


Abstract
Antibiotics methymycin (MTM) and pikromycin (PKM), co-produced by Streptomyces venezuelae, represent minimalist macrolide protein synthesis inhibitors. Unlike other macrolides, which carry several side chains, a single desosamine sugar is attached to the macrolactone ring of MTM and PKM. In addition, the macrolactone scaffold of MTM is smaller than in other macrolides. The unusual structure of MTM and PKM and their simultaneous secretion by S. venezuelae bring about the possibility that two compounds would bind to distinct ribosomal sites. However, by combining genetic, biochemical and crystallographic studies, we demonstrate that MTM and PKM inhibit translation by binding to overlapping sites in the ribosomal exit tunnel. Strikingly, while MTM and PKM readily arrest the growth of bacteria, ∼40% of cellular proteins continue to be synthesized even at saturating concentrations of the drugs. Gel electrophoretic analysis shows that compared to other ribosomal antibiotics, MTM and PKM prevent synthesis of a smaller number of cellular polypeptides illustrating a unique mode of action of these antibiotics.
INTRODUCTION
Ribosome-targeting macrolide antibiotics inhibit bacterial growth by binding in the nascent peptide exit tunnel (NPET) of the large ribosomal subunit and interfering with protein synthesis (reviewed in (1,2)). Macrolides are built of a macrolactone ring decorated with several side chains. They interact with rRNA nucleotides in the NPET and inhibit translation in a context-specific manner by interfering with polymerization of certain amino acid sequences (3,4). Some proteins that lack the ‘problematic’ sequences continue to be synthesized in macrolide-treated cells (5). The number and spectrum of the ‘resistant’ proteins depend on the structure of the antibiotic. Only a few proteins are synthesized in cells treated with erythromycin (ERY) whose structure contains C3-cladinose (Figure 1). However, synthesis of up to 25% of proteins continues in the cells exposed to ketolides solithromycin (SOL) or telithromycin (TEL) (5), which represent the more potent drugs of the newer generation, in which C3 cladinose is replaced with a keto group (Figure 1).
Figure 1.

Chemical structures of natural ketolides methymycin and pikromycin, semi-synthetic ketolides telithromycin and solithromycin and cladinose-containing macrolide erythromycin. The atom numbering of the macrolactone ring is indicated on the ERY structure and the cladinose and desosamine sugars are marked. Keto group, in which ketolides replaces cladinose, is marked by a dotted oval in the corresponding structures.
The majority of the natural 14-member macrolactone ring macrolides carry either cladinose or other sugars at the C3 position of the ring. The antibiotics secreted by Streptomyces venezuelae strain ATCC 15439 are a notable exception (6). Pikromycin (PKM), the main 14-member macrolactone compound secreted by this strain, carries a C5 desosamine and a C3 keto group (7) and, therefore, represents a minimalist natural ketolide (Figure 1). Furthermore, due to an alternative translation initiation site within the polyketide synthase gene, a second, even smaller and unusual 12-membered ring ketolide, methymycin (MTM), is generated via the same biosynthetic pathway (8,9) (Figure 1). A number of actinomycete species produce more than one antibiotic (e.g. streptogramin A and streptogramin B, or lankacidin and lankamycin), whose action upon sensitive bacteria is commonly additive or even synergistic (10). If MTM and PKM bind to the same conventional macrolide-binding site in the ribosome, they would be competing with each other and thus, act as antagonistic inhibitors, which would be a seemingly wasteful strategy for the producer. A possible solution was offered by crystallographic studies of the Deinococcus radiodurans large ribosomal subunit complexed with MTM, which showed additional electron density in the peptidyl transferase center (PTC), which was attributed to MTM (11). However, no biochemical or genetic data were available to substantiate this claim.
Here, by using a combination of genetic, biochemical and structural approaches, we show that both MTM and PKM bind in the NPET of the ribosomes from Gram-negative and Gram-positive bacteria. Strikingly, even at concentrations that exceed by many fold those required for cell growth inhibition, MTM and PKM abolished synthesis of only a limited number of proteins, revealing them as highly selective inhibitors of bacterial protein synthesis.
MATERIALS AND METHODS
Antibiotics, enzymes and chemicals
MTM and PKM were synthesized chemically as previously described (12–14), or generated chemoenzymatically (14). The compounds were repurified as necessary by high pressure (or high performance) liquid chromatography (HPLC) using a Phenomenex Luna 5u C18 250 × 21.2 mm column (serial 444304–4) monitored at 250 nm at a flow rate of 9 ml/min with an isocratic mobile phase of H2O/MeCN (45/55) and a 0.1% NEt3 modifier. SOL and TEL were obtained from Cempra, Inc., ERY and chloramphenicol (CHL) were purchased from Sigma-Aldrich. Enzymes used for DNA cloning were from Fermentas, ThermoFisher Scientific. [γ32P]-adenosine triphosphate (ATP) (specific activity 6000 Ci/mmol) was from MP Biomedicals. Other reagents and chemicals were purchased from either ThermoFisher Scientific or Sigma-Aldrich. All oligonucleotides used in the study were synthesized by Integrated DNA Technologies.
Selection and characterization of resistant mutants
The Escherichia coli SQ110DTC strain [Δ(rrsH-aspU)794(::FRT) Δ(rrfG-rrsG)791(::FRT) Δ(rrfF-rrsD)793(::FRT) Δ(rrsC-trpT)795(::FRT) Δ(rrsA-rrfA)792(::FRT) Δ(rrsB-rrfB)790(::FRT) rph-1 λ−; ptRNA67; tolC::kan] (15), which contains a single rrn allele and lacks the tolC gene, was used for selection of resistant mutants. SQ110DTC cells were grown overnight at 37°C in Luria-Bertani (LB) medium containing 50 μg/ml of spectinomycin and 50 μg/ml of kanamycin. Cells were then diluted 100-fold and grown at 37°C in the presence of spectinomycin and kanamycin until they reached mid-log phase (A600 ∼ 0.5). Approximately 109 cells (A600 of 1.25) were plated onto an LB agar plate supplemented with 4-fold the minimal inhibitory concentration (MIC) of MTM or PKM. Plates were incubated for 36 h at 37°C. The rrlB segment corresponding to domains V and VI of 23S rRNA was amplified either directly from the resistant colonies or using isolated genomic DNA as a template using primers L1880D (TCTTGATCGAAGCCCCGGTAA) and L2750 (CAAGTTTCGTGCTTAGATGC). The polymerase chain reaction (PCR) products were purified and sequenced by Sanger capillary sequencing using the same primers.
Cell growth inhibition
An exponentially growing culture of the tolC-lacking E. coli strain BWDK [F−, Δ(araD-araB)567, ΔlacZ4787(::rrnB-3), λ−, rph-1, Δ(rhaD-rhaB)568, hsdR514, ΔtolC(::FRT)] (5) was diluted to A600 ∼ 0.01, placed in the wells of a 96-well plate (120 μl per well) and grown at 37°C with shaking in a 96-well plate reader (Tecan). When the cell culture density reached the density A600 ∼ 0.16, the plate was taken out from the reader and MTM or PKM was added to the final concentration of 100-fold MIC (400 μg/ml). Equal volume of the solvent (ethanol) was added to the ‘no antibiotic’ control culture. Plates were immediately returned to the reader and the measurements continued for additional 6 h.
Possible synergy of MTM and PKM was tested by a checkerboard MIC experiment using the MTM- and PKM- sensitive strain Streptococcus pyogenes NZ131 (16). After a pilot MIC determination for each antibiotic individually, a 96-well checkerboard plate was set up with 2-fold serial dilutions of MTM in columns and of PKM in rows. The starting optical density of the bacterial cells was A600 = 0.004. The plate was incubated overnight at 30°C for 18 h and the cell growth was visualized by Alamar Blue staining. A checkerboard MIC experiment for a synergistic pair of streptogramin antibiotics quinupristin and dalfopristin was used as a positive control.
Ribosome preparation
Escherichia coli ribosomes were prepared from the SQ110DTC strain or the SQ110DTC(A2503C) mutant (15) following the procedure described by (17).
Staphylococcus aureus ribosomes were isolated from the RN4220 strain. Cells were grown to exponential phase in 1 l brain-heart infusion (BHI) medium, collected by centrifugation and flash frozen. Cell pellets were thawed, resuspended in 25 ml of buffer containing 10 mM HEPES–KOH (pH 7.6), 50 mM KCl, 10 mM Mg(OAc)2, 7 mM β-mercaptoethanol, 0.1 mg/ml lysostaphin (Sigma-Aldrich) and disrupted by two passes through a French press at 20 000 psi. Cell debris was removed by centrifugation at 20 000 × g for 30 min. Ribosomes were then purified from the cell lysates following the same protocol that was used for isolation of the E. coli ribosomes (17).
Competition binding of MTM and PKM to the E. coli ribosome
E. coli ribosomes (10 nM) were pre-incubated with 10 nM [14C]-ERY (55 mCi/mmol; American Radiolabeled Chemicals, Inc.) in 1 ml of binding buffer (20 mM Tris–HCl, pH 7.6, 10 mM MgCl2, 150 mM NH4Cl and 6 mM β-mercaptoethanol) at 37°C for 10 min. Competing non-labeled antibiotics (MTM or PKM) were added at various concentrations and the incubation continued for 5 h at 37°C. At the end of incubation, 10 μl of 50 mg/ml suspension of DEAE magnetic beads (BioClone) were added to the binding reactions and incubated for 15 min at room temperature. The beads with immobilized ribosomes were captured using a magnetic stand. The supernatant was aspirated and beads were washed twice with 0.7 ml of ice-cold binding buffer by rapidly resuspending them, flash-spinning the tubes in a microcentrifuge, capturing beads on the magnetic stand and aspirating the supernatant. The ribosome-bound [14C]-ERY was eluted by resuspending the magnetic beads in 100 μl of 10 mM ethylenediaminetetraacetic acid (EDTA), incubating tubes for 10 min at room temperature, capturing the beads and transferring the supernatant to scintillation vials. After measuring the radioactivity in a scintillation counter, the binding data were analyzed using Prism software (GraphPad). The Ki for MTM or PKM was calculated assuming an ERY equilibrium binding constant of 10.8 nM (18).
Chemical probing
rRNA probing was performed following published protocols (19) with minor modifications. Briefly, E. coli or S. aureus ribosomes were dissolved at 200 nM concentration in 50 μl of buffer B (400 mM HEPES-KOH, pH 7.8, 50 mM MgCl2, 500 mM NH4Cl) supplemented with 20 U of Ribolock RNase inhibitor (40 U/μl, Sigma). After addition of 2 μl of the antibiotic solution (or of ethanol in the ‘no drug’ control reactions), samples were incubated at 37°C for 10 min, followed by additional 10 min at 20°C. Antibiotics were present at the final concentration of 50 μM. After antibiotic binding, dimethylsulfate (DMS) was added and tubes were incubated for 10 min at 37°C. The reactions were quenched with β-mercaptoethanol, ribosomes were ethanol-precipitated and rRNA was extracted. Modifications of the E. coli 23S rRNA nucleotides in the PTC and NPET were assessed by primer extension using the primers L2081 (GGGTGGTATTTCAAGGTCGG), L2563 (TCGCGTACCACTTTA), L2667 (GGTCCTCTCGTACTAGGAGCAG) or L2750 (CAAGTTTCGTGCTTAGATGC). The primer SaL2230 (TAGTATCCCACCAGCGTCTC) was used in experiments with S. aureus ribosomes.
Crystallographic structure determination
Ribosome complexes with mRNA and tRNAs were formed by programming 5 μM Thermus thermophilus 70S ribosomes with 10 μM mRNA and incubation at 55°C for 10 min, followed by addition of 20 μM P-site (tRNAiMet) and 20 μM A-site (tRNAPhe) substrates (20). Each of the last two steps was allowed to reach equilibrium for 10 min at 37°C. In addition, because the E site of the 70S ribosome has a high affinity for deacylated elongator tRNA, in all our structures it is occupied by the elongator tRNAPhe. For co-crystallization of MTM and PKM with the ribosome, each antibiotic was added to a final concentration of 500 μM, a relatively high concentration due to their low affinity, and the samples were kept at room temperature for additional 20 min prior to crystallization. Thermus thermophilus 70S ribosome complexes were formed in a buffer containing 5 mM HEPES–KOH (pH 7.6), 50 mM KCl, 10 mM NH4Cl and 10 mM Mg(CH3COO)2, and then crystallized in a buffer containing 100 mM Tris–HCl (pH 7.6), 2.9% (w/v) PEG-20K, 7–12% (v/v) 2-methyl-2,4-pentanediol, 100–200 mM arginine and 0.5 mM β-mercaptoethanol. Crystals were grown by the vapor diffusion method in sitting drops at 19°C and stabilized as described previously (20). Diffraction data were collected using beamline 24ID-C at the Advanced Photon Source. All crystals belonged to the primitive orthorhombic space group P212121 with approximate unit cell dimensions of 210 Å × 450 Å × 620 Å and contained two copies of the 70S ribosome per asymmetric unit. Each structure was solved by molecular replacement using PHASER from the CCP4 program suite (21). The search model was generated from the previously published structure of T. thermophilus 70S ribosome with bound mRNA and tRNAs (PDB code: 4Y4P from (20)). The initial molecular replacement solutions were refined in several steps. First, rigid body refinement was performed with the ribosome split into multiple domains, followed by positional and individual B-factor refinement. The final models of the 70S ribosome in complex with MTM or PKM and mRNA/tRNAs were generated by multiple rounds of model building in COOT (22), followed by refinement in PHENIX (23). The statistics of data collection and refinement are compiled in Supplementary Table S1.
Inhibition of bulk protein synthesis
All the manipulations were performed in a 37°C room. Escherichia coli BWDK cells were grown overnight in the M9 minimal medium containing 0.003 mM thiamine and supplemented with 40 μg/ml of all natural amino acids except methionine (M9AA-M) (24). Cells were diluted 100-fold into fresh M9AA-M medium, grown until the culture density reached A600 of 0.2 and an aliquot of 350 μl was transferred to an Eppendorf tube. Antibiotics were added to the final concentration of 100-fold MIC. At each of the specified time points (0, 1, 2.5, 5, 15 and 30 min), 28 μl aliquots of the cultures were withdrawn and transferred to tubes, each containing 0.3 μCi [35S] L-methionine (specific activity 1175 Ci/mmol) in 2 μl of M9AA-M medium. After 1 min incubation, 25 μl of the tubes contents were spotted onto Ø 25 mm Whatman 3MM paper disks, which were pre-wetted with 25 μl of 5% trichloroacetic acid (TCA). The disks were then immediately immersed into a beaker containing 500 ml of 5% TCA. Following collection of all samples, the beaker with the paper disks was boiled for 5 min. TCA was discarded and the beaker was refilled with 500 ml of 5% TCA. After boiling for additional 5 min, the TCA was discarded, and the disks were rinsed with 200 ml acetone. Disks were air-dried, placed in vials, and following the addition of 5 ml of scintillation cocktail, the amount of radioactivity retained was determined by scintillation counting.
The measurements of residual translation in the presence of CHL were carried out following essentially the same protocol except that each sample contained 1 μCi [35S] L-methionine (specific activity 1175 Ci/mmol) and the incubation time was 3 min.
2D-gel electrophoresis analysis of radiolabeled proteins
Pulse labeling of proteins was carried out as described (5). Specifically, E. coli strain BWDK was grown overnight at 37°C in M9AA-M medium. The cells were diluted 1:200 in fresh M9AA-M medium and grown to exponential phase (A600 ≈ 0.2). One milliliter aliquots of the exponential cultures were incubated with either no drug, or 100-fold MIC of MTM (400 μg/ml), PKM (400 μg/ml) or SOL (50 μg/ml) for 10 min. One microliter (10 μCi) of [35S] L-methionine (specific activity 1175 Ci/mmol) was then added and the cells were incubated for 3 min followed by the addition of unlabeled L-methionine to the final concentration of 80 μg/ml and further incubation for 7 min. Cells were pelleted by centrifugation at 3000 rpm for 15 min at 4°C. Pellets were washed twice with 1.5 ml of M9AA-M and flash-frozen in liquid nitrogen. Protein isolation and 2D-gel electrophoresis analyses were performed by the Kendrick Labs.
Generation of templates for in vitro translation and toeprinting
The ermBL and ermDL DNA templates for toeprinting were generated by a four-primer PCR using AccuPrime DNA Polymerase (Thermo Fisher Scientific) as described previously (25). Toeprinting reactions were carried out in 5 μl of PURExpress transcription–translation system (New England Biolabs) as previously described (25,26). The reverse transcription was carried out using primer NV1 (GGTTATAATGAATTTTGCTTATTAAC) (26). The final concentrations of MTM, PKM and TEL in the reactions were 150 μM; concentration of retapamulin (Sigma-Aldrich) was 50 μM; borrelidine (Santa Cruz Biotech) or L-PSA (kindly provided by Dr Rogelio Cruz-Vera, University of Alabama-Huntigton), the inhibitors or ThrRS or ProRS, respectively, were present in the corresponding reactions at 50 μM.
In vitro translation of the fusA gene
In vitro translation was carried out in the E. coli S30 cell-free transcription–translation system for linear templates (Promega) in the presence of 50 μM TEL or varying concentrations of MTM. The PCR-generated DNA template (0.2 pmol) encoding the fusA gene under the control of the Ptac promoter (5) was expressed in a 5 μl reaction containing 1 μCi [35S] L-methionine (specific activity 1175 Ci/mmol) following the manufacturer’s protocol. After 30 min incubation at 37°C, the reactions were treated with 1 M NaOH for 1 min at room temperature and precipitated with eight volumes of ice-cold acetone. Proteins were fractionated in a 12% Tricine–sodium dodecyl sulphate (SDS) polyacrylamide gel. The gel was dried and exposed overnight to a phosphorimager screen.
RESULTS
MTM and PKM bind in the NPET of the bacterial ribosome
In order to locate the binding site of the minimalist ketolides, we crystallized T. thermophilus 70S ribosomes, carrying mRNA and A-, P- and E-site tRNAs, with either MTM or PKM and solved the structures of the obtained complexes at 2.7 and 2.6 Å resolution, respectively (Figure 2 and Supplementary Table S1). The structures were solved by molecular replacement using the atomic coordinates of the T. thermophilus 70S ribosome (PDB code: 4Y4P) (20). The unbiased difference Fourier maps revealed unique positive electron density peaks carrying characteristic features of the MTM and PKM chemical structures (Figure 2A and B). Binding sites of MTM and PKM overlapped with those of other macrolide antibiotics (Figure 2C, D and G) and like with other macrolides, binding of MTM or PKM is largely mediated by the interactions of the drugs with the 23S rRNA residues A2058, A2059 and A2062 (Figure 2 E, F, H and I).
Figure 2.

Crystallographic structures of MTM and PKM in complex with 70S ribosome and A- and P-tRNAs. (A and B) Chemical structures and difference Fourier maps of MTM (A) and PKM (B) in complex with the Thermus thermophilus 70S ribosome (blue mesh). The refined model of each compound is displayed in its respective electron density before the refinement. The unbiased (Fobs − Fcalc) difference electron density map is contoured at 3.0 σ. Carbon atoms are colored yellow for MTM and green for PKM, nitrogens are in blue, oxygens are in red. (C) Structural comparison of the ribosome-bound MTM (yellow), PKM (green) and ERY (magenta). Structure of ERY is from PDB code: 4V7X (37). All three structures of ribosome-bound antibiotics were aligned based on domain V of the 23S rRNA. (D and G) Overview of the MTM (D) and PKM (G) binding sites in the T. thermophilus 70S ribosome viewed as a cross-cut through the peptide exit tunnel. 30S subunit is shown in light yellow, 50S subunit is in light blue, mRNA is shown in magenta and tRNAs are displayed in green for the A-site, and in dark blue for the P-site. E-site tRNA is omitted for clarity. (E, F, H and I) Close-up views of the MTM (E and F) or PKM (H and I) binding site shown in panels (D and G). Escherichia coli numbering of the nucleotides in the 23S rRNA is used.
Chemical probing confirmed that MTM and PKM bind in the NPET not only in the crystalline state, but also in solution. Similar to other macrolides (27), MTM and PKM protected only NPET nucleotides A2058 and A2059 from DMS modification in the ribosomes of Gram-negative (E. coli) and Gram-positive (S. aureus) species (Figure 3A and B). Furthermore, although the affinity of MTM and PKM for the ribosome was much weaker than ERY, both inhibitors could compete with ERY for binding to the ribosome (Figure 3C).
Figure 3.

In vitro binding of MTM and PKM to the NPET. (A and B) DMS probing of interactions of antibiotics with (A) Escherichia coli or (B) Staphylococcus aureus ribosomes. Cladinose-containing macrolide ERY, ketolide TEL or PTC-binding antibiotic CHL were used as controls. The arrows indicate the 23S rRNA residues A2058 and A2059, located in the macrolide-binding site in the NPET. Samples in the lanes marked ‘none’ contained no antibiotic. (C) Competitive binding of non-radioactive ERY, PKM or MTM with [14C] ERY to E. coli 70S ribosomes. The MTM and PKM binding experiments were done in triplicates, the binding of the control antibiotic, ERY, was analyzed in duplicate samples. Assuming ERY Kd being 10.8 nM (18), the estimated Ki of MTM and PKM were 13.1 ± 3.5 and 3.2 ± 1.6 μM, respectively.
Finally, in order to verify that inhibition of cell growth by MTM and PKM is mediated by their binding in the NPET, we isolated MTM- and PKM-resistant mutants and mapped the location of the mutations. A number of resistant clones appeared when 109 cells of the E. coli strain SQ110DTC with a single chromosomal rrn allele (15,28) were plated on LB/agar plates supplemented with 4-fold MIC of MTM or PKM. All the isolated mutants carried alterations of the nucleotides clustered in the NPET in the known site of macrolide action (Supplementary Table S2) and all the mutations conferred cross-resistance to either antibiotic. Thus, the mutational analysis data suggested that the two antibiotics bind to the same ribosomal site in the NPET.
The small size and simplicity of the MTM and PKM molecules (Figure 1) compelled us to still consider the possibility that, when present simultaneously, MTM and PKM could bind to distinct ribosomal sites. PKM, more closely resembling conventional ketolides, would likely bind in the NPET but the 12-member ring MTM could potentially bind in the PTC, as suggested by previous structural studies of MTM bound to the D. radiodurans large ribosomal subunit (11). If this were the case, MTM and PKM could arrest bacterial cell growth in a synergistic manner, in a way reminiscent to that of streptogramins, which bind to the same two neighboring sites in the PTC and NPET. However, the combinations of MTM and PKM showed minimal synergy in comparison with the classic synergistic streptogramins quinupristin and dalfopristin (Supplementary Figure S1), providing no support for the concurrent association of MTM and PKM to separate ribosomal sites.
Altogether, the results of crystallographic, biochemical and genetic experiments showed that MTM and PKM exert their inhibitory action by binding to the same binding site located in the NPET of the ribosome, likely interfering with synthesis of proteins.
PKM and MTM interfere with cell growth by inhibiting translation of an unusually small fraction of the cellular proteome
Clinical ketolides equipped with an extended alkyl–aryl side chain, such as TEL or SOL, bind to the ribosome with a higher affinity than ERY (29) but allow for translation of more proteins (3–5). We wondered how would weakly binding ketolides lacking any side chains other than the C5 desosamine affect cellular growth and protein synthesis.
Addition of 100-fold MIC (400 μg/ml) of MTM or PKM to exponentially growing E. coli cells caused rapid growth arrest (Figure 4A and B). However, the arrested cells continued to synthesize proteins at an unexpectedly high level (Figure 4C). While E. coli cells treated with 100-fold MIC of ERY or SOL continued translation at ∼5 and ∼20% level, respectively, compared to the untreated control, protein synthesis persisted at more than 40% level in the cells exposed to 100-fold MIC of MTM or PKM. Thus, even at concentrations exceeding by two orders of magnitude those that are required for halting cells from growing, MTM and PKM allowed for protein synthesis to continue at nearly half the rate of uninhibited cells. This result becomes even more surprising when considering that E. coli cells continue to actively grow and proliferate when their translation is slowed down by 2-fold in nutrient-poor medium (30) or when cells are exposed to sub-MIC concentrations of PTC-targeting antibiotic CHL (Supplementary Figure S3). A straightforward explanation for this apparent discrepancy is that MTM and PKM stop cell growth by selectively affecting the production of certain proteins, some of which could be critical for bacterial proliferation.
Figure 4.

MTM and PKM abolish cell growth but allow translation to continue at high level. (A and B) Addition of MTM (A) or PKM (B) at 100-fold MIC causes rapid arrest of cell growth. Cell culture densities were recorded at 600 nm in a 96-well plate reader. Gray dots and curves represent cultures grown without antibiotics, black dots and curves show the growth of the cultures to which antibiotics were added at the time points marked by arrows. The inlets show the results of similar experiments but performed in 15 ml culture tubes with more frequent time-points measurements; the antibiotics were added at the time point marked as zero. (C) Residual translation in Escherichia coli cells treated for the indicated duration of time with 100-fold MIC of MTM (open circles), PKM (open diamonds), ERY (filled circles) or SOL (filled diamonds).
This suggestion was supported by the results of 2D gel electrophoresis analysis, which examined the protein-specific inhibitory action of MTM and PKM in vivo. For this analysis, E. coli cells were exposed for 10 min to 100-fold MIC of antibiotics followed by addition of [35S]-L-methionine and the radiolabeled proteins, those whose synthesis persisted in the antibiotic-treated cells, were resolved by 2D-gel electrophoresis (Figure 5). Consistent with the metabolic labeling data (Figure 4C), cells treated with MTM and PKM continued synthesis of a large set of proteins (Figure 5C and D). In fact, among the actively expressed proteins, we could detect only a small number of polypeptides whose synthesis was notably inhibited by MTM and PKM (some of them are indicated by red circles in Figure 5A, C and D). In contrast, and in agreement with the metabolic labeling data (Figure 4C) and our published results (5), the reference ketolide SOL inhibited synthesis of a larger subset of proteins (Figure 5B); as a consequence, some proteins inhibited by SOL continue to be synthesized in cells treated with high concentrations of MTM or PKM (blue circles in Figure 5B–D). The results of the 2D gel electrophoresis analysis suggest that MTM and PKM rapidly and efficiently arrest cell growth (Figure 4A and B), by preferentially interfering with translation of only specific polypeptides. Thus, MTM and PKM emerge as highly protein-selective inhibitors of translation.
Figure 5.

MTM and PKM inhibit synthesis of a limited subset of proteins. 2D gel electrophoresis of [35S]-labeled proteins synthesized in the Escherichia coli cells treated with no antibiotics (A) or exposed for 10 min to 100-fold MIC of a control ketolide SOL (B), MTM (C) or PKM (D). Spots representing examples of proteins whose translation is notably inhibited by MTM or PKM are indicated in the control sample (A) by solid red circles and corresponding sites are shown by dotted red circles in panels (C) and (D). Proteins that are efficiently inhibited by SOL, but not by MTM or PKM are marked by dotted blue circles (panel B); the corresponding sites are shown by solid blue circles in panels (C) and (D).
Protein-selective action of macrolides stems from their ability to inhibit translation of only specific amino acid sequences (3,4). Analysis of ribosome stalling induced by ketolides during synthesis of the well-characterized ‘problematic’ sequences VDK (in the ermBL open reading frame (ORF)) and RLR (in the ermDL ORF), showed that TEL caused a near-complete arrest while MTM and PKM only paused a fraction of the translating ribosomes (Supplementary Figure S2A and B). This result is in line with our previous observations of incomplete ribosome stalling induced by MTM or PKM at the sites of programmed translation arrest within the regulatory ORFs preceding the pikR1 and pikR2 resistance genes present in the MTM and PKM producer (31). In agreement with these data, in vitro translation of the model protein EF-G, encoded by the fusA gene, is readily inhibited by SOL, which efficiently arrests translation at codon 358 within the RER motif (4,5), but it is barely affected by high concentrations of MTM (Supplementary Figure S2C). Thus, even at saturation, binding of MTM or PKM in the NPET does not completely arrest translation at the sites where clinical ketolides halt progression of the ribosomes. This effect likely accounts for a more efficient residual translation of cellular proteins in the cells treated with 100-fold MIC concentrations of MTM and PKM compared to clinical ketolides.
DISCUSSION
We have shown that two natural ketolides, PKM and MTM, co-produced by S. venezuelae ATCC 15439, bind to the same site in the ribosomal exit tunnel at a location overlapping with the site of action of clinically important macrolides. Therefore, we believe that the previously proposed binding of MTM at the A site of the PTC of D. radiodurans large ribosomal subunit (11) either is species specific or results from misinterpretation of crystallographic data.
Although MTM and PKM bind in the conventional macrolide site, the minimalist structure of these inhibitors distinguishes them from other studied macrolides and results in an unconventional mode of action. It has been shown previously that due to context-specific action of clinical macrolides, a selected number of proteins continue to be synthesized in the cells treated with these drugs (3–5). MTM and PKM represent an extreme of this trend: cells exposed to MTM or PKM rapidly halt their growth (Figure 4A and B) but even at saturating concentrations of the inhibitors (exceeding MIC by 100-fold), translation continues at nearly half the level of the untreated control (Figure 4C) and synthesis of most of the actively expressed proteins persists at a high level (Figure 5C and D). Therefore, we suggest that the ability of MTM and PKM to stop cell proliferation stems primarily from selective inhibition of translation of a limited number of proteins.
Several factors could contribute to the unique ability of MTM and PKM to inhibit synthesis of only a subset of proteins. First, because MTM and PKM are notably smaller compared to conventional ribosome-targeting macrolides, they leave more room in the NPET (Figure 6) allowing the unimpeded passage of a larger variety of nascent peptides. Second, weaker interactions with the NPET and the nascent chain could diminish the ability of MTM and PKM to induce arrest of translation of the problematic sequences. In fact, we noticed that MTM and PKM arrest the ribosome progression only weakly at some of the motifs where other ketolides (e.g. TEL or SOL) halt translation. Therefore, if the gene contains one or a few sites problematic for ketolides, SOL or TEL would block translation of the encoded protein, while MTM or PKM would only marginally diminish its production. For example, the EF-G-encoding gene fusA, which contains a single strong site of ketolide-induced arrest (4,5) is likely to be actively translated in the MTM- or PKM-treated cells (Supplementary Figure S2C). However, proteins with a high content of troublesome motifs will likely still remain sensitive to MTM and PKM. Last, due to their reduced affinity for the ribosome, some nascent chains could potentially displace MTM and PKM from their binding site in a mode reminiscent of that reported previously for some macrolide resistance peptides (32–34). It should be emphasized, however, that in spite of their weaker binding to the ribosome, MTM and PKM are still able to interfere with translation and cell growth and that the residual translation we observed in our experiments was taken place when cells were exposed to saturating concentrations of the drug, significantly exceeding the concentrations required for inhibition of cell growth.
Figure 6.
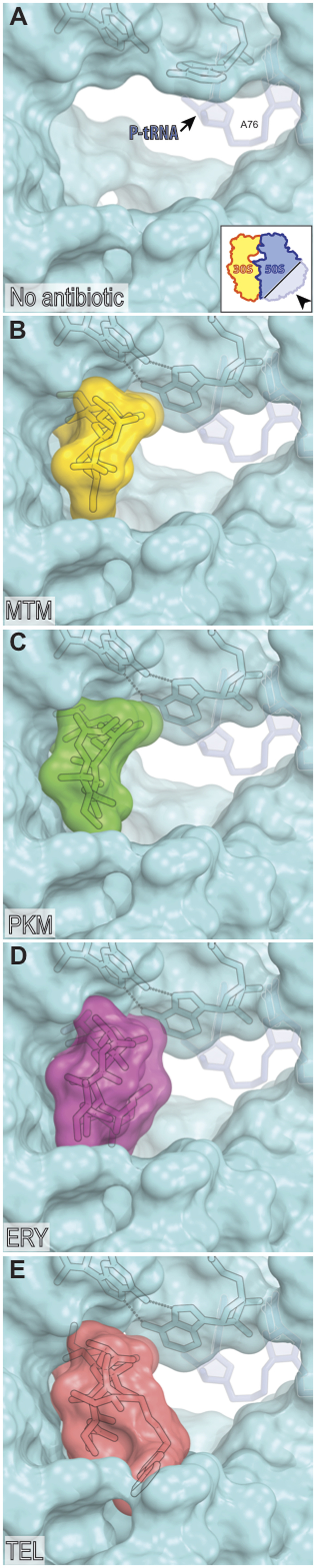
Partial occlusion of the NPET by MTM and PKM. (A) Lumen of the NPET of the drug-free Thermus thermophilus ribosome (PDB code: 4Y4P (20)). The view is from the inside of the tunnel towards the PTC. A76 of the P-site tRNA, which normally carries formyl-methionine or a growing peptide chain, is shown in gray. A-site tRNA is not visible in this view. (B–E) Occlusion of the NPET by MTM (B), PKM (C), cladinose-containing macrolide ERY (D) and clinical ketolide TEL (E). Structures of ERY and TEL are from PDB codes 4V7X and 4V7S, respectively (37).
In our toeprinting experiments (Supplementary Figure S2A and B), we observed that MTM and PKM could weakly arrest translation at the motifs (e.g. RLR or VDK) which have been previously shown to be problematic for clinically-relevant ketolides (3,4). It is conceivable that MTM and PKM could potentially inhibit translation also at motifs distinct from those. However, we have not observed MTM- or PKM-induced ribosome stalling at ‘new’ sites not observed with TEL or SOL at several tested templates (data not shown). Unfortunately, genome-wide ribosome profiling studies of context-specific action of MTM and PKM are currently not feasible because of the unavailability of sufficiently large quantities of these natural compounds.
Because of their ability to stop cell growth by inhibiting only a fraction of cellular proteins, MTM and PKM emerge as highly selective ribosome-targeting antibacterials. This finding illuminates an attractive possibility of developing new therapeutics that would achieve the desired effect (e.g. inhibition of proliferation of pathogenic bacteria or malicious cells) by interfering with synthesis of few or even a single specific protein. A recent example of a highly selective inhibitor likely targeting a eukaryotic ribosome (35) indicates that this principle is applicable across evolutionary kingdoms and could provide new valuable tools for medicine, biotechnology and synthetic biology.
MTM and PKM are generated via a single biosynthetic pathway in S. venezuelae ATCC 15439. The switch from production of PKM to MTM is controlled by a poorly understood molecular circuit, which utilizes an alternative translation start site within the polyketide synthase gene (36). The existence of such a sophisticated control mechanism strongly argues that production of two different inhibitors via the same pathway is a result of evolutionary selection rather then a peculiar ‘genetic accident’. The question why the producer benefits from generating two inhibitors that compete for the same ribosomal site and thus, they could be formally viewed as antagonistic antibiotics still remains unanswered. It could be that the spectrum of proteins sensitive to MTM and PKM differ in diverse bacterial species. As a result, some microorganisms co-existing with S. venezuelae ATCC 15439 could be more susceptible to MTM, whereas others could be more readily inhibited by PKM. If this scenario is correct, the switch in production of the two drugs could be controlled by environmental cues, which would tune the production of the most effective antibiotic in response to the composition of the microbiome in the biological niche.
ACCESSION NUMBERS
Coordinates and structure factors were deposited in the RCSB Protein Data Bank with accession code 5WIS for the Tth 70S ribosome in complex with MTM, mRNA, A-, P- and E-site tRNAs, and 5WIT for the Tth 70S ribosome in complex with PKM, mRNA, A-, P- and E-site tRNAs.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Prabha Fernandes (Cempra, Inc.) for providing SOL and TEL.
Footnotes
Present address: Mashal M. Almutairi, Pharmacology and Toxicology Department, College of Pharmacy, King Saud University, Riyadh 11451, Saudi Arabia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [R01 GM106386 to A.S.M., N.V.-L.], NIH [R35 GM118101 to D.H.S.]; Hans W. Vahlteich Professorship (to D.H.S.); University of Illinois startup funds (to Y.S.P.); King Saud University Scholarship for Graduate Studies (to M.A.); University of Michigan Rackham Merit Pre-doctoral Fellowship (to D.A.H.). This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the NIH [P41 GM103403 to NE-CAT]. The Pilatus 6M detector on 24ID-C beamline is funded by the NIH-ORIPHEI [S10 RR029205 to NE-CAT]. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory [DE-AC02–06CH11357]. Funding for open access charge: NIH [R01 106386].
Conflict of interest statement. The research in the ASM/NV-L laboratory is supported in part by grants from pharmaceutical companies developing unrelated antibiotics.
REFERENCES
- 1. Gaynor M., Mankin A.S.. Macrolide antibiotics: binding site, mechanism of action, resistance. Curr. Top. Med. Chem. 2003; 3:949–961. [DOI] [PubMed] [Google Scholar]
- 2. Wilson D.N. The A-Z of bacterial translation inhibitors. Crit. Rev. Biochem. Mol. Biol. 2009; 44:393–433. [DOI] [PubMed] [Google Scholar]
- 3. Davis A.R., Gohara D.W., Yap M.N.. Sequence selectivity of macrolide-induced translational attenuation. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:15379–15384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kannan K., Kanabar P., Schryer D., Florin T., Oh E., Bahroos N., Tenson T., Weissman J.S., Mankin A.S.. The general mode of translation inhibition by macrolide antibiotics. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:15958–15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kannan K., Vázquez-Laslop N., Mankin A.S.. Selective protein synthesis by ribosomes with a drug-obstructed exit tunnel. Cell. 2012; 151:508–520. [DOI] [PubMed] [Google Scholar]
- 6. Vazquez D. Inhibitors of Protein Biosynthesis. 1979; Berlin: Springer. [DOI] [PubMed] [Google Scholar]
- 7. Muxfeldt H., Shrader S., Hansen P., Brockman H.. The structure of pikromycin. J. Am. Chem. Soc. 1968; 90:4748–4749. [DOI] [PubMed] [Google Scholar]
- 8. Xue Y., Zhao L., Liu H.W., Sherman D.H.. A gene cluster for macrolide antibiotic biosynthesis in Streptomyces venezuelae: architecture of metabolic diversity. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:12111–12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kittendorf J.D., Sherman D.H.. The methymycin/pikromycin pathway: a model for metabolic diversity in natural product biosynthesis. Bioorg. Med. Chem. 2009; 17:2137–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Newman D.J., Cragg G.M.. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012; 75:311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Auerbach T., Mermershtain I., Bashan A., Davidovich C., Rozenberg C. H., Sherman D. H., Yonath A.. Structural basis for the antibacterial activity of the 12-membered-ring mono-sugar macrolide methymycin. Biotechnologia. 2009; 1:24–35. [Google Scholar]
- 12. Oh H.S., Kang H.Y.. Total synthesis of pikromycin. J. Org. Chem. 2012; 77:1125–1130. [DOI] [PubMed] [Google Scholar]
- 13. Oh H.S., Xuan R., Kang H.Y.. Total synthesis of methymycin. Org. Biomol. Chem. 2009; 7:4458–4463. [DOI] [PubMed] [Google Scholar]
- 14. Hansen D.A., Rath C.M., Eisman E.B., Narayan A.R., Kittendorf J.D., Mortison J.D., Yoon Y.J., Sherman D.H.. Biocatalytic synthesis of pikromycin, methymycin, neomethymycin, novamethymycin, and ketomethymycin. J. Am. Chem. Soc. 2013; 135:11232–11238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Orelle C., Carlson S., Kaushal B., Almutairi M.M., Liu H., Ochabowicz A., Quan S., Pham V.C., Squires C.L., Murphy B.T. et al. . Tools for characterizing bacterial protein synthesis inhibitors. Antimicrob. Agents Chemother. 2013; 57:5994–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McShan W.M., Ferretti J.J., Karasawa T., Suvorov A.N., Lin S., Qin B., Jia H., Kenton S., Najar F., Wu H. et al. . Genome sequence of a nephritogenic and highly transformable M49 strain of Streptococcus pyogenes. J. Bacteriol. 2008; 190:7773–7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ohashi H., Shimizu Y., Ying B.W., Ueda T.. Efficient protein selection based on ribosome display system with purified components. Biochem. Biophys. Res. Commun. 2007; 352:270–276. [DOI] [PubMed] [Google Scholar]
- 18. Lovmar M., Tenson T., Ehrenberg M.. Kinetics of macrolide action: the josamycin and erythromycin cases. J. Biol. Chem. 2004; 279:53506–53515. [DOI] [PubMed] [Google Scholar]
- 19. Merryman C., Noller H.F.. Smith CWJ. RNA: Protein Interactions, A Practical Approach. 1998; Oxford: Oxford University Press; 237–253. [Google Scholar]
- 20. Polikanov Y.S., Melnikov S.V., Soll D., Steitz T.A.. Structural insights into the role of rRNA modifications in protein synthesis and ribosome assembly. Nat. Struct. Mol. Biol. 2015; 22:342–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 23. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al. . PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miller J.H. A Short Course in Bacterial Genetics: a Laboratory Manual. 1992; NY: CSHL Press. [Google Scholar]
- 25. Orelle C., Szal T., Klepacki D., Shaw K.J., Vazquez-Laslop N., Mankin A.S.. Identifying the targets of aminoacyl-tRNA synthetase inhibitors by primer extension inhibition. Nucleic Acids Res. 2013; 41:e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vazquez-Laslop N., Thum C., Mankin A.S.. Molecular mechanism of drug-dependent ribosome stalling. Mol. Cell. 2008; 30:190–202. [DOI] [PubMed] [Google Scholar]
- 27. Moazed D., Noller H.F.. Chloramphenicol, erythromycin, carbomycin and vernamycin B protect overlapping sites in the peptidyl transferase region of 23S ribosomal RNA. Biochimie. 1987; 69:879–884. [DOI] [PubMed] [Google Scholar]
- 28. Quan S., Skovgaard O., McLaughlin R.E., Buurman E.T., Squires C.L.. Markerless Escherichia coli rrn deletion strains for genetic determination of ribosomal binding sites. G3. 2015; 5:2555–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Douthwaite S., Hansen L.H., Mauvais P.. Macrolide-ketolide inhibition of MLS-resistant ribosomes is improved by alternative drug interaction with domain II of 23S rRNA. Mol. Microbiol. 2000; 36:183–193. [DOI] [PubMed] [Google Scholar]
- 30. Forchhammer J., Lindahl L.. Growth rate of polypeptide chains as a function of the cell growth rate in a mutant of Escherichia coli 15. J. Mol. Biol. 1971; 55:563–568. [DOI] [PubMed] [Google Scholar]
- 31. Almutairi M.M., Park S.R., Rose S., Hansen D.A., Vazquez-Laslop N., Douthwaite S., Sherman D.H., Mankin A.S.. Resistance to ketolide antibiotics by coordinated expression of rRNA methyltransferases in a bacterial producer of natural ketolides. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:12956–12961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tenson T., Xiong L., Kloss P., Mankin A.S.. Erythromycin resistance peptides selected from random peptide libraries. J. Biol. Chem. 1997; 272:17425–17430. [DOI] [PubMed] [Google Scholar]
- 33. Tripathi S., Kloss P.S., Mankin A.S.. Ketolide resistance conferred by short peptides. J. Biol. Chem. 1998; 273:20073–20077. [DOI] [PubMed] [Google Scholar]
- 34. Vimberg V., Xiong L., Bailey M., Tenson T., Mankin A.. Peptide-mediated macrolide resistance reveals possible specific interactions in the nascent peptide exit tunnel. Mol. Microbiol. 2004; 54:376–385. [DOI] [PubMed] [Google Scholar]
- 35. Petersen D.N., Hawkins J., Ruangsiriluk W., Stevens K.A., Maguire B.A., O’Connell T.N., Rocke B.N., Boehm M., Ruggeri R.B., Rolph T. et al. . A small-molecule anti-secretagogue of PCSK9 targets the 80S ribosome to inhibit PCSK9 protein translation. Cell Chem. Biol. 2016; 23:1362–1371. [DOI] [PubMed] [Google Scholar]
- 36. Xue Y., Sherman D.H.. Alternative modular polyketide synthase expression controls macrolactone structure. Nature. 2000; 403:571–575. [DOI] [PubMed] [Google Scholar]
- 37. Bulkley D., Innis C.A., Blaha G., Steitz T.A.. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:17158–17163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.