


Abstract
As part of the HIV infection cycle, viral DNA inserts into the genome of host cells such that the integrated DNA encoding the viral proteins is flanked by long terminal repeat (LTR) regions from the retrovirus. In an effort to develop novel genome editing techniques that safely excise HIV provirus from cells, Tre, an engineered version of Cre recombinase, was designed to target a 34-bp sequence within the HIV-1 LTR (loxLTR). The sequence targeted by Tre lacks the symmetry present in loxP, the natural DNA substrate for Cre. We report here the crystal structure of a catalytically inactive (Y324F) mutant of this engineered Tre recombinase in complex with the loxLTR DNA substrate. We also report that 17 of the 19 amino acid changes relative to Cre contribute to the altered specificity, even though many of these residues do not contact the DNA directly. We hypothesize that some mutations increase the flexibility of the Cre tetramer and that this, along with flexibility in the DNA, enable the engineered enzyme and DNA substrate to adopt complementary conformations.
INTRODUCTION
Through the introduction of combination antiretroviral therapy, HIV was transformed from a death sentence to a manageable disease (1). For many infected individuals, however, long-term treatment has complications due to the toxicity of the antiretroviral medications. The cost of drugs, which must be taken daily, is also a factor, as is non-compliance with the treatment regimen. Thus, although modern HIV medications are remarkably effective in keeping the virus in check, there is a clear need for better therapies and, ideally, a means to permanently remove the retrovirus from infected individuals.
Proviral HIV DNA is integrated into the human genome as part of the normal life cycle of the virus. This integration represents a major obstacle to eradicating the virus from infected individuals. In recent years, a number of genome editing approaches have been investigated as a means to remove and/or inactivate the integrated provirus (reviewed in (2–4)). These methods include the RNA-based CRISPR/Cas9 system, zinc finger nucleases and transcription activator-like effector nucleases. While the convenience of the CRISPR/Cas9 system possesses certain advantages over the other technologies, unpredictable lesions introduced by the cellular DNA repair machinery represent a potential drawback of this system (5). In particular, the CRISPR/Cas9 approach is reported to increase viral escape through mutations associated with cellular DNA repair (6,7). An alternative genome editing approach to excise the HIV virus involves engineered versions of tyrosine site-specific recombinases, specifically the tyrosine recombinase Cre from bacteriophage P1 (reviewed in (8–10)). Cre (Causes Recombination) is one of the simpler members of the tyrosine recombinase family and is routinely employed as a research tool to catalyze gene recombination both in vitro and in vivo (reviewed in (11)). Unlike nuclease-based genome modification methods, which rely on endogenous, error-prone pathways to rejoin cleaved chromosomes, Cre acts with single nucleotide precision and does not require accessory proteins or co-factors to effect the recombination.
Cre naturally targets a DNA sequence known as loxP (locus of crossing (x) of P1) (Figure 1). It was demonstrated early on that Cre could remove intervening chromosomal DNA from loxP-flanked sequences in mice and that Cre is highly specific for its DNA target (12,13). Recombination by Cre occurs in an ordered, stepwise fashion (detailed schematically in Supplementary Figure S1) (reviewed in (14,15)). The multi-step recombination reaction involves ordered DNA cleavage and ligation at four distinct sites, and all four copies of Cre, bound to two loxP sites, participate in catalysis. Cre has been the subject of many structural studies, and the resulting structures have helped elucidate the details of Cre's interaction with the highly bent loxP DNA (reviewed in (14,15)). Within the Cre tetramer, two Cre molecules adopt a ‘cleaving’ conformation and the other two a ‘non-cleaving’ conformation. The reaction proceeds through a quasi-planar Holliday junction (HJ) intermediate with pseudo four-fold symmetry, and the complex undergoes isomerization such that the cleaving Cre protomers adopt the non-cleaving conformation and vise-versa. In this process, two molecules of Cre bind each loxP site. The loxP sequence is 34-nt long, and the first and last 13 nt form a perfect, inverted repeat. The 8 nt in the middle of loxP, commonly referred to as the spacer, have no sequence symmetry. Most of the contacts between Cre and loxP occur within the 13-bp regions of the DNA, but strand exchange occurs within the asymmetric, central nucleotides of loxP (Supplementary Figure S1).
Figure 1.

Sequence of loxP and loxLTR recombination sites and schematic comparison of protein–DNA interactions for Cre/loxP and Tre/loxLTR. DNA sequences are black (conserved between loxP and loxLTR) or red (not conserved). Standard loxP numbering is shown. Lower case letters indicate the central 8-bp spacer region. The horizontal arrows indicate the orientation of the inverted repeat sequences in loxP and highlight the symmetry in the loxP target. The scissile phosphates are indicated by pink and black circles in the active and inactive arms, respectively. Red arrows indicate site of cleavage. Phosphate backbone interactions are indicated by a red +. Only residues involved in base-specific interactions are shown and are placed adjacent to the interacting base, with a solid line drawn to the base for clarity. Residues in blue letters on yellow oval indicate amino acid sequence difference between Cre and Tre. The ┴ indicates the loss of an interaction. The black lower case letters at the ends of the loxLTR sequence show the additional nucleotides used for crystallization of the Tre/loxLTR complex. The PDB ID: 3C29 was used for the Cre/loxP analysis (except for the K201 interaction which was 1NZB).
When the HIV retrovirus integrates into human chromosomal DNA, it does so such that the protein-coding regions are flanked by long terminal repeats (LTRs). These LTR regions are important for transcriptional control of the HIV genes (16). A Cre variant known as Tre was previously generated by substrate-linked protein evolution (17). Tre has 19 amino acid substitutions relative to Cre and targets a 34-bp sequence called loxLTR from the LTR of an HIV primary isolate (18). The targeted loxLTR sequence, differs at 17 of the 34 positions relative to loxP, and it lacks the inverted repeats of Cre's target (Figure 1). Activity of Tre for loxLTR is comparable to tyrosine recombinases on their targets found in nature, and recombination is loxLTR specific, as little to no activity is observed on other target sequences (8,18). Expression of Tre in HIV-1 infected cells containing the loxLTR sequence can remove the integrated provirus and cure cultured cells from the infection (18). The anti-HIV activity of Tre was also demonstrated in vivo, using a lentivirus-based delivery system, where Tre successfully excised proviral DNA from human CD4+ T-cells and CD34+ hematopoietic stem and progenitor cells in a humanized mouse model. Recombination resulted in the expected genomic scar, and there were no cytopathic effects (19). This technology represents a promising new approach to tackling the problem of persistent HIV infection mediated by infected, but dormant, cells.
Within loxLTR, only 5 of the 13 bases in the ‘inverted-repeat’ region are identical. Thus, unlike Cre, Tre must specifically recognize two very different DNA sequences to successfully catalyze recombination. To understand how Tre interacts with its asymmetric target, we have solved the X-ray crystal structure of the tetrameric protein–DNA complex using a catalytically inactive point mutant (TreY324F). We have also analyzed the functional significance of the individual Cre-to-Tre sequence changes through mutagenesis and activity studies. Given the growing importance of designer recombinases, this work provides a foundation for accelerating the generation of new designer enzymes with therapeutic potential.
MATERIALS AND METHODS
Cloning, expression and purification of TreY324F recombinase
A catalytically inactive mutant of Tre, containing a Y324F mutation, was subcloned into the Escherichia Coli expression vector pLIC-His (20). This vector encodes a fusion protein with an N-terminal hexahistidine tag followed by a tobacco etch virus (TEV) protease recognition sequence. The TreY324F-expressing plasmid was transformed into RosettaTM(DE3) cells. Each liter of 2 × YT medium containing 100 μg/ml ampicillin and 34 μg/ml chloramphenicol was inoculated with an overnight culture (1:100 dilution). The cells were grown at 37°C with shaking until an OD600 of 0.7–0.8. At that point the temperature was dropped to 18°C for 30 min, after which isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 100 μM. The cells were harvested 16–20 h later. Each 1 l of cell pellet was resuspended in ∼50 ml of resuspension buffer (RB) (50 mM Tris pH 7.5, 1 M NaCl, 0.5% Triton-X-100, 10% glycerol, 0.01% β-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride (PMSF)). The cells were stored at −20°C until the protein purification. All purification steps were performed at 4°C. The cells were lysed with four passes through an Emulsiflex-C5 microfluidizer (Avestin). The lysate was ultracentrifuged at 125 000 g for 30 min, and the supernatant was loaded onto a Ni-NTA (Qiagen) column pre-equilibrated in RB with 10 mM imidazole pH 8.0. The protein was eluted using an imidazole gradient. The fractions containing TreY324F were pooled and TEV protease was added in a 1:100 mass ratio and allowed to incubate overnight. The digested TreY324F was diluted 3- to 4-fold into buffer S0 (10 mM Tris pH 7.5, 10% glycerol, 0.1% β-mercaptoethanol) and then immediately loaded onto a cation exchange column (Source15 S, GE Healthcare) pre-equilibrated in buffer SA (20 mM BisTris pH 5.6, 200 mM NaCl, 10% glycerol, 0.1% β-mercaptoethanol). The column was developed using a NaCl gradient to 2M. The fractions containing TreY324F were pooled and flash-frozen in liquid nitrogen and stored at −80°C.
Purification of DNA for crystallization
Oligonucleotides (one micromole scale) for crystallization were synthesized by IDT (Integrated DNA Technologies) and purified by high-pressure anion-exchange chromatography, as previously described (21). The purified oligonucleotides were resuspended in 500 μl annealing buffer (10 mM Tris pH 7.5, 50 mM NaCl) and mixed together in a 1:1 molar ratio. The mixture was heated to 94°C for 3 min, then allowed to cool slowly to room temperature. The concentration of the oligomers was calculated using the extinction coefficients from IDT’s website (https://www.idtdna.com/calc/analyzer). The DNA sequence used in the crystallization of the TreY324F/loxLTR complex is shown in Figure 1.
Crystallization
The loxLTR DNA duplex (sequence shown in Figure 1) used for crystallization was diluted to 150 μM with (20 mM BisTris pH 5.6, 750 mM NaCl, 0.1% β-mercaptoethanol). The duplex contains an extra G-C base pair on either side of loxLTR and 5′ Cyt or 5′ Gua base overhangs to facilitate crystal formation. The TreY324F/loxLTR complex was made by mixing the protein TreY324F (10 μM) and the DNA (150 μM) in a molar ratio of 1:1.5 of protein:DNA. This mixture was incubated for 15′ at RT and concentrated to ∼50 μM using a Vivaspin 500 5 kD spin concentrator (GE Healthcare). Crystallization trials using previous Cre/loxP conditions as a starting point were unsuccessful despite the fact that many Cre/loxP crystal structures are isomorphous to each other and previous Cre/loxP crystals were obtained under a fairly narrow set of crystallization conditions. High-throughput crystallization methods were used with commercially available crystallization screens to identify initial crystallization conditions. The best diffracting crystals of TreY324F/loxLTR were grown at RT using the vapor diffusion method in Linbro trays by mixing 2 μl of the TreY324F/loxLTR complex with 2 μl of the reservoir solution (10 mM sodium citrate, 25–32.5% Peg 6000). Crystals appeared in about 3–4 weeks, and micro-seeding, when successful, decreased the crystal growth time to 4–7 days.
X-ray data collection
TreY324F/loxLTR crystals were harvested using a cryoloop (Hampton Research Inc), transferred to a cryogenic solution (10 mM sodium citrate, 35% Peg 6000, 15% ethylene glycol), flash frozen in liquid nitrogen (LN2) and stored in LN2. X-ray data were collected at 100 K at the Advanced Light Source Beamline 8.22 (Lawrence Berkeley National Laboratory, Berkeley, CA, USA). X-ray data were reduced with HKL2000 (22) and MOSFLM (23). The crystals grew in the space group P21 (a = 70.26 Å, b = 193.39 Å, c = 89.02 Å, β = 111.25°), with a complete tetrameric complex (four copies of TreY324F and two copies of loxLTR dsDNA) in the asymmetric unit cell.
Structure determination, refinement
The TreY324F/loxLTR structure was solved by molecular replacement using the program PHASER (24), within the CCP4 suite (25) of crystallographic programs. The CRE/loxP structure (PDB ID: 1Q3U) was used as the initial search model. Both the program Refmac5 (26), within the CCP4 suite, and the program Phenix (27) were used to refine the structure at different stages. The molecular graphics program Coot (28) was used for manual rebuilding between successive rounds of refinement. In addition, near the end of the refinement, the coordinates were submitted to the PDB_REDO server (29) to further optimize the model. The final refined coordinates were deposited to the RCSB data bank (PDB ID: 5U91).
Structural analysis
Figures of molecular structures were made using PyMOL (The PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, Limited Liability Company). Figures of protein–DNA interaction surface areas were calculated using the PyMol plugin PDVis1.2 (30). Superpositions of structures were performed using the program ALIGN within PyMOL, SSM and LSQ within Coot. Superposition of the active site residues (Arg 173, Lys 201, His 289, Arg 292, Trp 315 and Tyr 324) was performed within PyMOL using the command pair_fit. To calculate relative rotations between Tre and Cre tetramers, one Tre monomer was superimposed upon one Cre monomer. Then, the amount of rotation required to superimpose each remaining monomer, was calculated. Sequence alignments were performed using the programs Esprit (31) (http://espript.ibcp.fr) and JalView (32). RMSDs between Tre and all Cre structures were calculated using PDBePISA (33) and PDBeFOLD (34). Surface areas were calculated using the program PISA (33). Nucleotide conformation were analyzed using the program 3DNA (35). Interaction diagrams were based upon analysis using the programs PDBSUM (36), CCP4 and PISA. Nucleotide–protein interactions of Cre-DNA structures deposited in the PDB database were also analyzed using the NPIDB database (37) (PDB IDs used include 3C29, 2HOI, 1CRX, 1Q3U, 1NZB).
Molecular modeling
A 3D structural model of a cleaving conformation of Tre bound to the right arm loxLTR sequence was generated. This model was made using the coordinates of the cleaving Tre conformer and modeling the right arm loxLTR sequence onto the left arm loxLTR sequence, by altering the necessary bases, but not altering the DNA backbone coordinates, using the molecular graphics program COOT.
Mutagenesis and activity assays
For activity tests in E. coli, recombinases were expressed from the pEVO vectors containing the target sites loxLTR or loxP, respectively (described in (18)). To generate the described Tre mutants, site-directed mutageneses were performed using the Q5® Site-Directed Mutagenesis Kit (NEB) following the manufacturer's instructions.
To assay recombination efficiency of the Tre mutants, plasmid DNA (pEVO-mutant-target) was isolated from L-arabinose (100 μg/ml) induced overnight cultures and digested with BsrGI and XbaI (NEB), resulting in different fragment sizes for recombined versus non-recombined substrate on an agarose gel.
RESULTS AND DISCUSSION
Mutational analysis of Tre on loxLTR
To understand the functional importance of the individual amino acid changes in Tre, each change in Tre was mutated back to its Cre counterpart and tested for activity in bacteria, using arabinose to induce expression of the mutated Tre enzymes (Figure 2A). This is the same type of assay used to identify the mutations leading to Tre and other designer recombinases (17,18). Of the 19 altered residues in Tre, 17 caused a strong to moderate decrease in activity against loxLTR when mutated back. Interestingly two mutations (at positions 30 and 35) increased activity. As discussed in detail below, many of the residues that cause the greatest decrease in activity contact the DNA directly (positions 262, 263, 317 and 320), and one (position 93) makes a water-mediated DNA contact. These residues are directly involved in changing specificity. Interestingly, changes near the N-terminus, which is not traced in Tre or earlier Cre structures, also appear to be important for the altered specificity, suggesting that the N-terminus has a yet unidentified role for target-site recognition. This result is surprising, as deletion of the first 12 AA from Cre has shown no effect on recombination on loxP (38). However, these results were obtained in in vitro recombination assays only. Future work is required to investigate the role of the N-terminus of Cre-like recombinases for target site selectivity.
Figure 2.

Recombination efficiency and specificity of Tre mutants on loxLTR and loxP. Agarose gel showing the activity of Tre mutants on loxLTR (A) and loxP (B), respectively, in comparison to Tre. Escherichia coli cells harboring the pEVO vector containing the respective recombinase coding sequence and indicated target site were grown at 100 μg/ml L-arabinose. Recombination was assayed by restriction enzyme digest, resulting in a smaller fragment for recombined (one triangle) and a larger fragment for non-recombined substrate (two triangles). M, DNA marker.
To determine whether Tre-to-Cre mutations cause the enzyme to revert its specificity back to loxP, we also examined the activity of a subset of these mutations against the Cre substrate (Figure 2B). As discussed below, mutations at positions 35 and 93 showed some activity against loxP. Individual mutations of DNA-contacting residues (262, 263, 317 and 320) were not sufficient to revert Tre's activity to loxP. Collectively these data show that the overwhelming majority of the Cre-to-Tre changes contribute to the activity of Tre against loxLTR.
Structural overview of Tre/loxLTR
In our crystal structure, Tre binds loxLTR, and forms a synaptic assembly wherein four copies of Tre and two copies of the intact, double-stranded DNA form a tight complex. The active site Y324F mutation traps the complex at this stage. The crystals are of space group P21, and the tetrameric model of TreY324F/loxLTR has been refined to 3.1 Å resolution (Table 1). The 19 mutations in Tre relative to Cre are distributed throughout the sequence (Figure 3A and Supplementary Figure S2). The location of these mutations is shown on the TreY324F structure as red spheres in Figure 3B–D. The Tre/loxLTR crystals are not isomorphous with those seen in earlier Cre/DNA structures, but the antiparallel arrangement of the tetrameric recombinase and DNA is similar. Two molecules of Tre bind each loxLTR duplex; one interacts with the ‘left’ arm, and the other with the ‘right’ arm (Figure 3D). Like Cre, Tre forms a C-clamp shape around each arm of the DNA (Figure 3C), with an N-terminal domain (AA 20–129) and C-terminal domain (AA 132–341) forming each end of the C-clamp. As in Cre/loxP, both copies of the loxLTR DNA are bent (∼108°), and most of this bending occurs in the central, 8 bp region of the DNA, a region with few protein contacts. Alignments with earlier Cre structures reveals that the Tre subunit bound to the left arm is in the cleaving conformation, and the Tre bound to the right arm is in the non-cleaving conformation. (Catalytic residue 324 of the non-cleaving conformer is 1.6 Å farther from the scissile phosphate than the cleaving conformer).
Table 1. X-ray data collection and refinement statistics.
| Tre/loxLTR (PDB ID: 5U91) | |
|---|---|
| Data collection | |
| Space group | P21 |
| Wavelength (Å) | 0.9792 |
| Cell dimensions | |
| a, b, c (Å) | 70.26, 193.39, 89.02 |
| α, β, γ (°) | 90.00, 111.25, 90.00 |
| Resolution (Å) | 96.7–3.1 (3.27–3.1)a |
| R merge b | 0.239 (0.293) |
| R pim c | 0.116 (0.232) |
| I/σ(I) | 3.9 (1.6) |
| CC 1/2 | 0.951 (0.798) |
| Completeness (%) | 90.7 (62.4) |
| Redundancy | 4.2 (2.1) |
| Refinement | |
| Resolution (Å) | 63.0–3.10 (3.21–3.10) |
| No. reflections | 36191 (2280) |
| R work/Rfree (%) | 20.9/25.3 |
| No. atoms | 13 259 |
| Protein | 10 202 |
| DNA | 3022 |
| Water | 35 |
| B factors | 57.0 |
| Protein | 55.4 |
| DNA | 62.5 |
| Water | 30.0 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.004 |
| Bond angles (º) | 0.64 |
aValues in parentheses are for highest-resolution shell.
b
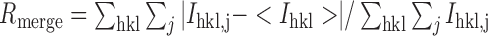
c
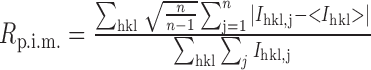
Figure 3.

Overview of the Tre/loxLTR structure. (A) Schematic of secondary structure of Tre. The amino acid sequence of Tre is shown as a gray box and numbered below. Helices are indicated by blue boxes. Location of mutations relative to Cre are shown as blue lines. Protein–DNA phosphate interactions, and base-specific interactions are shown as pink triangles and stars, respectively. The location of the nucleophile 324 is indicated by a yellow line and yellow star. (B) Ribbon diagram of Tre/loxLTR tetrameric complex. Mutations in Tre relative to Cre are shown as red spheres centered at the C-α atom, and labeled. The mutated catalytic nucleophile Y324F is shown as orange sticks. The cleaving and non-cleaving conformers are colored cyan and yellow, respectively. The angular rotation required to superimpose Tre molecules is indicated. (C) Ribbon diagram of Tre monomer. Two views are shown for clarity. Mutations and α-helices are labeled. N and C termini indicated. A small view of the monomer on DNA is shown below. The loxLTR DNA is colored gray where its sequence matches loxP, and pink, where its sequence differs. The location of the scissile phosphates (blue spheres) is indicated. (D) Ribbon diagram of a Tre/loxLTR dimer.
Within the crystal, the second pair of Tre molecules, bound to the second copy of the DNA, are arranged in an antiparallel orientation relative to the first (Figure 3B). The Tre molecules related by this non-crystallographic symmetry are very similar. Superposition of the two cleaving monomers or the two non-cleaving monomers gives a root mean square deviation (RMSD) of ∼0.3 Å over Cα’s from AA 21–340. The cleaving and non-cleaving Tre monomers superimpose less well as evidenced by an RMSD of 2.3 Å. In this superposition, the N-terminal domains superimpose more closely (RMSD 0.7 Å) than the C-terminal domains (RMSD 2.7 Å) (Figure 4A). Relative to the N-terminal domain, the C-terminal domain is rotated ∼6° between the cleaving and non-cleaving conformers. These observations are consistent with the differences observed between the cleaving and non-cleaving conformations of Cre (39).
Figure 4.

Superposition of cleaving and non-cleaving conformers of Tre. (A) The N-terminal domains of cleaving conformer (cyan) and the non-cleaving conformer (yellow) are superimposed (RMSD ∼0.4 Å). The C-terminal domains differ by ∼6° rotation, and superimpose less well (RMSD ∼2.7 Å). The mutations relative to Cre are shown as red sticks. The conformational changes result in a displacement of the helices M and N at the C-terminus. The loop region (AA 197–209) also exhibits a large conformational change (∼14 Å distance between Thr 206). This loop is colored orange (non-cleaving conformer), and blue (cleaving conformer) for clarity. (B) Comparison of non-cleaving Tre and Cre conformers. Non-cleaving conformer of Tre (yellow) and Cre (orange) were superimposed. The location of Tre mutations are shown as small red spheres. (C) Comparison of cleaving Tre and Cre conformers. Cleaving conformer of Tre (cyan) and Cre (orange) were superimposed. The PDB ID used in this superposition was 3C29. (D) Superposition of Tre and other Cre structures. For each Cre tetramer, a Cre cleaving conformer was superimposed onto the Tre cleaving conformer. The results are displayed as a ribbon diagram with Cre/loxP colored gray and Tre cleaving and non-cleaving conformers colored blue and purple, and loxLTR colored red. The PDB IDs of the Cre/loxP structures used in this superposition are 1NZB, 4CRX, 5CRX, 3C28, 3C29, 1Q3U, 2HOF, 2HOI, 1Q3V, 1OUQ, 1CRX, 3MGV, 1PVP, 1PVQ, 1PVR, 1MA7. Note that Tre is often at the extreme ‘edge’ of the range of variability seen for the other Cre structures.
Global changes of Tre/loxLTR relative to Cre/loxP
Conformational comparison of Tre to Cre
Twenty-three crystal structures of Cre/loxP representing various states along the reaction pathway are available from the Protein Data Bank. Although, there are many sidechain differences, the backbone structures of Tre and Cre monomers are generally similar, as evidenced by low RMSDs (0.7–2. 0 Å on α carbons) from the superposition of Tre monomers with all published Cre/loxP structures (Supplementary Table S1). The 2.2 Å structure of a synaptic complex (PDB ID: 3C29, K201A mutation) was selected for our comparisons because both the protein backbone and the DNA backbone seemed closest to the structure of TreY324F/loxLTR reported here. Unless indicated otherwise, comparisons discussed below are relative to this Cre/loxP structure. Superposition of the non-cleaving Tre and Cre conformers results in an RMSD of ∼0.94 Å (over 322 Cαs), Figure 4B. The largest differences between Tre and Cre are in the bending loop region (AA 198–208), a flexible region that contains no mutations and is disordered in some Cre/loxP structures. Superposition of the cleaving Tre and Cre conformers results in an RMSD of ∼0.8 Å over 322 Cαs (Figure 4C). Regions of larger differences (RMSDs > 1.3 Å) of the Cα backbone occur at amino acids 189–191, 244–247, 276–279, 330–340. Only one of these regions (AA 244–245) contains amino acid changes in Tre. Superposition of the C-terminal domains (AA 130–310, omitting helices M and N) results in a very low RMSD of 0.45 Å. The residues that move the most are the C-terminal residues that are involved in protein–protein interactions. These modest changes propagate throughout the complex, as seen by the superposition of the Tre tetramer with previously published Cre structures (Figure 4D).
Changes in DNA conformation
In Cre/loxP structures, DNA bending has been shown to occur primarily in the central region, but not always in the same location. As in 3C29, our structure shows a kink adjacent to the non-cleaving scissile phosphate (Figure 5). Both structures exhibit a similar, large negative roll (−30°) adjacent to the scissile phosphate in the inactive arm of the complex. A negative roll was also observed in the Cre/loxP structure of a synaptic complex (PDB ID: 2HOI) (40) as well as in the presence of loxS (41), a symmetrized loxP sequence having an even larger negative roll (−49°).
Figure 5.

Tre/loxLTR DNA conformational analysis. (A) loxLTR DNA parameters Roll versus loxLTR sequence. The loxLTR sequence is shown below the graph and colored pink where there is a mutation relative to loxP. The letters in bold indicate the location of the 13 bp ‘arms.’ The pink and blue circles indicate the location of the active and inactive scissile phosphates, respectively. The lower case letters indicate the nucleotides present in the crystal structure. (B) A stick representation of the loxLTR DNA with a line representing the helical axis. The strands are colored beige and gray and are colored pink where there is a mutation relative to loxP. The scissile phosphates are depicted as spheres colored as in A. (C) (left) A close-up view of the boxed region from B, highlighting the kink adjacent to the inactivated phosphate. (right) A surface representation of the central region highlighting the width of the major and minor grooves, and the location of scissile phosphates shown as spheres.
Detailed Tre-loxLTR analysis
The structural analyses detailed below demonstrate that Tre's recognition of loxLTR is achieved by an assortment of strategies including altered interactions of non-mutated residues with protein or DNA, loss of base-specific interactions, novel interactions with the mutated bases and changes in DNA conformation. In addition, we see protein–protein interface differences that are likely responsible for large-scale domain movements. We also see changes near the active site that may help compensate for these large-scale movements.
Of the 19 mutations in Tre, only nine contact the DNA in some way. Among these, three make sequence-specific contacts (AA 94, 244, 259); the others interact with the DNA backbone. Three of the DNA-contacting residues also interact with a neighboring Tre molecule (AA 131, 317, 320). The apparent role of each mutated residue is summarized in Supplementary Table S2. Protein–DNA interactions for both Cre/loxP and Tre/loxLTR are summarized in Figure 1, and a detailed schematic is shown in Supplementary Figure S3. The extensive interactions between the protein and the phosphate backbone (red plus signs) are highlighted in Figure 1. Six of the DNA-contacting mutations in Tre map to the ‘recognition helices’ (αB, αD and αJ) that lie within the major groove of the DNA: K43E on αB, G93C and Q94R on αD, and R259Y, E262Q, and G263R on αJ. Some of these mutations (e.g. Q94R, K43E and K244R) form or prevent direct, base-specific contacts. The others contact the phosphate and/or ribose backbone. As in Cre/loxP (reviewed in (10)) some of these interactions, notably those at positions Arg 94 and Glu 43, are buttressed by well-ordered water molecules. The text below describes the most important specificity determinants from the perspective of the DNA. Because the two copies of the Tre/loxLTR in the asymmetric unit are not completely identical, the interactions noted do not always appear in both complexes. In addition, the cleaving conformer of Tre interacts more closely with the left arm of the loxLTR, hence there are more sequence-specific protein–DNA interactions with the left arm, than the right arm, therefore, we have organized the following section by left arm loxLTR location. We have added information of the analogous position in the right arm where interesting.
loxLTR mutated base position 5: Ade-Thy to Thy-Ade
Altered specificity in recognition helix D: K86, Q90, Q94R
loxLTR contains a mutated base-pair at position 4′ on the right arm (Ade-Thy) and at position 5 on the left arm (Thy-Ade). Helix αD (AA 85–102) lies in the major groove near the scissile phosphate which is located between positions 4 and 3, or 4′ and 3′, respectively, of the DNA. Within helix αD, the sidechains of Lys 86, Gln 90 and Gln 94 are especially noteworthy. In Cre/loxP, Lys 86 makes base-specific interactions in both conformers to bases at position 4 or 4′. In the left arm, Lys 86 forms an H-bond with N7 of Ade 4 (Figure 1). In the right arm, it forms an H-bond with the O6 of Gua 4′. In the cleaving-conformer of Tre, however, Lys 86 no longer contacts the DNA at all and forms a hydrogen bond with Gln 89 sidechain (Figure 6). Superposition of Tre and Cre helix αD show that in Tre, Lys 86 is farther from the DNA, and no longer forms a hydrogen bond with the N7 of Ade 4. In one copy of the non-cleaving Tre conformer, there is no sidechain electron density for Lys 86. In the other copy, Lys 86 again preferentially interacts with Gln 89, and not the DNA. Therefore, altered DNA conformation and the addition of a hydrogen bond between Gln 89 and Lys 86 results in the loss of a Cre-DNA specific interaction at positions 4 and 4′.
Figure 6.

Comparison of Tre/loxLTR and Cre/loxP protein–DNA interactions. (A) Overview of selected Tre segments on loxLTR shown as a ribbon diagram. Shown is the left arm of the LoxLTR colored gray (conserved) and magenta (mutated). The scissile phosphates shown as blue spheres. The Tre regions shown are labeled, and the location of the mutated residues present in these regions is shown as red spheres. (B) (Left) Close-up of the Tre Q94R/loxLTR interaction. Helix D from the cleaving Tre monomer (cyan, mutations colored red) bound to loxLTR (colored magenta where sequence differs from loxP) is shown. (Right) This shows the same region of Cre (light brown)/loxP. Hydrogen bond or polar interactions are indicated by dashed lines as determined by PyMOL or distance measurements. Below is the loxLTR or loxP sequence with the relevant base in a larger font to indicate where this interaction occurs in the DNA target.
Residues 90 and 94 appear to work in concert. Gln 90 makes base-specific interactions in both Cre/loxP and Tre/loxLTR, but these interactions differ. In Cre/loxP, the Gln 90 sidechain is positioned in the major groove where it is locked in place by interactions both with DNA (forming hydrogen bonds with O4 of Thy 6 and N6 of Ade 5, and with the opposite DNA strand) and with the sidechain of Gln 94, a residue mutated to Arg in Tre. In contrast, in Tre, Gln 90 must alter its sidechain conformation to prevent a steric collision that would otherwise occur with Arg 94. Therefore Gln 90–DNA interactions shift by one base pair relative to loxP (from positions 6–5 to positions 5–4).
In Cre, the sidechain of Gln 94 is too short to make any direct interaction with the DNA. The longer sidechain of the Q94R mutation adds a new a base-specific interaction with the N7 of Ade 5 (altered from Thy 5 in loxP). In Tre, Gln 90 also helps lock the Q94R sidechain into position through a hydrogen bond. In the right arm of loxLTR, Arg 94 is too far from Ade 4′ and instead interacts with the phosphate backbone, again, buttressed by Gln 90. Together, the altered conformation of Gln 90 and the Arg 94 mutation recognize the altered base pair Thy-Ade at position 5.
loxLTR mutated base position 9: Gua-Cyt to Cyt-Gua
Altered specificity in recognition helix B: K43E
In Cre/loxP Lys 43, located on helix B, forms a hydrogen bond with N7 of Gua 9 (Figure 7). The mutation of Gua 9 to Cyt 9 in the left arm of loxLTR replaces a hydrogen bond acceptor (N7 of Gua) with a hydrogen bond donor (NH2 of Cyt). In addition, superposition of Tre and Cre helix B, shows that the DNA conformation differs and that the sidechain of Cre's Lys 43 would sterically collide with the Cyt 9 of loxLTR. Therefore Lys 43 cannot form a hydrogen bond with Cyt at this position. In Tre, the K43E mutation is simply too far to interact with the DNA. Our mutational data, however, indicate that this change is of little consequence, as changing Tre residue 43 back to Lys has only a very mild effect on activity (Figure 2A).
Figure 7.

Close-up of the Tre K43E/loxLTR interaction. Colored as in Figure 6. On the right panel, Tre/loxLTR are shown as transparent sticks.
loxLTR conserved base position 10: Cyt-Gua
Altered specificity in recognition helix J: R259Y, E262Q, G263R
The loxLTR sequence is the same as loxP at position 10 in the left arm, but the Gua and Cyt bases are flipped in the right arm at position 10′. The DNA recognition helix αJ contains three Tre mutations: R259Y, E262Q and G263R. In Cre, the Arg 259 sidechains form strong bidentate interactions with Gua 10 (left arm) and Gua 10′ (right arm): the Nε with O6 and the Nη with N7 and this residue has been identified as a primary determinant of (42) specificity. The R259Y substitution results in the loss of these hydrogen bonds (Figure 8). Superposition of helix J shows that the loxLTR DNA has shifted away from Tre. In Tre, in general, Y259 is not correctly orientated to form a strong hydrogen bond with loxLTR, although there are van der Waals contacts. In one of the cleaving conformers, a polar interaction between the OH group of Tyr 259 and the N7 of Gua 10 is observed. In the other copies, the Tyr 259 sidechain is too far to form a hydrogen bond, and more closely resembles an edge-to-face (T-shaped) interaction with the DNA. This type of pi–pi interaction is stabilizing and may be involved in nucleic acid recognition (43). The G263R mutation appears to stabilizes the Tyr 259 by forming favorable interactions along the sidechain. Arg 263 is further locked into place via a hydrogen bond from Glu 266.
Figure 8.

Close-up of the Tre R259Y/loxLTR interaction. Colored as in Figure 6. On the right panel, Tre/loxLTR are shown as transparent sticks. In addition, Y324F from each of the two copies of the cleaving Tre is shown to highlight the conformational variability, where one is within hydrogen bonding distance. Also the loxLTR G 10 shifts relative to loxP ∼ 1.3 Å.
Cre/loxP structures reveal that the negatively charged sidechain of Glu 262 has an unusual direct interaction with the phosphate backbone (between position 7 and 8 on the bottom strand). In Tre, the Gln 262 forms a ‘traditional’ phosphate interaction at the analogous position as in Cre. In Cre, Glu 262 was previously identified as a ‘gatekeeper’ residue that relaxes specificity when mutated to almost any other amino acid (44,45). In Cre, the E262Q mutation appears to lower specificity and increase overall affinity for the DNA (45). Interestingly, mutations at position 262 allowed Cre to recombine loxP sites mutated at position 11 and 12, positions where no direct base interaction occurs (44). The authors proposed that the Glu 262 mutation allows helix J flexibility to reorient itself within the major groove, although this comes at the price of lower selectivity ((44) and reviewed in (11)). These data reinforce the idea that altered selectivity often proceeds through a state of relaxed specificity. Indeed, for the evolution of Tre, Glu 262 was altered early in the selection process. Later, as the DNA targets were closer to loxLTR, Arg 259 and Gly 263 were observed to co-evolve for increased specificity, usually to R259Y and G263R (18). Interestingly, Gln 262 is required for Tre activity, and its mutation back to Glu does not relax specificity, (Figure 2). Thus it appears necessary for selectivity in the context of the other Tre mutations.
loxLTR mutated base position 16: Thy-Ade to Cyt-Gua
Altered specificity in minor groove: K244R, N245Y
loxLTR contains mutated base pairs at position 16, 16′ and 17′. Residues in the loop preceding recognition helix J (AA 241–245) make base-specific interactions in the minor groove in this region. Tre contains two mutations in this region: K244R and N245Y. In Cre, Lys 244 interacts with both strands and forms hydrogen bonds with O2 of Thy 16 and Thy 17, and Asn 245 only interacts with the phosphate backbone at position 17 (Figure 9). In Tre, the two amino acid substitutions force a widening of the minor groove to avoid steric collisions. The K244R mutation is too far to interact with the O2 of the Thy at position 17, but it does recognize the O2 of Cyt 16. In the right arm, Arg 244 moves away from the NH2 of Gua 16′, and instead interacts with the O2 of Cyt 16′ and with the O2 of Cyt 17′ in loxLTR. The Tyr 245 sidechain packs alongside and stabilizes the sidechain of Arg 244. In Tre, the N245Y mutation results in a loss of the Asn 245-phosphate interaction and the DNA backbone is farther from the protein, resulting in a wider minor groove at this point.
Figure 9.

Close-up of the Tre K244R, N245Y/loxLTR interaction. Colored as in Figure 6. On the right panel, Tre/loxLTR is shown as transparent sticks to highlight the conformation differences in this region.
Tre mutations not involved in DNA sequence-specific interactions
Altered protein–protein interface: M30V, Q35P, A131T
Three Cre to Tre mutations (M30V, Q35P and A131T) occur at the protein–protein interfaces of the tetrameric recombinase complex. Disruption of these interfaces are known to have functional consequences in Cre (46). In Cre/loxP, Gln 35 of the non-cleaving monomer and Glu 123 on the adjacent, cleaving monomer form a hydrogen bond. Prolines often add rigidity to a structure, and the Q35P mutation can no longer form the intermolecular hydrogen bond (Figure 10A). Interestingly, comparison of this interface to Cre/loxP shows the loss of another inter-protein salt bridge between Arg 32 (on the non-cleaving monomer) with Glu 69 (cleaving monomer) (Figure 10B), neither of which was mutated. Both of the M30V and Q35P mutations result in a loss of hydrogen bonds, and hence a weaker protein–protein interface. We expect that these changes allow the monomers to make minor conformational adjustments to improve the protein–DNA interactions. Such changes may have been beneficial during the evolution process leading to Tre and might not be possible when the protomers are locked in a particular orientation as in Cre. Indeed, earlier work proposed that Cre mutants R32V and R32M would lose the inter-molecular salt bridge of Arg 32 with Glu 69, and that this weakened interface would reduce the cooperativity and enhance accuracy to loxP (46). The authors propose this as a general method to improve specificity of dimeric DNA-binding proteins such as Cre. The crystal structure presented here only partially supports this hypothesis since in the fully evolved recombinase, mutating either of these back to Cre (V30M or P35Q) increased Tre's ability to recombine loxLTR. In the case of M30V, we believe this is partially because the smaller sidechain leaves a small void in Tre (Figure 10B). The P35Q mutation likewise relaxed specificity such that the mutated enzyme also exhibited increased activity against loxP. Interestingly, the M30V mutation also appeared in another evolved recombinase, Brec1 (47), suggesting that it serves an important purpose. These mutations highlight the importance of the protein-protein interface in both recognition and catalysis.
Figure 10.

Tre mutations not involved in sequence-specific DNA interactions. (A) Close-up of Tre Q35P mutation. This view highlights loss of an intermolecular Gln 35 and Gln 123 hydrogen bond in the Q35P mutation. The mutated residues are colored red. Tre (yellow and cyan) and Cre (tan) are superimposed. Hydrogen bonds and salt bridges are shown by yellow dashed lines. (B) Close-up of Tre M30V mutation at protein–protein interface. This view highlights loss of salt bridge between Arg 32 in Glu 69 in the Tre/loxLTR complex. A close-up of the boxed portion shows the cavity formed by Val 30 (red spheres) versus Met 30 (tan spheres) (C) Close-up of Tre A131T mutation. This view highlights additional inter-molecular hydrogen bond in Tre between Thr 131 and Leu 203. (D) Close-up of N317T, I320S Tre mutation. This view highlights new intra- and intermolecular polar interactions.
The A131T mutation sits at a protein–protein interface, in the region of extended structure (AA 128–133) between the N-terminal and the C terminal domains. A131T interacts with residues in the flexible loop region (AA 198–208) that adopt different conformations, hence the interactions are non-identical. In the non-cleaving conformer, A131T sidechain is close to both the phosphate backbone at position Ade 5′ on the right arm and the sidechain of Thr 206 of an adjacent monomer from the opposite loxLTR (Figure 10C). These extra contacts cannot form in the Cre/loxP structure.
Tre mutations impacting active site orientation: N317T, I320S
Finally, Cre utilizes residues in the C-terminus to communicate allosterically with the neighboring protomer. Two residues in this region have mutations in Tre: N317T and I320S. Three new hydrogen bonds between I320S and N317T and the phosphate of Ade 2 (next to the scissile phosphate at Cyt 3) are formed, locking the position of the loop (Figure 10D). The N317T mutation also results in the formation of a new intermolecular hydrogen bond between Asn 319 and Thr 316 of the adjacent monomer. These mutations may be important for correctly orienting the catalytic nucleophile, Tyr 324 and the active site residues. Superposition of the active sites of Tre and Cre (PDB ID: 3C29) reveal a striking similarity in the positions of the catalytic residues (RMSD = 0.21 Å over 6 Cαs). This suggests that these substitutions compensate for unfavorable positional changes induced by other specificity-determining substitutions. Changing either position 317 or 320 to their Cre counterparts causes a complete loss of activity against loxLTR. The changes in Tre at these positions may compensate for movements near the active site induced by the other residues necessary for altered specificity.
Comparison of loxP and loxLTR half-sites
To assess if the DNA conformation between the cleaving and non-cleaving conformers plays a different role in the Tre/loxLTR structure, we first superimposed the left loxP half-site (or arm) onto the right loxP half-site (PDB ID: 3C29). This analysis reveals that the loxP repeats have a nearly identical conformation and the DNA backbone differ from each other most within the central spacer region, starting near the scissile phosphates. Nonetheless, there are differences in how Cre interacts with each arm. For example, the sidechain of Lys 43 interacts specifically with Gua 9 in the left arm, but points away from the DNA in the right arm. Next, we superimposed the left loxLTR arm onto the right loxLTR arm (182 backbone atoms, RMSD = 0.83 Å). Unlike the loxP half-sites, the conformation of the loxLTR arms begin to differ from each other earlier than loxP at position 6 and then more dramatically as it approaches the scissile phosphates (between positions 3 and 4). These differences result in different protein–DNA interactions. For example, in Tre, Arg 94 interacts specifically with altered base Ade 5 in the active arm, but with the phosphate backbone in the non-specific arm.
Modeling the cleaving conformation on the right arm
The two loxLTR arm sequences are identical at only 5 of 13 positions (bases 6,7,11,5,16) (Supplementary Figure S4). Since our structure only shows the cleaving conformation of Tre bound to the left arm, and during the recombination cycle, the cleaving conformation must interact with the DNA sequence of the right arm, we generated a 3D model of Tre in a cleaving conformation with the right arm of loxLTR (see ‘Materials and Methods’ section). In this model, three sequence-specific contacts are conserved between both arms: Arg 244-Cyt 16, Arg 243-Thy 15 and Arg 282-Ade 7. Tyr 259 appears to provide van der Waals stabilization, but not high specificity at position 10 (Cyt-Gua or Gua-Cyt). loxLTR positions 4 and 5 are not conserved: Thy5pAde4 versus Ade5pThy4. This location involves sequence-specific interactions with Gln 90, Arg 94 and Lys 201. Gln 90 can easily rotate its sidechain to interact with either a Thy-Ade or Ade-Thy at position 5. Similarly, Lys 201 can easily interact with either an Ade or Thy at position 4. Arg 94 which interacts with Ade 5 in the left arm, must adjust its sidechain to avoid a clash with the Thy 5, but could interact with Ade 4′ in the right arm. These observations suggest that Tre interacts specifically at positions near the ends of each ‘arm’, and less so in the central portion of the arms (i.e. nucleotide and position 12 in Figure 1). This is consistent with the finding that these regions of the DNA target are important in Cre/loxP specificity (48).
CONCLUSION AND FUTURE DIRECTIONS
The structure of Tre in complex with its DNA target, loxLTR, details the molecular basis for the dual specificity required by engineered, Cre-like recombinases that target highly asymmetrical DNA sequences. Despite the 13-bp sequence identity present within the two halves of loxP, Cre recombinase interacts with the 13-bp sequences differently: one via the cleaving conformation, the other via the non-cleaving conformation. This inherent property of Cre, along with the intrinsic flexibility of both the protein and the DNA, helps explain the ability of Tre and other engineered recombinases to recognize asymmetric targets. Given the size of the dimer footprint on the DNA substrate, it is also important that there are relatively few direct base contacts made by Cre and Tre to loxP and loxLTR, respectively. This and other work also demonstrate that not every position in the DNA substrate is specifically targeted. The presence of so few direct protein-base contacts highlight this point. Thus, it is important to identify the key positions on the DNA substrate when choosing targets and designing new recombinases. Not every mutation that arose in the evolution of Tre can be explained in the context of altered specificity. We speculate that some of these may have been structurally important during the evolution process, but are now dispensable. This may explain why mutating positions 30 and 35 back to their original, Cre amino acids enhances Tre activity. In addition, this work shows how both mutated and non-mutated residues in Cre can interact with an altered DNA target like loxLTR.
Mutagenesis shows that the almost all of the 19 mutated residues in Tre contribute to the altered enzyme's activity against loxLTR. Thus, it is difficult to derive a single ‘take-home message’ from this work. Previous structural studies of mutant Cre/mutant loxP have demonstrated that Cre can utilize multiple mechanisms to recognize altered sequences ((49,50) and reviewed in (10)). Our findings expand on these themes. From a structural perspective, it appears that the specificity change in Tre was largely achieved, because Tre/loxLTR loses three important, base-specific contacts (at K43E, K86 and R259Y), alters a base-specific interaction (K244R) and gains a new base-specific contact at Q94R. K43E, Q94R and K244R appear at modified base positions 9, 5 and 16 respectively. The structure strongly suggests that conformational flexibility of the protein and DNA are also important. For example, mutated bases at position 5 in loxLTR and changes in nearby amino acids alter the conformations of non-mutated amino acid Lys 86 and Gln 90. Another specificity-altering strategy we observe involves disruption of the protein–protein interfaces by mutations such as M30V and Q35P. It is unclear why the Q35P and M30V mutations arose, since the structure and the behavior of the single revertants indicate that these substitutions were detrimental to Tre function. One explanation, shown in earlier work (46) is that weakening these interfaces allows the monomers to more tightly engage the DNA. This may be beneficial during the molecular evolution process, and it would explain why similar mutations are found in another engineered recombinase (47). Mutations which do not seem important for selectivity, but may be necessary for correct orientation of the active site, specifically N317T and I320S, also occur. In addition, mutagenesis demonstrates that residues 7, 9 and 10, which are in a region not seen in Tre or any Cre structures are important for activity against loxLTR. Tre is an enzyme involved in a multistep reaction that includes complex structural changes. Only one step in this pathway is illustrated by our structure. Thus, additional specificity likely occurs at other points in the reaction pathway.
The number of changes in the DNA arms relative to loxP and the considerable asymmetry of the loxLTR substrate (8 of 13 nt in the arms differ) distinguish Tre from earlier Cre mutants and naturally-occurring homologs. For instance, recent characterization of two new Cre-like recombinases called Nigri and Panto allowed identification of key residues that alter specificity (51). Like Cre, however, these enzymes act on symmetrical substrates. Interestingly, Cre mutations of R259P and G263K were sufficient to recombine rox, a substrate that differs at positions 8–10 within the arms (51). Similarly, CreA174-L258-S259-H262-G266 acts on loxM7, a substrate where the arms differ at positions 7–9. In the crystal structure, CreALSHG recognizes loxM7 by altering the hydration network to create new protein–DNA contacts (50). In contrast, the Cre variant CreL174-N258-S259-G262-G266 which recognizes both loxP and loxM7 (and presumably also acts on asymmetrical combination of these substrates) created novel base contacts through a structural shift of the DNA (50). The Tre/loxLTR structure builds upon these themes and reveals a combination of strategies for recognition of an altered DNA target. Many of these structural changes also involve disruption or alteration of extensive hydrogen bonding networks.
Like Tre, XerH tyrosine recombinase from Helicobacter pylori also acts on an asymmetrical DNA substrate; one arm contains an added base pair near the middle relative to the other. In contrast to Tre, where the same amino acids must engage different bases on each side of the complex, the crystal structure of XerH in complex with its DNA substrate shows largely similar contacts with both arms (52). As in Tre/loxLTR, flexibility in both the DNA and the protein are important for recognizing of the asymmetrical substrate.
The holy grail of genome editing is the ability to target any DNA sequence for insertion, deletion or inversion. Because they act with high efficiency and with single-base resolution, engineered variants of Cre have certain intrinsic advantages over nuclease-based approach such as the CRISPR/Cas9 system. Our work with Tre demonstrates that dual specificity recombinases can be used for genome editing, but it is important to note that an alternative approach, using combinations of individually-engineered Cre recombinases has also shown promise. Biochemical studies of Cre-variants that are designed to preferentially form 2:2 heterotetramers represent a step toward this goal (53,54). The Tre/loxLTR structure highlights an important challenge with this alternative approach, namely that the protein–protein interface may change significantly during the evolution of the altered specificity. Remodeling of these interfaces is an important consideration in the design of Cre-like heterotetramers.
The primary value of this structure is the insight it provides into rational design of improved recombinases with altered specificities. Indeed, Tre has already been the subject of such work, using structure-based modeling (based on Cre/loxP) and molecular dynamics simulations to predict structural changes (55). X-ray structures are able to provide better accuracy, particularly with respect to DNA conformation and water molecule participation in DNA recognition. For instance, the crystal structure of Tre/loxLTR described here reveals a significant widening of the major groove in the central region which was not anticipated by previous modeling studies (55). Also, our structure explains why mutating Val 30 back to Met results in an improved Tre recombinase.
In recent years, a growing number of recombinases have been engineered (10). Of particular importance in the HIV field is the recently Cre-based engineered recombinase Brec1 (47). Like Tre, Brec1 targets a sequence in the HIV-1 LTR. Whereas the loxLTR sequence is fairly rare in HIV-1 positive patients, the sequence targeted by Brec1 is present in at least 80% of HIV-1 isolates. Moreover, using a mouse model and a lentiviral delivery scheme, Brec1 has been shown to efficiently excise the HIV provirus from patient-derived cells. We anticipate that the continued combination of clever genetic selection schemes, molecular modeling and experimental X-ray crystal structures will further enhance the utility and power of these potentially life-changing therapeutics. This structure has shown that a combination of factors including DNA and protein flexibility, the fact that not every position of the DNA is recognized and the inherent structural asymmetry of Cre–DNA complexes are all important. Our structure, along with the mutagenesis, shows that almost all of the Cre-to-Tre mutations contribute to the activity of the evolved enzyme against its LTR substrate, and that the individual mutations work via a variety of different mechanisms. More crystal structures of these engineered recombinases will further improve our understanding of the complex rules governing recombinase–DNA interactions and will facilitate our ability to tailor recombinase specificity as needed.
ACCESSION NUMBER
Atomic coordinates and structure factors for the reported crystal structure of TreY324F/loxLTR have been deposited with the Protein Data bank under accession number 5U91.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to acknowledge the staff at the NSLS (Vivian Stojanoff) and SSRL (BL 14.1) synchrotrons for help with X-ray data collection.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Department of Developmental, Molecular and Chemical Biology at Tufts University School of Medicine internal funding (to A.B); German Research Foundation (DFG) [BU 1400/7–1]; Federal Ministry of Education and Research (BMBF) [GO-Bio FKZ 0315090]. Funding for open access charge: Developmental, Molecular and Chemical Biology at Tufts University School of Medicine internal funding.
Conflict of interest statement. None declared.
REFERENCES
- 1. Rathbun R.C., Lockhart S.M., Stephens J.R.. Current HIV treatment guidelines–an overview. Curr. Pharm. Des. 2006; 12:1045–1063. [DOI] [PubMed] [Google Scholar]
- 2. Buchholz F., Hauber J.. Antiviral therapy of persistent viral infection using genome editing. Curr. Opin. Virol. 2016; 20:85–91. [DOI] [PubMed] [Google Scholar]
- 3. van Lunzen J., Fehse B., Hauber J.. Gene therapy strategies: can we eradicate HIV. Curr. HIV/AIDS Rep. 2011; 8:78–84. [DOI] [PubMed] [Google Scholar]
- 4. Khalili K., Kaminski R., Gordon J., Cosentino L., Hu W.. Genome editing strategies: potential tools for eradicating HIV-1/AIDS. J. Neurovirol. 2015; 21:310–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Govindan G., Ramalingam S.. Programmable site-specific nucleases for targeted genome engineering in higher eukaryotes. J. Cell. Physiol. 2016; 231:2380–2392. [DOI] [PubMed] [Google Scholar]
- 6. Wang G., Ming M., Ye Y., Xi J.. [High-throughput functional screening using CRISPR/Cas9 system]. Yi Chuan Hered. 2016; 38:391–401. [DOI] [PubMed] [Google Scholar]
- 7. Wang Z., Pan Q., Gendron P., Zhu W., Guo F., Cen S., Wainberg M.A., Liang C.. CRISPR/Cas9-derived mutations both inhibit HIV-1 replication and accelerate viral escape. Cell Rep. 2016; 15:481–489. [DOI] [PubMed] [Google Scholar]
- 8. Buchholz F., Hauber J.. In vitro evolution and analysis of HIV-1 LTR-specific recombinases. Methods. 2011; 53:102–109. [DOI] [PubMed] [Google Scholar]
- 9. Gaj T., Sirk S.J., Barbas C.F.. Expanding the scope of site-specific recombinases for genetic and metabolic engineering. Biotechnol. Bioeng. 2014; 111:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meinke G., Bohm A., Hauber J., Pisabarro M.T., Buchholz F.. Cre recombinase and other tyrosine recombinases. Chem. Rev. 2016; 116:12785–12820. [DOI] [PubMed] [Google Scholar]
- 11. Van Duyne G.D. Cre recombinase. Microbiol. Spectr. 2015; 3:1–19. [DOI] [PubMed] [Google Scholar]
- 12. Kühn R., Schwenk F., Aguet M., Rajewsky K.. Inducible gene targeting in mice. Science. 1995; 269:1427–1429. [DOI] [PubMed] [Google Scholar]
- 13. Orban P.C., Chui D., Marth J.D.. Tissue- and site-specific DNA recombination in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:6861–6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Van Duyne G.D. A structural view of cre-loxp site-specific recombination. Annu. Rev. Biophys. Biomol. Struct. 2001; 30:87–104. [DOI] [PubMed] [Google Scholar]
- 15. Grindley N.D.F., Whiteson K.L., Rice P.A.. Mechanisms of site-specific recombination. Annu. Rev. Biochem. 2006; 75:567–605. [DOI] [PubMed] [Google Scholar]
- 16. Luciw P. Fields BN, Howley PM. Human Immunodefiecency Viruses and Their Replication. Fields Virology. 1996; 3rd edn, Philadelphia: Lippincott-Raven Publishers; 1881–1952. [Google Scholar]
- 17. Buchholz F., Stewart A.F.. Alteration of Cre recombinase site specificity by substrate-linked protein evolution. Nat. Biotechnol. 2001; 19:1047–1052. [DOI] [PubMed] [Google Scholar]
- 18. Sarkar I., Hauber I., Hauber J., Buchholz F.. HIV-1 proviral DNA excision using an evolved recombinase. Science. 2007; 316:1912–1915. [DOI] [PubMed] [Google Scholar]
- 19. Hauber I., Hofmann-Sieber H., Chemnitz J., Dubrau D., Chusainow J., Stucka R., Hartjen P., Schambach A., Ziegler P., Hackmann K. et al. Highly significant antiviral activity of HIV-1 LTR-specific tre-recombinase in humanized mice. PLoS Pathog. 2013; 9:e1003587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cabrita L.D., Dai W., Bottomley S.P.. A family of E. coli expression vectors for laboratory scale and high throughput soluble protein production. BMC Biotechnol. 2006; 6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meinke G., Phelan P.J., Harrison C.J., Bullock P.A.. Analysis of the costructure of the simian virus 40 T-antigen origin binding domain with site I reveals a correlation between GAGGC spacing and spiral assembly. J. Virol. 2013; 87:2923–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Otwinowski Z., Minor W.. Carter CW Jr, Sweet RM. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Part A, Methods in Enzymology. 1997; 276, NY: Academic Press; 307–326. [DOI] [PubMed] [Google Scholar]
- 23. Leslie A.G.W., Powell H.R.. Read RJ, Sussman JL. Processing diffraction data with mosflm. Evolving Methods for Macromolecular Crystallography. 2007; 245, Dordrecht: Springer; 41–51. [Google Scholar]
- 24. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G.W., McCoy A. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011; 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murshudov G.N., Skubák P., Lebedev A.A., Pannu N.S., Steiner R.A., Nicholls R.A., Winn M.D., Long F., Vagin A.A.. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011; 67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Joosten R.P., Long F., Murshudov G.N., Perrakis A.. The PDB_REDO server for macromolecular structure model optimization. IUCrJ. 2014; 1:213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ribeiro J., Melo F., Schüller A.. PDIviz: analysis and visualization of protein–DNA binding interfaces. Bioinformatics. 2015; 31:2751–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Robert X., Gouet P.. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014; 42:W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Waterhouse A.M., Procter J.B., Martin D.M.A., Clamp M., Barton G.J.. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009; 25:1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Krissinel E., Henrick K.. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007; 372:774–797. [DOI] [PubMed] [Google Scholar]
- 34. Krissinel E., Henrick K.. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004; 60:2256–2268. [DOI] [PubMed] [Google Scholar]
- 35. Colasanti A.V., Lu X.-J., Olson W.K.. Analyzing and building nucleic acid structures with 3DNA. J. Vis. Exp. 2013; 74:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. de Beer T.A.P., Berka K., Thornton J.M., Laskowski R.A.. PDBsum additions. Nucleic Acids Res. 2014; 42:D292–D296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zanegina O., Kirsanov D., Baulin E., Karyagina A., Alexeevski A., Spirin S.. An updated version of NPIDB includes new classifications of DNA-protein complexes and their families. Nucleic Acids Res. 2016; 44:D144–D153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rongrong L., Lixia W., Zhongping L.. Effect of deletion mutation on the recombination activity of Cre recombinase. Acta Biochim. Pol. 2005; 52:541–544. [PubMed] [Google Scholar]
- 39. Guo F., Gopaul D.N., van Duyne G.D.. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature. 1997; 389:40–46. [DOI] [PubMed] [Google Scholar]
- 40. Ghosh K., Guo F., Duyne G.D.V.. Synapsis of loxP Sites by Cre Recombinase. J. Biol. Chem. 2007; 282:24004–24016. [DOI] [PubMed] [Google Scholar]
- 41. Guo F., Gopaul D.N., Van Duyne G.D.. Asymmetric DNA bending in the Cre-loxP site-specific recombination synapse. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:7143–7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim S., Kim G., Lee Y., Park J.. Characterization of Cre–loxP interaction in the major groove: Hint for structural distortion of mutant Cre and possible strategy for HIV-1 therapy. J. Cell. Biochem. 2001; 80:321–327. [PubMed] [Google Scholar]
- 43. Wilson K.A., Kellie J.L., Wetmore S.D.. DNA-protein π-interactions in nature: abundance, structure, composition and strength of contacts between aromatic amino acids and DNA nucleobases or deoxyribose sugar. Nucleic Acids Res. 2014; 42:6726–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rüfer A.W., Sauer B.. Non-contact positions impose site selectivity on Cre recombinase. Nucleic Acids Res. 2002; 30:2764–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gelato K.A., Martin S.S., Wong S., Baldwin E.P.. Multiple levels of affinity-dependent DNA discrimination in Cre-LoxP recombination. Biochemistry. 2006; 45:12216–12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eroshenko N., Church G.M.. Mutants of Cre recombinase with improved accuracy. Nat. Commun. 2013; 4:2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Karpinski J., Hauber I., Chemnitz J., Schäfer C., Paszkowski-Rogacz M., Chakraborty D., Beschorner N., Hofmann-Sieber H., Lange U.C., Grundhoff A. et al. Directed evolution of a recombinase that excises the provirus of most HIV-1 primary isolates with high specificity. Nat. Biotechnol. 2016; 34:401–409. [DOI] [PubMed] [Google Scholar]
- 48. Sheren J., Langer S.J., Leinwand L.A.. A randomized library approach to identifying functional lox site domains for the Cre recombinase. Nucleic Acids Res. 2007; 35:5464–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Santoro S.W., Schultz P.G.. Directed evolution of the site specificity of Cre recombinase. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:4185–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Baldwin E.P., Martin S.S., Abel J., Gelato K.A., Kim H., Schultz P.G., Santoro S.W.. A specificity switch in selected cre recombinase variants is mediated by macromolecular plasticity and water. Chem. Biol. 2003; 10:1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Karimova M., Splith V., Karpinski J., Pisabarro M.T., Buchholz F.. Discovery of Nigri/nox and Panto/pox site-specific recombinase systems facilitates advanced genome engineering. Sci. Rep. 2016; 6:30130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bebel A., Karaca E., Kumar B., Stark W.M., Barabas O.. Structural snapshots of Xer recombination reveal activation by synaptic complex remodeling and DNA bending. Elife. 2016; 5:e19706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gelato K.A., Martin S.S., Liu P.H., Saunders A.A., Baldwin E.P.. Spatially directed assembly of a heterotetrameric Cre-Lox synapse restricts recombination specificity. J. Mol. Biol. 2008; 378:653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang C., Myers C.A., Qi Z., Mitra R.D., Corbo J.C., Havranek J.J.. Redesign of the monomer–monomer interface of Cre recombinase yields an obligate heterotetrameric complex. Nucleic Acids Res. 2015; 43:9076–9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Abi-Ghanem J., Chusainow J., Karimova M., Spiegel C., Hofmann-Sieber H., Hauber J., Buchholz F., Pisabarro M.T.. Engineering of a target site-specific recombinase by a combined evolution- and structure-guided approach. Nucleic Acids Res. 2013; 41:2394–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.