

Summary
The transition from the fed to the fasted state necessitates a shift from carbohydrate to fat metabolism, which is thought to be mostly orchestrated by reductions in plasma insulin concentrations. Here we show in awake rats that insulinopenia per se does not cause this transition, but that both hypoleptinemia and insulinopenia are necessary. Furthermore, we show that hypoleptinemia mediates a glucose-fatty acid cycle through activation of the hypothalamic-pituitary-adrenal axis, resulting in increased white adipose tissue (WAT) lipolysis rates and increased hepatic acetyl-CoA content, which are essential to maintain gluconeogenesis during starvation. We also show that in prolonged starvation substrate limitation due to reduced rates of glucose-alanine cycling lowers rates of hepatic mitochondrial anaplerosis, oxidation and gluconeogenesis. Taken together these data identify a leptin-mediated glucose-fatty acid cycle that integrates responses of the muscle, WAT, and liver to promote a shift from carbohydrate to fat oxidation and maintain glucose homeostasis during starvation.
Graphical abstract
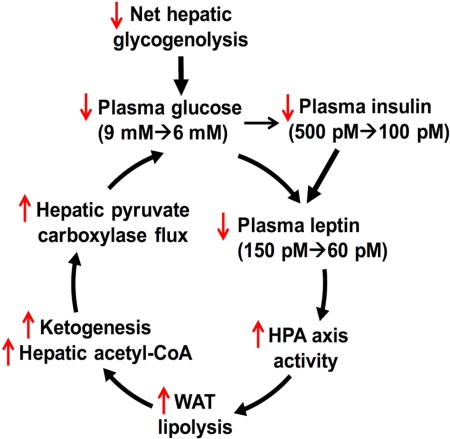
Introduction
The neocortex is one of the crowning achievements of evolution and mammals have evolved robust mechanisms to maintain sufficient substrate supply to meet the prodigious energy demands of the brain during starvation. Chief among them is a shift from carbohydrate to fat metabolism to preserve essential protein stores necessary for survival, which would otherwise be catabolized for gluconeogenesis (Cahill, 1970). In the transition from the fed to the early fasted state, there is a shift from substrate absorption to hepatic glycogenolysis and de novo synthesis of glucose from non-carbohydrate precursors such as lactate, alanine and glycerol (gluconeogenesis) (Benedict, 1907; Felig et al., 1969; Rothman et al., 1991). In contrast in the prolonged fasted state, when hepatic glycogen stores have been depleted, hepatic gluconeogenesis and ketogenesis supply substrate to the brain and other obligate glucose utilizers such as erythrocytes and the renal medulla. The shift from glucose metabolism to fat and ketone metabolism is thought to be primarily orchestrated by a decrease in plasma insulin concentrations and to a lesser extent an increase in plasma glucagon concentrations, which in turn are thought to regulate hepatic gluconeogenesis principally by the transcriptional regulation of key unidirectional enzymes such as phosphoenolpyruvate carboxykinase (PEPCK), fructose-1,6-biphosphatase and glucose-6-phosphatase by forkhead box protein O1 (FOXO1), peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), hepatocyte nuclear factor-4 alpha (HNF-4α) and other factors.
Prolonged starvation has also been shown to reduce plasma leptin concentrations in both humans and mice, which in turn has been shown to alter reproductive, thyroid and adrenal function (Ahima et al., 1996; Chan et al., 2003). However, the effect of starvation-induced hypoleptinemia on glucose and fat metabolism has not been explored. In this regard, we hypothesized that hypoleptinemia may be a critical signal to increase WAT lipolysis through activation of the HPA axis, resulting in increased hepatic acetyl-CoA content and pyruvate carboxylase activity, thereby maintaining glucose homeostasis and increasing hepatic ketogenesis during starvation.
In order to address this hypothesis we utilized a novel positional isotopomer NMR tracer analysis (PINTA) method (Perry et al., 2017b) to assess in vivo rates of mitochondrial pyruvate carboxylase flux (VPC) and mitochondrial oxidation (VCS) along with stable isotope infusions to assess rates of whole body glucose turnover, WAT lipolysis, hepatic ketogenesis, glucose-alanine cycling and glucose-lactate cycling in awake rats during the transition from the fed to the prolonged fasted state. Using this comprehensive metabolic approach, we show here that hypoleptinemia drives a glucose-fatty acid cycle, mediated by activation of the hypothalamic-pituitary-adrenal axis, resulting in increased rates of WAT lipolysis, β-oxidation, ketogenesis and hepatic acetyl-CoA content, which is necessary to maintain glucose homeostasis and adequate substrate supply to the brain during prolonged starvation.
Results
Depletion of hepatic glycogen during starvation induces hypoleptinemia which stimulates the HPA axis and promotes the transition from whole body glucose oxidation to fat/ketone oxidation
Following food removal from the cage, plasma glucose concentrations progressively declined despite minimal differences in body weight and rats were found to be post-absorptive at 8 hours of fasting as indicated by the absence of a glucose concentration gradient between the portal vein and the jugular vein (Fig. S1A–B). This progressive reduction in plasma glucose concentrations in the early postabsorptive state (8 hr–16 hr) could be entirely attributed to reductions in rates of net hepatic glycogenolysis, as reflected by the strong correlation between plasma glucose concentrations and rates of net hepatic glycogenolysis (R2=0.96, P=0.003) (Fig. 1B, Fig. S1C–F). In contrast plasma glucose concentrations were dissociated from rates of hepatic gluconeogenesis, which increased between 8 and 16 hr but decreased between 16 and 48 hr (Fig. 1B). In addition, the progressive reduction in rates of net hepatic glycogenolysis during the fast was associated with a shift from glucose to fat oxidation, demonstrated by fasting duration-dependent reductions in the ratio of pyruvate dehydrogenase flux to citrate synthase flux (VPDH/VCS) in liver, kidney (cortex), skeletal muscle, WAT and brown adipose tissue (BAT), heart, and brain (Fig. 1C).
Figure 1. A decreased rate of hepatic glycogenolysis is the primary determinant of the switch from glucose to fat oxidation during starvation.
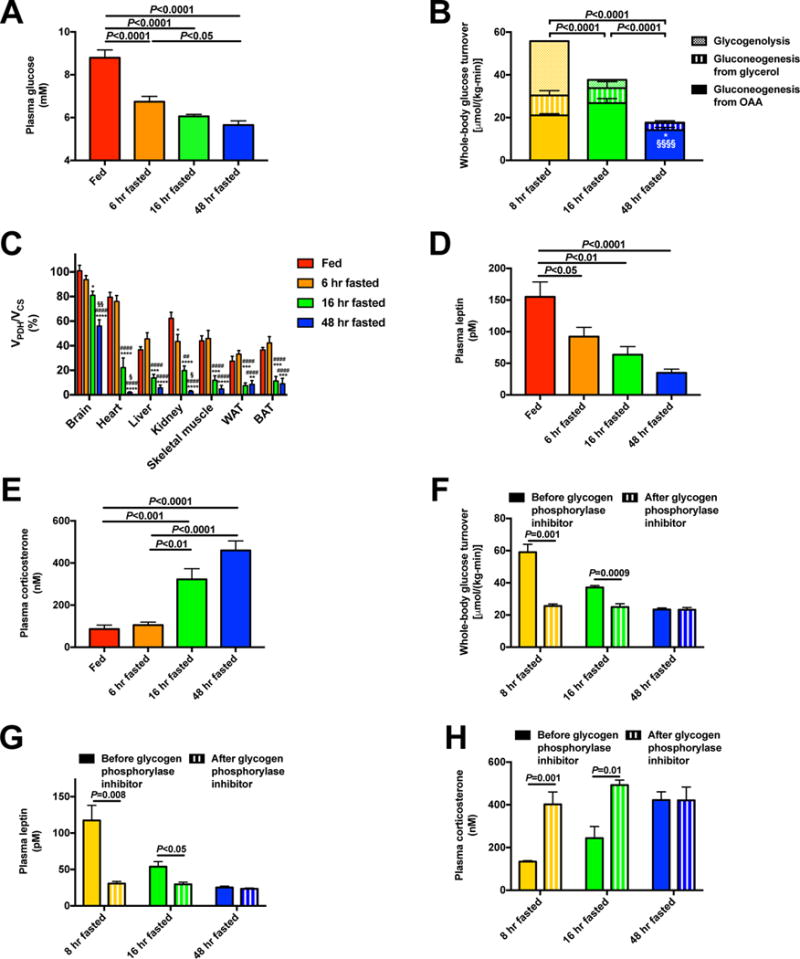
(A) Plasma glucose. (B) Hepatic glucose production from net hepatic glycogenolysis, gluconeogenesis from oxaloacetate (i.e. VPC), and gluconeogenesis from glycerol. *P<0.05 vs. 8 hr fasted rats and §§§§P<0.0001 vs. 16 hr fasted rats, in both cases comparing gluconeogenesis from oxaloacetate. (C) Percent glucose oxidation in the TCA cycle [pyruvate dehydrogenase flux (VPDH)/citrate synthase flux (VCS)]. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs. fed rats; ####P<0.0001 vs. 6 hr fasted rats, §P<0.05, §§P<0.01 vs. 16 hr fasted rats. (D)–(E) Plasma leptin and corticosterone. (F) Endogenous glucose production before and 2 hrs after treatment with a glycogen phosphorylase inhibitor. (G)–(H) Plasma leptin and corticosterone in glycogen phosphorylase inhibitor-treated rats. In panels (F)–(H), data from the same rats before and after treatment with the inhibitor were compared by the paired Student’s t-test; in all other panels, ANOVA with Bonferroni’s multiple comparisons test was used. Data are the mean±S.E.M. of n=6–8 per group. See also Fig. S1 and S2.
Plasma non-esterified fatty acid (NEFA), glycerol, and β–OHB concentrations increased progressively throughout the fast (Fig. S1G–I). Interestingly, during starvation plasma triglyceride (TAG) concentrations were reduced at 48 hr of fasting, whereas liver TAG and diacylglycerol (DAG) content were increased. This increase in hepatic DAG content was associated with increased protein kinase C-epsilon (PKCε) translocation (Fig. S1J–M), which in turn was associated with reduced percent suppression of endogenous glucose production during a hyperinsulinemic-euglycemic clamp (Fig. S1N–P). The dissociation between plasma and liver triglyceride can likely be explained in part by increased hepatic esterification of fatty acids into hepatic triglycerides driven by increased WAT lipolysis, combined with reduced hepatic triglyceride export, which may be in part attributed to reductions in liver ApoB concentrations (Fig. S1Q). Similarly, skeletal muscle triglyceride content increased in fasted rats as the fast progressed (Fig. S1R). In addition, there was no change in peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1α) expression with starvation, and gluconeogenic enzyme (phosphoenolpyruvate carboxykinase, pyruvate carboxylase, glucose-6-phosphatase) expression did not correlate with the measured changes in gluconeogenic flux (Fig. S1S–W).
Not surprisingly the reduction in plasma glucose concentrations that occurs as the fast progresses was accompanied by reductions in plasma insulin concentrations and increases in plasma glucagon concentrations (Fig. S2A–B). Plasma leptin concentrations decreased by 80% between fed and 48 hr fasted rats, which was associated with activation of the HPA axis as indicated by increases in plasma corticotropin releasing hormone (CRH), adrenocorticotropic hormone (ACTH), and corticosterone concentrations (Fig. 1D–E, Fig. S2C–D). Corticosteroid binding globulin also increased modestly, but there were no differences in plasma epinephrine or norepinephrine concentrations and a transient increase in plasma growth hormone concentration after a short-term but not a long-term fast (Fig. S2E–H). Consistent with previous reports starvation was also associated with an increase in plasma fibroblast growth factor-21 (FGF-21) concentrations (Kim and Lee, 2014; Kliewer and Mangelsdorf, 2010) (Fig. S2I) and a reduction in plasma triiodothyronine (T3) concentrations (Flier et al., 2000) (Fig. S2J). In contrast to lean chow fed rats, plasma leptin, corticosterone, NEFA, and ketone concentrations did not change significantly with a 48 hr fast in hyperleptinemic, diet-induced obese rats (Fig. S2K–N).
Next to more specifically investigate the impact of reductions in net hepatic glycogenolysis on rates of endogenous glucose production, plasma leptin concentrations and activation of the HPA axis, we treated post-absorptive rats fasted for 8, 16, and 48 hr with a small molecule inhibitor of glycogen phosphorylase and found that inhibition of hepatic glycogenolysis lowered plasma glucose, lactate, and insulin concentrations and whole-body glucose turnover in the short-term (8 hr and 16 hr) fasted rats but not in the 48 hr fasted rats, consistent with the negligible contributions that hepatic glycogenolysis would be expected to have on plasma glucose concentrations in this glycogen-depleted state (Fig. 1F, Fig. S2O–R). Interestingly the reductions in plasma glucose and insulin concentrations in 8 and 16 hr fasted rats treated with the glycogen phosphorylase inhibitor were associated with reductions in plasma leptin concentrations and increases in the HPA axis as reflected by increases in plasma corticosterone and ACTH concentrations in these groups (Fig. 1G–H, Fig. S2S). In contrast, plasma glucagon and catecholamine concentrations were not altered by treatment with the glycogen phosphorylase inhibitor, despite increased plasma FGF-21 concentrations in 8 and 16 hr fasted rats (Fig. S2T–W).
Physiologic leptin replacement suppresses lipolysis and reduces plasma glucose, but supraphysiologic leptin stimulates lipolysis and increases plasma glucose
Next, we sought to understand the physiologic role of the reductions in plasma leptin concentrations associated with prolonged starvation on the HPA axis, WAT lipolysis and hepatic gluconeogenesis. To address this question, we infused leptin in 48 hr fasted rats at three infusion rates designed to increase plasma leptin to physiologic concentrations observed in a 16 hr fast (~60 pM), to concentrations similar to those observed in obese rats (Fig. S2K) (~150 pM), and to supraphysiologic concentrations (~1250 pM) similar to or lower than those measured with leptin infusion in previous reports (Morton et al., 2015; Ravussin et al., 2014; Shek et al., 1998) (Fig. 2A). We found that physiologic leptin replacement (~60 pM) lowered plasma glucose concentrations and hepatic glucose production requiring an infusion of glucose to prevent hypoglycemia and counter-regulation; however, infusions of leptin to raise plasma leptin concentrations to ~150 pM and ~1250 pM were unable to lower either parameter (Fig. 2B–C, Fig. S3A). The reductions in plasma glucose concentrations with physiologic leptin replacement (plasma leptin concentration ~60 pM) and increases in plasma glucose concentrations with supraphysiologic leptin treatment (plasma leptin concentrations ~150 pM and ~1250 pM) were mirrored in suppression of plasma NEFA, glycerol, β–OHB, and lactate concentrations with physiologic leptin replacement and increases with supraphysiologic leptin infusions (Fig. S3B–E). Consistent with leptin’s effect to suppress HPA axis activity, plasma corticosterone was suppressed by all three infusion rates of leptin. However, plasma catecholamine concentrations increased dose-dependently with increased leptin infusion, with the highest infusion rate of leptin increasing plasma epinephrine and norepinephrine concentrations by 2–3 fold (Fig. 2D–F). Plasma insulin concentrations also increased at the highest dose of leptin, most likely secondary to the increased rates of hepatic glucose production and resultant hyperglycemia; in contrast, there was no difference in plasma growth hormone concentrations (Fig. S3F–G). However, leptin infusion caused a dose-dependent reduction in plasma FGF-21 concentrations, suggesting that the increase in plasma FGF-21 concentrations observed in fasted rats is secondary to hypoleptinemia and the resulting changes in the HPA axis, WAT lipolysis and hepatic fat oxidation (Fig. S3H). As predicted by the changes in plasma corticosterone and catecholamines, both fatty acid and glycerol turnover were suppressed with physiologic leptin replacement in 48 hr fasted rats and increased with supraphysiologic leptin treatment, resulting in increases in hepatic acetyl-CoA content and β–OHB turnover (Fig. 2G–J, Fig. S3I).
Figure 2. Physiologic leptin replacement reduces WAT lipolysis and hepatic gluconeogenesis in 48 hr fasted rats, but supraphysiologic leptin reverses this effect by stimulation of catecholamine secretion.
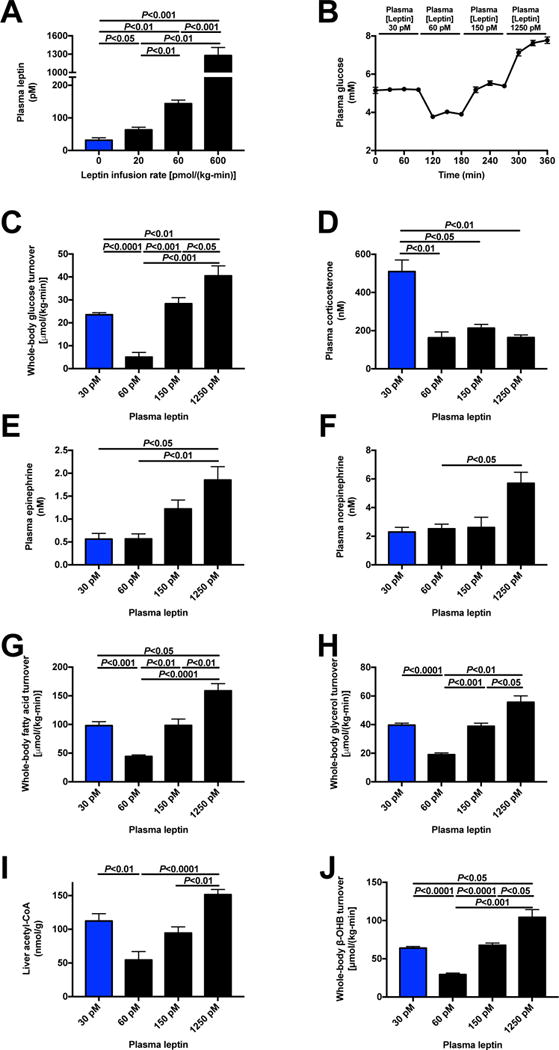
(A)–(B) Plasma leptin and glucose concentrations during a 6 hr infusion of stepwise increasing doses of leptin in 48 hr fasted rats. (C) Whole-body glucose turnover. (D)–(F) Plasma corticosterone, epinephrine and norepinephrine concentrations. (G)–(H) Whole-body fatty acid and glycerol turnover. (I) Liver acetyl-CoA content. (J) Whole-body β-OHB turnover. In all panels, n=8. Paired comparisons (ANOVA with Bonferroni’s multiple comparisons test) were performed. Data are the mean±S.E.M. See also Fig. S3.
To examine the potential insulin dependence of the ability of leptin to suppress HPA-axis mediated WAT lipolysis under physiologic conditions but promote WAT lipolysis under supraphysiologic conditions due to increases in plasma catecholamine concentrations, we performed leptin dose response studies in a severely insulinopenic streptozotocin-induced rat model of type 1 diabetes (T1D), which almost entirely lack plasma leptin (Perry et al., 2016). Similar to what we observed in the starved normal rats, we found that physiologic leptin replacement (~60 pM) in the T1D rats promoted a marked reduction in plasma glucose concentrations and endogenous glucose production rates but supraphysiologic leptin concentrations (~1300 pM) promoted increases in both plasma glucose concentrations and endogenous glucose production (Fig. S3J–M). Both doses of leptin suppressed HPA axis activity, but supraphysiologic leptin concentrations (~1300 pM) were associated with a four-fold increase in plasma catecholamine concentrations without any change in growth hormone concentrations (Fig. S3N–R). These alterations in HPA axis activity and catecholamine concentrations were associated with suppression of WAT lipolysis in T1D rats infused with leptin to achieve physiologic replacement leptin concentrations (~60 pM), an effect which was abrogated in rats infused with leptin to reach supraphysiologic plasma leptin concentrations (~1300 pM) (Fig. S3S–U).
Increases in leptin/HPA axis-mediated WAT lipolysis and hepatic acetyl-CoA content maintain euglycemia during the early stages (6–>16h) of fasting
Having observed that variations in plasma leptin concentrations are associated with alterations in plasma glucocorticoid and catecholamine concentrations as well as WAT lipolysis, we next asked how WAT lipolysis changes during the fast. Using stable isotope ([13C16]palmitate, [2H5]glycerol, and [13C4]β–OHB) infusions, we found a progressive increase in turnover of fatty acids, glycerol, and ketones (Fig. 3A–C, Fig. S4A). As predicted by the increase in β–OHB turnover observed with increasing duration of fasting (Perry et al., 2017c), hepatic acetyl-CoA concentrations also increased as the fast progressed, whereas hepatic malonyl-CoA content was reduced by 75% between 6 and 16 hr of fasting, but did not change between the 16 and 48 hr time points (Fig. 3D–E), suggesting that inhibition of carnitine palmitoyltransferase-1 (CPT-1) activity by hepatic malonyl-CoA content was likely minimized by 16 hr of fasting (McGarry et al., 1978; Saggerson and Carpenter, 1981). Next to directly assess the impact of increases in hepatic acetyl-CoA content on hepatic glucose metabolism, we treated rats fasted for 0, 6, 16, and 48 hr with etomoxir, a small-molecule inhibitor of CPT-1 and found that in fasted rats, CPT-1 inhibition lowered plasma glucose and insulin concentrations and whole-body glucose turnover associated with suppression of hepatic acetyl-CoA content and β–OHB turnover despite increased WAT lipolysis, which was likely a consequence of the reductions in plasma insulin concentrations observed with inhibition of CPT-1, and unchanged plasma lactate and hepatic malonyl-CoA concentrations (Fig. 3F–H, Fig. S4B–J). To examine whether the reductions in plasma glucose and insulin measured with CPT-1 inhibition were associated with alterations in HPA axis activity, we measured plasma leptin and corticosterone concentrations and found that etomoxir treatment was associated with 50% reductions in plasma leptin concentrations as well as increases in plasma corticosterone concentrations in 16 and 48 hr fasted rats (Fig. 3I–J). In contrast, there were no differences in plasma epinephrine or norepinephrine concentrations (Fig. S4K–L).
Figure 3. Increased hepatic acetyl-CoA content maintains euglycemia during starvation.
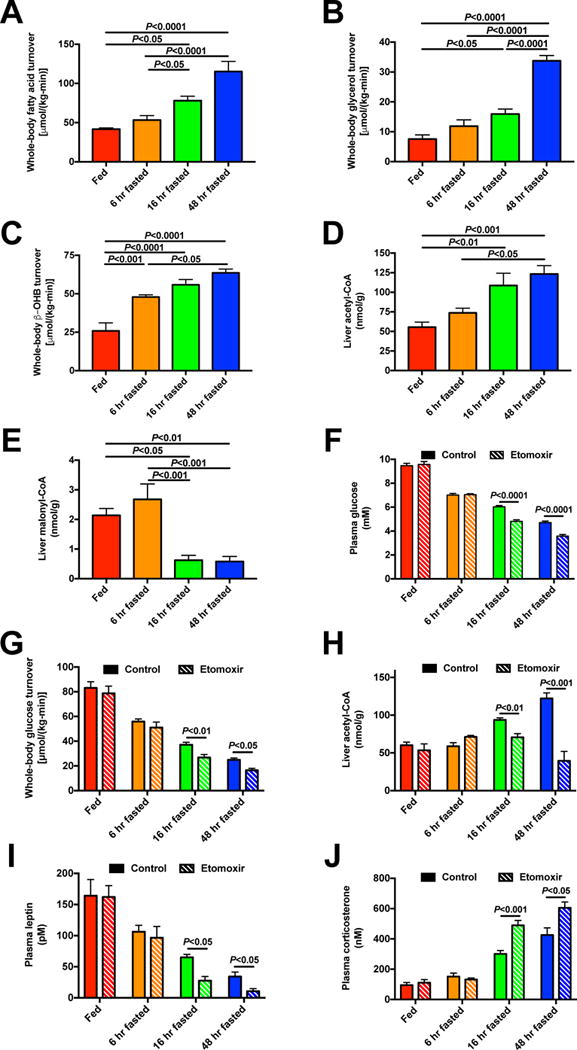
(A)–(C) Whole-body fatty acid, glycerol, and β-OHB turnover. (D)–(E) Hepatic acetyl- and malonyl-CoA content. In panels (A)-(E), n=7 per time point, and data were compared by ANOVA with Bonferroni’s multiple comparisons test. (F)–(G) Plasma glucose and whole-body glucose turnover 2 hr after treatment with etomoxir. (H) Liver acetyl-CoA content. (I)–(J) Plasma leptin and corticosterone concentrations. In panels (F)–(J), data are the mean±S.E.M. of n=6 per group. Data were compared by ANOVA with Bonferroni’s multiple comparisons test (control vs. etomoxir vs. atglistatin, with these groups compared separately at each time point; control data are duplicated between Fig. 3 and Fig. 4). See also Fig. S4.
Next to more directly assess the role of increased WAT lipolysis in the maintenance of hepatic gluconeogenesis by hepatic acetyl-CoA in starvation we treated rats with atglistatin, a small molecule inhibitor of adipose triglyceride lipase (ATGL). Atglistatin treatment rapidly decreased plasma glucose concentrations in 16 hr and 48 hr fasted rats and an infusion of glucose was required to avoid severe hypoglycemia due to suppression of WAT lipolysis and hepatic acetyl-CoA content in the latter group despite unchanged plasma lactate, epinephrine and norepinephrine concentrations and hepatic malonyl-CoA content (Fig. 4A–F, Fig. S5A–I). As in etomoxir-treated rats, 16 and 48 hr fasted atglistatin-treated rats exhibited reductions in plasma leptin and increases in plasma corticosterone concentration (Fig. 4G–H), consistent with plasma glucose/insulin regulation of leptin release from the adipocyte. Next to assess whether increased plasma corticosterone concentrations are necessary to maintain euglycemia in the starved state, we treated rats with a glucocorticoid receptor antagonist, mifepristone. Despite infusion of glucose to avoid hypoglycemia provoking a counter-regulatory response, mifepristone treatment decreased plasma glucose concentrations, whole-body glucose turnover, WAT lipolysis, and ketogenesis in 48 hr fasted rats but not in recently fed rats (Fig. 5A–G, Fig. S6A–H). Finally, consistent with data in rats treated with etomoxir and atglistatin, the lower plasma glucose and insulin concentrations caused by treating 48 hr fasted rats with mifepristone were associated with a 60% reduction in plasma leptin concentrations (Fig. 5H). Taken together these data demonstrate the key role for hepatic acetyl-CoA in supporting hepatic gluconeogenesis during starvation. Furthermore, they demonstrate the critical role for hypoleptinemia-mediated increases in HPA axis activity in promoting increased WAT lipolysis and hepatic ketogenesis in this process.
Figure 4. Increased WAT lipolysis is necessary to increase hepatic acetyl-CoA content and maintain euglycemia in the starved state.
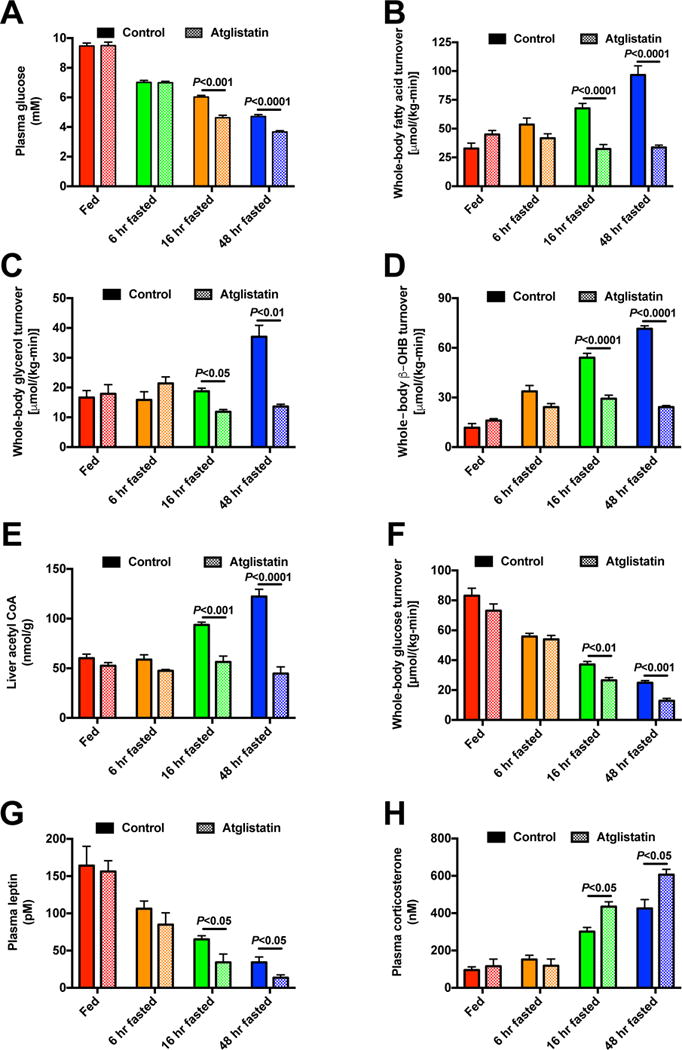
(A) Plasma glucose concentrations in control and atglistatin-treated rats. (B)–(D) Whole-body fatty acid, glycerol, and β-OHB turnover. (E) Hepatic acetyl-CoA content. (F) Whole-body glucose turnover. (G)–(H) Plasma leptin and corticosterone concentrations. In all panels, data are the mean±S.E.M. of 5–6 rats per group, and control data are duplicated from Fig. 3. Data were compared by ANOVA with Bonferroni’s multiple comparisons test (control vs. etomoxir vs. atglistatin, with these groups compared separately at each time point. See also Fig. S5.
Figure 5. Glucocorticoid activity is required to maintain WAT lipolysis and euglycemia in starvation.
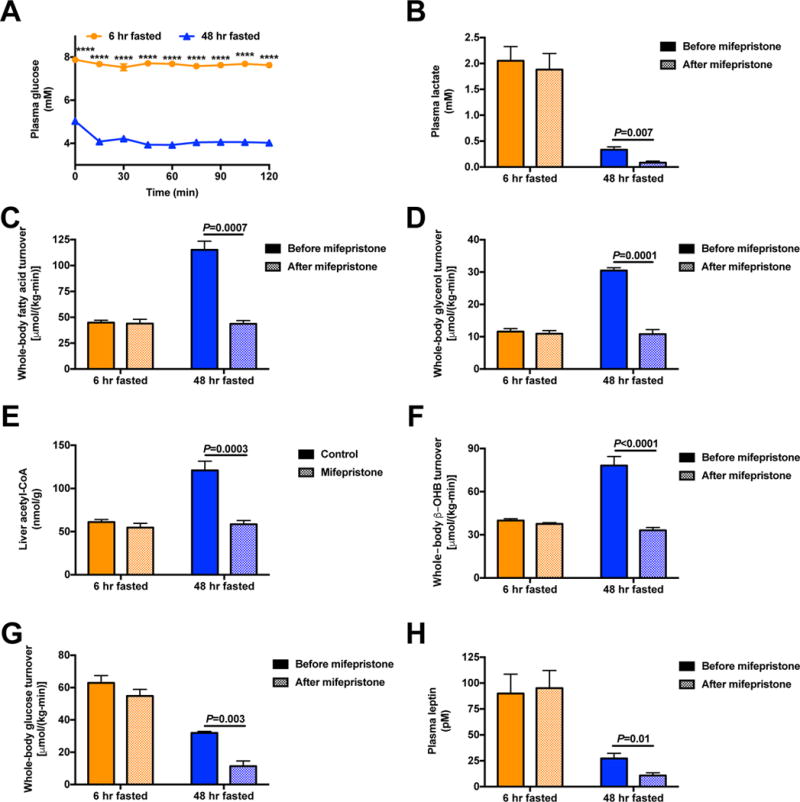
(A)–(B) Plasma glucose and lactate concentrations before and after treatment with mifepristone. (C)–(D) Whole-body fatty acid and glycerol turnover. (E) Hepatic acetyl-CoA content. (F)–(G) Whole-body β-OHB and glucose turnover. (H) Plasma leptin concentrations. In all panels, data are the mean±S.E.M. of n=6, with data from before and after mifepristone treatment compared by the 2-tailed paired Student’s t-test. See also Fig. S6.
Reductions in muscle glucose-alanine cycling promote decreased rates of hepatic mitochondrial oxidation and gluconeogenesis in the prolonged (48 h) fasted state
Given the progressive increases in WAT lipolysis and hepatic acetyl-CoA content with increasing duration of the fast, it might be expected that VPC flux would also continue to increase with an extended fast. Surprisingly we observed a ~50% reduction in pyruvate carboxylase flux between 16 and 48 hr fasted rats (Fig. 1B) despite a 15% increase in hepatic acetyl-CoA content during this time interval (Fig. 3D). To understand the reasons for this discordance between VPC and hepatic acetyl-CoA content, we assessed plasma concentrations and turnover rates of the two predominant gluconeogenic substrates (lactate and alanine) and observed 65% reductions in rates of whole-body alanine turnover as well as 35% reductions in hepatic alanine concentrations between 16 and 48 hr fasted rats, associated with smaller reductions in lactate turnover and plasma amino acid concentrations (Fig. 6A, Fig. S7A–L). Surprisingly, PINTA analysis of hepatic mitochondrial oxidation rates revealed a 50% reduction in rates of hepatic mitochondrial VCS flux associated with reductions in liver citrate, malate, and succinate concentrations. Taken together these data suggest that prolonged (48 hr) starvation leads to reductions in glucose-alanine cycling which in turn results in decreased hepatic mitochondrial anaplerosis via pyruvate carboxylase, resulting in decreased rates of mitochondrial VCS flux (Fig. 6B–C, Fig. S7M–O). To test this hypothesis, we infused alanine in 48 hr fasted rats to match alanine turnover rates and hepatic alanine concentrations measured in 16 hr fasted rats (Fig. S7P–Q). This intervention increased plasma glucose, insulin, and lactate concentrations, endogenous glucose production, and VPC flux, associated with a three-fold increase in plasma leptin concentrations but independent of any change in plasma glucagon (Fig. 6D–F, Fig. S7R–U). In addition, normalizing alanine turnover increased liver tricarboxylic acid cycle intermediate concentrations and hepatic VCS flux in 48 hr fasted rats independent of changes in plasma T3 concentrations (Fig. 6G–H, Fig. S7V–Y).
Figure 6. Reductions in glucose-alanine cycling with prolonged starvation lead to suppression of hepatic gluconeogenesis and hepatic mitochondrial oxidation.
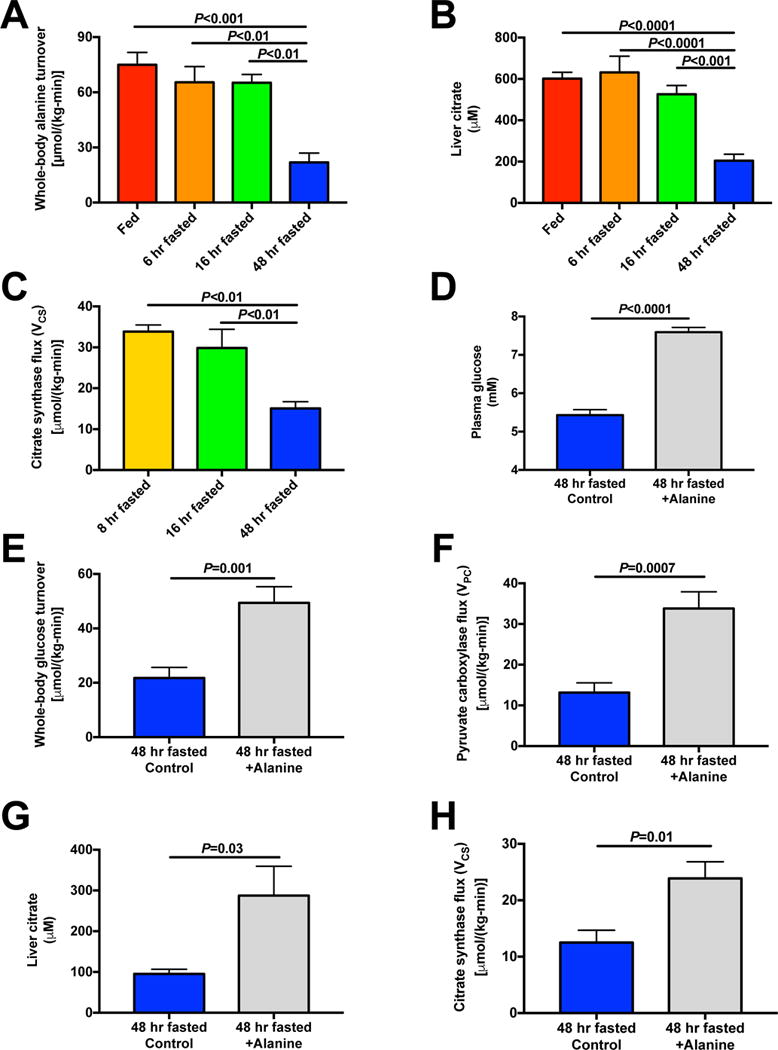
(A) Whole-body alanine turnover. (B) Hepatic citrate concentrations. (C) Liver VCS flux. (D) Plasma glucose concentrations after a 2 hr infusion of alanine [45 μmol/(kg-min)] in 48 hr fasted rats. (E)–(F) Whole-body glucose turnover and liver VPC flux. (G) Liver citrate. (H) Hepatic mitochondrial citrate synthase (VCS) flux. In all panels, data are the mean±S.E.M. of n=6–8 per group, with data compared by ANOVA [panels (A)–(C)] or by the 2-tailed unpaired Student’s t-test [panels (D)–(H)]. See also Fig. S7.
Small reductions in plasma glucose (6 –> 5 mM) suppress muscle glucose-alanine cycling during prolonged (48 h) starvation
Finally, we sought to test the hypothesis that subtle reductions in plasma glucose concentrations from 6 mM to 5 mM promote hypoleptinemia, increased HPA axis activity, and reductions in glucose-alanine cycling as well as glucose-lactate cycling in the starved state. To that end, we performed a low-dose glucose infusion in 48 hr fasted rats to increase plasma glucose concentrations from 5 mM to 6 mM, as measured in 16 hr fasted rats (Fig. S8A–B). This intervention increased plasma insulin concentrations by 75% and doubled plasma lactate concentrations while reducing plasma FGF-21 concentrations by 50%. Likely due to the increase in plasma insulin concentrations, rates of endogenous glucose production were also reduced by 30% (Fig. 7A–B, S8C–E). Increasing plasma glucose from 5 to 6 mM doubled both glucose uptake into skeletal muscle and whole-body alanine turnover (Fig. 7C–D), increasing the latter to rates similar to those observed in 16 hr fasted rats. In addition, 48 hr fasted rats with plasma glucose concentrations ~6 mM exhibited a two-fold increase in WAT glucose uptake as compared to 48 hr fasted rats with plasma glucose ~5 mM, doubling plasma leptin concentrations and suppressing HPA axis activity as reflected by 50–60% reductions in plasma ACTH and corticosterone concentrations (Fig. 7E–H). Similarly, we found that treatment of short-term fasted rats (8 and 16 hr) with an inhibitor of glycogen phosphorylase, resulted in hypoleptinemia, increased HPA axis activity, as well as increased rates of WAT lipolysis and decreased rates of alanine turnover (Fig. 7I–J, Fig. S8F–J).
Figure 7. Reductions in plasma glucose concentrations from ~6 mM to ~5 mM during prolonged starvation lead to reduced glucose-alanine cycling, hypoleptinemia and HPA axis activation.
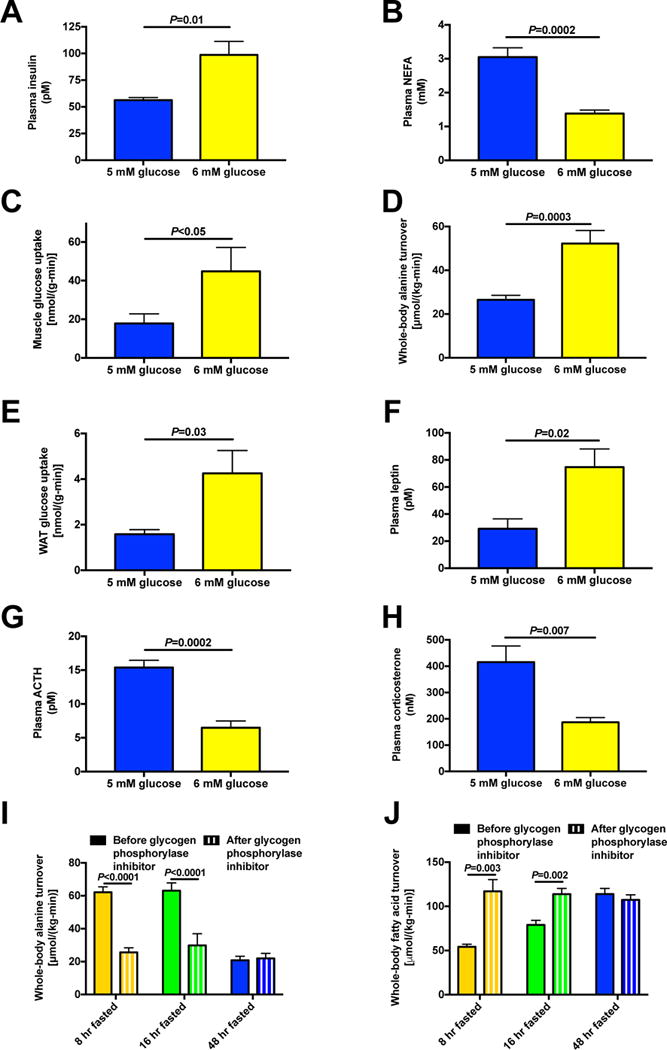
(A)–(B) Plasma insulin and NEFA concentrations. (C) Muscle glucose uptake in 48 hr fasted rats with and without an infusion of glucose to increase plasma glucose concentrations from 5 to 6 mM. (D) Whole-body alanine turnover. (E) White adipose tissue glucose uptake. (F)–(H) Plasma leptin, ACTH, and corticosterone concentrations. (I)–(J) Whole-body alanine and fatty acid turnover in rats treated with an inhibitor of glycogen phosphorylase. In panels (C) and (E) data were compared by the 2-tailed unpaired Student’s t-test; otherwise the paired t-test was used. Data are the mean±S.E.M. of n=6–7 per group. See also Fig. S8.
Discussion
To prolong survival during an extended fast, mammals must shift from a primary reliance upon carbohydrate metabolism to a primary reliance upon fat and ketone metabolism in order to maintain adequate substrate supply to the brain, heart, and other organs, thus preserving essential protein stores that would otherwise be catabolized to support gluconeogenesis (Cahill, 1970). While insulin and to a lesser extent glucagon have long been thought to be the major orchestrators of this transition from glucose to fat/ketone metabolism, here we demonstrate a leptin-mediated glucose-fatty acid cycle that is essential to support this process.
Consistent with previous in vivo 13C nuclear magnetic resonance studies of hepatic glycogen metabolism in humans (Rothman et al., 1991), we found that the fraction of glucose production from net hepatic glycogenolysis decreases over the course of a 48 hr fast in lean rats, while gluconeogenesis rates remain relatively constant until rats reach a state of prolonged starvation at 48 hrs whereupon gluconeogenic flux decreases by ~50%. Similar to data from fasting humans, hepatic glycogen concentrations in rats followed a pattern of exponential decay during fasting (Fig. S1D) and the reduction in rates of net hepatic glycogenolysis was the major determinant of the reduction in the rate of endogenous glucose production and plasma glucose concentration observed during a prolonged fast (Fig. 1A–C). To specifically test the physiologic impact of reductions in rates of hepatic glycogenolysis on leptin secretion, HPA axis activity and WAT lipolysis, we treated rats with a small molecule inhibitor of glycogen phosphorylase. Consistent with a key role for hepatic glycogenolysis as the major determinant of endogenous glucose production and fasting plasma glucose concentrations in the early stages (0 to 16 hr) of the fast, 8 and 16 hr fasted rats treated with the glycogen phosphorylase inhibitor manifested rates of endogenous glucose production that were reduced by 60 and 30%, respectively (Fig. 1F). In contrast, 48 hr fasted rats treated with the glycogen phosphorylase inhibitor exhibited no difference in rates of endogenous glucose production consistent with rates of net hepatic glycogenolysis being negligible by 48 hr of fasting.
In order to examine the ability of reductions in hepatic glycogenolysis to drive the HPA activation observed in the starved state, we assessed plasma lipolytic hormone concentrations and WAT lipolysis in rats treated with the glycogen phosphorylase inhibitor. We observed a reduction in plasma leptin concentrations and increases in plasma corticosterone concentrations and WAT lipolysis, bringing each parameter to concentrations measured in 48 hr fasted rats despite unchanged plasma catecholamine concentrations in short-term fasted rats treated with the glycogen phosphorylase inhibitor (Fig. 1G–H, Fig. 7J, Fig. S2S,U–V, Fig. S8F–J). These results suggest that hypoleptinemia-driven HPA axis activation could promote increased hepatic gluconeogenic flux by increasing WAT lipolysis, resulting in increased fatty acid delivery to liver and hepatic acetyl-CoA content to maintain euglycemia during prolonged (≥16 hrs) fasting. Consistent with this hypothesis, we found that both WAT lipolysis and hepatic acetyl-CoA content increased with increasing duration of the fast. Interestingly, these increases in WAT lipolysis were also associated with the development of severe hepatic steatosis, increases in hepatic DAG content, hepatic PKCε activation and impaired percent suppression of endogenous glucose production during a hyperinsulinemic-euglycemic clamp (Fig. S1K–P). These results are consistent with prior studies demonstrating that increased fatty acid delivery to the liver can promote increased hepatic esterification and hepatic triglyceride synthesis in an insulin-independent fashion (Vatner et al., 2015) as well as well as a causal role for hepatic DAG activation of PKCε in hepatic insulin resistance, through increased insulin receptor kinase threonine1160 (murine 1150) phosphorylation (Petersen et al., 2016; Samuel et al., 2004; Samuel et al., 2007).
In contrast to ectopic lipid content, which increased progressively through the fast, hepatic malonyl-CoA decreased between 6 and 16 hr of the fast, facilitating mitochondrial β-oxidation by relieving inhibition of CPT-1. To further examine the potential role of increasing hepatic acetyl-CoA concentrations in the maintenance of hepatic gluconeogenesis and endogenous glucose production, we treated rats with two agents to directly modulate hepatic acetyl-CoA content: ATGL inhibitor atglistatin, and CPT-1 inhibitor etomoxir. Despite different mechanisms of action both agents suppressed hepatic acetyl-CoA content, hepatic gluconeogenesis, and plasma glucose and insulin concentrations, thus demonstrating a critical role for hepatic acetyl-CoA content in the maintenance of hepatic gluconeogenesis during starvation.
Taken together these data challenge the canonical view of the primacy of insulinopenia-mediated transcriptional and/or translational regulation of gluconeogenic enzymes in increasing hepatic gluconeogenesis and maintaining euglycemia in starvation. Although PEPCK protein expression increased progressively during the fast, hepatic gluconeogenesis from oxaloacetate was reduced by 50% from 16 to 48 hrs due to substrate limitation (Fig. 1B, Fig. 6E–F). These data raise the question of whether insulinopenia per se promotes WAT lipolysis and increases in hepatic acetyl-CoA content in the starved state, or whether other factors such as HPA axis activation (Fig. 1D) are also required.
To answer that question, we first administered mifepristone, a selective glucocorticoid receptor antagonist, to rats fasted for 6 and 48 hrs. Although mifepristone had no impact on WAT lipolysis, whole body glucose turnover, or plasma glucose or insulin concentrations in recently fed rats (6 hr), inhibition of glucocorticoid action markedly suppressed each of these parameters, necessitating glucose infusion to avoid severe hypoglycemia and counter-regulation in prolonged (48 hrs) fasted rats (Fig. 5). Because mifepristone treatment suppressed WAT lipolysis in the setting of reductions in plasma insulin concentrations, these data suggest that severe insulinopenia per se is not sufficient to promote increased WAT lipolysis and hepatic gluconeogenesis in starved rats. To further test the role of HPA axis activation and specifically the role of hypoleptinemia in driving this process, we infused leptin in a stepwise fashion to increase plasma leptin concentrations in 48 hr fasted rats to: (1) physiologic concentrations typical of overnight fasted, chow-fed rats (~60 pM), (2) modestly elevated concentrations similar to those of high fat fed rats (Fig. S2K) (~150 pM), and (3) supraphysiologic concentrations (~1250 pM) similar to or lower than those measured in previous systemic leptin infusion studies in the literature (Morton et al., 2015; Ravussin et al., 2014; Shek et al., 1998). This study demonstrated that physiologic leptin replacement suppressed HPA axis activity, WAT lipolysis, hepatic acetyl-CoA content, and rates of hepatic gluconeogenesis in 48 hr fasted rats without altering plasma insulin concentrations, thereby demonstrating that insulinopenia per se is not sufficient to increase WAT lipolysis, hepatic acetyl-CoA content, and hepatic gluconeogenesis during starvation.
To further examine the role of insulin in mediating leptin’s effect on WAT lipolysis, we performed leptin dose response studies in a severely insulinopenic streptoztocin-induced rat model of T1D that is also severely leptinopenic (Perry et al., 2017a; Perry et al., 2014). These data demonstrate the impact of the absence of leptin: with plasma leptin concentrations only ~12 pM (92% lower than fed rats) and severe insulinopenia (99% lower than fed rats), both WAT lipolysis and endogenous glucose production rates were markedly increased, but again we found that physiologic leptin replacement suppressed endogenous glucose production and WAT lipolysis in T1D rats (Fig. S3J–U) consistent with previous studies (Perry et al., 2017a; Perry et al., 2014). However, in both fasting control rats and T1D rats, supraphysiologic leptin concentrations promoted increased catecholamine release, increasing WAT lipolysis and abrogating leptin’s effect to lower rates of endogenous glucose production and plasma glucose concentrations. These data are consistent with prior studies demonstrating that leptin activates catecholamine synthesis (Shibuya et al., 2002), catecholamine signaling (Luan et al., 2014), and energy expenditure (Levin et al., 1996; Pelleymounter et al., 1995). Further, the ability of leptin to stimulate the sympathetic nervous system, WAT lipolysis, and hepatic glucose production at plasma concentrations ≥150 pM may explain the failure of previous studies (Denroche et al., 2015; Denroche et al., 2011; Morton et al., 2015) to observe an effect of leptin to reduce hyperglycemia in fasted insulinopenic diabetic rodents through suppression of HPA axis activity. In addition, hypoleptinemia-induced activation of HPA axis activity may not occur universally in starvation as we observed in 4 week high fat fed (HFD) obese rats (Fig. S2K–N). In contrast to regular chow fed lean control rats, we found that HFD obese hyperleptinemic rats do not manifest reductions in plasma leptin concentrations and increases in HPA activity during 48 hr of fasting (Fig. S2K–N). This may explain why starvation-induced reductions in plasma leptin concentrations and increases in HPA activity have not always been observed in human studies: in contrast to young lean rodents, which have minimal fat mass after a 48 hr fast, most normal weight human subjects will still have substantial fat mass even after several days of starvation, which would not be expected to decrease plasma leptin concentrations to the critical threshold that maybe required to activate the HPA axis. In contrast patients with anorexia nervosa often develop severe hypoleptinemia, due to depleted fat stores, which is associated with activation of the HPA axis and hypercortisolemia (Schorr and Miller, 2017), suggesting that under conditions of severe depletion of adipose tissue mass leptin may decrease sufficiently with starvation to activate HPA axis-driven WAT lipolysis. In contrast in overweight subjects or those undergoing caloric restriction instead of fasting, in whom leptin may not be reduced sufficiently to produce increased HPA activity, this mechanism may not be as relevant or may take a longer time to occur in order to deplete the larger fat mass in these overweight/obese individuals. Consistent with this possibility several recent studies do show an association between hypoleptinemia and increased plasma cortisol concentrations in high-normal weight humans during caloric deprivation (Karl et al., 2016; Pasiakos et al., 2011; Villareal et al., 2016). In addition, it is possible that the relative importance of insulinopenia and hypoleptinemia-induced HPA axis activation in driving the shift from glucose to fat metabolism may differ between rodents and humans; in contrast to rodents, insulinopenia may be sufficient to produce most or all of the effect of starvation to increase WAT lipolysis in humans independent of increases in plasma cortisol concentrations (Jensen et al., 1989).
Several reports suggest that in lean and obese human subjects, plasma leptin concentrations decrease during caloric restriction out of proportion to the decline in total body weight or fat mass and increase with refeeding or long-term hyperinsulinemia (Ahima and Lazar, 2008; Hintze et al., 2017), consistent with our data (Fig. 1D, Fig. S1A, Fig. 7F). These data pose the question of whether starvation regulates plasma leptin concentrations by an alternative mechanism in addition to reductions in total fat mass. Consistent with this possibility, acute treatment with inhibitors of glycogen phosphorylase, CPT-1, ATGL, and the glucocorticoid receptor all reduced plasma glucose, insulin, and leptin concentrations within just two hours, a time frame incompatible with similar reductions in body weight. We hypothesized that a reduction in plasma glucose concentrations from 6 to 5 mM and consequent reductions in plasma insulin concentrations from ~100 pM to ~45 pM during prolonged fasting – which we have shown results primarily from depletion of hepatic glycogen stores (Fig. 1F, Fig. S2P–Q) – reduce plasma leptin concentrations from ~65pM to ~35pM. Consistent with this hypothesis, we found that a small increase in plasma glucose concentrations from 5 mM to 6 mM in 48 hr fasted rats doubled glucose uptake into WAT, doubled plasma leptin concentrations, and reduced plasma corticosterone concentrations by 50% (Fig. 7E–H). These data demonstrate that progressive reductions in plasma glucose concentrations during the fed to fasted transition in normal chow fed rats signals the depletion of stored hepatic carbohydrate (glycogen) reserves and results in reduced WAT leptin secretion, which in turn stimulates the HPA axis activity to promote increased WAT lipolysis and the transition from whole body glucose oxidation to fat/ketone oxidation.
Surprisingly, despite increased or unchanged WAT lipolysis and hepatic acetyl-CoA content between 16 and 48 hrs of fasting, rats exhibit a ~50% reduction in rates of hepatic gluconeogenesis after 48 hrs of starvation, which could entirely be attributed to reductions in rates of hepatic pyruvate carboxylase (VPC) flux between these time points (Fig. 1B). We hypothesized that reductions in alanine turnover, which we observed in 48 hr starved rodents (Fig. 6A) and which are consistent with previous studies demonstrating a reduction in alanine release from the forearm in fasted humans (Felig et al., 1970), may be responsible for reduced hepatic gluconeogenesis due to substrate limitation. To test that hypothesis, we infused rats with alanine to increase total alanine turnover to match rates of alanine turnover measured in 16 hr fasted rats. This intervention increased plasma glucose and insulin concentrations, hepatic glucose production, and hepatic pyruvate carboxylase flux similar to or greater than what was measured in 16 hr fasted rats. In addition, rates of hepatic mitochondrial oxidation (citrate synthase flux, VCS), assessed by PINTA analysis, revealed suppression of hepatic mitochondrial oxidation flux (VCS) with 48 hr of fasting, but alanine replacement normalized rates of hepatic mitochondrial oxidation (VCS) independent in changes in plasma T3 concentrations (Fig. S2J, Fig. S7Y). Taken together these data reveal that reductions in alanine turnover during prolonged starvation cause suppression of hepatic mitochondrial oxidation rates. The reductions in alanine turnover observed in the starved state would be expected to promote further increases in hepatic ketogenesis due to an expanding mismatch between rates of hepatic β-oxidation and hepatic mitochondrial oxidation (VCS), thereby providing increased ketones to serve as a substrate for brain mitochondrial oxidative metabolism, supplying ~45% of the brain’s energy needs after 48 hrs of starvation (Fig. 1C). The reductions in alanine turnover observed in the starved state were confirmed to result from reductions in plasma glucose concentrations from 6 mM to 5 mM: infusion of glucose to raise plasma glucose concentrations from 5 mM to 6 mM in 48 hr fasted rats increased whole-body alanine turnover to rates similar to those measured in 16 hr fasted rats (Fig. 7D).
In summary, these data reveal several new concepts regarding leptin biology and the regulation of whole-body and tissue-specific substrate metabolism from the transition from the fed to fasted state in normal lean rats. Specifically: 1) Progressive decreases in plasma glucose (9→6 mM) and insulin (500→100 pM) concentrations during early starvation (6–16 hr) can mostly be ascribed to reduced rates of net hepatic glycogenolysis [25→4 μmol/(kg-min)], since rates of hepatic gluconeogenesis during this period remain relatively constant, thus providing a systemic index of remaining stored hepatic carbohydrate (glycogen) reserves. This in turn promotes progressive reductions in plasma leptin concentrations (150→60 pM). 2) Reductions in plasma leptin concentrations (150→60 pM) stimulate the HPA axis thus increasing plasma corticosterone concentrations (100→450 nM), which in turn results in stimulation of WAT lipolysis and the shift from whole body carbohydrate oxidation to fat/ketone oxidation, 3) Increases in WAT lipolysis increase hepatic acetyl-CoA content and allosterically stimulate hepatic pyruvate carboxylase flux, which is essential for the maintenance of hepatic glucose production and euglycemia during starvation, 4) Insulinopenia is necessary but not sufficient for increased rates of WAT lipolysis, increased hepatic acetyl-CoA content, increased rates of hepatic ketogenesis, and the shift from carbohydrate oxidation to fat/ketone oxidation during starvation, 5) Decreased glucose-alanine cycling, due to hepatic glycogen depletion, results in marked (~50%) reductions in rates of hepatic pyruvate carboxylase flux (VPC flux) and hepatic mitochondrial oxidative metabolism (VCS flux), 6) Reductions in rates of hepatic mitochondrial oxidation (VCS flux) during prolonged (48 hrs) starvation can be attributed to reductions in rates of hepatic anaplerosis (VPC flux), 7) Physiologic replacement of plasma leptin concentrations (30→60 pM) during prolonged (48 hrs) starvation inhibits WAT lipolysis and results in decreased rates of hepatic gluconeogenesis through reductions in HPA axis activity. In contrast, supraphysiologic plasma leptin concentrations (>150 pM) stimulate WAT lipolysis and result in increased rates of hepatic gluconeogenesis and hyperglycemia through activation of the sympathetic nervous system and increased catecholamine secretion and 8) Increased rates of WAT lipolysis promote increased hepatic fat (DAG) accumulation and PKCε→activation during prolonged (48 hr) starvation. Regarding this last point it is interesting to speculate that fasting-induced hepatic steatosis and lipid-induced hepatic insulin resistance may also play an important role in promoting survival during famine by minimizing hepatic glucose uptake and energy storage as glycogen, therefore sparing any ingested carbohydrate for the central nervous system and other obligate glucose-requiring tissues, thus providing an evolutionary basis for DAG-PKCε induced hepatic insulin resistance.
Taken together these data show that both insulinopenia and hypoleptinemia are necessary for maintenance of euglycemia during short-term starvation (6–16 hr) in lean rats, with insufficient anaplerosis from glucose-alanine cycling limiting both hepatic gluconeogenesis and mitochondrial oxidation in prolonged starvation. These data further identify a novel leptin-mediated glucose-fatty acid cycle that integrates responses of the muscle, white adipose tissue, and liver to maintain adequate substrate supply to the brain to promote survival during starvation.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Dr. Gerald I. Shulman, gerald.shulman@yale.edu.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All rodent studies were approved by the Yale University Institutional Animal Care and Use Committee. Male Sprague-Dawley rats were ordered from Charles River Laboratories (Wilmington, MA) at ~250 g and were group housed (3 per cage) for 1–2 weeks until they underwent surgery under general isoflurane anesthesia for placement of polyethylene catheters in the common carotid artery (PE50 tubing, Instech Solomon, Plymouth Meeting, PA), and the jugular vein (PE90 tubing, Instech), and/or the portal vein (PE50 tubing, Instech), after which they were singly housed until sacrifice. Unless otherwise specified, rats were fed a regular chow diet (Harlan Teklad #2018, Indianapolis, IN) throughout, and fasted as described below. High fat fed rats were given ad lib access to a safflower oil-based high fat diet containing 60% calories from fat (Dyets, Inc. #112245, Bethlehem, PA) for 4 weeks prior to sacrifice, before which they were fasted as described above. In order to induce T1D, rats were injected with streptozotocin (65 mg/kg IP, Sigma Aldrich, St. Louis, MO) 24 hr prior to the start of a leptin infusion study and were fasted overnight 12 hr after streptozotocin injection. T1D was confirmed by the combination of hyperglycemia (>17 mM) and insulinopenia (<30 pM) after the overnight fast; all rats that did not meet both criteria (~25% of those injected with streptozotocin) were excluded from analysis.
Rats were randomly assigned to fasting time points following a 24 hr fast-refeeding protocol designed to maximize whole-body glycogen content prior to the fast, as occurs following a large meal. The feeding and fasting times for each group of rats are listed in Table S1.
Following the fasting period designated, rats underwent studies as described in the following sections. They were euthanized with IV pentobarbital at the conclusion of each study, and their tissues rapidly freeze-clamped (liver freeze-clamped in situ) in tongs pre-cooled in liquid nitrogen.
METHOD DETAILS
Animal Studies
In all studies, rats were sacrificed with tissues and plasma collected for analysis at 14:00 hr, removing any impact of diurnal variation on the measurements taken. All tracers were infused through a catheter placed ~1 week prior in the carotid artery, and blood was obtained from a catheter in the jugular vein. Measurements of portal vein glucose concentrations were performed on blood samples taken from a catheter in the portal vein; unless otherwise specified, all blood was drawn from the jugular vein. All studies began 1 hr after catheters were connected, reducing any impact of stress from handling on the physiology assessed.
Tracers
Primed-continuous infusions of tracers were performed at the following rates. In each case, the prime was for 5 min, with the continuous infusion spanning 5–120 min. Tracer infusion rates are shown in Table S2.
A bolus of [6,6-14C]2-deoxyglucose was administered in 48 hr fasted control rats and 48 hr fasted rats infused with glucose to increase plasma glucose concentrations to 6 mM and gastrocnemius muscle and epididymal white adipose tissue were harvested and processed to determine basal glucose uptake in both tissues by comparing the plasma [14C] specific activity decay curve to tissue [14C] specific activity, both measured using a scintillation counter (Jurczak et al., 2012).
Leptin Infusions
In order to examine the physiologic impact of varying doses of leptin on glucose, hormones, and lipolysis, rats were infused with stepwise increasing doses of leptin (Sigma) during a continuous infusion of [1,2,3,4,5,6,6-2H7]glucose, [U-13C16]palmitate, [1,1,2,3,3-2H5]glycerol, and [U-13C4]β–OHB starting at hour 42 of a 48 hr fast. From 42–43.5 hrs of the fast rats were infused with tracer only, from hours 43.5–45 they were infused with 20 pmol/(kg-min) leptin, from hours 45–46.5 they were infused with 60 pmol/(kg-min) leptin, and from 46.5–48 hrs the infusion rate was 600 pmol/(kg-min). Blood samples were taken at 43.5, 45, 46.5, and 48 hr for measurement of substrate/hormone concentrations and turnover of each tracer, as described below.
In the T1D leptin dose response studies, overnight (12 hr) fasted rats were infused with stepwise increasing dose of leptin during the same tracer infusion as described above. From 12–14 hrs of the fast rats were infused with tracer only, from 14–16 hrs they were infused with 50 pmol/(kg-min) leptin, and from hours 16–18 they were infused with 600 pmol/(kg-min) leptin. Blood samples were taken at 14, 16, and 18 hr of the fast for measurement of substrate/hormone concentrations and turnover of each tracer, as described below.
Pharmacologic Manipulation of Glycogen and Acetyl-CoA
8, 16, and 48 hr fasted rats were injected with a small molecule inhibitor of glycogen phosphorylase, 1-(3-(3-(2-Chloro-4,5-difluorobenzoyl)ureido)-4-methoxyphenyl)-3-methylurea (Sigma #361515; 5 mg/kg IV) and a tracer infusion of [2,3,3,3-2H4]alanine (15 μmol/[kg-min] prime for 5 min and 5 μmol/[kg-min] continuous) and [3-3H]glucose (0.3 μCi/min prime for 5 min and 0.3 μCi/min continuous) as well as [13C4]β–OHB, [1,1,2,3,3-2H5]glycerol, and [13C16]palmitate at the rates listed in Table S2, for 120 min. Blood samples were obtained before (time zero) and 120 min after treatment with the glycogen phosphorylase inhibitor for measurement of turnover rates and plasma hormone/substrate concentrations.
Hepatic acetyl-CoA content was altered by treatment with a CPT-1 inhibitor (etomoxir, 8 mg/kg) or an ATGL inhibitor (atglistatin, 200 μmol/kg) in 0, 6, 16, and 48 hr fasted rats. Both agents were solubilized by suspending the drug in 4% ethanol/96% normal saline, heating to 60°C, and sonicating, and were cooled to room tempe rature before they were injected intraperitoneally. Intra-arterial infusion of a small amount of glucose was required to avoid hypoglycemia in 48 hr fasted, atglistatin-treated rats. In these animals plasma glucose concentrations were measured every 15 minutes and a variable infusion of 20% glucose (Pfizer Hospira, Lake Forest, IL) was used to maintain plasma glucose concentrations ~3.6–3.7 mM.
The physiologic role of glucocorticoids was assessed in 6 and 48 hr fasted rats by treatment with a glucocorticoid receptor antagonist, mifepristone (Sigma). Following a 2 hr tracer infusion at the rates described above to assess fatty acid, glycerol, glucose, and β-OHB turnover, rats were injected with mifepristone (40 mg/kg) IV. The tracer infusion was continued and the tracer of each of the above substrates was again measured 2 hr after treatment with mifepristone.
Glucose Clamps
Alanine turnover was determined in 48 hr fasted rats following a 2 hr infusion of [2,3,3,3-2H4]alanine as described below. Then, in order to match plasma glucose concentrations to those of 16 hr fasted rats, a variable infusion of glucose was begun through an arterial catheter in the same rats, with plasma glucose concentrations checked every 15 min and adjusted to maintain plasma glucose concentrations ~6.0 mM. Finally, with the glucose infusion continuing, a tracer dose of [6,6-14C2]2-deoxyglucose was administered through a venous catheter and tissue glucose uptake was determined as described below.
In the hyperinsulinemic-euglycemic clamps, glucose turnover was measured during a basal infusion of [1,2,3,4,5,6,6-2H7]glucose as described above. A primed-continuous infusion of Regular insulin (prime 40 mU/kg, continuous infusion rate 4 mU/[kg-min]) was initiated for 150 min, during which plasma glucose concentrations were measured every 10–15 min and a variable infusion of 20% glucose (2.5% [1,2,3,4,5,6,6-2H7] enriched) was infused to maintain euglycemia (~6 pM). The rats were euthanized with IV pentobarbital immediately at the conclusion of the clamp.
Alanine Replacement
48 hr fasted rats were infused with alanine [45 μmol/(kg-min)] during an infusion of [2H7]glucose and [3-13C]lactate at the infusion rates listed above. Hepatic fluxes (whole-body glucose turnover, VPC and VCS), liver TCA cycle intermediate concentrations, and plasma metabolites and hormones were measured as described below.
Flux Measurements
All whole-body substrate turnover rates were calculated using the equation Turnover = ([Tracer APE/Plasma APE]-1)*Infusion rate, where APE designates the atom percent enrichment measured by mass spectrometry as described below. All chemicals used for flux analysis were obtained from Sigma. Turnover of glucose (Perry et al., 2017a), glycerol (Perry et al., 2015), and β–OHB (Perry et al., 2017a) were determined by gas chromatography-mass spectrometry (GC-MS) as we have described. Briefly, both glucose and glycerol were deprotonized with 5 volumes of methanol, derivitized with 3 volumes of 1:1 acetic anhydride:pyridine, heated to 65°C for 20 min, and 1 volume of methanol was added, then GC-MS was used in CI mode to measure glucose enrichment and EI mode to measure glycerol enrichment, with turnover calculated using the equation above. β–OHB was deprotonized with 5 volumes of methanol and derivitized with 3 volumes of b-butanol 4N HCl, after which the samples were heated to 65°C for 60 min, evaporated under N2 gas, and resuspented in 100 μL of trifluoroacetic acid:methylene chloride (1:7). Fatty acid turnover was measured by determining the plasma palmitate enrichment by GC-MS. Samples were prepared for measurements of palmitate enrichment by evaporating 25 μL plasma, dissolving in 750 μL 1:1 chloroform:methanol and derivitizing with 250 μL boron trifluoride/methanol, then heating to 100°C for 5 min, adding 2 mL pentane and 1 mL water, and centrifuging at low speed for 5 min. The supernatant was transferred to another vial, dissolved, and resuspended in 100 μL hexane for measurement of palmitate enrichment by GC-MS (CI mode). We then corrected for the percent fatty acids (30–40% in each study) comprised by palmitate to measure total fatty acid turnover (Vatner et al., 2015). Alanine turnover was determined in rats infused with [2,3,3,3-2H4]alanine by preparing samples to measure alanine enrichment by GC/MS (CI mode) using the same protocol as was employed to measure β–OHB enrichment. GC-MS was then used to determine the m+4 alanine enrichment (retention time ~4.1 min, m/z 242 [m0], 243 [m+1], 244 [m+2], 245 [m+3], 246 [m+4]). Lactate turnover was determined in rats infused with [3-13C]lactate by performing the same extraction as for alanine and measuring the m+1 lactate enrichment (retention time ~2.8 min, m/z 243 [m0] and 244 [m+1]). VPC and VCS flux rates were determined by PINTA (Perry et al., 2017b) in rats infused with [3-13C]lactate at the rates described above. Flux analysis was performed in rats confirmed post-absorptive (≥8 hr fasted) based on the absence of a portal-systemic glucose gradient (Fig. S1B). The equations used for calculation of flux ratios, which use the positional enrichment of glucose and isotopomers of total and C4C5C6 glucose to calculate these relative fluxes, are as follows:
| (1) |
| (2) |
where G2 denotes glucose [m+2] arising from the condensation of two [m+1] trioses as follows:
| (3) |
| (4) |
where G1 denotes glucose [m+1] and G2 is as calculated in equation (3).
The derivations of these equations can be found in our previous report (Perry et al., 2017b). Absolute VPC flux was measured by multiplying the VPC/VEGP ratio by the VEGP (whole-body glucose turnover) measured by dilution of [2H7]glucose. Absolute VCS flux was calculated by dividing VPC by the ratio VPC/VCS. Rates of net hepatic glycogenolysis were calculated by measuring liver glycogen concentrations at the time points indicated and at one hour before or after the relevant time point. Assuming a constant rate of glycogenolysis during that hour, glycogenolysis rates were calculated by measuring the difference in liver glycogen between these time points and dividing by 60 min. The contribution of glycerol to gluconeogenesis was calculated as the difference between EGP and the sum of VPC and the rate of net hepatic glycogenolysis.
The VPDH/VCS flux was measured as the ratio of [4,5-13C2]glutamate / [13C3]alanine in liver, brain, heart, skeletal muscle (gastrocnemius), kidney, WAT, and BAT after a 2 hr infusion of [1,2,3,4,5,6-13C6]glucose (16.7 μmol/[kg-min] prime for 5 min, 5.6 μmol/[kg-min] continuous infusion) based on the assumptions we have described previously (Shulman et al., 1987). Alanine enrichment was measured by GC-MS as described above, and glutamate by LC-MS/MS: the samples were homogenized in 500 μL ice-cold methanol using a TissueLyser and filtered through a Nanosep filter. LC-MS/MS (AbSCIEX 6500 QTRAP with a Shimadzu ultrafast liquid chromatography system, negative ion mode) was used to monitor the relevant ion pairs: [m0] C4-5 glutamate, 146/41, [m+1] C4-5 glutamate, 147/47, and [m+2] C4-5 glutamate, 148/48.
Biochemical Analysis
Plasma glucose was measured enzymatically using the YSI Glucose Analyzer (Yellow Springs, OH). Plasma lactate, triglyceride, and β–OHB concentrations were measured by COBAS (Roche Diagnostics, Indianapolis, IN). Plasma NEFA were measured enzymatically using a Wako reagent (Wako Diagnostics, Mountain View, CA). Plasma glycerol and alanine concentrations were measured by GC-MS: samples were spiked with a 2H (alanine) or 13C (glycerol) internal standard and prepared for GC-MS using the protocols described above, with the ratio of labeled to unlabeled substrate compared to a standard curve to measure absolute concentrations. Plasma amino acid concentrations (glycine, alanine, serine, leucine, isoleucine, aspartate+asparagine, phenylalanine, glutamate+glutamine) were measured by GC-MS after spiking with 2H or 13C internal standards and derivitizing using the protocol described above for β–OHB measurements (Leimer et al., 1977). Plasma insulin, leptin, CRH, ACTH, corticosterone, corticosteroid binding globulin, epinephrine, norepinephrine, growth hormone, T3, and FGF-21 concentrations were measured by ELISA (Mercodia, Winston-Salem, NC; Abcam, Cambridge, MA; MyBioSource, San Diego, CA; MyBioSource; Alpco, Salem, NH; MyBioSource; Abnova; Abnova; Millipore, Billerica, MA; MyBioSource; and R&D Systems, Minneapolis, MN, respectively). Plasma insulin concentrations in the hyperinsulinemic-euglycemic clamps (and corresponding basal plasma) were measured by radioimmunoassay by the Yale Diabetes Research Center Radioimmunoassay Core. Samples used for measurement of epinephrine and norepinephrine were collected into prechilled EDTA-coated tubes. Plasma glucagon was measured in plasma samples collected in prechilled tubes containing aprotinin (0.5 μg/ml), by RIA through the Yale Diabetes Research Center Radioimmunoassay Core.
Tissue Analysis
Liver and muscle glycogen concentrations were assessed using amyloglucosidase digestion (Jurczak et al., 2011). Liver TAG content was measured using the method of Bligh and Dyer (Bligh and Dyer, 1959): lipids were extracted from livers using 2:1 chloroform:methanol, and the Sekisui triglyceride-SL reagent was used to measure triglyceride content spectrophotometrically. Liver and muscle DAG content (Yu et al., 2002) and liver acetyl- and malonyl-CoA concentrations (Perry et al., 2015; Perry et al., 2017a) were measured by LC-MS/MS as we have described. Concentrations of TCA cycle intermediates (citrate, malate, succinate) were measured by LC-MS/MS: ~100 mg of tissue were weighed out, and a solution containing 0.1 μmol of internal standards ([13C6]citrate, [13C4]succinate, and [13 C4]L-malate, all from Sigma) was added. The samples were homogenized in 500 μL ice-cold methanol using a TissueLyser and filtered through a Nanosep filter. LC-MS/MS (AbSCIEX 6500 QTRAP with a Shimadzu ultrafast liquid chromatography system, negative ion mode) was again used to monitor the relevant ion pairs: succinate and [13C4]succinate, 117/99 and 121/103, respectively; malate and [13C4]malate, 133/115 and 137/119, respectively; and citrate and [13C6]citrate, 191/173 and 197/179, respectively. Liver alanine concentrations were measured by GC/MS after spiking liver samples with an internal standard ([13C3]alanine). Hepatic PKCε→translocation (Samuel et al., 2004), gluconeogenic enzyme protein expression (Perry et al., 2015), ApoB (antibody from Meridian Life Science, Memphis, TN), and PGC1α (antibody from Santa Cruz Biotechnology, Dallas, TX) protein expression were assessed by Western blot. Expression of each of these proteins was normalized to GAPDH.
QUANTIFICATION AND STATISTICAL ANALYSIS
Comparisons were performed using the 2-tailed Student’s t-test (if two groups were compared) or ANOVA (if more than two groups were compared), paired or unpaired as specified in the figure legends, with significance defined as a p-value <0.05. GraphPad Prism 7.0 (San Diego, CA) was used for all statistical analysis. In most cases, n=6–8 rats per group, unless otherwise indicated in the figure legends. Data are presented as the mean±S.E.M.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ApoB | Meridian Life Science | CAT#K23300R |
| GAPDH | Cell Signaling | CAT#2118 |
| Pyruvate carboxylase | Santa Cruz | CAT#sc-271493 |
| Cytosolic PEPCK | Santa Cruz | CAT#sc-32879 |
| Glucose-6-phosphatase | Santa Cruz | CAT#sc-27198 |
| PGC1α | Santa Cruz | CAT#sc-13067 |
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Glycogen phosphorylase inhibitor | Sigma Aldrich | CAT#361515 |
| Regular insulin | Eli Lilly | CAT#NDC 0002-8215-01 |
| 20% glucose | Hospira | CAT#NDC 0409-7935-19 |
| Leptin | Sigma Aldrich | CAT#L5037 |
| Streptozotocin | Sigma Aldrich | CAT#S0130 |
| Etomoxir | Sigma Aldrich | CAT#E1905 |
| Atglistatin | Sigma Aldrich | CAT#SML1075 |
| Mifepristone | Sigma Aldrich | CAT#M8046 |
| L-alanine | Sigma Aldrich | CAT#A7469 |
| [1,2,3,4,5,6-13C6] glucose | Cambridge Isotopes | CAT#CLM-1396 |
| [1,2,3,4,5,6,6-2H7] glucose | Cambridge Isotopes | CAT# DLM-2062 |
| Sodium L-[3-13C] lactate | Sigma Aldrich | CAT#721336 |
| [U-13C16] potassium palmitate | Cambridge Isotopes | CAT#CLM-3943 |
| [1,1,2,3,3-2H5] glycerol | Sigma Aldrich | CAT#661473 |
| [U-13C4] sodium D-3-hydroxybutyrate | Cambridge Isotopes | CAT#CLM-3853 |
| [2,3,3,3-2H4] alanine | Sigma Aldrich | CAT#485845 |
| Critical Commercial Assays | ||
| Rat insulin ELISA | Mercodia | CAT#10-1250 |
| Rat leptin ELISA | Abcam | CAT#ab100773 |
| Rat CRH ELISA | MyBioSource | CAT#MBS703710 |
| Rat ACTH ELISA | MyBioSource | CAT#MBS2502683 |
| Mouse/rat corticosterone ELISA | Alpco | CAT#55-CORMS-E01 |
| Rat CBG ELISA | MyBiosource | CAT#MBS2601237 |
| Epinephrine/norepinephrine ELISA | Abnova | CAT#KA1877 |
| Rat/mouse growth hormone ELISA | Millipore | CAT#EZRMGH-45K |
| Mouse, rat triiodothyronine (T3) ELISA | MyBioSource | CAT#MBS580039 |
| Mouse/rat FGF-21 quantikine ELISA | R&D Systems | MF2100 |
| HR Series NEFA-HR(2) | Wako | CAT#999-34691 |
| HR Series NEFA-HR(2) | Wako | CAT#995-34791 |
| HR Series NEFA-HR(2) | Wako | CAT#991-34891 |
| HR Series NEFA-HR(2) | Wako | CAT#993-35191 |
| Triglyceride-SL reagent | Sekisui | Cat#236-99 |
| Deposited Data | ||
| N/A | ||
| Experimental Models: Cell Lines | ||
| N/A | ||
| Experimental Models: Organisms/Strains | ||
| Sprague-Dawley rats | Charles River | Strain code: 400 |
| Oligonucleotides | ||
| N/A | ||
| Recombinant DNA | ||
| N/A | ||
| Software and Algorithms | ||
| N/A | ||
| Other | ||
| Regular chow diet | Harlan Teklad | CAT#2018 |
Supplementary Material
Figure S1 (related to Figure 1). Depletion of hepatic glycogen content lowers plasma glucose and insulin concentrations, causes hypoleptinemia, and activates the HPA axis in starvation. (A) Body weight. In panels (A), (C), (E), (G)–(T), data were compared by ANOVA with Bonferroni’s multiple comparisons test. (B) Plasma glucose concentrations in the jugular vein and portal vein. Jugular vein concentrations are duplicated from Fig. 1A. *P<0.05, **P<0.01 by the 2-tailed unpaired Student’s t-test. (C) Rates of whole-body glucose turnover. (D) Liver glycogen concentrations. (E) Muscle glycogen content. (F) Correlation between plasma glucose concentrations and hepatic glycogenolysis rates. (G)–(I) Plasma non-esterified fatty acid, glycerol, and β-OHB concentrations. (J)–(K) Plasma and liver triglyceride content. (L) Liver membrane diacylglycerol. (M) Hepatic membrane/cytosolic PKC epsilon content. (N) Liver ApoB protein expression. (O) Muscle triglyceride content. (P) Hepatic PGC-1α→protein expression. (Q)–(T) Liver gluconeogenic protein expression. (U) Plasma insulin concentrations before (basal) and after a 150 min hyperinsulinemic-euglycemic clamp. In panels (U)–(V), basal and clamp data within the same group were compared by the 2-tailed paired Student’s t-test. (V) Basal and clamp endogenous glucose production. (W) Suppression of endogenous glucose production during the clamp, with groups compared by the 2-tailed unpaired Student’s t-test. In all panels, data are the mean±S.E.M. of n=6-8 per group.
Figure S2 (Related to Figure 1). Depletion of hepatic glycogen content lowers plasma glucose and insulin concentrations, causes hypoleptinemia, and activates the HPA axis in starvation. (A)–(B) Plasma insulin and glucagon concentrations. (C)–(J) Plasma CRH, ACTH, CBG, epinephrine, norepinephrine, growth hormone, FGF-21, and T3 concentrations. (K)–(L) Plasma leptin and corticosterone concentrations in 4-week high fat fed rats. (M)–(N) Plasma NEFA and β-OHB concentrations in 4-week high fat fed rats. (O) Liver glycogen in rats treated with a glycogen phosphorylase inhibitor as compared to controls (control data are duplicated from Fig. S1D). The 2-tailed unpaired Student’s t-test was used to compare glycogen concentrations with or without the inhibitor at each time point. (P)–(W) Plasma glucose, insulin, lactate, ACTH, glucagon, epinephrine, norepinephrine, and FGF-21 concentrations. In panels (P)–(W), data were compared before and after phosphorylase inhibitor treatment by the 2-tailed paired Student’s t-test. In all panels, data are the mean±S.E.M. of n=6–8 per group.
Figure S3 (Related to Figure 2). Physiologic leptin replacement reduces WAT lipolysis and hepatic glucose production, but supraphysiologic leptin reverses this effect by stimulation of catecholamine secretion in 48 hr fasted normal chow fed rats [(A)–(I)] and type 1 diabetic rats [(J)–(U)]. (A) Glucose infusion rate during a stepwise leptin infusion study. (B)–(E) Plasma non-esterified fatty acid, glycerol, β-OHB, and lactate concentrations. (F)–(H) Plasma insulin, growth hormone, and FGF-21 concentrations. (I) Liver malonyl-CoA content. (J)–(L) Plasma leptin, glucose, and insulin concentrations. (M) Whole-body glucose turnover. (N)–(R) Plasma lipolytic hormone concentrations: plasma ACTH, corticosterone, epinephrine, norepinephrine, and growth hormone. (S)–(U) Whole-body fatty acid, glycerol, and β-OHB turnover. In all panels, data were compared by paired ANOVA with Bonferroni’s multiple comparisons test. The mean±S.E.M. of n=8 per dose are shown. Each rat was studied stepwise with increasing leptin infusion rates.
Figure S4 (Related to Figure 3). Increased hepatic acetyl-CoA content maintains euglycemia in an extended fast. (A) Whole-body palmitate turnover during a fast. (B)–(F) Plasma insulin, lactate, NEFA, glycerol, and β-OHB concentrations in rats treated with the CPT-1 inhibitor etomoxir. (G)–(I) Whole-body fatty acid, glycerol, and β-OHB turnover. (J) Liver malonyl-CoA. (K)–(L) Plasma epinephrine and norepinephrine. Data are the mean±S.E.M. of n=6 per group. In panels (B)–(L), data were compared by ANOVA with Bonferroni’s multiple comparisons test (control vs. etomoxir vs. atglistatin, with these three groups compared separately at each time point; control data are duplicated between Fig. S4 and Fig. S5).
Figure S5 (Related to Figure 4). Increased WAT lipolysis is necessary to increase hepatic acetyl-CoA content and maintain euglycemia in the starved state. (A) Glucose infusion rate required to maintain euglycemia in 48 hr fasted rats treated with ATGL inhibitor atglistatin (rats fasted for 0, 6, and 16 hrs did not require glucose to avoid symptomatic hypoglycemia). (B)–(E) Plasma NEFA, glycerol, lactate, and β-OHB concentrations. (F)–(H) Plasma insulin, epinephrine, and norepinephrine concentrations. (I) Liver malonyl-CoA content. In all panels, data are the mean±S.E.M. of n=5–6 per group. In panels (B)–(I), data were compared by ANOVA with Bonferroni’s multiple comparisons test (control vs. etomoxir vs. atglistatin, with these three groups compared separately at each time point; control data are duplicated between Fig. S4 and Fig. S5).
Figure S6 (Related to Figure 5). Glucocorticoid activity is required to maintain WAT lipolysis and euglycemia in the starved state. (A) Glucose infusion rate required to avoid symptomatic hypoglycemia. **P<0.01, ***P<0.001, ****P<0.0001 by the 2-tailed unpaired Student’s t-test. (B)–(G) Plasma insulin, NEFA, glycerol, and β-OHB concentrations. (H) Hepatic malonyl-CoA. In all panels, data are the mean±S.E.M. of n=6 per group, with the 2-tailed paired Student’s t-test used to compare before vs. after mifepristone treatment at each time point separately in panels (B)–(G).
Figure S7 (Related to Figure 6). Reductions in glucose-alanine cycling with starvation lead to suppression of hepatic glucose production and mitochondrial oxidation. (A)–(B) Plasma and liver alanine concentrations. In panels (A)–(O), comparisons were performed using ANOVA with Bonferroni’s multiple comparisons test. (C) Plasma lactate. (D) Whole-body lactate turnover. (E) Total amino acid concentrations. (F)–(L) Plasma leucine, isoleucine, phenylalanine, glycine, aspartate/asparagine, glutamate/glutamine, and serine concentrations. (M) VPC/VCS ratio. (N)–(O) Liver succinate and malate concentrations. (P)–(Q) Plasma and liver alanine concentrations in 48 hr fasted rats infused with alanine (45 μmol/[kg-min]). In panels (P)–(Y) data were compared by the 2-tailed unpaired Student’s t-test, with the exception of panel (S) in which data were compared by the 2-tailed paired Student’s t-test. (R)–(U) Plasma lactate, insulin, glucagon, and leptin concentrations. (V)–(W) Liver succinate and malate concentrations. (X) VPC/VCS. (Y) Plasma T3. Data are the mean±S.E.M. of n=6–8 per group.
Figure S8 (Related to Figure 7). Reductions in plasma glucose concentrations from 6 to 5 mM in the starved state leads to reduced muscle glucose-alanine cycling and hypoleptinemia. (A)–(B) Plasma glucose and glucose infusion rate during a clamp to increase plasma glucose concentrations in 48 hr fasted rats to 6 mM. (C) Plasma lactate. In panels (C)–(E), rats with 5 mM (time zero of the clamp) and 6 mM (120 min of the clamp) plasma glucose were compared by the 2-tailed paired Student’s t-test. (F)–(H) Plasma NEFA, glycerol, and β-OHB concentrations in rats treated with a glycogen phosphorylase inhibitor. In panels (F)–(K), data at each time point were compared by the 2-tailed paired Student’s t-test. (G)–(H) Plasma glycerol and β-OHB concentrations. (I)–(J) Whole-body glycerol and β-OHB turnover. In all panels, data are the mean±S.E.M. of n=6 per group.
Highlights.
Hypoleptinemia promotes HPA axis-driven WAT lipolysis to maintain euglycemia
Increased hepatic acetyl-CoA content maintains euglycemia during starvation
Substrate limitation reduces mitochondrial VCS and VPC flux during a prolonged fast
Acknowledgments
The authors thank Jianying Dong, Mario Kahn, Dongyan Zhang, Gina Butrico, Irina Smolgovsky, Maria Batsu, and Codruta Todeasa for their valuable technical contributions. This study was funded by grants from the United States Public Health Service (R01 DK113984, R01 DK40936, P30 DK059635, R01 AG23686, T32 DK101019, K99 CA215315, R01 NS087568, UL1TR000142).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Experiments were performed and data analyzed by R.J.P., Y.W., G.W.C., A.R.-C., J.D.S., S.D., and X.-M. Z. The study was designed and the manuscript written by R.J.P., K.F.P., and G.I.S.
References
- Ahima RS, Lazar MA. Adipokines and the peripheral and neural control of energy balance. Molecular endocrinology. 2008;22:1023–1031. doi: 10.1210/me.2007-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, Flier JS. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- Benedict FG. A study of prolonged fasting. Carnegie Inst Wash Pub. 1907;77:66. [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Canadian journal of biochemistry and physiology. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Cahill GF., Jr Starvation in man. The New England journal of medicine. 1970;282:668–675. doi: 10.1056/NEJM197003192821209. [DOI] [PubMed] [Google Scholar]
- Chan JL, Heist K, DePaoli AM, Veldhuis JD, Mantzoros CS. The role of falling leptin levels in the neuroendocrine and metabolic adaptation to short-term starvation in healthy men. The Journal of clinical investigation. 2003;111:1409–1421. doi: 10.1172/JCI17490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denroche HC, Kwon MM, Quong WL, Neumann UH, Kulpa JE, Karunakaran S, Clee SM, Brownsey RW, Covey SD, Kieffer TJ. Leptin induces fasting hypoglycaemia in a mouse model of diabetes through the depletion of glycerol. Diabetologia. 2015;58:1100–1108. doi: 10.1007/s00125-015-3529-4. [DOI] [PubMed] [Google Scholar]
- Denroche HC, Levi J, Wideman RD, Sequeira RM, Huynh FK, Covey SD, Kieffer TJ. Leptin therapy reverses hyperglycemia in mice with streptozotocin-induced diabetes, independent of hepatic leptin signaling. Diabetes. 2011;60:1414–1423. doi: 10.2337/db10-0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felig P, Marliss E, Owen OE, Cahill GF., Jr Blood glucose and cluconeogenesis in fasting man. Arch Intern Med. 1969;123:293–298. [PubMed] [Google Scholar]
- Flier JS, Harris M, Hollenberg AN. Leptin, nutrition, and the thyroid: the why, the wherefore, and the wiring. The Journal of clinical investigation. 2000;105:859–861. doi: 10.1172/JCI9725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintze LJ, Mahmoodianfard S, Auguste CB, Doucet E. Weight Loss and Appetite Control in Women. Curr Obes Rep. 2017 doi: 10.1007/s13679-017-0273-8. [DOI] [PubMed] [Google Scholar]
- Jensen MD, Caruso M, Heiling V, Miles JM. Insulin regulation of lipolysis in nondiabetic and IDDM subjects. Diabetes. 1989;38:1595–1601. doi: 10.2337/diab.38.12.1595. [DOI] [PubMed] [Google Scholar]
- Jurczak MJ, Lee AH, Jornayvaz FR, Lee HY, Birkenfeld AL, Guigni BA, Kahn M, Samuel VT, Glimcher LH, Shulman GI. Dissociation of inositol-requiring enzyme (IRE1alpha)-mediated c-Jun N-terminal kinase activation from hepatic insulin resistance in conditional X-box-binding protein-1 (XBP1) knock-out mice. The Journal of biological chemistry. 2012;287:2558–2567. doi: 10.1074/jbc.M111.316760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurczak MJ, Lee HY, Birkenfeld AL, Jornayvaz FR, Frederick DW, Pongratz RL, Zhao X, Moeckel GW, Samuel VT, Whaley JM, et al. SGLT2 deletion improves glucose homeostasis and preserves pancreatic beta-cell function. Diabetes. 2011;60:890–898. doi: 10.2337/db10-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karl JP, Smith TJ, Wilson MA, Bukhari AS, Pasiakos SM, McClung HL, McClung JP, Lieberman HR. Altered metabolic homeostasis is associated with appetite regulation during and following 48-h of severe energy deprivation in adults. Metabolism: clinical and experimental. 2016;65:416–427. doi: 10.1016/j.metabol.2015.11.001. [DOI] [PubMed] [Google Scholar]
- Kim KH, Lee MS. FGF21 as a Stress Hormone: The Roles of FGF21 in Stress Adaptation and the Treatment of Metabolic Diseases. Diabetes Metab J. 2014;38:245–251. doi: 10.4093/dmj.2014.38.4.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Mangelsdorf DJ. Fibroblast growth factor 21: from pharmacology to physiology. The American journal of clinical nutrition. 2010;91:254S–257S. doi: 10.3945/ajcn.2009.28449B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leimer KR, Rice RH, Gehrke CW. Complete mass spectra of N-trifluoroacetyl-n-butyl esters of amino acids. J Chromatogr. 1977;141:121–144. doi: 10.1016/s0021-9673(00)99131-3. [DOI] [PubMed] [Google Scholar]
- Levin N, Nelson C, Gurney A, Vandlen R, de Sauvage F. Decreased food intake does not completely account for adiposity reduction after ob protein infusion. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:1726–1730. doi: 10.1073/pnas.93.4.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan B, Goodarzi MO, Phillips NG, Guo X, Chen YD, Yao J, Allison M, Rotter JI, Shaw R, Montminy M. Leptin-mediated increases in catecholamine signaling reduce adipose tissue inflammation via activation of macrophage HDAC4. Cell metabolism. 2014;19:1058–1065. doi: 10.1016/j.cmet.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry JD, Stark MJ, Foster DW. Hepatic malonyl-CoA levels of fed, fasted and diabetic rats as measured using a simple radioisotopic assay. The Journal of biological chemistry. 1978;253:8291–8293. [PubMed] [Google Scholar]
- Morton GJ, Meek TH, Matsen ME, Schwartz MW. Evidence against hypothalamic-pituitary-adrenal axis suppression in the antidiabetic action of leptin. The Journal of clinical investigation 2015. 2015 doi: 10.1172/JCI82723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasiakos SM, Caruso CM, Kellogg MD, Kramer FM, Lieberman HR. Appetite and endocrine regulators of energy balance after 2 days of energy restriction: insulin, leptin, ghrelin, and DHEA-S. Obesity. 2011;19:1124–1130. doi: 10.1038/oby.2010.316. [DOI] [PubMed] [Google Scholar]
- Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM, et al. Hepatic Acetyl CoA Links Adipose Tissue Inflammation to Hepatic Insulin Resistance and Type 2 Diabetes. Cell. 2015;160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Peng L, Abulizi A, Kennedy L, Cline GW, Shulman GI. Mechanism for leptin’s acute insulin-independent effect to reverse diabetic ketoacidosis. The Journal of clinical investigation. 2017a;127:657–669. doi: 10.1172/JCI88477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Peng L, Cline GW, Butrico GM, Wang Y, Zhang XM, Rothman DL, Petersen KF, Shulman GI. Non-invasive assessment of hepatic mitochondrial metabolism by positional isotopomer NMR tracer analysis (PINTA) Nature communications. 2017b;8:798. doi: 10.1038/s41467-017-01143-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Peng L, Cline GW, Petersen KF, Shulman GI. A Non-invasive Method to Assess Hepatic Acetyl-CoA In Vivo. Cell metabolism. 2017c;25:749–756. doi: 10.1016/j.cmet.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Petersen KF, Shulman GI. Pleotropic effects of leptin to reverse insulin resistance and diabetic ketoacidosis. Diabetologia. 2016;59:933–937. doi: 10.1007/s00125-016-3909-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Zhang XM, Zhang D, Kumashiro N, Camporez JP, Cline GW, Rothman DL, Shulman GI. Leptin reverses diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nature medicine. 2014 doi: 10.1038/nm.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MC, Madiraju AK, Gassaway BM, Marcel M, Nasiri AR, Butrico G, Marcucci MJ, Zhang D, Abulizi A, Zhang XM, et al. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. The Journal of clinical investigation. 2016;126:4361–4371. doi: 10.1172/JCI86013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravussin Y, LeDuc CA, Watanabe K, Mueller BR, Skowronski A, Rosenbaum M, Leibel RL. Effects of chronic leptin infusion on subsequent body weight and composition in mice: Can body weight set point be reset? Molecular metabolism. 2014;3:432–440. doi: 10.1016/j.molmet.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman DL, Magnusson I, Katz LD, Shulman RG, Shulman GI. Quantitation of hepatic glycogenolysis and gluconeogenesis in fasting humans with 13C NMR. Science. 1991;254:573–576. doi: 10.1126/science.1948033. [DOI] [PubMed] [Google Scholar]
- Saggerson ED, Carpenter CA. Effects of fasting and malonyl CoA on the kinetics of carnitine palmitoyltransferase and carnitine octanoyltransferase in intact rat liver mitochondria. FEBS letters. 1981;132:166–168. doi: 10.1016/0014-5793(81)81152-0. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. The Journal of biological chemistry. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Liu ZX, Wang A, Beddow SA, Geisler JG, Kahn M, Zhang XM, Monia BP, Bhanot S, Shulman GI. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. The Journal of clinical investigation. 2007;117:739–745. doi: 10.1172/JCI30400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorr M, Miller KK. The endocrine manifestations of anorexia nervosa: mechanisms and management. Nature reviews Endocrinology. 2017;13:174–186. doi: 10.1038/nrendo.2016.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shek EW, Brands MW, Hall JE. Chronic leptin infusion increases arterial pressure. Hypertension. 1998;31:409–414. doi: 10.1161/01.hyp.31.1.409. [DOI] [PubMed] [Google Scholar]
- Shibuya I, Utsunomiya K, Toyohira Y, Ueno S, Tsutsui M, Cheah TB, Ueta Y, Izumi F, Yanagihara N. Regulation of catecholamine synthesis by leptin. Annals of the New York Academy of Sciences. 2002;971:522–527. doi: 10.1111/j.1749-6632.2002.tb04517.x. [DOI] [PubMed] [Google Scholar]
- Shulman GI, Rossetti L, Rothman DL, Blair JB, Smith D. Quantitative analysis of glycogen repletion by nuclear magnetic resonance spectroscopy in the conscious rat. The Journal of clinical investigation. 1987;80:387–393. doi: 10.1172/JCI113084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatner DF, Majumdar SK, Kumashiro N, Petersen MC, Rahimi Y, Gattu AK, Bears M, Camporez JP, Cline GW, Jurczak MJ, et al. Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:1143–1148. doi: 10.1073/pnas.1423952112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villareal DT, Fontana L, Das SK, Redman L, Smith SR, Saltzman E, Bales C, Rochon J, Pieper C, Huang M, et al. Effect of Two-Year Caloric Restriction on Bone Metabolism and Bone Mineral Density in Non-Obese Younger Adults: A Randomized Clinical Trial. J Bone Miner Res. 2016;31:40–51. doi: 10.1002/jbmr.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. The Journal of biological chemistry. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (related to Figure 1). Depletion of hepatic glycogen content lowers plasma glucose and insulin concentrations, causes hypoleptinemia, and activates the HPA axis in starvation. (A) Body weight. In panels (A), (C), (E), (G)–(T), data were compared by ANOVA with Bonferroni’s multiple comparisons test. (B) Plasma glucose concentrations in the jugular vein and portal vein. Jugular vein concentrations are duplicated from Fig. 1A. *P<0.05, **P<0.01 by the 2-tailed unpaired Student’s t-test. (C) Rates of whole-body glucose turnover. (D) Liver glycogen concentrations. (E) Muscle glycogen content. (F) Correlation between plasma glucose concentrations and hepatic glycogenolysis rates. (G)–(I) Plasma non-esterified fatty acid, glycerol, and β-OHB concentrations. (J)–(K) Plasma and liver triglyceride content. (L) Liver membrane diacylglycerol. (M) Hepatic membrane/cytosolic PKC epsilon content. (N) Liver ApoB protein expression. (O) Muscle triglyceride content. (P) Hepatic PGC-1α→protein expression. (Q)–(T) Liver gluconeogenic protein expression. (U) Plasma insulin concentrations before (basal) and after a 150 min hyperinsulinemic-euglycemic clamp. In panels (U)–(V), basal and clamp data within the same group were compared by the 2-tailed paired Student’s t-test. (V) Basal and clamp endogenous glucose production. (W) Suppression of endogenous glucose production during the clamp, with groups compared by the 2-tailed unpaired Student’s t-test. In all panels, data are the mean±S.E.M. of n=6-8 per group.
Figure S2 (Related to Figure 1). Depletion of hepatic glycogen content lowers plasma glucose and insulin concentrations, causes hypoleptinemia, and activates the HPA axis in starvation. (A)–(B) Plasma insulin and glucagon concentrations. (C)–(J) Plasma CRH, ACTH, CBG, epinephrine, norepinephrine, growth hormone, FGF-21, and T3 concentrations. (K)–(L) Plasma leptin and corticosterone concentrations in 4-week high fat fed rats. (M)–(N) Plasma NEFA and β-OHB concentrations in 4-week high fat fed rats. (O) Liver glycogen in rats treated with a glycogen phosphorylase inhibitor as compared to controls (control data are duplicated from Fig. S1D). The 2-tailed unpaired Student’s t-test was used to compare glycogen concentrations with or without the inhibitor at each time point. (P)–(W) Plasma glucose, insulin, lactate, ACTH, glucagon, epinephrine, norepinephrine, and FGF-21 concentrations. In panels (P)–(W), data were compared before and after phosphorylase inhibitor treatment by the 2-tailed paired Student’s t-test. In all panels, data are the mean±S.E.M. of n=6–8 per group.
Figure S3 (Related to Figure 2). Physiologic leptin replacement reduces WAT lipolysis and hepatic glucose production, but supraphysiologic leptin reverses this effect by stimulation of catecholamine secretion in 48 hr fasted normal chow fed rats [(A)–(I)] and type 1 diabetic rats [(J)–(U)]. (A) Glucose infusion rate during a stepwise leptin infusion study. (B)–(E) Plasma non-esterified fatty acid, glycerol, β-OHB, and lactate concentrations. (F)–(H) Plasma insulin, growth hormone, and FGF-21 concentrations. (I) Liver malonyl-CoA content. (J)–(L) Plasma leptin, glucose, and insulin concentrations. (M) Whole-body glucose turnover. (N)–(R) Plasma lipolytic hormone concentrations: plasma ACTH, corticosterone, epinephrine, norepinephrine, and growth hormone. (S)–(U) Whole-body fatty acid, glycerol, and β-OHB turnover. In all panels, data were compared by paired ANOVA with Bonferroni’s multiple comparisons test. The mean±S.E.M. of n=8 per dose are shown. Each rat was studied stepwise with increasing leptin infusion rates.
Figure S4 (Related to Figure 3). Increased hepatic acetyl-CoA content maintains euglycemia in an extended fast. (A) Whole-body palmitate turnover during a fast. (B)–(F) Plasma insulin, lactate, NEFA, glycerol, and β-OHB concentrations in rats treated with the CPT-1 inhibitor etomoxir. (G)–(I) Whole-body fatty acid, glycerol, and β-OHB turnover. (J) Liver malonyl-CoA. (K)–(L) Plasma epinephrine and norepinephrine. Data are the mean±S.E.M. of n=6 per group. In panels (B)–(L), data were compared by ANOVA with Bonferroni’s multiple comparisons test (control vs. etomoxir vs. atglistatin, with these three groups compared separately at each time point; control data are duplicated between Fig. S4 and Fig. S5).
Figure S5 (Related to Figure 4). Increased WAT lipolysis is necessary to increase hepatic acetyl-CoA content and maintain euglycemia in the starved state. (A) Glucose infusion rate required to maintain euglycemia in 48 hr fasted rats treated with ATGL inhibitor atglistatin (rats fasted for 0, 6, and 16 hrs did not require glucose to avoid symptomatic hypoglycemia). (B)–(E) Plasma NEFA, glycerol, lactate, and β-OHB concentrations. (F)–(H) Plasma insulin, epinephrine, and norepinephrine concentrations. (I) Liver malonyl-CoA content. In all panels, data are the mean±S.E.M. of n=5–6 per group. In panels (B)–(I), data were compared by ANOVA with Bonferroni’s multiple comparisons test (control vs. etomoxir vs. atglistatin, with these three groups compared separately at each time point; control data are duplicated between Fig. S4 and Fig. S5).
Figure S6 (Related to Figure 5). Glucocorticoid activity is required to maintain WAT lipolysis and euglycemia in the starved state. (A) Glucose infusion rate required to avoid symptomatic hypoglycemia. **P<0.01, ***P<0.001, ****P<0.0001 by the 2-tailed unpaired Student’s t-test. (B)–(G) Plasma insulin, NEFA, glycerol, and β-OHB concentrations. (H) Hepatic malonyl-CoA. In all panels, data are the mean±S.E.M. of n=6 per group, with the 2-tailed paired Student’s t-test used to compare before vs. after mifepristone treatment at each time point separately in panels (B)–(G).
Figure S7 (Related to Figure 6). Reductions in glucose-alanine cycling with starvation lead to suppression of hepatic glucose production and mitochondrial oxidation. (A)–(B) Plasma and liver alanine concentrations. In panels (A)–(O), comparisons were performed using ANOVA with Bonferroni’s multiple comparisons test. (C) Plasma lactate. (D) Whole-body lactate turnover. (E) Total amino acid concentrations. (F)–(L) Plasma leucine, isoleucine, phenylalanine, glycine, aspartate/asparagine, glutamate/glutamine, and serine concentrations. (M) VPC/VCS ratio. (N)–(O) Liver succinate and malate concentrations. (P)–(Q) Plasma and liver alanine concentrations in 48 hr fasted rats infused with alanine (45 μmol/[kg-min]). In panels (P)–(Y) data were compared by the 2-tailed unpaired Student’s t-test, with the exception of panel (S) in which data were compared by the 2-tailed paired Student’s t-test. (R)–(U) Plasma lactate, insulin, glucagon, and leptin concentrations. (V)–(W) Liver succinate and malate concentrations. (X) VPC/VCS. (Y) Plasma T3. Data are the mean±S.E.M. of n=6–8 per group.
Figure S8 (Related to Figure 7). Reductions in plasma glucose concentrations from 6 to 5 mM in the starved state leads to reduced muscle glucose-alanine cycling and hypoleptinemia. (A)–(B) Plasma glucose and glucose infusion rate during a clamp to increase plasma glucose concentrations in 48 hr fasted rats to 6 mM. (C) Plasma lactate. In panels (C)–(E), rats with 5 mM (time zero of the clamp) and 6 mM (120 min of the clamp) plasma glucose were compared by the 2-tailed paired Student’s t-test. (F)–(H) Plasma NEFA, glycerol, and β-OHB concentrations in rats treated with a glycogen phosphorylase inhibitor. In panels (F)–(K), data at each time point were compared by the 2-tailed paired Student’s t-test. (G)–(H) Plasma glycerol and β-OHB concentrations. (I)–(J) Whole-body glycerol and β-OHB turnover. In all panels, data are the mean±S.E.M. of n=6 per group.