

Abstract
Ionic liquids are used to dewater a suspension of birch Kraft pulp cellulose nanofibrils (CNF) and as a medium for water‐free topochemical modification of the nanocellulose (a process denoted as “WtF‐Nano”). Acetylation was applied as a model reaction to investigate the degree of modification and scope of effective ionic liquid structures. Little difference in reactivity was observed when water was removed, after introduction of an ionic liquid or molecular co‐solvent. However, the viscoelastic properties of the CNF suspended in two ionic liquids show that the more basic, but non‐dissolving ionic liquid, allows for better solvation of the CNF. Vibrio fischeri bacterial tests show that all ionic liquids in this study were harmless. Scanning electron microscopy and wide‐angle X‐ray scattering on regenerated samples show that the acetylated CNF is still in a fibrillar form. 1 D and 2 D NMR analyses, after direct dissolution in a novel ionic liquid electrolyte solution, indicate that both cellulose and residual xylan on the surface of the nanofibrils reacts to give acetate esters.
Keywords: acetylation, ionic liquids, cellulose, nanofibers, topochemistry
Introduction
There is intense interest in the conversion of chemical cellulosic pulps and even wood into nanostructured materials, to revalorize our forest feedstocks. Nanocellulose, in which the native nanoscaled ultrastructure of wood is preserved, offers the opportunity to produce high mechanical strength materials based on the high aspect ratio of native elementary fibrils and fibrillar bundles.1 Cellulose nanofibrils (CNF) are of particular interest as aspect ratios are so much higher. Cellulosic nano‐ and microfibrils can be micrometers long but only tens to hundreds of nanometers in diameter, depending on the degree of fibrillation/aggregation.2 Conversion of microscale pulps into CNF is achieved primarily through mechanical shearing of the pulp fibers.3 A particularly low‐cost method for production of finely dispersed CNF from pulp is through the use of a high‐pressure homogenizer or microfluidizer.4 This equipment allows for repeated passing of aqueous solutions of pulp through microchannels at extremely high pressure (typically 2000 bar). As the microchannels change direction, shear forces fibrillate the fibers along the length of the fibers into smaller and smaller dimensions, thus rendering the material down to the desired degree of fibrillation, depending on the number of passes.2b (2,2,6,6‐Tetramethylpiperidin‐1‐yl)oxyl (TEMPO)‐catalyzed oxidation of cellulose C6 to carboxylic acids, based on the work of Isogai and co‐workers,5 can be applied to reduce the energy demand for production of stable nanofibers, but this also has the added effect of increasing the water absorptivity. The solutions, which are typically thixotropic gels,6 are then stored for further application after fibrillation. However, when these materials are stored for a period of time, they have the tendency to reaggregate and precipitate from solution, depending on the degree of surface charging. Evaporation of water from the mixtures is also highly problematic. As the cellulose concentration increases typically above roughly 3 wt %, cellulose aggregates much more rapidly. Aggregation can be prevented by installing surface charge,7 or by modification as, for example, acyl esters, which would inhibit reaggregation through a hydrogen‐bonding mechanism. However, carrying out this kind of acylation chemistry (and most of organic chemistry in general) in the presence of water is not sustainable, as water reacts with the vast majority of activated acylates. Removal of water from the nanocellulose gels is possible but is energy intensive if one wishes to preserve the nanostructure. It can be achieved on a laboratory scale by, for example, solvent exchange, typically with acetone or dipolar aprotic solvents. For large‐scale work, this is not economical as often four solvent‐exchange passes (including mixing and centrifugation) are required to remove water down to the low levels required for wide application of organic chemistry. Likewise, neither freeze‐drying nor spray‐drying are economical for large‐scale work and moreover do not reach low enough water contents for water‐free surface chemistry. Spray drying is, however, shown to largely preserve the fibrillar structure, whereas supercritical and freeze drying are less effective.8 Thus, there is a major obstacle to cost‐effective dewatering of nanocellulose suspensions, specifically for application in water‐sensitive chemical modification steps. Surfactants have been proposed to stabilize nanocellulose surfaces, to allow for dispersion in organic media.9 An overview of surfactant–nanocellulose interactions was given in a review by Tardy et al.9c
In one early case, surfactant impregnation, pH adjustment, and centrifugation were required to obtain redispersible nanocelluloses.9a However, this demonstrates potential for surface stabilization, preventing aggregation during dewatering. In another case, cetyltrimethylammonium bromide (CTAB) was adsorbed onto highly TEMPO‐oxidized nanocelluloses, allowing for increased fibrillation and also hydrophobicity of the CNF.9b The negatively charged surface carboxylates were thought to bind the CTAB cation to the nanocellulose surface, with increasing charge offering increased effect. Clearly, in this case the binding is dependent on charge and, although a high degree of fibrillation is achieved, the strong cation–anion interaction and surface structuring will prevent application of certain types of chemistry. Thus, a more general procedure is needed for broad application. Therefore, we present herein our results in this area that take advantage of both the cellulose surface‐stabilizing nature of some polar ionic liquids and also the fact that they do not exhibit significant vapor pressures, as molten salts, allowing for selective water removal through control of the temperature and pressure. These combined features of some ionic liquids allows for a process which we call ‘water‐free topochemical modification of nanocellulose’ (“WtF‐Nano”).
To prove the validity and future potential of this approach for process development or as an analytical method, we have attempted to provide a holistic study including: 1) Application of acetylation as a quantitative model reaction to assess the efficiency of surface stabilization after different drying sequences; 2) application of novel analytics (including liquid‐state NMR spectroscopy of modified nanocelluloses) to prove the regioselectivity and rough degree of topochemical (surface) reaction; 3) attempts at understanding the fundamentals of cellulose surface stabilization through Kamlet–Taft parameterization and rheometry studies of CNF dispersed in ionic liquids; 4) discussion of technoeconomics for future development; 5) preliminary assessment of ionic liquid toxicity by using a standard bacterial assay.
Results and Discussion
Ionic liquid choice
Some ionic liquids are known to be excellent solvents for cellulose, allowing for complete dissolution.10 Their abilities to dissolve, in terms of changing acidity and polarity, are reasonably well understood.10b–10e The Kamlet–Taft β parameter or the “net basicity” (β−α) are commonly used as predictors of the cellulose‐dissolving capability of an ionic liquid, with β>0.84 in the imidazolium series generally capable of dissolving cellulose.10d However, nanocellulose stabilization requires the avoidance of dissolution of cellulose, otherwise the high surface area and aspect ratio of the nanocellulose will be destroyed.
To achieve our goals of cellulose surface stabilization while avoiding dissolution (destruction of nanostructure), the stabilization of CNF during water evaporation was first tested by adding two non‐dissolving ionic liquids in roughly equal quantities to an aqueous CNF solution (1.7 wt % dry pulp content). This pulp still contained 24 wt % xylan (surface adsorbed) and was not pre‐ or post‐oxidized to improve dispersibility. The two initial ionic liquids were IoLiLyte 221PG (Figure 1) and [emim][OTf] (Figure 1; emim=1‐ethyl‐3‐methylimidazolium). Acetylation regioselectivity and product morphology were originally tested by using a 2.4 wt % solution of CNF in [emim][OTf]. For comparative acetylation studies the additional imidazolium‐based ionic liquids [emim][OMs], [emim][DCA], and [emim][TCM] were also tested (Figure 1). They were chosen as they all have Kamlet–Taft β values below 0.84 (Figure 2, Table 1). They are also all relatively thermally stable compared to cellulose‐dissolving ionic liquids such as 1,5‐diazabicyclo[4.3.0]non‐5‐enium propionate ([DBNH][CO2Et])10b or 1‐ethyl‐3‐methylimidazolium acetate ([emim][OAc]),11 which have Kamlet–Taft β parameters of 1.11 and 1.09 respectively.10b In addition to the ionic liquids studied, γ‐valerolactone (GVL; Figure 1) was also chosen as an alternative to other common dipolar aprotic solvents. It is preferred over other solvents such as acetone, DMF, or DMSO as it has a higher boiling point and flash point,12a is potentially bio‐based,12b and to date has not shown any significant toxicity or biological solvent effects, which are known for some of the aforementioned alternatives.
Figure 1.
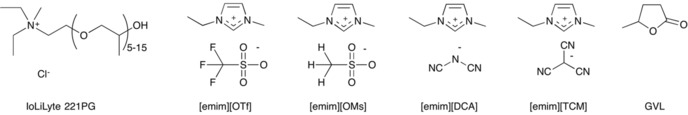
Ionic liquids and molecular solvent used in the study.
Figure 2.
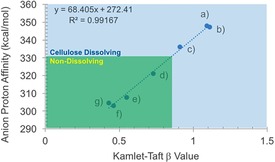
Correlation between the Kamlet–Taft β values and the anion proton affinities11 for a) [emim][OAc], b) [DBNH][CO2Et], c) IoLiLyte 221PG, d) [emim][OMs], e) [emim][DCA], f) [emim][OTf], and g) [emim][TCM].
Table 1.
Kamlet–Taft parameters, anion proton affinities (Anion PA), and pH values of the unpurified solvents used in this study.
| Ionic Liquid | Kamlet–Taft Parameters[a] | Anion PA | pH[c] | ||
|---|---|---|---|---|---|
| π* | α | β | [kcal mol−1][b] | ||
| IoLiLyte 221PG | 0.85 | –[d] | 0.91 | 336.06 | 3.64 |
| [emim][OMs] | 1.09 | –[d] | 0.73 | 321.18 | 3.75 |
| [emim][DCA] | 1.09 | 0.55 | 0.55 | 307.68 | 7.53 |
| [emim][OTf] | 1.00 | –[d] | 0.46 | 303.12 | 5.86 |
| [emim][TCM] | 1.08 | –[d] | 0.43 | 304.68 | 5.95 |
| GVL12c | 0.88 | 0 | 0.56 | –[e] | –[e] |
[a] 20 °C values from the 20–100 °C range (see the Supporting Information, S13). [b] Proton affinities for the unconjugated anions in the gas‐phase at the MP2/6‐311+G(d,p)//MP2/6‐311+G(d,p) level.11 [c] pH was measured for each sample by diluting 1 g of ionic liquid into 9 mL of distilled water. [d] In these cases, we could not find the correct absorption for Reichardt's dye, possibly due to protonation by trace acidic impurities in the ionic liquid. [e] A comparable proton affinity cannot be calculated for GVL, as it is not stable as an anion in the gas phase.
Initial stabilization phenomenon
The dewatering was performed by adding a roughly equal weight of ionic liquid (initially IoLiLyte 221PG and [emim][OTf]) to the total mass of CNF gel. The solutions were thoroughly mixed and then dried using a rotary evaporator down to 15 mbar at 80 °C. The water content of the IoLiLyte 221PG‐CNF solution was 0.6 %, (determined by Karl Fischer analysis) and 2.1 wt % dry pulp content. The water content of the CNF–[emim][OTf] solution was 0.1 wt % (by Karl Fischer analysis) and 1.9 wt % dry pulp content. Both solutions were thick gels. As IoLiLyte 221PG contained the hydrogen‐bond basic chloride ion, it was expected that this would stabilize the nanocellulose best and potentially even dissolve the sample under harsh enough conditions. The triflate anion is very non‐basic and thus [emim][OTf] was not expected to stabilize nanocellulose as well as IoLiLyte 221PG. DMF was added to both samples and they were allowed to stand for one month. By this point neither had precipitated to any significant degree. The IoLiLyte 221PG‐CNF solution was then centrifuged and the solvent was exchanged a further two times by washing with DMF and centrifugation to remove all traces of the ionic liquid. The CNF was then solvent‐cast from DMF to prepare a film for AFM analysis (Figure 3 a). The original CNF solution was also directly solvent exchanged with DMF and cast as a control for AFM analysis (Figure 3 b). Both showed nanofibrillar structures but the IoLiLyte 221PG‐dried sample had a more “swollen” appearance. This may be due to actual swelling of the fiber surface or partial aggregation. However, from Figure 3 it can be clearly seen that IoLiLyte 221PG has the ability to preserve the fibrillar structure of CNF during the drying procedure over the long term. The Kamlet–Taft β value for IoLiLyte 221PG is 0.91, which is above the 0.84 threshold for cellulose dissolution.10d However, this threshold was defined mainly by using imidazolium ionic liquids. The 221PG cation is quite bulky and hydrophobic. This is thought to have a negative effect on cellulose dissolution but clearly such a basic anion will interact strongly with surface hydroxy groups. Water contents for this CNF‐IoLiLyte 221PG mixture could be easily lowered to 0.6 wt %. 0.6 wt % water with 1.9 wt % of cellulose equates to a H2O/AGU (anhydroglucose unit) molar ratio of 2.8:1. This would incur significant reagent losses, which would end up being costly. In any case, IoLiLyte 221PG also has a terminal OH in its structure, having a core choline cation structure with polypropylene oxide oligomers grafted onto the β‐OH (Figure 1). This essentially means that IoLiLyte 221PG would not be recovered, as the terminal OH would also be reactive under cellulose modification conditions. Thus, nonprotic ionic liquids are preferred for most chemistries and our studies continued with the [emim] series.
Figure 3.
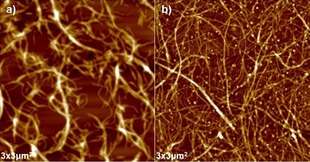
AFM images: a) CNF dried in IoLiLyte 221PG and solvent exchanged (DMF). b) CNF film prepared directly from the water dispersion.
Acetylation regioselectivity
Therefore, we proceeded with acetylation of the [emim][OTf]–CNF solution to demonstrate proof of concept for chemical modification. Acetylation was chosen as a model reaction, as acetylated cellulose and xylan have already been thoroughly characterized by 2 D NMR spectroscopy.13 Previously CNF, isolated as a dry solid by four solvent exchanges with acetone, was acetylated in the ionic liquid 1‐butyl‐3‐methylimidazolium hexafluorophosphate ([bmim][PF6]), by Missoum et al.14a However, the difference between that report and the present study is that their CNF was isolated by solvent exchange and then introduced, as a solid suspension in acetone, into [bmim][PF6] before batch distillation of most the acetone out and introduction of different acylating reagents. In the present method, the CNF suspension is dewatered by rotary evaporation and acetylated directly in [emim][OTf], without the need for preliminary solvent exchanges. We initially acetylated a 2.4 wt % CNF–[emim][OTf] gel by addition of acetic anhydride (6 wt equiv to nanocellulose) with catalytic DMAP (4‐dimethylaminopyridine; see the Supporting Information, S4). The acetylated CNF (AcCNF) film was analyzed directly by SEM (Figure 4). Clearly, a nanofibrillar structure is still observable, despite [emim][OTf] being a “non‐basic ionic liquid,” that is, one with less ability to form hydrogen bonds to polysaccharide surfaces. Some aggregation of fibers still occurs, which is typical for CNF, and some more dense regions are also visible, lacking fibrillar structure, which are due to uneven distribution of hemicellulose. Clearly no swelling or dissolution of the fibrils has occurred. To confirm that acetylation had occurred, 1H and 13C NMR spectroscopy (see the Supporting Information, S6 and S7) were applied after dissolution of the film in the ionic liquid [P4444][OAc] (tetrabutylphosphonium acetate)/[D6]DMSO (1:4 w/v ratio). Acetylation, characterized by the acetate signal at 2 ppm, was not completely visible, owing to overlap with the [P4444][OAc] signals. However, acetylation can most clearly be demonstrated by measuring a diffusion‐ordered spectroscopy (DOSY) gradient array. This was completed by using the bipolar pulse pair stimulated echo (BPPSTE)16a,16b pulse sequence. Stacking and normalization of the array to the largest peak displays the disappearance of the low molecular weight, fast‐diffusing, species (DMSO and [P4444][OAc]) while emphasizing the slow‐diffusing polymeric material therein, with the acetate resonance at 2 ppm (Figure 5). The signal intensity in the acetate chemical‐shift range is clearly exaggerated as the acetate 1H T2 values are much longer than those for the polysaccharide backbone CH groups and, thus, acetate signals are attenuated less, owing to to T2 relaxation effects in the course of the BPPSTE pulse sequence. However, this result could indicate modification of cellulose or merely the surface adsorbed xylan, which may even dissolve and reprecipitate, during the modification procedure. Therefore, we also recorded a multiplicity‐edited 1H–13C heteronuclear single quantum correlation (HSQC)16c–16f NMR spectrum for the AcCNF film (Figure 6). Expansion of the cellulose biopolymer region clearly shows that at least cellulose C6 and C2 and xylan C2 and C3 are acetylated, using previously reported assignments for acetylated cellulose13a,13b,13d and xylan,13c for comparison. Assignments for the acetylated xylan resonances were also performed by taking the AcCNF film and heating in [D6]DMSO at 80 °C for 1 h (see the Supporting Information, S11). This allowed for extraction of the surface‐adsorbed acetylated xylan, thus, allowing for better discrimination between the cellulosic and hemicellulosic resonances.
Figure 4.
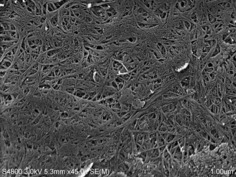
SEM image of AcCNF film, showing fibrillar structure of down to roughly 10–15 nm diameter (ca. 5–10 AGU fibril diameter, based on an 8.2 Å value for the distance between adjacent H‐bonded AGU units in the H‐bonding plane of the cellulose Iβ structure15 and approximately 5 nm for the sputtered layer).
Figure 5.
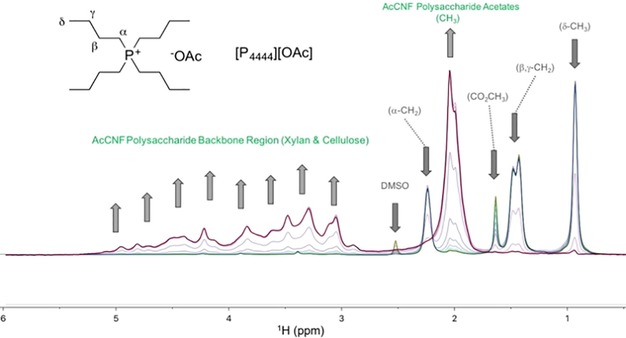
Normalized DOSY gradient array (BPPSTE) showing decrease in the fast‐diffusing ionic liquid ([P4444][OAc]) signals and increase in the slow‐diffusing AcCNF signals, corresponding to acetylated cellulose and hemicellulose.
Figure 6.
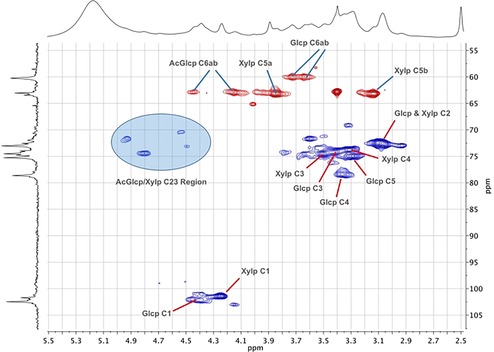
Multiplicity‐edited 1H–13C HSQC (CH2 in red and CH/CH3 in blue) of AcCNF film, derived from birch pulp, in 1:4 [P4444][OAc]/[D6]DMSO with corresponding assignments for acetylated cellulose (Glcp=glucopyranose) and xylan (Xylp=xylopyranose).
Comparison of drying procedures upon acetylation
All data strongly suggest that we are able to dewater CNF gel in [emim][OTf] to allow for high surface‐area modification. However, it is not clear if there is actually any stabilizing effect from the ionic liquid during the dewatering that contributes to a higher degree of acetylation, that is, ionic liquid preventing aggregation before modification. To assess this, we tested a series of drying sequences prior to acetylation as a model reaction, including replacing [emim][OTf] with different ionic liquids and GVL. A new acetylation procedure was used with slightly increased amounts of acetic anhydride (9 equiv per AGU, assuming pure cellulose) to minimize error due to consumption with residual water. A new batch of CNF gel (1.2 wt %) was used. Three different drying sequences were tested: Sequence A) Evaporation of water from the aqueous CNF solution directly; sequence B) the procedure reported by Missoum et al.14a (initial acetone solvent exchange); sequence C) dewatering of the CNF gels with either GVL, [emim][OTf], [emim][OMs], [emim][DCA], or [emim][TCM] (50:50 w/w).
The regenerated films were analyzed by attenuated total reflectance infrared (ATR‐IR) spectroscopy to derive the relative degrees of acetylation (ratio of the carbonyl peak heights: ≈1745 cm−1 vs. pyranose backbone: ≈1035 cm−1 stretches, after baseline correction) These were then correlated against a quantitative 13C NMR spectrum (acetate ester carbonyl: δ=168–169 ppm vs. total C1: δ=96–106 ppm) of the original AcCNF film prepared from [emim][OAc]. The result is a comparative measure of the modification of surface hydroxy groups by acetate groups for the different drying and acetylation sequences (Figure 7). The degree of acetylation in this case was assessed by the percentage of total hydroxy groups in the bulk CNF sample that were acetylated. From comparison with the vacuum dried CNF reference, it is clear that all sequences where cosolvents are used for solvent exchange (by evaporation) offer high degrees of acetylation at almost the same value. Owing to the low concentrations of CNF used (not significantly limited by mass transfer), this seems to correspond to a total surface coverage value of 14 % of the bulk hydroxy groups. It is not unexpected that vacuum drying the CNF would lead to “hornification” of the sample. Introduction of the vacuum dried CNF into [emim][OTf] for acetylation did not yield any significant increase in acetylation against the unacetylated material. Thus, solvent stabilization is definitely required during dewatering.
Figure 7.
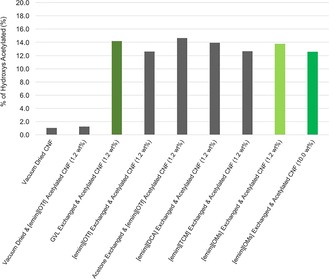
Degrees of surface acetylation, as determined by a combination of quantitative 13C NMR and ATR‐IR spectroscopy for the different drying and acetylation sequences, as represented by the percentage of total hydroxy groups in the bulk CNF samples that are acetylated.
Following sequence B, simulating the procedure reported by Missoum et al.,14a the acetylation degrees were the highest by a small margin. However, not all acetone was removed as the viscosity of the mixture was slightly lower than that of the pure CNF–[emim][OTf] solution. It may be that the presence of acetone in ionic liquid has a synergistic stabilizing effect. Viscosity or basicity of the pure ionic liquids, at these concentrations, do not seem to have any pronounced effect as there is no visible correlation between the acetylation degree and spread of β values or changes in ionic liquid viscosity (Figure 8). Surprisingly, dewatering in GVL allowed for very efficient acetylation. It was possible to remove water down to 0.4 % by rotary evaporation (80 °C down to 15 mbar). Vacuum drying allowed for a further reduction to 0.11 %. Whereas most dialkylimidazolium ionic liquids have the advantage that they have very low vapor pressures, even approaching 200 °C, clearly water can be almost completely separated from GVL during batch distillation (with low theoretical plate number). Other common dipolar aprotic solvents, such as DMSO or DMF, would certainly require more expensive distillation designs to reach such low water contents.
Figure 8.
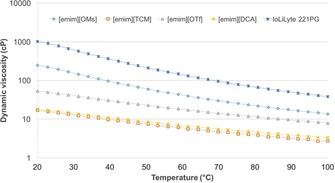
Dynamic viscosities as a function of temperature for all ionic liquids.
Dissolution versus surface modification
It could be argued that the different ionic liquids still have power to dissolve the cellulose or that acetylation occurs on the interior of the fibrils. Therefore, WAXS (wide‐angle X‐ray scattering) was performed on all regenerated samples (Figure 9). This shows that all samples retained peaks corresponding to the main Miller indices for cellulose Iβ.15 This is also consistent with the fact that we see that a maximum of 14 % of bulk hydroxy groups are acetylated. Therefore, we can conclusively say that only cellulose surface stabilization is occurring with no dissolution. Extraction of hemicelluloses during the procedure is also a possibility, as hemicelluloses, in particular acetylated hemicelluloses, are soluble in a wide range of solvents. Therefore, we subjected the acetylated samples to heating at 120 °C for 5 min in [D6]DMSO (20 mg, 600 μL) as this allows for selective dissolution of surface‐adsorbed hemicellulose over cellulose. 1H NMR spectroscopy showed in all cases the presence of hemicelluloses (see the Supporting Information, S15). Only the material prepared from [emim][DCA] showed reduced amounts of hemicellulose, but this procedure is not robust enough to draw any solid conclusions about if, or where, losses occur.
Figure 9.
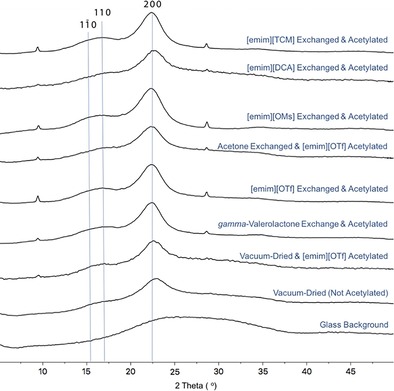
WAXS diffractograms for the AcCNF recovered from the different dewatering and acetylation sequences.
Degradation during drying and acetylation
Table 1 shows that the pH (aqueous, 10 wt % in deionized water) of some of the ionic liquids is rather low. There is potential for degradation, although (aqueous) pH is not an entirely suitable acidity scale to assess the acidity and associated reactivity of pure ionic liquids. Nevertheless, degradation and, in particular, changes in molecular weight, of the cellulosic fraction, could have an effect on potential applications. As such, we measured the molecular weight distribution of the samples and references, acetylated at 1.2 wt %, to assess depolymerization. This was achieved by using gel‐permeation chromatography with the cellulose‐dissolving lithium chloride/N,N‐dimethylacetamide (LiCl/DMA) mobile phase (Figure 10). Figure 10 a shows the drying sequences and ionic liquids that did not undergo any apparent degradation. Figure 10 b shows those that did undergo degradation and possibly loss of a portion of the hemicellulose fraction. The data are tabulated in the Supporting Information. What is most interesting is that the samples dried and/or acetylated in both [emim][OTf] and [emim][OMs] underwent little degradation of the cellulose fraction (M w=221 500 g mol−1 for the vacuum‐dried reference) and very little, if any, loss of the hemicellulose fraction (M w=30 600 g mol−1 for the vacuum‐dried reference). The other samples, including those involving molecular solvents (acetone and GVL) showed clear losses of molecular weight for the cellulosic fraction and also removal of varying quantities of hemicellulose. Loss of the hemicellulose fraction may be a consequence of not having an optimized workup, as acetylated hemicelluloses are known to be water soluble to some extent. Loss of cellulose molecular weight, with corresponding increase in polydispersity, is clearly due to breakage of the glycosidic linkages, at least to a small degree. There was no clear correlation between the (aqueous) pH and the degradation. [emim][OMs], which demonstrated the lowest (aqueous) pH, did not demonstrate any degradation after drying and acetylation. One fact to account for this is that catalytic DMAP was used, which should scavenge any highly acidic species. Considering this, depolymerization of the cellulosic fraction may be related more to degradation of the solvent itself and not to the presence of acidic impurities. For now, the highly thermally and chemically stable [emim][OTf] and [emim][OMs]11 offer suitable solutions, which avoid both cellulose and hemicellulose degradation in the acetylated nanocelluloses, when drying and acetylation is applied.
Figure 10.
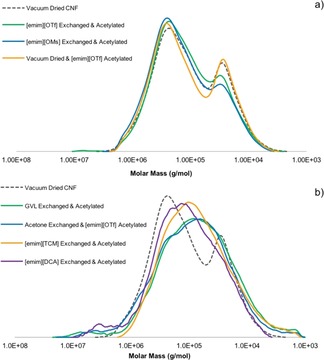
Molecular weight distribution of samples after drying and acetylation that show a) little degradation and b) clear degradation.
Optimum solvent for surface stabilization
Although the concentrations of CNF are too low to really observe differences in the stabilizing effect of different ionic liquids, based on the acetylation degrees, a look at the viscoelastic properties of the ionic liquid–CNF gels should give better indications as to the abilities of the ionic liquids to stabilize the surface. We chose to look at the CNF solutions in [emim][OTf] vs. [emim][OMs], as the mesylate and triflate anions are analogous, with fluorines replacing hydrogens in the triflate (Figure 1). The main difference between these anions is that the electronegativity of the fluorines results in a reduction in electron density on the sulfonate oxygens and hence a reduction in hydrogen‐bond basicity of the ionic liquid.
Complex viscosities and dynamic moduli were recorded for 1.2 wt % CNF ionic liquid solutions (Figure 11). Both suspensions show a strong gel character indicated by the power‐law behavior of the complex viscosity over the entire angular frequency range.6c The dynamic moduli are almost frequency independent (G′>G′′), with the [emim][OTf] suspension showing a stronger gel network than [emim][OMs]. The viscoelastic properties are thus not dependent on the viscosity of the neat ionic liquids ([emim][OTf] would have the lowest viscosity) but are connected to the ability of the ionic liquids to stabilize the suspension. This is in line with the optical appearance of the gels. The aggregate size in the [emim][OMs] gel is smaller, reflected by its more transparent character (Figure 12 a–d). The increased grain size in the case of [emim][OTf] is also clearly visible through the birefringence pattern in the polarized light microscopy images (Figure 12 e and f). Both ionic‐liquid gels are more viscous than the initial aqueous CNF suspension. This can be attributed to the substantially lower viscosity of water superposing the contribution of the increased aggregate size.
Figure 11.
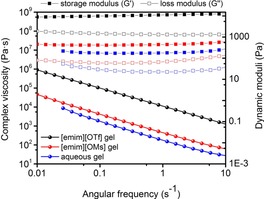
Complex viscosities and dynamic moduli of 1.2 wt % CNF in [emim][OTf], [emim][OMs], and water.
Figure 12.

Visual appearance and optical microscopy images (with cross‐polarizing filters) for the CNF–[emim][OTf] (a,c,e) and CNF–[emim][OMs] solutions (b,d,f).
These results strongly suggest that the increased hydrogen‐bonding capability of the mesylate has a positive effect on the surface stabilization of nanocellulose. The CNF–[emim][OTf] must take more gel‐like characteristics as the surface hydroxy groups are stabilized to a lesser degree, allowing for a stronger interaction between fibrils. In the mesylate case, the better stabilization of hydroxy groups reduces the interfibrillar interaction, resulting in more liquid‐like properties.
Technoeconomics and future work
If we look at previous literature reports for economical and “green” methods of acylation of nanocelluloses, there is only one competing and seemingly efficient process for esterification of cellulose nanocrystals (CNCs). This is the “SolReact” process, reported by Bras and co‐workers.14b However, it seems to be limited to Fischer esterification of cellulose nanocrystals by organic acids with certain properties (water miscible, specific melting and boiling range, and not too acidic). To date, this procedure has not been applied to CNF, possibly due to the potential for acid degradation of its accessible amorphous content, although it is not specifically stated in this study that this would be the case. All other reported studies, to our knowledge, have used volatile, flammable, or toxic solvents, such as DMF, DMA and acetone, for the solvent‐exchange steps. Herein, we demonstrate esterification on CNF, not CNCs, with acetic anhydride, under water‐free conditions. A process scheme is proposed whereby the ionic liquid can be easily recycled (Figure 13). This makes the assumption that 100 % atom‐efficient and quantitative reactions are used to avoid accumulation of byproducts in the ionic liquid. However, for simpler reactions where byproducts are volatile, they can potentially be recovered by fractional distillation from the ionic liquid after the separation step. Catalysts may be cycled during the process, providing they are stable under aqueous conditions. It is possible that the byproducts may also be cycled in the process and regenerated to the reactive reagent before or after the dewatering step. It still requires a little ingenuity and engineering knowhow to design the sustainable process. However, being water‐free, this drying procedure may allow access to a much wider range of chemistries other than acylation. As a demonstration of the potential for solvent recycling, high purity ionic liquids were recovered after regeneration from AcCNF by solvent evaporation in a rotary evaporator (80 °C down to 15 mbar) and characterized by1H NMR spectroscopy (see the Supporting Information, S16). No degradation of the ionic liquids was apparent but more detailed studies after continued cycling are required.
Figure 13.
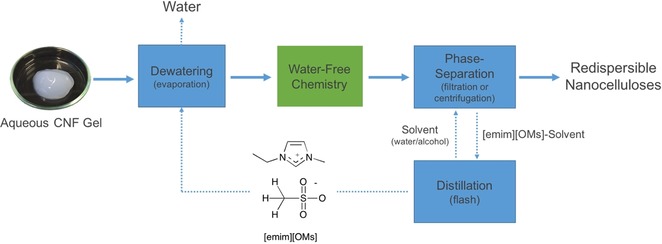
Proposed process scheme for dewatering and water‐free chemical modification in [emim][OMs].
With regard to future process technoeconomics, there are two major drawbacks of the procedure presented thus far. The first is that we have been working with very low cellulose loadings for the initial proof of concept (1.2–2.4 wt %). However, it has been demonstrated that there is an excellent stabilizing effect from [emim][OMs] as a basic, but non‐dissolving, ionic liquid. To test this further, we prepared a 10.0 wt % CNF solution in [emim][OMs] and acetylated this time by using only 3 equivalents of acetic anhydride and no catalytic DMAP (see Experimental Section, procedure 3). After ATR‐IR spectroscopy, the degree of acetylation was almost equivalent to the alternative drying and acetylation procedures, all using 1.2 wt % CNF (Figure 7). This is a clear indication of the potential for increasing biomass loading above technical concentrations. Full optimization of this will be completed in future studies.
The other major drawback is in the removal of water or other regeneration solvents. A very crude estimate can be calculated by simply looking at the amount of water to be evaporated (kg), multiplied by the heat of evaporation (J kg−1) and cost of energy (€ J−1). The latent heat of evaporation of water is about 2300 kJ kg−1 and the electricity price for industry about 0.1 € kWh−1 (3600 kJ). This would lead to about 0.07 € kg−1 of water evaporated. On the large scale, multi‐effect evaporators would save energy and energy derived from steam is cheaper than purchasing electricity but other investment and consumables equally add to the cost. Based on removal of water from a 2 wt % solution, this adds 3.5 € kg−1 of dry weight CNF per drying cycle. Two cycles (initial drying step and CNF regeneration under more concentrated conditions) may push the cost up to 5 € kg−1, not including additional purification steps, capital expenditure, or running costs. One clear method of minimizing these costs would be to carry out the fibrillation of the pulp directly in the ionic liquid. To our knowledge, this has not been reported for ionic liquids but the viscoelastic properties of CNF in [emim][OMs] (Figure 11) are very promising in this regard.
An additional cost of 5 € kg−1, added to rough costs for Kraft pulp of 500 € tonne−1, would give a rough value of 5500 € tonne−1 for modified nanocellulose. Energy savings, as mentioned above, could significantly reduce this cost.
Environmental impact
Finally, to really consider the use of ionic liquids on an industrial scale, ecotoxicity of such structures must be assessed. As such, an initial assessment of the toxicity of the ionic liquids presented herein was carried out against the Vibrio fischeri marine bacteria using a Microtox apparatus. EC50 values were calculated at 5 and 15 min (Figure 14). The results show that all the ionic liquids in the study come above the “harmless” bracket (>1000 mg L−1) and [emim][OMs] is the least toxic toward V. fischeri. GVL was not tested in this series but its use as a food flavoring and in cosmetics or perfumes is well known. The oral toxicity (LD50) against rats (8800 mg kg−1) and rabbits (2480 mg kg−1) was reported by Deichmann.17a An overview of its biological effects from the period of 1962–1980 is given in a monograph report from 1982.17b These results are by far inconclusive regarding ecotoxicity but offer good indications of low toxicity for continuation. By contrast, dipolar aprotic solvents used for conventional (centrifugation‐based) solvent exchange, for removal of water from nanocelluloses, demonstrate some concerning toxic effects or other hazards; Carboxamides (DMF, DMA, and N‐methyl‐2‐pyrrolidone) are suspected teratogens, or reproductive toxins. DMSO is a biological solvent, allowing for very rapid transport of solutes across cell membranes. Acetone is relatively low in toxicity, but is highly flammable with a low flashpoint. By contrast, GVL is of low toxicity and is flammable but with a high flashpoint. It also allows for selective water evaporation, which acetone does not. This is entirely more suitable for solvent exchange. Figure 10 indicates that some degradation and hemicellulose loss may be occurring during the drying and acetylation sequences when using GVL, but this requires further study, as GVL would provide a molecular (distillable and potentially low‐cost) alternative to ionic liquids.
Figure 14.
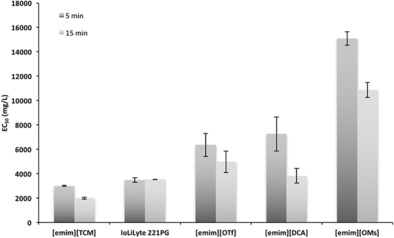
EC50 values for the ionic liquids in this study against a Vibrio fischeri bacterial line.
Conclusions
It has been demonstrated that it is possible to surface stabilize Kraft pulp CNF (24 % surface adsorbed xylan) during water removal, by addition of a roughly similar quantity of ionic liquid to the original bulk of water. During water removal, the surface of the nanocellulose is stabilized, allowing for highly efficient water‐free surface modification, through acetylation as a model reaction. We call this process “WtF‐Nano”. The surface stabilization seems to be dependent on choosing an ionic liquid with a basic anion but which is not basic enough to facilitate the significant swelling or dissolution of cellulose. Naturally, dissolution of the nanocellulose would destroy the fibrillar high aspect‐ratio structure, thus, rendering the material into a dysfunctional form. GVL also seems to allow for almost complete water removal and CNF surface stabilization. GVL is a potential bio‐based solvent and suitable replacement for the toxic DMF and DMA and the flammable acetone. It also allows for complete separation (by distillation) of water from CNF gels, which has not previously been demonstrated for the lower boiling dipolar aprotic solvents. However, ionic liquids have the advantage that they have much lower vapor pressures, allowing for essentially flashing of solvent from the mixture. Although the ionic liquids in the current study are definitely higher in cost than GVL, the ionic liquid should be recyclable at close to 100 %, depending on chemistry performed. Flashing of solvent, as opposed to fractional distillation, also would require lower process and capital costs. [emim][OTf] and [emim][OMs], when applied as media for water evaporation and acetylation, also demonstrated no reduction in cellulose molecular weight or removal of the hemicellulose fraction. Due to the seemingly high degree of surface modification, WtF‐Nano may be ideal to form the basis of analytical procedures, although dewatering and acetylation of dissolving pulp CNF and CNC (without significant hemicellulose fractions) should be optimized to avoid reactive dissolution of cellulose. In this regard, it should be stated that it is unknown if hemicellulose has some additional stabilizing effect, facilitating easier processing of Kraft pulp CNF. This will be the subject of future work. It has also been demonstrated that this procedure can be applied to technical concentrations (10 wt % CNF) and using less acetic anhydride, with no significant loss in degree of acetylation. This manuscript is not intended to demonstrate the production of acetylated CNF (AcCNF) in a materials science concept, but rather acetylation is used to illustrate the potential for chemical modification. Therefore, different options for sustainable and water‐free chemistry must now also be explored, including the material properties of novel products.
Experimental Section
Materials
CNF solutions (1.2 wt % and 1.7 wt % dry pulp contents) were produced by six passes in a microfludizer, from birch Kraft pulp (UPM Kymmene, Pietarsaari, Finland). This pulp still contained 24 wt % xylan (surface adsorbed) and was not pre‐ or post‐oxidized to improve dispersibility. Ionic liquids IoLiLyte 221PG, 1‐ethyl‐3‐methylimidazolium triflate ([emim][OTf]), 1‐ethyl‐3‐methylimidazolium mesylate ([emim][OMs]), 1‐ethyl‐3‐methylimidazolium dicyanamide ([emim][DCA]), and 1‐ethyl‐3‐methylimidazolium tricyanomethanide ([emim][TCM]), were purchased from Iolitec GmbH (Heilbronn, Germany) and used without further purification. Tetrabutylphosphonium chloride ([P4444]Cl) (80 wt % aq.) was purchased from TCI Europe (product #T1649) for conversion into tetrabutylphosphonium acetate ([P4444][OAc]), for NMR studies. The sources of other chemicals are given in the Supporting Information (S1).
Preparation of [P4444][OAc] for NMR studies
[P4444][OAc] was prepared by anion metathesis from [P4444]Cl and potassium acetate in isopropanol (see the Supporting Information, S2). Purity was assessed by NMR spectroscopy and was found to be comparable with a previously reported method using silver acetate.10e
Solvent exchange sequences
Water removal was carried out by rotary evaporation at 80 °C at 15 mbar for 30 min. The evaporation was carried out without added ionic liquid and with added ionic liquid or GVL. Typically, an equal volume of ionic liquid or GVL was added to the mixtures. The solutions were allowed to mix for 20 min before the evaporation step. Finally, the samples were put under vacuum for 30 min and the water contents were measured by Karl Fischer titration, before and after the final vacuum step (see the Supporting Information, S3). At this point, all water contents were below 0.3 wt %. This final step was necessary to avoid consuming the acetylating reagent, to ensure a comparative procedure between different drying sequences. Three different drying sequences were tested:
Sequence A) Evaporation of water from the CNF solution directly, without any ionic liquid. This material was introduced to [emim][OTf] for acetylation.
Sequence B) A procedure previously reported by Missoum et al.14a was emulated, where the CNF was acetone exchanged and the acetone suspension introduced to [emim][OTf] before mild rotary evaporator evaporation and acetylation.
Sequence C) Mixing of the CNF gels with either GVL, [emim][OTf], [emim][OMs], [emim][DCA] or [emim][TCM] (50:50 w/w), rotary evaporation of the water, vacuum drying and acetylation.
CNF acetylation procedures
Three procedures were used for acetylation of the CNF solutions. An initial procedure was tested with [emim][OTf] to determine if it was possible to stabilize nanocellulose enough during water removal to allow for water‐free surface modification:
Procedure 1) In this procedure, we acetylated the CNF–[emim][OTf] gel (2.4 wt % CNF in [emim][OTf] by addition of acetic anhydride (6 wt equiv to dry matter CNF) and catalytic DMAP (see the Supporting Information, S4). The mixture was heated at 80 °C overnight and the material regenerated by centrifugation with methanol to give a semi‐transparent acetylated cellulose nanofbrillar (AcCNF) thin film.
Procedure 2) A second procedure was practically the same as procedure 1, but in this case 1.2 wt % CNF solutions were prepared and a slightly higher amount of acetic anhydride was used (9 mol equiv to AGU, assuming 100 % cellulose). Regeneration was completed by extended shaking and centrifugation both water and methanol to ensure complete removal of ionic liquid. All samples were dried by rotary evaporation, ready for further analysis (see the Supporting Information, S4).
Procedure 3) A third procedure was practically the same as procedure 1, but in this case a 10 wt % CNF solution in [emim][OMs] was prepared and a lower amount of acetic anhydride was used (3 mol equiv to AGU, assuming 100 % cellulose), without catalytic DMAP. This was to simulate a more technical and green solution for large‐scale synthesis. Regeneration was completed by extended shaking and centrifugation in both water and ethanol (not methanol as before). The sample was dried under vacuum, ready for further analysis (see the Supporting Information, S4).
NMR analysis of acetylated CNF
The initial sample, prepared from [emim][OTf] according to sequence C/procedure 1, was dissolved in [P4444][OAc]/[D6]DMSO (ca. 6 wt % CNF in a 1:4 w/v solvent ratio)13a,13b at 100 °C for 30 min and was introduced to a 5 mm NMR tube for analysis. The NMR measurements included 1H, 13C, and quantitative 13C NMR spectroscopy, as well as two‐dimensional heteronuclear single quantum correlation (HSQC)16c–16f and diffusion‐ordered spectroscopy (DOSY), utilizing the bipolar pulse pair stimulated echo (BPPSTE)16a,16b pulse sequence (see the Supporting Information, S6–S11). Quantitative 13C NMR spectroscopy was used to determine the degree of acetylation, by integration of the carbonyl peak (δ=168–169 ppm) against the polysaccharide C1 region (96–106 ppm). To obtain an adequate quality result, the quantitative 13C experiment took three days of measurement time using a 5 mm broadband probe‐head, so this procedure was not performed for every acetylated sample. Instead, this was used as an absolute value for the degree of acetylation and the remaining samples were correlated against the ratio of carbonyl to polysaccharide backbone IR absorption, by using ATR‐IR spectrscopy (see the Supporting Information, S12). The presence of acetylated hemicelluloses on the AcCNF samples, prepared from different ionic liquids, was confirmed by either 1H or HSQC NMR analysis after extraction of the samples with [D6]DMSO (see the Supporting Information, S11).
Microscopy
Atomic force microscopy (AFM) was performed on CNF regenerated from IoLiLyte 221PG and the aqueous solution, as reference. An approximately 2 wt % IoLiLyte 221PG CNF solution was prepared. Four volumes of DMF were added and the mixture was solvent exchanged three times, with centrifugation. The recovered CNF was suspended in DMF and then solvent cast onto a silicon cantilever. Solvent was removed by drying the sample overnight in a vacuum‐oven. The dried sample was analyzed and compared against a CNF film prepared directly from water dispersion. Topographical changes on the surfaces of substrates were then characterized using a Nanoscope V Multimode scanning probe microscope from Digital Instruments Inc. (Santa Barbara, CA, USA).
Scanning electron microscopy (SEM) was performed on the acetylated CNF (AcCNF) film, obtained by procedure 1. The original film was prepared by evaporation from methanol in a rotary evaporator. The SEM images were acquired with a Hitachi S‐4800 field emission scanning electron microscope. The film was coated with 5 nm of Au–Pd alloy prior to imaging.
Optical microscopy, with cross‐polarizing filters, was performed on 1.2 wt % CNF in [emim][OTf] and [emim][OMs]. A few milligrams of the solutions were placed on a glass slide and covered with a glass cover‐slip. The cross‐polarizing filters were adjusted to give good contrast between the different orientations of the gel particle.
WAXS analysis
Dried regenerated nanocelluloses were swollen by addition of a few mass equivalents of water and/or methanol. The gels were then thinly spread onto glass microscope slides and allowed to dry, before being placed on the reflection stage. In some cases, where we already had an irregular film, the dry films were gently pressed in a hydraulic press to form a thin film, which could then be placed on a glass microscope slide for calibration on the reflection stage. These were measured in reflection mode using Bragg–Brentano geometry, (λ=1.54 Å, 45 kV and 40 mA) in a 2θ range between 5°and 50°.
Kamlet–Taft parameterization
Kamlet–Taft parameters were determined as previously reported by Parviainen et al.10b In short, each dye (N,N‐diethyl‐4‐nitroaniline, 4‐nitroaniline, and Reichardt's dye) was mixed with 1 mL of ionic liquid. Instead of using 200 μL of Reichardt's dye in acetone stock solution, as described earlier, in the present study 1000 μL was used to increase the intensity of the absorbance maximum (385 nm for [emim][DCA]). The absorption spectrum was recorded from 800 to 200 nm at 20, 40, 60, 80, and 100 °C (see the Supporting Information, S13). The wavelengths at the absorbance maxima were used to calculate the Kamlet–Taft parameters, as described previously.10b
Toxicity analysis
The acute aquatic toxicities of the ionic liquids were assessed by determining their EC50 values, by using Vibrio fischeri bacteria in a Microtox luminometer/thermostate apparatus (Modern Water, USA). The recorded response is the decay of bioluminescence produced by the bacteria. The bacteria were exposed to four differing toxicant concentrations in 2 % (w/v) NaCl solution. Based on the concentration‐dependent decay of the bioluminescence, the EC50 values were determined at set time intervals of 5 and 15 minutes. Two independent measurements were performed for each ionic liquid as duplicates.
Rheology
Shear rheology of the neat ionic liquids and dewatered CNF gels were measured on an Anton Paar MCR 300 rheometer with a plate and plate geometry (25 mm plate diameter, 1 mm gap size). The ionic liquids were subjected to a temperature sweep (20–100 °C) at a shear rate of 50 s−1. The viscoelastic properties of the gels were determined in oscillatory mode by a dynamic frequency sweep at 25 °C, over an angular frequency range of 100–0.01 s−1. More experimental details are given in the Supporting Information, S13.
Gel‐permeation chromatography
Molar mass determination was performed by using an Agilent Infinity 1260 LC‐system, with guard column and three PLgel MIXED 10 μm columns, connected in series. The mobile phase was 0.5 % LiCl/DMA and calibration correction was performed using pullulan standards. Correction factors, as reported by Berggren et al.,18a were applied. Sample preparation was performed as reported by Timpa,18b with the modifications reported by Kakko et al.18c More experimental details and the tabulated data are given in the Supporting Information, S17.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We would like to acknowledge our funding partner, CLIC Innovation, for support on the general topic of biomass processing with ionic liquids. We would like to acknowledge the Academy of Finland for funding under the projects “WTF‐Click‐Nano” (311255) and “ILBioMem” (266342). We would also like to acknowledge Prof. Ville Alopaeus (Aalto University) for discussions regarding the very rough calculation of process costs for evaporation of water.
T. Laaksonen, J. K. J. Helminen, L. Lemetti, J. Långbacka, D. Rico del Cerro, M. Hummel, I. Filpponen, A. H. Rantamäki, T. Kakko, M. L. Kemell, S. K. Wiedmer, S. Heikkinen, I. Kilpeläinen, A. W. T. King, ChemSusChem 2017, 10, 4879.
Contributor Information
Dr. Ilari Filpponen, Email: ilari.filpponen@auburn.edu.
Dr. Alistair W. T. King, Email: alistair.king@helsinki.fi.
References
- 1.
- 1a. Klemm D., Kramer F., Moritz S., Lindström T., Ankerfors M., Gray D., Dorris A., Angew. Chem. Int. Ed. 2011, 50, 5438–5466; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 5550–5580; [Google Scholar]
- 1b. Moon R. J., Martini A., Nairn J., Simonsen J., Youngblood J., Chem. Soc. Rev. 2011, 40, 3941–3994. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Abdul Khalil A. P. S., Bhat A. H., Ireana Yusra A. F., Carbohydr. Polym. 2012, 87, 963–979; [Google Scholar]
- 2b. Taheri H., Samyn P., Cellulose 2016, 23, 1221–1238. [Google Scholar]
- 3. Taniguchi T., J. Soc. Mater. Sci. Jpn. 1996, 45, 472–473. [Google Scholar]
- 4.
- 4a. Pääkkö M., Ankerfors M., Kosonen H., Nykänen A., Ahola S., Österberg M., Ruokolainen J., Laine J., Larsson P. T., Ikkala O., Lindström T., Biomacromolecules 2007, 8, 1934–1941; [DOI] [PubMed] [Google Scholar]
- 4b. Turbak A. F., Snyder F. W., Sandberg K. R., J. Appl. Polym. Sci. Appl. Polym. Symp. 1983, 37, 815–827; [Google Scholar]
- 4c. Herrick F. W., Casebier R. L., Hamilton J. K., Sandberg K. R., J. Appl. Polym. Sci. Appl. Polym. Symp. 1983, 37, 797–813. [Google Scholar]
- 5.
- 5a. Isogai A., Saito T., Fukuzumi H., Nanoscale 2011, 3, 71–85; [DOI] [PubMed] [Google Scholar]
- 5b. Hirota M., Furihata K., Saito T., Kawada T., Isogai A., Angew. Chem. Int. Ed. 2010, 49, 7670–7672; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 7836–7838. [Google Scholar]
- 6.
- 6a. Lasseuguette E., Roux D., Nishiyama Y., Cellulose 2008, 15, 425–443; [Google Scholar]
- 6b. Iotti M., Gregersen Ø., Moe S., Lenes M., J. Polym. Environ. 2011, 19, 137–145; [Google Scholar]
- 6c. Benhamou K., Dufresne A., Magninc A., Mortha G., Kaddami H., Carbohydr. Polym. 2014, 99, 74–83; [DOI] [PubMed] [Google Scholar]
- 6d. Saito T., Nishiyama Y., Putaux J., Vignon M., Isogai A., Biomacromolecules 2006, 7, 1687–1691. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Salas C., Nypelö T., Rodríguez-Abreu C., Carrillo C., Rojas O. J., Current Opinion Colloid Interface Sci. 2014, 19, 383–396; [Google Scholar]
- 7b. Pei A., Butchosa N., Berglund L. A., Zhou Q., Soft Matter 2013, 9, 2047–2055. [Google Scholar]
- 8. Peng Y., Gardner D. J., Han Y., Cellulose 2012, 19, 91–102. [Google Scholar]
- 9.
- 9a. Heux L., Chauve G., Bonini C., Langmuir 2000, 16, 8210–8212; [Google Scholar]
- 9b. Syverud K., Xhanari K., Chinga-Carrasco G., Yu Y., Stenius P., J. Nanopart. Res. 2011, 13, 773–782; [Google Scholar]
- 9c. Tardy B. L., Yokota S., Ago M., Xiang W., Kondo T., Bordes R., Rojas O. J., Curr. Opin. Colloid Interface Sci. 2017, 29, 57–67. [Google Scholar]
- 10.
- 10a. Swatloski R. P., Spear S. K., Holbrey J. D., Rogers R. D., J. Am. Chem. Soc. 2002, 124, 4974–4975; [DOI] [PubMed] [Google Scholar]
- 10b. Parviainen A., King A. W. T., Mutikainen I., Hummel M., Selg C., Hauru L. K. J., Sixta H., Kilpeläinen I., ChemSusChem 2013, 6, 2161–2169; [DOI] [PubMed] [Google Scholar]
- 10c. King A. W. T., Asikkala J., Mutikainen I., Järvi P., Kilpeläinen I., Angew. Chem. Int. Ed. 2011, 50, 6301–6305; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6425–6429; [Google Scholar]
- 10d. Hauru L. K. J., Hummel M., King A. W. T., Kilpeläinen I., Sixta H., Biomacromolecules 2012, 13, 2896–2905; [DOI] [PubMed] [Google Scholar]
- 10e. Holding A. J., Heikkilä M., Kilpeläinen I., King A. W. T., ChemSusChem 2014, 7, 1422–1434. [DOI] [PubMed] [Google Scholar]
- 11. King A. W. T., Parviainen A., Karhunen P., Matikainen J., Hauru L. K. J., Sixta H., Kilpeläinen I., RSC Adv. 2012, 2, 8020–8025. [Google Scholar]
- 12.
- 12a. Horváth I. T., Mehdi H., Fábos V., Boda L., Mika L. T., Green Chem. 2008, 10, 238–242; [Google Scholar]
- 12b. Luterbacher J. S., Rand J. M., Alonso D. M., Han J., Youngquist J. T., Maravelias C. T., Pfleger B. F., Dumesic J. A., Science 2014, 343, 277–280; [DOI] [PubMed] [Google Scholar]
- 12c. Duereh A., Sato Y., Smith R. L., Inomata H., J. Phys. Chem. B 2016, 120, 4467–4481. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Holding A. J., Mäkelä V., Tolonen L., Sixta H., Kilpeläinen I., King A. W. T., ChemSusChem 2016, 9, 880–892; [DOI] [PubMed] [Google Scholar]
- 13b. Deb S., Labafzadeh S. R., Liimatainen U., Parviainen A., Hauru L. K. J., Azhar S., Lawoko M., Kulomaa T., Kakko T., Fiskari J., Borrega M., Sixta H., Kilpeläinen I., King A. W. T., Green Chem. 2016, 18, 3286–3294; [Google Scholar]
- 13c. Teleman A., Tenkanen M., Jacobs A., Dahlman O., Carbohydr. Res. 2002, 337, 373–377; [DOI] [PubMed] [Google Scholar]
- 13d. Kono H., Hashimoto H., Shimizu Y., Carbohydr. Polym. 2015, 118, 91–100. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Missoum K., Belgacem M. N., Barnes J.-P., Brochier-Salon M.-C., Bras J., Soft Matter 2012, 8, 8338–8349; [Google Scholar]
- 14b. Espino-Pérez E., Domenek S., Belgacem N., Sillard C., Bras J., Biomacromolecules 2014, 15, 4551–4560. [DOI] [PubMed] [Google Scholar]
- 15. Nishiyama Y., Langan P., Chanzy H., J. Am. Chem. Soc. 2002, 124, 9074–9082. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Wu D. H., Chen A. D., Johnson C. S., J. Magn. Reson. Ser. A 1995, 115, 260–264; [Google Scholar]
- 16b. Jerschow A., Muller N., J. Magn. Reson. 1997, 125, 372–375; [Google Scholar]
- 16c. Kay L. E., Bax A., J. Magn. Reson. 1989, 84, 598–603; [Google Scholar]
- 16d. Davis D. G., J. Magn. Reson. 1991, 91, 665–672; [Google Scholar]
- 16e. Schmieder P., Domke T., Norris D. G., Kurz M., Kessler H., Leibfritz D., J. Magn. Reson. 1991, 93, 430–435; [Google Scholar]
- 16f. Willker W., Leibfritz D., Kerssebaum R., Bermel W., Magn. Reson. Chem. 1993, 31, 287–292. [Google Scholar]
- 17.
- 17a. Deichmann W., Hirose R., Witherup S., J. Ind. Hyg. Tox. 1945, 27, 263–268; [PubMed] [Google Scholar]
- 17b. Fragrance raw materials monographs, Food Chem. Toxicol 1982, 20, 637–852. [DOI] [PubMed]
- 18.
- 18a. Berggren R., Berthold F., Sjöholm E., Lindström M., J. Appl. Polym. Sci. 2003, 88, 1170–1179; [Google Scholar]
- 18b. Timpa J. D., J. Agric. Food Chem. 1991, 39, 270–275; [Google Scholar]
- 18c. Kakko T., King A. W. T., Kilpeläinen I., Cellulose 2017, 24, 5341–5354. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary