goal was to apply a range of injury magnitudes, from minimal cell death (M1) to cell death without surface cracking (M2) to subcritical damage (i.e., impacts that produced surface fissuring but not full thickness defects; M3 and M4, Table 1.)
Table 1.
Mechanical Parameters of Impact by Experimental Group
| Experimental Group | Impact Magnitude
|
|
|---|---|---|
| Mean Peak Stress; MPa (±s.d.) | Mean Peak Stress Rate; GPa/s (±s.d.) | |
| Control | n/a | n/a |
| M1 | 5.6 (0.4) | 6.7 (1.3) |
| M2 | 7.5 (0.4) | 9.3 (1.5) |
| M3 | 14.1 (0.7) | 28.1 (1.8) |
| M4 | 16.2 (0.7) | 32.0 (1.6) |
Following impact, two cartilage disks (3 mm diameter × 500 um thickness from the articular surface) were from prepared (Supplement 1) and immediately loaded into a randomly assigned well of a 24-well tissue capture microplate (Seahorse Biosciences, North Billerica, MA) containing assay media (bicarbonate-free DMEM supplemented with 2.5 mM glucose, 2 mM L-glutamine, 2 mM pyruvate, and 1% FBS). Each respirometry assay (n = 7 assays) utilized cartilage explants from a single animal, and three experimental groups (n = 6–7 wells per group); i) control; (ii) low impact (M1 or M2); and (iii) high impact (M3 or M4). Following a calibration cycle, glycolysis, and oxidative phosphorylation were quantified every ~8 min for a minimum of 225 min by measuring extracellular acidification (ECAR) and oxygen consumption rates (OCR) within each well, respectively using an XF24 Extracellular Flux Analyzer (Seahorse Biosciences). After baseline respiration was measured for at least 40 min, a MT stress test was performed according to standard protocols, as previously described.9,25,28 Briefly, OCR was measured in response to the automated sequential addition of i) oligomycin (2 μM), an ATP synthase inhibitor; ii) carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP; 1.0 μM), a proton circuit uncoupler; and iii) a combination of rotenone (0.5 μM) + antimycin A (1.0 μM), inhibitors of MT complexes I and III, respectively (Seahorse Biosciences).
The remainder of each explant was used to determine chondrocyte density and viability, as described below, in order to normalize respirometry data to viable cell number on an individual explant basis (Supplement S1). Data were normalized to viable cell number by dividing OCRs measured in each well containing a single cartilage plug, by the number of viable cells in that well. MT functional indices were calculated as previously described9,25 and visually represented in Figure 1b, using OCR values as follows: Basal OCR (bOCR) = initial OCR-nonMT respiration (NMR); oOCR = oligomycin-inhibited OCR; maximal (uncoupled) respiration (mOCR) = FCCP stimulated OCR-NMR; spare respiratory capacity (SRC) = (mOCR)-(bOCR); Proton leak = (oOCR-NMR)/bOCR; ATP turnover = (bOCR-oOCR).
Chondrocyte Viability and Cell Membrane Damage Assays
In order to determine cell density and quantify chondrocyte viability, cartilage was placed in PBS containing calcein AM (2 μM) and ethidium homodimer (1 μM) for 30 min at 37°C in the dark, to stain live and dead cells, respectively. Explants were then rinsed in PBS and imaged on a Leica SP5 confocal microscope. The number of live, dead, and total cells in each image was quantified using custom ImageJ macros optimized for each imaging channel (Supplement 1). The explant volume and chondrocyte density were calculated for each explant and used to normalize respirometry data to viable cell number (Supplement 1).
As a measure of cell membrane damage, lactate dehydrogenase (LDH) activity was assayed in cartilage-conditioned media from each well of the XF assay plate, according to manufacturer’s instructions (Sigma–Aldrich, St. Louis, MO) and absorbance was measured at 450 nm by a spectrophotometric microplate reader (Tecan Safire; Männedorf, Switzerland). To established a post-impact LDH release time-course and validate the use of media obtained from the XF assay plates following microrespirometry assays, cartilage explants (n = 16) were impacted at the magnitudes described above (M1–M4: Table 1), and incubated for 24 h in cartilage explant media. LDH assay was performed on cartilage-conditioned media at 1, 5, 7, and 24 h after impact.
Comparison of Chondrocyte Response to Injury Between Two Locations Within the Same Joint
The goal of the second set of experiments was to compare the response to injury between two anatomical regions within the same joint that are subjected to distinct loading regimens in vivo. Three explants were harvested from two locations within each joint; the MFC and distal patellofemoral groove (PFG), as described above (n = 40 total). The MFC is the main weight-bearing surface of the knee, while the distal PFG is a non-weight bearing articular surface. One explant from each area was randomly assigned to one of three impact treatments; lower magnitude (M1; 5.6 ± 0.4 MPa mean peak stress, 6.7 ± 1.3 GPa/s mean peak stress rate), higher magnitude impact (M2; 7.5 ± 0.4 MPa, 9.3 ± 1.5 GPa/s) or non-impacted control. Microrespirometry was performed (n = 8/group, 3–4 wells/group/assay) and data was normalized as described above. Impact magnitudes (M1 and M2) were chosen based on preliminary data, which revealed that impact above ~8 MPa peak stress (~11 GPa/s peak stress rate) in the PFG resulted in extensive cell death, preventing comparisons to the MFC.
MT Membrane Polarity Assay
The functional integrity of the inner MT membrane was assessed in situ using confocal imaging of fluorescent MT probes. Following impact and sectioning, samples (n = 36) were placed in PBS containing tetramethylrhodamine methyl ester perchlorate (TMRM;10 nM, Molecular Probes, Eugene, OR), MitoTracker Green (MTrG; 200 nM, Molecular Probes), and Hoechst 33342 (1 μg/ml, Molecular Probes) for 40 min and protected from light. TMRM is a polarity-sensitive MT probe, and red fluorescence indicates active transport of the dye across a polarized (functional) MT membrane. MTrG (green) is a polarity-insensitive MT probe, which stains all MT regardless of MT membrane potential. Hoechst acts as a nuclear counterstain, and preferentially stains cells with compromised plasma membranes. Explants were imaged on a Leica SP5 confocal microscope, and red:green florescent intensity (R:G) ratios for each image were determined using custom ImageJ macros (Supplement 1, Detailed Methods).
Statistical Analysis
Response variables were analyzed using a linear mixed effects model with fixed effects of treatment group and site (MFC or PFG; when applicable), and random effect of trial (animal) and limb (left or right). For analysis of bOCR and mOCR by impact magnitude, data were log-transformed. The unit of study was a cartilage explant. Post-hoc pairwise comparisons between treatment groups were performed using Tukey’s HSD method to control for multiple comparisons. Residual analyses were performed to ensure the assumptions of normality and homogeneous variance were met. The relationship between impact magnitude (stress and stress rate) and chondrocyte death was analyzed using a linear regression model. Differences were considered statistically significant when p <0.05. All statistical analyses were performed using JMP Pro Version 11.0 (SAS Inc., Cary, NC) software.
RESULTS
Chondrocyte Respiration After Cartilage Injury
MT respiratory function in MFC cartilage was assessed by measuring oxygen consumption rate (OCR) in the acute phase (from 2 to 6 h) after injury. Representative curves for OCR are shown in Figure 2a, and demonstrate differences in respiratory function between low impacted, high impacted, and control cartilage. MT respiration declines with increasing injury magnitude (Fig. 2); There was a significant effect of treatment (impact) group on bOCR (p = 0.0009) and mOCR (p = 0.026). Cartilage injury resulted in a 20–32% decrease in bOCR in explants from impact groups M2–M4 (Fig. 2b), and a 26–44% decrease in mOCR in groups M3 and M4 compared to un-impacted controls (Fig. 2c). Parameters of respiratory control calculated using oOCR by injury group could not be reliably determined because steady state OCR following oligomycin treatment was not reached in the majority of samples (Fig. 2a.)
Figure 2.

Respirometry reveals acute impact-induced respiratory impairment. Cartilage from the medial femoral condyle was impacted at various magnitudes (M1–M4) and MT respiration was quantified by measuring oxygen consumption rate (OCR), then normalizing data to live cell number for each explant. (A) Representative curves for OCR versus time for control, low impact (M2), and high impact (M4) groups demonstrate differences in MT respiration between groups. Note that oligomycin-inhibited respiration (oOCR) does not reach steady state (121 min), but Rot + AA inhibited respiration does (225 min). (B) Basal OCR (bOCR) and (C) max respiration (mOCR) decreased with increasing impact magnitude (M1–M4). Groups that do not share a letter are significantly different at p <0.05. Box and whisker plots represent data quartiles.
To gain additional information related to MT dysfunction, individual MT stress-test curves for each sample were plotted with treatment group blinded. Samples not reaching steady state after oligomycin administration were excluded, then impact groups were collapsed, resulting in two groups; control (n = 14), injury (n = 16). Data were re-analyzed using a mixed effect model as previously described, with the random effect of trial to account for variability between experiments. This analysis revealed that, in addition to decreased bOCR (p <0.001) and mOCR (p <0.0001), the injury group had decreased oOCR (p = 0.03) and NMR (p <0.05; Fig. 3a). The injury group also displayed increased proton leakage (p = 0.01) and decreased ATP turnover (p <0.001; Fig. 3b). Neither injury nor treatment with MT inhibitors had a significant effect on ECAR (p = 0.66, Fig. S1).
Figure 3.

Mitochondrial dysfunction occurs after cartilage injury. (A) Values for bOCR, oOCR, mOCR, and non-MT respiration (NMR) and (B) spare respiratory capacity (SRC), proton leak (as a % of bOCR), and ATP turnover in injured versus control samples. Error bars = ±s.d.; *p <0.05.
Chondrocyte death in MFC cartilage was positively correlated with impact magnitude, with the strongest correlations associated with peak impact stress (r2 = 0.70, p <0.0001; Fig. 4a) and stress rate (r2 = 0.64, p <0.001; Fig. S2). A significant increase in cell death was observed above 7 MPa peak stress, establishing a threshold for acute chondrocyte death in this model system. Cell membrane damage was assessed in cartilage-conditioned media obtained from wells following the respirometry assay, and revealed 2–3 fold increase in LDH activity for explants impacted at higher peak stresses (M3, M4) compared to lower impacts (M1, M2) and controls (Fig. S3a). Based on the time-course experiment, LDH activity peaked at approximately 5 h following cartilage injury at all impact magnitudes (Fig. S3b), corresponding to the time when cell viability assays were performed.
Figure 4.

Impact-induced chondrocyte death differs by location within the joint. (A) Chondrocyte death is correlated with impact magnitude. Cell death was positively correlated with peak impact stress for both the MFC (r2 = 0.70, p <0.0001) and the PFG (r2 = 0.79, p <0.001). Data presented is for all impacts performed throughout the study. (B) Chondrocytes from the patellofemoral groove (PFG) were more sensitive to impact-induced cell death than the medial femoral condyle (MFC). At lower impact magnitudes (M1) MFC viability was not affected. Groups that do not share a letter are significantly different at p <0.05. Error bars, ±s.d. (C and D) Representative images of PFG cartilage stained for live cells (green) with calcein AM and dead cells (red) with ethidium homodimer and imaged in cross-section using confocal microscopy. The articular surface is toward the top of the images. (C) Un-impacted control cartilage had less dead (red) staining than (D) lower impacted (M1) and (E) higher impacted (M2) in PFG explants.
Comparison of Chondrocyte Response to Injury in MFC Versus PFG Cartilage
Similar to MFC explants, chondrocyte death in PFG explants was positively correlated with peak impact stress (r2 = 0.79, p <0.001; Fig. 4a) and stress rate (r2 = 0.82, p <0.001; Fig. S2). However, chondrocytes from PFG cartilage, a non-weight-bearing articular surface, were more sensitive than the MFC to impact-induced cell death; PFG explants experienced an approximately twofold and fivefold increase in cell death over controls at the lower (M1) and higher (M2) impact magnitudes, respectively (Fig. 4b). At lower impact magnitudes (M1), MFC viability was not affected.
Cartilage from the PFG was more sensitive to impact-induced respiratory decline than the MFC (Fig. 5). The bOCR of viable PFG chondrocytes was significantly lower in groups impacted at the lowest (M1) and higher (M2) magnitudes compared to uninjured control cartilage, whereas in MFC cartilage, bOCR was only affected at the higher impact magnitude (M2). Maximal OCR showed similar trends, but differences between groups did not reach statistical significance (p = 0.06–0.1; data not presented), and additional measures of respiratory control using oOCR could not reliably calculated, given the small number of samples reaching steady state oOCR. Relative MT membrane potential (MT polarity) was used to assess the functional integrity of the inner MT membrane by calculating the R:G ratio, which represents the ratio of polarized to depolarized MT within each explant. In uninjured controls, MT polarity was similar in MFC and PFG cartilage. MT polarity was significantly decreased in both the lower (M1) and higher (M2) impacted explants from the PFG. Over this range of impact magnitudes, no statistically significant differences were detected between control and impacted samples from the MFC (Fig. 6).
Figure 5.
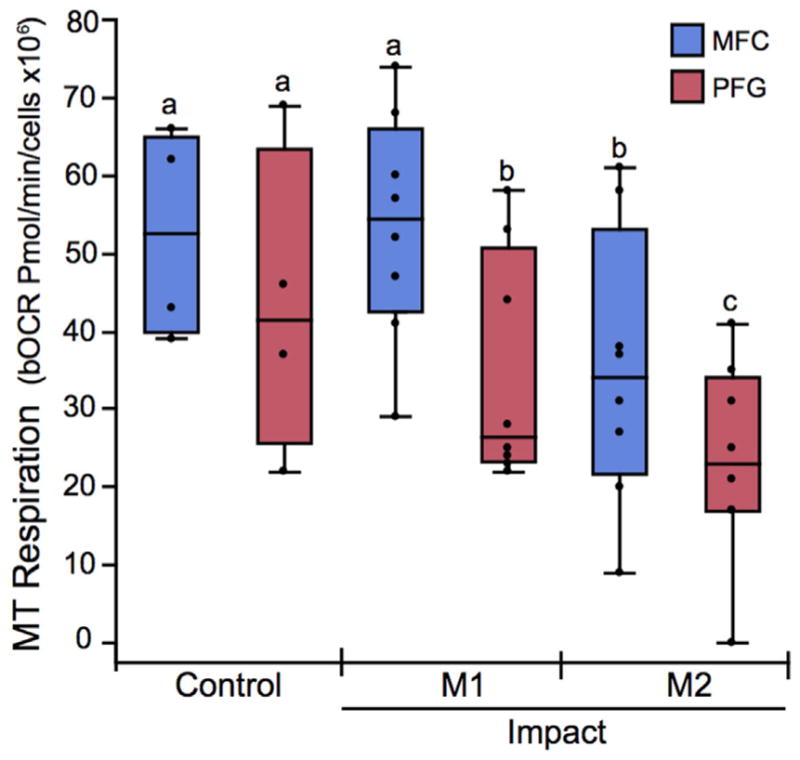
The patellofemoral groove (PFG) is more sensitive to impact-induced respiratory impairment than the medial femoral condyle (MFC). The basal oxygen consumption rate (bOCR) of viable chondrocytes was significantly lower in PFG cartilage (red box and whisker quartile plots) impacted at the lowest (M1) and higher (M2) magnitudes compared to un-injured control cartilage, whereas in MFC cartilage (blue plots), bOCR is only affected at the higher impact magnitude (M2). Groups that do not share a letter are significantly different at p <0.05.
Figure 6.

The patellofemoral groove (PFG) is more sensitive to impact-induced MT depolarization than the medial femoral condyle (MFC). (A) Representative confocal images of control and impacted (M2) PFG explants stained for MT polarity at low (top) and high (bottom) magnification. Cartilage is stained with Mitotracker Green (green; all MT), tetramethylrhodamine methyl ester perchlorate (red; polarized/functional MT), and Hoechst 33342 (blue; nuclear counterstain, higher affinity for cells with compromised cell membranes). Red:green fluorescent intensity ratios were calculated on an image-wide basis in multiple low magnification z-stacks for each explant (top) using a custom ImageJ macro. This high-throughput technique was validated by manually drawing ROIs around individual cells at higher magnification (bottom) to calculate R:G ratios on a single-cell basis. (B) MT depolarization occurred in PFG cartilage from both the lower (M1) and higher impact (M2) groups compared to PFG controls. Significant differences were not detected between impact groups from the MFC. Asterisks denote a significant difference compared to control at p <0.05. Error bars = ±s.d.
DISCUSSION
The goal of this work was to investigate the role of mitochondria in the very early response of cartilage to mechanical injury. The most important findings of this study were that MT dysfunction is an acute response of chondrocytes to cartilage trauma, and that the response of chondrocytes to injury differs based on location within a single joint.
Although several lines of investigation have supported the assumption that MT participate in catabolic events immediately following cartilage injury,21,29,30 and microscale respirometry has recently been used to assess MT function in cultured chondrocytes,17,25 this is the first report of assessing in situ chondrocyte MT respiratory function in the acute time frame following cartilage trauma. This distinction is important for several reasons. First, chondrocyte phenotype and gene expression change significantly when cells are isolated from the native extracellular matrix and cultured in monolayer.31–33 In one study, MT metabolism and MT biogenesis were drastically altered in both primary cultured (i.e., non-passaged) and first-passage chondrocytes.34 Second, accurate modeling of mechanical trauma to cartilage requires the native extracellular matrix. Finally, our primary objective was to investigate MT function within hours of mechanical injury, and the time required to isolate and perform primary chondrocyte culture is incompatible with this goal.
Recent evidence suggests that the main determinant for the development of PTOA is the magnitude of the initial cartilage/subchondral bone injury.35 A challenge in the field of early PTOA research has been to establish thresholds for mechanical injury in vitro and correlate these to the development of disease. Recently, our group fully characterized the mechanics of an in vivo impactor system and correlated impact magnitude (stress and stress rate) with structural damage in vitro.23 A threshold for superficial extracellular matrix cracking was established above a peak stress of 13 MPa. In the present study, the same impact system was used to determine the relationship between impact magnitude and tissue damage (i.e., chondrocyte death and cell membrane damage) in the acute phase after cartilage injury. We found that cell death was correlated to impact stress above a threshold of 7 MPa in MFC cartilage, and that bOCR and mOCR in viable cells declines with increasing injury magnitude.
Our finding that MT dysfunction occurs independent of cell death is in agreement with recent work by Coleman et al.17 which demonstrated MT dysfunction and oxidative stress after 1 week in chronically overloaded cartilage. The loading regimen consisted of load-controlled cyclic compression at 0.25 MPa (physiologic loading) or 1 MPa (supra-physiologic) amplitude and 0.5 Hz frequency for 3 h once daily for 1 week. Slow load rise times resulted in stress rates in the MPa/s range. In that study, no differences were observed in chondrocyte viability or mechanical behavior of the tissue between the physiologically loaded and overloaded chondrocytes after 7 days. Furthermore, no differences in MT function or oxidation status were observed between groups after 1 day. Therefore, their model may aptly be described as chronic, moderate overloading.
In contrast, the current study was designed to more closely model acute cartilage trauma, and utilized an energy-controlled spring-loaded device to deliver a single rapid impact, with impact times in the millisecond range and stress rates in the GPa/s range. In addition to distinct mechanobiology in these two systems, Coleman et al. models the early chronic time frame, with MT function assessed 1–2 weeks after start of loading, whereas the aim of this study was to assess the acute responses of chondrocytes after injury. This is an important distinction, because the subcellular mechanisms occurring within these time frames are likely different. Chronic MT dysfunction, as occurs in aging, can encompass many cellular processes including chronic oxidative stress, altered MT dynamics (MT biogenesis, turnover, and plasticity), metabolic reprogramming, and inflammation through a complex network of signaling pathways, while acute MT dysfunction occurs following traumatic injury before transcriptional changes can occur.36,37 Examples of disease processes mediated by acute MT dysfunction include ischemia reperfusion injury after acute myocardial infarction, neuronal damage following traumatic brain injury, and here we propose PTOA.38 Clinical myocardial infarction and traumatic brain injury research focuses on potentially treatable events in the early phase following injury. In this acute phase, MT participate in both the perpetuation of tissue damage, as well as protective mechanisms. For example, following injury, rapid calcium buffering occurs, whereby Ca++ flows into the cytosol due to changes in cell membrane permeability and is rapidly sequestered within MT. This process leads to MT swelling within 10 min of traumatic brain injury. MT swelling is reversible if cell repair mechanisms remain intact and the MT transition pore does not open.39 Opening of the MT transition pore results in further osmotic swelling of the MT, complete uncoupling of oxidative phosphorylation, rupture of the MT membrane and subsequent cell death. Calcium signaling is known to be involved in chondrocyte death after injury,21 and our data supports impaired efficiency of the electron transport chain after cartilage injury, consistent with acute swelling of the MT. Our findings suggest a population of unhealthy but viable chondrocytes undergoing impact-induced MT dysfunction and this sub-population may represent a target for therapeutics that would protect MT function, prevent cell death, and block perpetuation of tissue damage. To more specifically investigate this hypothesis, present studies are focused on improving spatial and temporal resolution of MT dysfunction in whole cartilage in the peracute (minutes to hours) time frame after injury and following the fate of depolarized cells for days to weeks after impact.
Recently, post-impact chondrocyte viability was found to differ between joints,40 but to our knowledge, this is the first report of regional differences in impact-induced cellular responses within the same joint. The MFC and PFG sites were selected because the cartilage in these locations is subjected to distinct physiological loading regimens and differs in susceptibility to disease. As the major weight-bearing surface of the knee, the MFC is exposed to the highest compressive and shear strains, and is a common site for focal cartilage lesions.41 In contrast, the PFG is a non-weight bearing articular surface and is often used as a “healthy” cartilage harvest site for autografting procedures.42 The inferior PFG articulates with the patella alone, and only when the knee is in maximal flexion, so it experiences relatively low compressive and shear forces.43 Our data indicate that MFC chondrocytes are less sensitive to impact-induced cell death, MT depolarization and respiratory impairment than the PFG, suggesting regional differences in acute MT-related mechanotransduction. MT are known to act as mechanotransducers, converting strain experienced by the tissue to chemical signals, but the mechanisms are not well understood.7,18,44 In cartilage, loading causes differential deformations of intracellular organelles, and cytoskeletal proteins can directly transfer strain to MT.45,46 Although we did not directly assess tissue mechanical properties in the current study, previous work by our group revealed that the shear modulus of the MFC is higher than the PFG, and that differences in depth-dependent modulus and energy dissipation between these regions coincide with depth-dependent heterogeneity in collagen density and fiber organization.47 Taken together, these findings suggest that observed differences between MFC and PFG in the current study may be related regional differences in mechanical properties of the extracellular matrix. Other factors could include differences in pericellular matrix properties, chondrocyte cytoskeletal organization, and/or cellular responses to injury. A better understanding of the mechanisms by which mechanical forces influence MT function may have important implications for development of disease-modifying OA drugs and orthopedic regenerative therapies. For example, recent work suggests that chondrocyte MT depolarization is correlated with local shear strain during cartilage sliding, and connects MT depolarization and chondrocyte apoptosis with increased frictional coefficients due to inadequate cartilage lubrication.48
While the use of cartilage explants in this study provides a number of advantages for improving the physiological relevance, it also presents several limitations. The time required for diffusion of solutes through the dense extracellular matrix of cartilage is significantly greater compared to cells in 2D culture. The diffusivity of glucose in cartilage is on the order of 2 × 10−6 cm2 s−1, which is ~1/3 of the diffusivity of glucose through water.49 With this in mind, the diffusion of glucose and smaller solutes were likely not limiting in our microrespirometry assays. Diffusivities of the pharmacological agents used in the MT stress test are expected to be lower due to larger molecular weights (glucose 180 Da, oligomycin 790 Da, FCCP 250 Da, rotenone 390 Da, Antimycin A 550 Da) however, as noted, steady state values were observed for all agents except oligomycin. Acknowledging the relatively small variations in MW, it is likely that diffusion limitations were predominantly responsible for the lack of oOCR steady state, with small variations in cartilage slice thickness, extracellular matrix composition, concentration of solute, and metabolic action contributing to the variability between samples. In the subset of respirometry assays where oOCR did reach steady state, impacted samples showed increased proton leak and decreased ATP turnover. These findings are similar to Goetz, et al. who found decreased bOCR, mOCR and increased proton leak in cultured chondrocytes 4 weeks after destabilization of the medial meniscus in the rabbit, and Coleman, et al. who found decreased bOCR, mOCR and SRC in cultured bovine chondrocytes after 7 days of repeated overloading.17,28
It should be noted that all experiments were performed at 21% O2 concentration, which is considered relative hyperoxia for cartilage.50 There is considerable disagreement in the literature regarding the effect of O2 concentration on chondrocyte metabolism.51–54 Most studies have investigated this question in isolated chondrocytes, after days or weeks in culture. One group assessed the OCR of freshly harvested cartilage (1 mm cubes) from young mature (18–24-month-old) bovids and found that at O2 concentrations between 5% and 21%, OCR was relatively constant at ~10 nM/106 cells/h,52 a value comparable to the bOCR values measured for un-injured cartilage in the present study. Furthermore, OCR in cultured chondrocytes was independent of O2 and glucose concentration in short term (48 h) culture.51 Therefore, the caveat of relative hyperoxia is unlikely to have bearing on our finding of acute impact-induced MT dysfunction. Other experimental conditions, including osmolarity and pH can also impact the results of respirometry studies. Heywood, et. al. found that osmolarity between 316–600mOsm had minimal effect on OCR of chondrocytes in 3D culture, but glucose availability was a significant factor; high glucose concentrations (5.3 and 15.5 mM) resulted in significantly lower OCRs than very low (1.3 mM) glucose. For the current experiments, standard protocols were used to prepare XF assay media, (see Detailed Methods), and these parameters remained constant between experiments. We used low glucose media (2.5 mM) to more closely approximate in vivo conditions for chondrocytes. Given the cell density per well, glucose should not be significantly depleted over the time frame of our experiments,55 although glucose concentration in the medial was not directly monitored.
A limitation of the current work is the use of immature cartilage. Young bovine cartilage is more metabolically active and more sensitive to injury-induced apoptosis than mature cartilage.56,57 Although the magnitudes differ between young and mature tissue, chondrocytes respond to injurious mechanical loading with increased cell death, intracellular ROS, increased catabolism, and a decrease in anabolic activity, regardless of donor age. The rationale for selecting young cartilage for this proof of concept study is that chondrocyte density in young cartilage is approximately twofold higher in the first 1 mm of depth from the articular surface than adult cartilage, maximizing our ability to measure impact-induced changes in OCR in situ, over a broad range of impact magnitudes.58 Further studies are warranted to investigate the effect of tissue maturity on the acute response of chondrocytes to injury.
In summary, this study demonstrates that MT dysfunction is an acute response of chondrocytes to cartilage impact and is the first report of adapting microrespirometry to study the subcellular mechanisms of impact-induced chondrocyte MT dysfunction in situ. Our results reveal impact-induced decline in bOCR and mOCR, as well as increased proton leak and decreased ATP turnover. These findings are consistent with the known phenomenon of MT membrane swelling after injury, which causes unfolding of the cristae leading to decreased efficiency of the electron transport chain.9 Cartilage from the MFC, a weight-bearing surface of the distal femur, is more resistant to impact-induced cell death, MT depolarization and respiratory decline than that of a non-weight bearing surface, the PFG. This suggests regional differences in mechanotransduction, likely due to local differences in tissue mechanics and/or site-specific cellular response to injury. Our findings suggests that direct targeting of MT respiratory function in the acute stages after cartilage injury may represent a viable therapeutic strategy in the prevention of PTOA.
Supplementary Material
Acknowledgments
Grant sponsor: Weill Cornell Medical College; Grant number: 5 UL1 TR000457-09; Grant sponsor: National Institutes of Health; Grant number: 5T32OD011000-20; Grant sponsor: Harry M. Zweig Memorial Fund for Equine Research; Grant sponsor: National Institute of Arthritis and Musculoskeletal and Skin Diseases; Grant number: 1K08AR068470.
The authors thank Hussni Mohammed and Lynn Johnson for statistical consulting. We also thank and Meg Goodale and Becky Hicks for technical and logistical support. This work was supported by the National Institutes of Health (5T32OD011000-20 and 1K08AR068470), the Weill Cornell Medical College Clinical & Translational Science Center Award/National Center for Advancing Translational Sciences (5 UL1 TR000457-09), and The Harry M. Zweig Memorial Fund for Equine Research. Funding sources did not participate in any aspect of this study.
Footnotes
AUTHORS’ CONTRIBUTIONS
MD and LF are responsible for conception and design of the study. MD developed the methodologies to assess MT function, performed the assays, and drafted the article. MD and EB were responsible for data acquisition. All authors contributed intellectual content, participated in analysis, and interpretation of the data, provided critical revision and approve the final submitted version of the manuscript.
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
References
- 1.Buckwalter JA. The role of mechanical forces in the initiation and progression of osteoarthritis. HSS Jrnl. 2012;8:37–38. doi: 10.1007/s11420-011-9251-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kramer WC, Hendricks KJ, Wang J. Pathogenetic mechanisms of posttraumatic osteoarthritis: opportunities for early intervention. Int J Clin Exp Med. 2011;4:285–298. [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng DS, Visco CJ. Pharmaceutical therapy for osteoarthritis. PM&R. 2012;4:S82–S88. doi: 10.1016/j.pmrj.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Anderson DD, Chubinskaya S, Guilak F, et al. Post-traumatic osteoarthritis: improved understanding and opportunities for early intervention. J Orthop Res. 2011;29:802–809. doi: 10.1002/jor.21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chubinskaya S, Wimmer MA. Key pathways to prevent posttraumatic arthritis for future molecule-based therapy. Cartilage. 2013;4:13S–21S. doi: 10.1177/1947603513487457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pagano G, Talamanca AA, Castello G, et al. Oxidative stress and mitochondrial dysfunction across broad-ranging pathologies: toward mitochondria-targeted clinical strategies. Oxid Med Cell Longev. 2014;2014:541230. doi: 10.1155/2014/541230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ali MH, Pearlstein DP, Mathieu CE, et al. Mitochondrial requirement for endothelial responses to cyclic strain: implications for mechanotransduction. Am J Physiol Lung Cell Mol Physiol. 2004;287:L486–L496. doi: 10.1152/ajplung.00389.2003. [DOI] [PubMed] [Google Scholar]
- 8.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee H-C, Wei Y-H. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol. 2005;37:822–834. doi: 10.1016/j.biocel.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 11.Dai D-F, Chiao YA, Marcinek DJ, et al. Mitochondrial oxidative stress in aging and healthspan. Longev Health-span. 2014;3:6. doi: 10.1186/2046-2395-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blanco FJ, Rego I, Ruiz-Romero C. The role of mitochondria in osteoarthritis. Nat Rev Rheumatol. 2011;7:161–169. doi: 10.1038/nrrheum.2010.213. [DOI] [PubMed] [Google Scholar]
- 13.Maneiro E, Martín MA, de Andres MC, et al. Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis Rheum. 2003;48:700–708. doi: 10.1002/art.10837. [DOI] [PubMed] [Google Scholar]
- 14.Ruiz-Romero C, Calamia V, Mateos J, et al. Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: a decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol Cell Proteomics. 2009;8:172–189. doi: 10.1074/mcp.M800292-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson K, Jung A, Murphy A, et al. Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. Arthritis Rheum. 2000;43:1560–1570. doi: 10.1002/1529-0131(200007)43:7<1560::AID-ANR21>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 16.Cillero-Pastor B, Rego-Pérez I, Oreiro N, et al. Mitochondrial respiratory chain dysfunction modulates metalloproteases –1, –3 and –13 in human normal chondrocytes in culture. BMC Musculoskelet Disord. 2013;14:235–235. doi: 10.1186/1471-2474-14-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coleman MC, Ramakrishnan PS, Brouillette MJ, et al. Injurious loading of articular cartilage compromises chondrocyte respiratory function. Arthritis Rheum. 2015;68:662–671. doi: 10.1002/art.39460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brouillette MJ, Ramakrishnan PS, Wagner VM, et al. Strain-dependent oxidant release in articular cartilage originates from mitochondria. Biomech Model Mechanobiol. 2014;13:565–572. doi: 10.1007/s10237-013-0518-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin JA, McCabe D, Walter M, et al. N-acetylcysteine inhibits post-impact chondrocyte death in osteochondral explants. J Bone Joint Surg Am. 2009;91:1890–1897. doi: 10.2106/JBJS.H.00545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garrido CP, Hakimiyan AA, Rappoport L, et al. Anti-apoptotic treatments prevent cartilage degradation after acute trauma to human ankle cartilage. Osteoarthr Cartil. 2009;17:1244–1251. doi: 10.1016/j.joca.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huser CAM, Davies ME. Calcium signaling leads to mitochondrial depolarization in impact-induced chondrocyte death in equine articular cartilage explants. Arthritis Rheum. 2007;56:2322–2334. doi: 10.1002/art.22717. [DOI] [PubMed] [Google Scholar]
- 22.Leary S, Underwood W, Anthony R, et al. AVMA guidelines for the euthanasia of animals, 2013 edition. Schaumburg, IL: American Veterinary Medical Association; 2013. [Google Scholar]
- 23.Bonnevie ED, Delco ML, Fortier LA, et al. Characterization of tissue response to impact loads delivered using a hand-held instrument for studying articular cartilage injury. Cartilage. 2015;6:226–232. doi: 10.1177/1947603515595071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexander PG, Song Y, Taboas JM, et al. Development of a spring-loaded impact device to deliver injurious mechanical impacts to the articular cartilage surface. Cartilage. 2012;4:52–62. doi: 10.1177/1947603512455195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gavriilidis C, Miwa S, Zglinicki von T, et al. Mitochondrial dysfunction in osteoarthritis is associated with down-regulation of superoxide dismutase 2. Arthritis Rheum. 2013;65:378–387. doi: 10.1002/art.37782. [DOI] [PubMed] [Google Scholar]
- 26.Stackley KD, Beeson CC, Rahn JJ, et al. Bioenergetic profiling of zebrafish embryonic development. In: Kowaltowski AJ, editor. PLoS ONE. Vol. 6. 2011. p. e25652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuh RA, Clerc P, Hwang H, et al. Adaptation of microplate-based respirometry for hippocampal slices and analysis of respiratory capacity. In: McKenna MC, Schousboe A, editors. J Neurosci Res. Vol. 89. 2011. pp. 1979–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goetz JE, Coleman MC, Fredericks DC, et al. Time-dependent loss of mitochondrial function precedes progressive histologic cartilage degeneration in a rabbit meniscal destabilization model. J Orthop Res. 2016;35:590–599. doi: 10.1002/jor.23327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goodwin W, McCabe D, Sauter E, et al. Rotenone prevents impact-induced chondrocyte death. J Orthop Res. 2010 doi: 10.1002/jor.21091. n/a-n/a. https://doi.org/10.1002/jor.21091. [DOI] [PMC free article] [PubMed]
- 30.Huser CAM, Peacock M, Davies ME. Inhibition of caspase-9 reduces chondrocyte apoptosis and proteoglycan loss following mechanical trauma. Osteoarthr Cartil. 2006;14:1002–1010. doi: 10.1016/j.joca.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 31.Diaz-Romero J, Gaillard JP, Grogan SP, et al. Immunophenotypic analysis of human articular chondrocytes: changes in surface markers associated with cell expansion in monolayer culture. J Cell Physiol. 2005;202:731–742. doi: 10.1002/jcp.20164. [DOI] [PubMed] [Google Scholar]
- 32.Schnabel M, Marlovits S, Eckhoff G, et al. Dedifferentiation-associated changes in morphology and gene expression in primary human articular chondrocytes in cell culture. Osteoarthr Cartil. 2002;10:62–70. doi: 10.1053/joca.2001.0482. [DOI] [PubMed] [Google Scholar]
- 33.Benya PD, Shaffer JD. Dedifferentiated chondrocytes reexpress the differentiated collagen phenotype when cultured in agarose gels. Cell. 1982;30:215–224. doi: 10.1016/0092-8674(82)90027-7. [DOI] [PubMed] [Google Scholar]
- 34.Champagne AM, Benel L, Ronot X, et al. Rhodamine 123 uptake and mitochondrial DNA content in rabbit articular chondrocytes evolve differently upon transfer from cartilage to culture conditions. Exp Cell Res. 1987;171:404–410. doi: 10.1016/0014-4827(87)90172-8. [DOI] [PubMed] [Google Scholar]
- 35.Delco ML, Kennedy JG, Bonassar LJ, et al. Post-traumatic osteoarthritis of the ankle: a distinct clinical entity requiring new research approaches. J Othop Res. 2017;35:440–453. doi: 10.1002/jor.23462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olsen RKJ, Cornelius N, Gregersen N. Redox signalling and mitochondrial stress responses; lessons from inborn errors of metabolism. J Inherit Metab Dis. 2015;38:703–719. doi: 10.1007/s10545-015-9861-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soubannier V, McBride HM. Positioning mitochondrial plasticity within cellular signaling cascades. Biochim Biophys Acta. 2009;1793:154–170. doi: 10.1016/j.bbamcr.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 38.Mazzeo AT, Beat A, Singh A, et al. Experimental neurology. Exp Neurol. 2009;218:363–370. doi: 10.1016/j.expneurol.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 39.Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999;160:226–234. doi: 10.1006/exnr.1999.7197. [DOI] [PubMed] [Google Scholar]
- 40.Novakofski KD, Berg LC, Bronzini I, et al. Joint-dependent response to impact and implications for post-traumatic osteoarthritis. Osteoarthr Cartil. 2015;23:1130–1137. doi: 10.1016/j.joca.2015.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Curl WW, Krome J, Gordon ES, et al. Cartilage injuries: a review of 31, 516 knee arthroscopies. Arthroscopy. 1997;13:456–460. doi: 10.1016/s0749-8063(97)90124-9. [DOI] [PubMed] [Google Scholar]
- 42.Cole BJ, Gomoll AH. Biologic Joint Reconstruction: alternatives to Arthroplasty. Thorofare, NJ: SLACK; 2009. [Google Scholar]
- 43.Madeti BK, Chalamalasetti SR, Bolla Pragada SKSSR. Biomechanics of knee joint—a review. Front Mech Eng. 2015;10:176–186. [Google Scholar]
- 44.Sauter E, Buckwalter JA, McKinley TO, et al. Cytoskeletal dissolution blocks oxidant release and cell death in injured cartilage. J Orthop Res. 2012;30:593–598. doi: 10.1002/jor.21552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szafranski JD, Grodzinsky AJ, Burger E, et al. Chondrocyte mechanotransduction: effects of compression on deformation of intracellular organelles and relevance to cellular biosynthesis. Osteoarthr Cartil. 2004;12:937–946. doi: 10.1016/j.joca.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 46.Ohashi T, Hagiwara M, Bader DL, et al. Intracellular mechanics and mechanotransduction associated with chondrocyte deformation during pipette aspiration. Biorheology. 2006;43:201–214. [PubMed] [Google Scholar]
- 47.Silverberg JL, Dillavou S, Bonassar L, et al. Anatomic variation of depth-dependent mechanical properties in neonatal bovine articular cartilage. J Orthop Res. 2012;31:686–691. doi: 10.1002/jor.22303. [DOI] [PubMed] [Google Scholar]
- 48.Bonnevie ED, Delco ML, Jasty N, et al. Chondrocyte death and mitochondrial dysfunction are mediated by cartilage friction and shear strain. Osteoarthr Cartil. 2016;24 IS -: S46EP–. [Google Scholar]
- 49.Maroudas A. Distribution and diffusion of solutes in articular cartilage. Biophys J. 1970;10:365–379. doi: 10.1016/S0006-3495(70)86307-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rajpurohit R, Koch CJ, Tao ZL, et al. Adaptation of chondrocytes to low oxygen tension: relationship between hypoxia and cellular metabolism. J Cell Physiol. 1996;168:424–432. doi: 10.1002/(SICI)1097-4652(199608)168:2<424::AID-JCP21>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 51.Ströbel S, Loparic M, Wendt D, et al. Anabolic and catabolic responses of human articular chondrocytes to varying oxygen percentages. Arthritis Res Ther. 2010;12:R34–R34. doi: 10.1186/ar2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schneider N, Mouithys-Mickalad A, Lejeune J-P, et al. Oxygen consumption of equine articular chondrocytes: influence of applied oxygen tension and glucose concentration during culture. Cell Biol Int. 2007;31:878–886. doi: 10.1016/j.cellbi.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 53.Zhou S, Cui Z, Urban JPG. Factors influencing the oxygen concentration gradient from the synovial surface of articular cartilage to the cartilage-bone interface: a modeling study. Arthritis Rheum. 2004;50:3915–3924. doi: 10.1002/art.20675. [DOI] [PubMed] [Google Scholar]
- 54.Grimshaw MJ, Mason RM. Bovine articular chondrocyte function in vitro depends upon oxygen tension. Osteoarthr Cartil. 2000;8:386–392. doi: 10.1053/joca.1999.0314. [DOI] [PubMed] [Google Scholar]
- 55.Heywood HK, Bader DL, Lee DA. Rate of oxygen consumption by isolated articular chondrocytes is sensitive to medium glucose concentration. J Cell Physiol. 2005;206:402–410. doi: 10.1002/jcp.20491. [DOI] [PubMed] [Google Scholar]
- 56.Kurz B, Lemke A, Kehn M, et al. Influence of tissue maturation and antioxidants on the apoptotic response of articular cartilage after injurious compression. Arthritis Rheum. 2004;50:123–130. doi: 10.1002/art.11438. [DOI] [PubMed] [Google Scholar]
- 57.Farnsworth NL, Antunez LR, Bryant SJ. Influence of chondrocyte maturation on acute response to impact injury in PEG hydrogels. J Biomech. 2012;45:2556–2563. doi: 10.1016/j.jbiomech.2012.07.035. [DOI] [PubMed] [Google Scholar]
- 58.Jadin KD. Depth-varying density and organization of chondrocytes in immature and mature bovine articular cartilage assessed by 3D imaging and analysis. J Histochem Cytochem. 2005;53:1109–1119. doi: 10.1369/jhc.4A6511.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.