

Abstract
Chronic infection is one of the major causes of cancer, and there are several mechanisms for infection‐mediated oncogenesis. Some pathogens encode gene products that behave like oncogenic factors, hijacking cellular pathways to promote the survival and proliferation of infected cells in vivo. Some of these viral oncoproteins trigger a cellular damage defense response leading to senescence; however, other viral factors hinder this suppressive effect, suggesting that cooperation of those viral factors is important for malignant transformation. Coinfection with multiple agents is known to accelerate cancer development in certain cases. For example, parasitic or bacterial infection is a risk factor for adult T‐cell leukemia‐lymphoma induced by human T‐cell leukemia virus type 1, and Epstein‐Barr virus and malaria are closely associated with endemic Burkitt lymphoma. Human immunodeficiency virus type 1 infection is accompanied by various types of infection‐related cancer. These findings indicate that these oncogenic pathogens can cooperate to overcome host barriers against cancer development. In this review, the authors focus on the collaborative strategies of pathogens for oncogenesis from two different points of view: (i) the cooperation of two or more different factors encoded by a single pathogen; and (ii) the acceleration of oncogenesis by coinfection with multiple agents.
Keywords: HTLV‐1 bZIP factor (HBZ), human T‐cell leukemia virus type 1 (HTLV‐1), oncogenic pathogen, superinfection, tax
Abbreviations
- AID
activation‐induced cytidine deaminase
- ATL
adult T‐cell leukemia‐lymphoma
- BCR
B‐cell receptor
- BL
Burkitt lymphoma
- DDR
DNA damage response
- eBL
endemic Burkitt lymphoma
- EBV
Epstein‐Barr virus
- EGFR
epidermal growth factor receptor
- GC
germinal center
- HBV
hepatitis B virus
- HBZ
HTLV‐1 bZIP factor
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HHV
human herpesviruses
- HIV
human immunodeficiency virus
- HL
Hodgkin lymphoma
- HPV
human papillomavirus
- HTLV‐1
human T‐cell leukemia virus type 1
- IARC
International Agency for Research on Cancer
- IDH
infective dermatitis associated with HTLV‐1
- IL
interleukin
- KS
Kaposi's sarcoma
- KSHV
Kaposi's sarcoma‐associated herpesvirus
- MCC
Merkel cell carcinoma
- NF‐κB
nuclear factor kappa B
- NHL
non‐Hodgkin lymphoma
- OIS
oncogene‐induced senescence
- PEL
primary effusion lymphoma
- ROS
reactive oxygen species
- TCR
T‐cell receptor
- TIGIT
T‐cell immunoreceptor with Ig and ITIM domains
- Treg
regulatory T cells
1. INTRODUCTION
Infection is an important cause of human malignant diseases. It is estimated that approximately 15% of cancer cases worldwide are attributable to infection with oncogenic pathogens.1 The IARC has classified 7 viruses, 1 bacterium, and 3 parasites as well established (Group 1) carcinogenic agents based on sufficient evidence in humans: HBV, HCV, HPV (high‐risk types), EBV, KSHV, HIV, HTLV‐1, Helicobacter pylori, Opisthorchis viverrini, Clonorchis sinensis, and Schistosoma haematobium (Table 1).2 These pathogens contribute to oncogenesis by several distinct actions. Pathogens that productively replicate in vivo, such as HBV, HCV, H. pylori, and the parasites, cause tissue inflammation, leading to the accumulation of damage in host cells.3, 4, 5, 6, 7 In contrast, EBV, KSHV, HPV, and HTLV‐1 establish latent infection, and most individuals infected with these viruses are asymptomatic.8, 9, 10, 11 Each of these latent viruses encodes its own oncogene(s); however, these oncogenes are tightly regulated and selectively expressed in cancer cells in vivo. Finally, HIV is unique among the pathogens categorized in IARC's Group 1, as it doesn't have oncogenic potential itself, but it increases cancer risk by allowing other oncogenic agents to propagate in coinfected subjects.1, 12 Collectively, infectious agents are thought to cause cancer by one or more of the following mechanisms: persistent inflammation, potent oncogenic activity, and promoting escape from host immunity. The first mechanism (inflammation) is reviewed elsewhere.13, 14 We will begin this discussion by focusing on the second mechanism (oncogenic factors produced by pathogens), and then we will discuss how various pathogens (including HIV) can interact with one another to further increase the risk of cancer.
Table 1.
Infectious agents categorized as Groups 1 and 2 by the International Agency for Research on Cancer (IARC) Monograph2
| IARC Group | Biological agent | Main malignant diseases | |
|---|---|---|---|
| 1 | Virus | Hepatitis B virus | HCC |
| 1 | Hepatitis C virus | HCC, NHL | |
| 1 | HIV‐1 | NHL, KS, HL, MCC | |
| 1 | HTLV‐1 | ATL | |
| 1 | EBV | BL, NHL, HL, NK/T lymphoma, nasopharyngeal ca, gastric ca | |
| 1 | KSHV | KS, PEL | |
| 1 | HPV 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59 | Cervical ca, anogenital ca, head and neck ca | |
| 1 | Bacterium | Helicobacter pylori | Gastric ca, MALT lymphoma |
| 1 | Parasite | Schistosoma haematobium | Bladder ca |
| 1 | Opisthorchis viverrini | Cholangiocarcinoma | |
| 1 | Clonorchis sinensis | Cholangiocarcinoma | |
| 2A | Virus | HPV type 68 | Cervical ca |
| 2A | Merkel cell polyomavirus (MCPyV) | MCC | |
| 2A | Parasite | Malaria (Plasmodium falciparum) | BL |
| 2B | Virus | HIV‐2 | |
| 2B | HPV 26, 30, 34, 53, 66, 67, 69, 70, 73, 82, 85, 97 | ||
| 2B | HPV 5 and 8 of genera beta | ||
| 2B | BK polyomavirus (BKV) | ||
| 2B | JC polyomavirus (JCV) | ||
| 2B | Parasite | Schistosoma japonicum | |
IARC has classified 7 viruses, 1 bacterium, and 3 parasites as Group 1, and another 7 viruses and 2 parasites as Group 2. Group 1, oncogenic to humans; Group 2A, probably oncogenic to humans; Group 2B, possibly oncogenic to humans.
ATL, adult T‐cell leukemia‐lymphoma; BL, Burkitt lymphoma; ca, cancer; EBV, Epstein‐Barr virus; HCC, hepatocellular carcinoma; HIV, human immunodeficiency virus; HL, Hodgkin lymphoma; HPV, human papillomavirus; HTLV‐1, human T‐cell leukemia virus type 1; IARC, International Agency for Research on Cancer; KS, Kaposi's sarcoma; KSHV, Kaposi's sarcoma‐associated herpesvirus; MALT, mucosa‐associated lymphoid tissue; MCC, Merkel cell carcinoma; NHL, non‐Hodgkin lymphoma; NK/T lymphoma, natural killer / T ‐cell lymphoma; PEL, primary effusion lymphoma.
2. COLLABORATION OF VIRAL FACTORS ALLOWS PROLIFERATION OF INFECTED CELLS WITH DNA DAMAGE: POSSIBLE MECHANISM OF MALIGNANT TRANSFORMATION
Viral oncogenes play important roles in the persistent proliferation and survival of infected cells, resulting in malignant transformation. It is known that the potent activity of any of several viral oncoproteins would normally induce the cellular DDR, leading to apoptosis or OIS. Subversion of the OIS pathway15 by the collaboration of two or more viral factors is a common mechanism that drives the increase in infected cells with genetic abnormalities.
2.1. HTLV‐1 Tax and HBZ
Human T‐cell leukemia virus type 1 is a retrovirus that induces a malignant disease of CD4+ T cells, ATL.16 The HTLV‐1 provirus is integrated into the host genome and encodes several regulatory and accessory genes that regulate viral replication.17, 18 Among them, Tax and HBZ (Figure 1A) are important for the persistence of HTLV‐1 in vivo, although they have different roles in its propagation. Viral replication depends on Tax, and Tax is thus crucial to de novo infection by the virus. In contrast, HBZ is critical for clonal proliferation of infected cells.19, 20 Both Tax and HBZ possess oncogenic potential;18, 21 however, they have opposite functions in many signaling pathways (Figure 1B). Tax strongly activates both the canonical and non‐canonical NF‐κB pathways mainly through activation of IKK complex,17, 22, 23, 24 whereas HBZ specifically suppresses the canonical NF‐κB pathway by inhibiting p65/RelA;25 Tax activates the CREB pathway and HBZ suppresses it by competitive binding to CREB family proteins;26 and Tax activates the Wnt/β‐catenin cascade, but HBZ inhibits this signaling pathway by suppressing the downstream transcription factors TCF1 and LEF1.27 These findings suggest that Tax and HBZ can fine‐tune the regulation of these pathways by counteracting one another.
Figure 1.
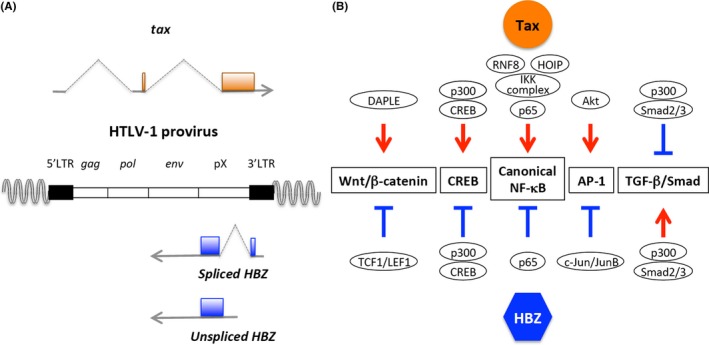
Human T‐cell leukemia virus type 1 (HTLV‐1) Tax and HTLV‐1 bZIP factor (HBZ) counteract one another. (A) HTLV‐1 Tax and HBZ are encoded in the plus and the minus strand of HTLV‐1 provirus, respectively. (B) These two factors counteract one another in many signaling pathways. Representative examples of cellular proteins targeted by Tax and/or HBZ are shown. NF‐κB, nuclear factor kappa B; TGF‐β, transforming growth factor beta
As Tax is a potent oncoprotein, it triggers an oncogenic stress response in the expressing cells and, consequently, induces cell cycle arrest and senescence in a way that is similar to OIS triggered by Ras28 and c‐Myc (Figure 2A).29 Kinjo et al30 reported that Tax induces DNA damage through the generation of intracellular ROS and, consequently, leads to senescence in primary human cells. It has also been shown that hyper‐activation of NF‐κB by Tax induces cellular senescence.31, 32 Importantly, HBZ can release Tax‐expressing cells from cell cycle arrest, probably by inhibition of canonical NF‐κB signaling.25, 31 In addition, both Tax and HBZ have a potential to inhibit DNA repair.11, 17 These observations suggest that these two viral factors facilitate the clonal expansion of infected cells even in the presence of genetic aberrations.
Figure 2.
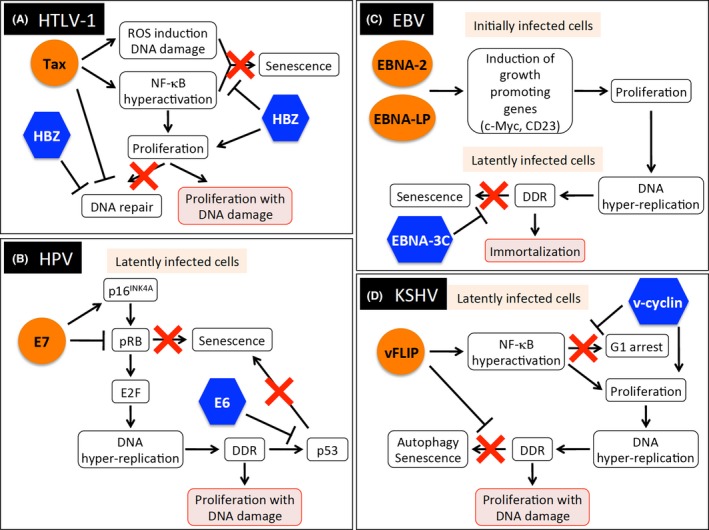
Induction of viral oncogene‐induced senescence and its subversion by multiple viral proteins. (A) Human T‐cell leukemia virus type 1 (HTLV‐1) Tax and HTLV‐1 bZIP factor (HBZ). (B) Human papillomavirus (HPV) E6 and E7. (C) Epstein‐Barr virus (EBV) EBNA‐2, EBNA‐LP, and EBNA‐3c. (D) Kaposi's sarcoma‐associated herpesvirus (KSHV) v‐cyclin and vFLIP. DDR, DNA damage response; NF‐κB, nuclear factor kappa B; ROS, reactive oxygen species
Tax is a highly immunogenic protein, and therefore it is a main target of CTL.33, 34, 35 In addition, Tax might be involved in the emergence of neoantigens in ATL cells, as it has clastogenic and genotoxic potential.17, 30, 36 Thus, Tax evokes a host immune reaction against HTLV‐1‐infected and ATL cells. In contrast, HBZ is closely associated with immune‐suppressive phenotypes of infected cells. HBZ potently enhances transcription of Foxp3, which is a master gene of Treg and, indeed, the number of Treg is significantly increased in HBZ‐transgenic mice and primary HTLV‐1‐infected and ATL cells.21, 37, 38 A recent study demonstrated that HBZ induces a co‐inhibitory receptor, TIGIT, on the surface of T cells, and suppresses the immune response through induction of IL‐10 from dendritic cells.39 In addition, vaccination with Tax protein is less efficient in HBZ‐transgenic mice than in wild‐type littermates.39 These results suggest that HBZ expressed in infected cells can suppress anti‐Tax immunity in vivo. Anti‐HBZ CTL are less frequent than anti‐Tax CTL in HTLV‐1‐infected subjects despite the constant expression of HBZ, implying that the immunogenicity of HBZ is low.40 In addition, HBZ suppresses the intracellular inhibitory signal through TIGIT by inactivation of SHP‐2 and, consequently, enhances cell proliferation upon TCR stimulation.41 Taken together, these reports indicate that Tax stimulates many oncogenic pathways while HBZ counterbalances the negative effects of Tax, leading to malignant transformation of infected cells.
2.2. HPV E6 and E7
High‐risk types of HPV are etiological agents of cervical, anal, and head and neck cancers. The IARC has classified 12 types of HPV (HPV16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, and 59) as Group 1 carcinogenic agents.1 The products of two early genes encoded in HPV, E6 and E7, are constitutively expressed in HPV‐positive tumor cells and play major roles in oncogenesis by HPV. It is well known that E6 and E7 inactivate the cellular tumor suppressors p53 and retinoblastoma tumor suppressor protein (pRb) respectively (Figure 2B).42 The transforming activity of E7 is strongly increased in the presence of E6. E7 triggers a carcinogenic cascade by binding to pRb and inactivating it; the resulting activation of E2F transcription factors promotes cell proliferation. Overactive E2Fs would normally cause p53 activation and induce apoptosis or senescence as a cellular defense response; however, the E6/E6‐associated protein (E6AP) complex ubiquitinates p53 for proteasomal degradation. Thus, the sequential effects of E7 and E6 trigger oncogenesis in HPV‐infected cells. It has also been reported that high‐risk, but not low‐risk, HPV E7 causes p16INK4A induction in a pRb‐independent way – a phenomenon that is also observed in Ras‐mediated OIS.43, 44 A recent genomic sequencing study showed that the sequence of HPV16 E7 is strictly conserved in precancerous and cancerous tissues, suggesting its importance in carcinogenesis.45 Collectively, these studies show that persistent replicative stresses from E7 and inhibition of cell cycle arrest and senescence by E6 induce the accumulation of genetic aberrations and, consequently, increase the probability of malignant transformation by HPV. It is also known that HPV E5 protein has oncogenic properties; it promotes cell proliferation by stimulation of EGFR, and suppresses TRAIL‐ and FasL‐mediated apoptosis.46, 47 In addition, E5 downregulates expression of MHC class I, class II, and CD1d on the surface of infected cells, enabling them to escape from CTL and NKT cells.46 Thus, E5 plays important collaborative roles in HPV‐mediated carcinogenesis, especially by modulation of host immune systems.
2.3. EBV oncoproteins
Human herpesviruses including EBV and KSHV establish latent infection in the host by expressing a limited number of gene products.48 This latency is reversible – the virus can resume productive viral replication (the so‐called lytic phase) – and, in both phases, HHV induce DDR to remodel the infected cells.49 After B cells become infected with EBV, the viral nuclear antigens EBNA2 and EBNA‐LP are immediately expressed, and they upregulate cellular growth‐promoting genes such as c‐Myc and CD23 (Figure 2C).50 Hyper‐proliferation of B cells induced by these viral factors triggers replication‐mediated DNA damage and, consequently, the cellular DDR would suppress cell proliferation. However, another EBV oncoprotein, EBNA3C, inhibits DDR signaling and promotes proliferation and immortalization of the infected cell, suggesting that EBNA2, EBNA‐LP, and EBNA3C cooperate with each other in cellular transformation.
Two latent membrane proteins, LMP‐1 and LMP‐2A, are major EBV‐encoded oncoproteins, and mimic CD40 and BCR signaling, respectively.51 LMP‐1 stimulates several oncogenic signaling pathways such as the NF‐κB and MAPK pathways.52 LMP‐2A constitutively activates the Ras/PI3K/Akt pathway, and mediates cellular transformation.53 Interestingly, a transgenic mouse model, which co‐expresses LMP‐1 and LMP‐2A in B cells, showed that LMP‐2A reduced hyperactivation of B cells induced by LMP‐1.54 It is possible that LMP‐2A curbs overstimulation of oncogenic signals by LMP‐1 to establish persistent infection in vivo.
2.4. KSHV v‐cyclin and vFLIP
Kaposi's sarcoma‐associated herpesvirus v‐cyclin and vFLIP are encoded in the same viral mRNA and expressed in latently infected cells.48 v‐cyclin accelerates cell cycle progression, triggers the DDR, and finally induces autophagy and senescence in primary human foreskin fibroblasts (Figure 2D). In contrast, a potent viral anti‐apoptotic factor, vFLIP, suppresses v‐cyclin‐induced senescence, contributing to the expansion of KSHV‐infected cells with DNA damage.55 vFLIP also induces G1 arrest/senescence in HeLa cells through hyperactivation of NF‐κB that is reminiscent of Tax‐mediated senescence. In this setting, v‐cyclin prevents cell cycle arrest by vFLIP.56 It is suggested that these two proteins work in concert to accelerate the transformation of KSHV‐infected cells.
In summary, investigations of a variety of oncogenic viruses have shown a common scheme: viral gene products collaborate to induce proliferation while blocking senescence.
3. ENHANCEMENT OF CANCER DEVELOPMENT BY COINFECTION WITH MULTIPLE AGENTS
Collaboration of viral gene products in oncogenesis is not limited to gene products that all come from the same virus. In some cases, coinfection with two or more pathogens is associated with a higher cancer risk than infection with either of the pathogens alone. Recent studies have shown a variety of molecular mechanisms for this enhanced oncogenesis. Here, we summarize several examples.
3.1. Strongyloidiasis and infective dermatitis as risk factors for ATL
Two types of superinfections are associated with an increased risk of ATL in people already infected with HTLV‐1. The first, strongyloidiasis, is a chronic parasitic infection of humans caused by Strongyloides stercoralis. HTLV‐1 proviral load is higher in S. stercoralis‐positive carriers than in non‐coinfected subjects,57 and the incidence of ATL is high in such superinfected subjects,58 suggesting that S. stercoralis facilitates the clonal proliferation of HTLV‐1‐infected cells. The second type of superinfection associated with increased ATL risk is IDH: a refractory skin infection with Staphylococcus aureus and/or beta‐hemolytic Streptococci observed in HTLV‐1‐infected children in developing countries.59 In IDH patients, HTLV‐1 proviral load is also higher60 and clonal expansion occurs at a higher rate than in subjects infected with HTLV‐1 only.61 A recent study showed that clonal expansion and turnover of HTLV‐1‐infected cells are increased in patients coinfected with S. stercoralis.61 These observations suggest that coinfection with S. stercoralis or IDH promotes both de novo infection and cell proliferation, which depend on Tax and HBZ, respectively. This idea is compatible with the fact that T‐cell activation enhances the activities of Tax and HBZ on cell proliferation in primary T cells.19, 41, 62 Thus, inflammation and T‐cell activation caused by the coinfective agent may increase the risk of ATL development in HTLV‐1 positive individuals.
3.2. HIV‐associated cancers
Human immunodeficiency virus 1 is rated a Group 1 oncogenic agent by the IARC.2 HIV‐1 induces AIDS and indirectly causes cancers by allowing other oncogenic pathogens to express. It is noteworthy that HIV‐1 infection is particularly closely associated with several subtypes of NHL. Most AIDS‐related NHL are of B‐cell origin; it is suggested that depletion of CD4+ T cells by HIV‐1 permits dysregulation of B‐cell expansion and the expression of B‐lymphotrophic viruses.63 EBV is detected in almost all cases of central nervous system lymphoma, ~40% of diffuse large‐cell lymphoma, and ~30% of BL. HL is also associated with coinfection by HIV‐1 and EBV. It has been reported that HIV‐1‐infected people have an approximately 10‐fold higher risk of developing HL than uninfected people.64 The rate of EBV‐positive HL is higher in HIV‐1 infected cases than in the general population,65 suggesting that EBV is closely involved in the genesis of HIV‐mediated HL. Importantly, the standard incidence rate of HL in HIV‐infected subjects has increased despite entering the era of combination antiretroviral therapy, whereas that of NHL has dropped.66 These findings suggest that there are distinct mechanisms of lymphomagenesis by EBV in the presence of immune modulation by HIV, and that these mechanisms vary between HL and NHL. KSHV67 and Merkel cell polyomavirus (MCPyV)68 were first identified from cancer cells of AIDS‐related Kaposi's sarcoma and Merkel‐cell carcinoma respectively, and are now recognized as causative agents of each cancer. KSHV is also associated with a rare subtype of B‐cell lymphoma, primary effusion lymphoma (PEL), which occurs predominantly in the pleural or peritoneal cavity. The risk of HPV‐associated cancers, such as cervical cancer, anal cancer, and oropharyngeal cancer, is also elevated among persons with AIDS.69
It is not surprising that immunosuppression by HIV allows malignant transformation of host cells infected with other oncogenic pathogens. However, a recent case study reported a rare “cancer‐like” malignant disease developed in an AIDS patient; tumors were derived from cells of the tapeworm Hymenolepis nana.70 Characteristics of this non‐human origin tumor fulfilled the working definitions of cancer: invasion and metastasis, hyperproliferative and monomorphic features, and genetic alterations (in H. nana's genes). This case demonstrated a novel mechanism for HIV‐related tumorigenesis triggered by another pathogen.
HIV‐1 is also associated with several cancers that have not been linked to other oncogenic pathogens. The risk of lung cancer is higher in HIV‐infected subjects than in the general population, and the higher smoking rate in the HIV‐infected population is one of the factors explaining this trend.71 In addition, recent studies show that immunosuppression by HIV and chronic inflammation are involved in oncogenesis.72 It is known that HIV infection is a risk factor for cancers of the conjunctiva and the lip.12 Impairment of anti‐cancer immunity by HIV is a possible mechanism for the emergence of these malignancies.
3.3. EBV and malaria
Burkitt lymphoma (BL) is an aggressive B‐cell malignancy originating from a GC, and it can be classified into three subtypes according to clinical features: endemic BL, sporadic BL, and HIV‐associated BL.73 eBL is the most common childhood cancer in Africa, and more than 95% of cases are associated with EBV. In addition, it occurs at higher incidence in areas in which malaria is endemic; therefore an additional role of malaria in the etiology has long been suspected.74 Malaria is caused by several species of Plasmodium. A recent study demonstrated that chronic infection with Plasmodium in mice promotes clonal expansion of GC cells and induces expression of AID, which is associated with genetic mutations and chromosomal translocations.75 It is suggested that Plasmodium superinfection promotes EBV‐mediated oncogenesis by inducing genomic aberrations, leading to the distinct B‐cell malignancy, eBL.
3.4. Helicobacter pylori and EBV
Most gastric cancers are associated with infection of gastric epithelial cells with H. pylori.12 Helicobacter pylori is a highly heterogeneous bacterium with a large genomic diversity. Helicobacter pylori strains possessing the cagA gene are more virulent than cagA‐negative strains, and the CagA protein is closely linked to severe diseases such as peptic ulcers and gastric cancer.76 EBV is also causally associated with gastric carcinoma, and approximately 10% of cases contain EBV in tumor cells.53 Recently, the mechanism of cooperative oncogenesis by these two pathogens was clarified.77 CagA is injected into gastric epithelial cells and interacts with the host tyrosine phosphatase SHP2, resulting in activation of SHP2, which is important for oncogenesis by H. pylori. However, the SHP2 homologue SHP1 inactivates CagA and thus prevents SHP2 activation. EBV infection reduces the expression of SHP1 by inducing DNA hypermethylation in the SHP1 promoter region. Thus, EBV cancels the negative effect of SHP1 on gastric carcinogenesis by H. pylori. This may be a distinct mechanism of gastric cancer induced by dual agents.
3.5. Schistosoma haematobium and bacterial infection
Schistosoma haematobium is endemic in sub‐Saharan Africa and the Middle East, and induces urinary bladder cancer.7, 78 Bacterial superinfection is suggested to be a risk factor for S. haematobium‐associated bladder cancer. Schistosoma haematobium migrate into the bladder tissue and cause chronic granulomatous inflammation in the mucosal and submucosal layers of the bladder. The inflammation leads to the development of squamous metaplasia and urine stasis, resulting in bacterial infection. Some of the bacteria are thought to be associated with the endogenous formation of carcinogenic N‐nitrosamines in the urinary tract. It is suggested that coinfection with S. haematobium and these bacteria thus initiates oncogenic processes in the bladder.
4. CONCLUDING REMARKS
In the present review, we summarized the mechanisms by which multiple pathogens can cooperate to cause cancer (Figure 3). Viral oncoproteins encoded by HTLV‐1, HPV, EBV, and KSHV promote cell proliferation and subvert senescence within each infected cell (Figures 2 and 3). Chronic inflammation triggered by persistent infection induces an accumulation of damage to tissue and DNA, and coinfection with certain pathogens accelerates oncogenesis. HIV infection modifies the immune status of hosts coinfected with other oncogenic agents, allowing malignant diseases to emerge. Thus, host immunity plays a critical role in controlling infection‐mediated cancers.
Figure 3.
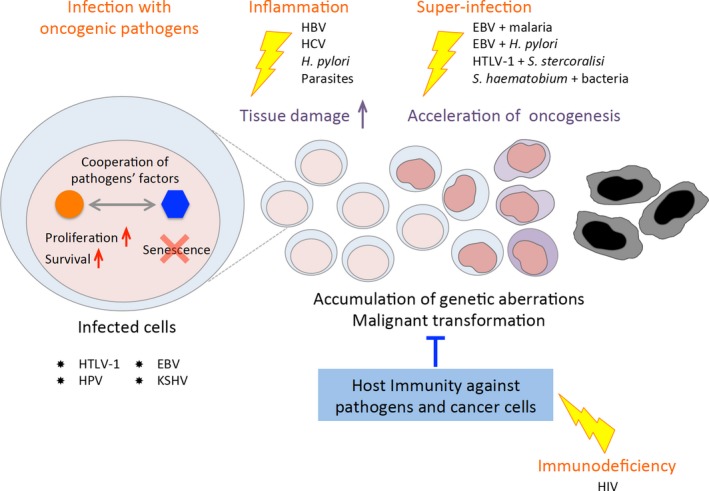
Schema of oncogenic processes mediated by collaboration of multiple pathogens. Cooperation of several pathogens can produce malignant transformation by four processes: cell proliferation promoted by oncogenic factors in infected cells, chronic inflammation, coinfection with certain combinations of pathogens, and immunodeficiency. EBV, Epstein‐Barr virus; HBV, hepatitis B virus; HCV, hepatitis C virus; HIV, human immunodeficiency virus; HPV, human papillomavirus; HTLV‐1, human T‐cell leukemia virus type 1; KSHV, Kaposi's sarcoma‐associated herpesvirus
Understanding and appreciating the role of infectious agents in oncogenesis is important for public health, as certain cancers are theoretically preventable by infection control and prevention. The majority of infection‐mediated cancers occur in developing countries (1.5 million in 1.9 million cases),12 suggesting that the number of cancer cases can be reduced by appropriate medical strategies, such as vaccination, antiviral treatments, antibiotics, antiparasitics, and health education. Future research is needed to clarify the molecular functions of the factors encoded in each pathogen, and to understand their interactions with factors from other pathogens. It will be beneficial to establish novel therapeutic/prophylactic strategies against both infection‐related cancers and chronic inflammatory diseases caused by their etiological agents.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
ACKNOWLEDGMENTS
We thank Dr Linda Kingsbury for proofreading. This study was supported by the Project for Cancer Research And Therapeutic Evolution (P‐CREATE) and the Research Program on Emerging and Re‐emerging Infectious Diseases from the Japan Agency for Medical Research and Development (M.M. and J.Y.), JSPS KAKENHI Grant Number JP17K07166 (J.Y.), a grant from Princess Takamatsu Cancer Research Fund (J.Y.), and a grant from The Yasuda Medical Foundation (J.Y.). This study was also supported in part by the JSPS Core‐to‐Core Program A, Advanced Research Networks. This brief review is intended to raise selected illustrative examples. We apologize, in view of space and format limitations, for our failure to cover other examples and issues.
Yasunaga J‐I, Matsuoka M. Oncogenic spiral by infectious pathogens: Cooperation of multiple factors in cancer development. Cancer Sci. 2018;109:24–32. https://doi.org/10.1111/cas.13443
Funding information
Princess Takamatsu Cancer Research Fund, Yasuda Memorial Medical Foundation, Japan Society for the Promotion of Science Grant/Award Number: “JP17K07166”, Japan Agency for Medical Research and Development (Grant/Award Numbers: “16fk0108127 h0001”, “16cm010630 h0001”).
REFERENCES
- 1. Plummer M, de Martel C, Vignat J, Ferlay J, Bray F, Franceschi S. Global burden of cancers attributable to infections in 2012: a synthetic analysis. Lancet Glob Health. 2016;4:e609‐e616. [DOI] [PubMed] [Google Scholar]
- 2. IARC . Malaria and some polyomaviruses (SV40, BK, JC, and Merkel cell viruses). IARC Monogr Eval Carcinog Risks Hum. 2013;104:1‐353. [PMC free article] [PubMed] [Google Scholar]
- 3. Shirvani‐Dastgerdi E, Schwartz RE, Ploss A. Hepatocarcinogenesis associated with hepatitis B, delta and C viruses. Curr Opinion Virol. 2016;20:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hunt RH, Camilleri M, Crowe SE, et al. The stomach in health and disease. Gut. 2015;64:1650‐1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kaewpitoon N, Kaewpitoon SJ, Pengsaa P, Sripa B. Opisthorchis viverrini: the carcinogenic human liver fluke. World J Gastroenterol. 2008;14:666‐674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim TS, Pak JH, Kim JB, Bahk YY. Clonorchis sinensis, an oriental liver fluke, as a human biological agent of cholangiocarcinoma: a brief review. BMB Rep. 2016;49:590‐597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mostafa MH, Sheweita SA, O'Connor PJ. Relationship between schistosomiasis and bladder cancer. Clin Microbiol Rev. 1999;12:97‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murata T. Regulation of Epstein‐Barr virus reactivation from latency. Microbiol Immunol. 2014;58:307‐317. [DOI] [PubMed] [Google Scholar]
- 9. Thakker S, Verma SC. Co‐infections and pathogenesis of KSHV‐associated malignancies. Front Microbiol. 2016;7:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cubie HA. Diseases associated with human papillomavirus infection. Virology. 2013;445:21‐34. [DOI] [PubMed] [Google Scholar]
- 11. Ma G, Yasunaga J, Matsuoka M. Multifaceted functions and roles of HBZ in HTLV‐1 pathogenesis. Retrovirology. 2016;13:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. IARC . Biological agents. IARC Monogr Eval Carcinog Risks Hum. 2012;100B:1‐475. [PMC free article] [PubMed] [Google Scholar]
- 13. Pesic M, Greten FR. Inflammation and cancer: tissue regeneration gone awry. Curr Opin Cell Biol. 2016;43:55‐61. [DOI] [PubMed] [Google Scholar]
- 14. Shalapour S, Karin M. Immunity, inflammation, and cancer: an eternal fight between good and evil. J Clin Invest. 2015;125:3347‐3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Adams PD. Healing and hurting: molecular mechanisms, functions, and pathologies of cellular senescence. Mol Cell. 2009;36:2‐14. [DOI] [PubMed] [Google Scholar]
- 16. Takatsuki K. Discovery of adult T‐cell leukemia. Retrovirology. 2005;2:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matsuoka M, Jeang KT. Human T‐cell leukaemia virus type 1 (HTLV‐1) infectivity and cellular transformation. Nat Rev Cancer. 2007;7:270‐280. [DOI] [PubMed] [Google Scholar]
- 18. Yasunaga J, Matsuoka M. Molecular mechanisms of HTLV‐1 infection and pathogenesis. Int J Hematol. 2011;94:435‐442. [DOI] [PubMed] [Google Scholar]
- 19. Satou Y, Yasunaga J, Yoshida M, Matsuoka M. HTLV‐I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci USA. 2006;103:720‐725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arnold J, Zimmerman B, Li M, Lairmore MD, Green PL. Human T‐cell leukemia virus type‐1 antisense‐encoded gene, Hbz, promotes T‐lymphocyte proliferation. Blood. 2008;112:3788‐3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Satou Y, Yasunaga J, Zhao T, et al. HTLV‐1 bZIP factor induces T‐cell lymphoma and systemic inflammation in vivo. PLoS Pathog. 2011;7:e1001274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boxus M, Twizere JC, Legros S, Dewulf JF, Kettmann R, Willems L. The HTLV‐1 tax interactome. Retrovirology. 2008;5:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Giam CZ, Semmes OJ. HTLV‐1 infection and adult T‐Cell leukemia/lymphoma‐A tale of two proteins: tax and HBZ. Viruses 2016;8:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shibata Y, Tokunaga F, Goto E, et al. HTLV‐1 tax induces formation of the active macromolecular IKK complex by generating Lys63‐ and Met1‐linked hybrid polyubiquitin chains. PLoS Pathog. 2017;13:e1006162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao T, Yasunaga J, Satou Y, et al. Human T‐cell leukemia virus type 1 bZIP factor selectively suppresses the classical pathway of NF‐kappaB. Blood. 2009;113:2755‐2764. [DOI] [PubMed] [Google Scholar]
- 26. Lemasson I, Lewis MR, Polakowski N, et al. Human T‐cell leukemia virus type 1 (HTLV‐1) bZIP protein interacts with the cellular transcription factor CREB to inhibit HTLV‐1 transcription. J Virol. 2007;81:1543‐1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ma G, Yasunaga J, Fan J, Yanagawa S, Matsuoka M. HTLV‐1 bZIP factor dysregulates the Wnt pathways to support proliferation and migration of adult T‐cell leukemia cells. Oncogene. 2013;32:4222‐4230. [DOI] [PubMed] [Google Scholar]
- 28. Lee AC, Fenster BE, Ito H, et al. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem. 1999;274:7936‐7940. [DOI] [PubMed] [Google Scholar]
- 29. Vafa O, Wade M, Kern S, et al. c‐Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene‐induced genetic instability. Mol Cell. 2002;9:1031‐1044. [DOI] [PubMed] [Google Scholar]
- 30. Kinjo T, Ham‐Terhune J, Peloponese JM Jr, Jeang KT. Induction of reactive oxygen species by human T‐cell leukemia virus type 1 tax correlates with DNA damage and expression of cellular senescence marker. J Virol. 2010;84:5431‐5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhi H, Yang L, Kuo YL, Ho YK, Shih HM, Giam CZ. NF‐kappaB hyper‐activation by HTLV‐1 tax induces cellular senescence, but can be alleviated by the viral anti‐sense protein HBZ. PLoS Pathog. 2011;7:e1002025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ho YK, Zhi H, DeBiaso D, Philip S, Shih HM, Giam CZ. HTLV‐1 tax‐induced rapid senescence is driven by the transcriptional activity of NF‐kappaB and depends on chronically activated IKKalpha and p65/RelA. J Virol. 2012;86:9474‐9483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jacobson S, Shida H, McFarlin DE, Fauci AS, Koenig S. Circulating CD8+ cytotoxic T lymphocytes specific for HTLV‐I pX in patients with HTLV‐I associated neurological disease. Nature. 1990;348:245‐248. [DOI] [PubMed] [Google Scholar]
- 34. Kannagi M, Harada S, Maruyama I, et al. Predominant recognition of human T cell leukemia virus type I (HTLV‐I) pX gene products by human CD8+ cytotoxic T cells directed against HTLV‐I‐infected cells. Int Immunol. 1991;3:761‐767. [DOI] [PubMed] [Google Scholar]
- 35. Kannagi M, Hasegawa A, Takamori A, Kinpara S, Utsunomiya A. The roles of acquired and innate immunity in human T‐cell leukemia virus type 1‐mediated diseases. Front Microbiol. 2012;3:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yasunaga J, Jeang KT. Viral transformation and aneuploidy. Environ Mol Mutagen. 2009;50:733‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao T, Satou Y, Sugata K, et al. HTLV‐1 bZIP factor enhances TGF‐beta signaling through p300 coactivator. Blood. 2011;118:1865‐1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Satou Y, Utsunomiya A, Tanabe J, Nakagawa M, Nosaka K, Matsuoka M. HTLV‐1 modulates the frequency and phenotype of FoxP3+CD4+ T cells in virus‐infected individuals. Retrovirology. 2012;9:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yasuma K, Yasunaga J, Takemoto K, et al. HTLV‐1 bZIP factor impairs anti‐viral immunity by inducing co‐inhibitory molecule, T cell immunoglobulin and ITIM domain (TIGIT). PLoS Pathog. 2016;12:e1005372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sugata K, Yasunaga J, Miura M, et al. Enhancement of anti‐STLV‐1/HTLV‐1 immune responses through multimodal effects of anti‐CCR4 antibody. Sci Rep. 2016;6:27150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kinosada H, Yasunaga JI, Shimura K, et al. HTLV‐1 bZIP factor enhances T‐cell proliferation by impeding the suppressive signaling of co‐inhibitory receptors. PLoS Pathog. 2017;13:e1006120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hoppe‐Seyler K, Bossler F, Braun JA, Herrmann AL, Hoppe‐Seyler F. The HPV E6/E7 oncogenes: key factors for viral carcinogenesis and therapeutic targets. Trends Microbiol 2017. https://doi.org/10.1016/j.tim.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 43. McLaughlin‐Drubin ME, Crum CP, Munger K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc Natl Acad Sci USA. 2011;108:2130‐2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Munger K, Jones DL. Human papillomavirus carcinogenesis: an identity crisis in the retinoblastoma tumor suppressor pathway. J Virol. 2015;89:4708‐4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mirabello L, Yeager M, Yu K, et al. HPV16 E7 genetic conservation is critical to carcinogenesis. Cell. 2017;170:1164‐1174.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. de Freitas AC, de Oliveira THA, Barros MR Jr, Venuti A. hrHPV E5 oncoprotein: immune evasion and related immunotherapies. J Exp Clin Cancer Res. 2017;36:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kabsch K, Alonso A. The human papillomavirus type 16 E5 protein impairs TRAIL‐ and FasL‐mediated apoptosis in HaCaT cells by different mechanisms. J Virol. 2002;76:12162‐12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Speck SH, Ganem D. Viral latency and its regulation: lessons from the gamma‐herpesviruses. Cell Host Microbe. 2010;8:100‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Leidal AM, Pringle ES, McCormick C. Evasion of oncogene‐induced senescence by gammaherpesviruses. Curr Opin Virol. 2012;2:748‐754. [DOI] [PubMed] [Google Scholar]
- 50. Nikitin PA, Yan CM, Forte E, et al. An ATM/Chk2‐mediated DNA damage‐responsive signaling pathway suppresses Epstein‐Barr virus transformation of primary human B cells. Cell Host Microbe. 2010;8:510‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kang MS, Kieff E. Epstein‐Barr virus latent genes. Exp Mol Med. 2015;47:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang LW, Jiang S, Gewurz BE. Epstein‐barr virus LMP1‐mediated oncogenicity. J Virol. 2017;91:e01718‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fukayama M, Hino R, Uozaki H. Epstein‐Barr virus and gastric carcinoma: virus‐host interactions leading to carcinoma. Cancer Sci. 2008;99:1726‐1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vrazo AC, Chauchard M, Raab‐Traub N, Longnecker R. Epstein‐Barr virus LMP2A reduces hyperactivation induced by LMP1 to restore normal B cell phenotype in transgenic mice. PLoS Pathog. 2012;8:e1002662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Leidal AM, Cyr DP, Hill RJ, Lee PW, McCormick C. Subversion of autophagy by Kaposi's sarcoma‐associated herpesvirus impairs oncogene‐induced senescence. Cell Host Microbe. 2012;11:167‐180. [DOI] [PubMed] [Google Scholar]
- 56. Zhi H, Zahoor MA, Shudofsky AM, Giam CZ. KSHV vCyclin counters the senescence/G1 arrest response triggered by NF‐kappaB hyperactivation. Oncogene. 2015;34:496‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gabet AS, Mortreux F, Talarmin A, et al. High circulating proviral load with oligoclonal expansion of HTLV‐1 bearing T cells in HTLV‐1 carriers with strongyloidiasis. Oncogene. 2000;19:4954‐4960. [DOI] [PubMed] [Google Scholar]
- 58. Nakada K, Yamaguchi K, Furugen S, et al. Monoclonal integration of HTLV‐I proviral DNA in patients with strongyloidiasis. Int J Cancer. 1987;40:145‐148. [DOI] [PubMed] [Google Scholar]
- 59. Lee R, Schwartz RA. Human T‐lymphotrophic virus type 1‐associated infective dermatitis: a comprehensive review. J Am Acad Dermatol. 2011;64:152‐160. [DOI] [PubMed] [Google Scholar]
- 60. Primo J, Siqueira I, Nascimento MC, et al. High HTLV‐1 proviral load, a marker for HTLV‐1 associated myelopathy/tropical spastic paraparesis, is also detected in patients with infective dermatitis associated with HTLV‐1. Braz J Med Biol Res. 2009;42:761‐764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gillet NA, Cook L, Laydon DJ, et al. Strongyloidiasis and infective dermatitis alter human T lymphotropic virus‐1 clonality in vivo. PLoS Pathog. 2013;9:e1003263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Swaims AY, Khani F, Zhang Y, et al. Immune activation induces immortalization of HTLV‐1 LTR‐Tax transgenic CD4+ T cells. Blood. 2010;116:2994‐3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Engels EA. Infectious agents as causes of non‐Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev. 2007;16:401‐404. [DOI] [PubMed] [Google Scholar]
- 64. Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta‐analysis. Lancet. 2007;370:59‐67. [DOI] [PubMed] [Google Scholar]
- 65. Tirelli U, Errante D, Dolcetti R, et al. Hodgkin's disease and human immunodeficiency virus infection: clinicopathologic and virologic features of 114 patients from the Italian Cooperative Group on AIDS and Tumors. J Clin Oncol. 1995;13:1758‐1767. [DOI] [PubMed] [Google Scholar]
- 66. Engels EA, Pfeiffer RM, Goedert JJ, et al. Trends in cancer risk among people with AIDS in the United States 1980‐2002. AIDS. 2006;20:1645‐1654. [DOI] [PubMed] [Google Scholar]
- 67. Chang Y, Cesarman E, Pessin MS, et al. Identification of herpesvirus‐like DNA sequences in AIDS‐associated Kaposi's sarcoma. Science. 1994;266:1865‐1869. [DOI] [PubMed] [Google Scholar]
- 68. Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096‐1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chaturvedi AK, Madeleine MM, Biggar RJ, Engels EA. Risk of human papillomavirus‐associated cancers among persons with AIDS. J Natl Cancer Inst. 2009;101:1120‐1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Muehlenbachs A, Bhatnagar J, Agudelo CA, et al. Malignant transformation of hymenolepis nana in a human host. N Engl J Med. 2015;373:1845‐1852. [DOI] [PubMed] [Google Scholar]
- 71. Giordano TP, Kramer JR. Does HIV infection independently increase the incidence of lung cancer? Clin Infect Dis. 2005;40:490‐491. [DOI] [PubMed] [Google Scholar]
- 72. Sigel K, Makinson A, Thaler J. Lung cancer in persons with HIV. Curr Opin HIV AIDS. 2017;12:31‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Brady G, MacArthur GJ, Farrell PJ. Epstein‐Barr virus and Burkitt lymphoma. J Clin Pathol. 2007;60:1397‐1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Burkitt DP. Observations on the geography of malignant lymphoma. East Afr Med J. 1961;38:511‐514. [PubMed] [Google Scholar]
- 75. Robbiani DF, Deroubaix S, Feldhahn N, et al. Plasmodium infection promotes genomic instability and AID‐dependent B cell lymphoma. Cell. 2015;162:727‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hatakeyama M. Helicobacter pylori CagA and gastric cancer: a paradigm for hit‐and‐run carcinogenesis. Cell Host Microbe. 2014;15:306‐316. [DOI] [PubMed] [Google Scholar]
- 77. Saju P, Murata‐Kamiya N, Hayashi T, et al. Host SHP1 phosphatase antagonizes Helicobacter pylori CagA and can be downregulated by Epstein‐Barr virus. Nat Microbiol. 2016;1:16026. [DOI] [PubMed] [Google Scholar]
- 78. Rambau PF, Chalya PL, Jackson K. Schistosomiasis and urinary bladder cancer in North Western Tanzania: a retrospective review of 185 patients. Infect Agent Cancer. 2013;8:19. [DOI] [PMC free article] [PubMed] [Google Scholar]