

Abstract
Signaling across cellular membranes, the 826 human G protein-coupled receptors (GPCR) govern a wide range of vital physiological processes, making GPCRs prominent drug targets. X-ray crystallography provided GPCR molecular architectures, which also revealed the need for additional structural dynamics data to support drug development. Here, NMR with the wild type-like A2A receptor (A2AAR) in solution provides a comprehensive characterization of signaling-related structural dynamics. All six tryptophan indole and eight glycine backbone 15N–1H NMR signals in A2AAR were individually assigned. These NMR probes provided insight into the role of Asp522.50 as an allosteric link between the orthosteric drug binding site and the intracellular signaling surface, revealing strong interactions with the toggle switch Trp 2466.48, and delineated the structural response to variable efficacy of bound drugs across A2AAR. The present data support GPCR signaling based on dynamic interactions between two semi-independent subdomains connected by an allosteric switch at Asp522.50.
Graphical Abstract
Monitoring dynamics of GPCR signaling using stable isotope NMR reveals the path of communication enabling an allosteric response to ligand binding.
Introduction
Drug binding in G protein-coupled receptors (GPCRs) initiates signaling across cell membranes over a distance of about 30 Ångstroms (Audet and Bouvier, 2012; Rosenbaum et al., 2009). From the molecular architecture of GPCRs, as determined by X-ray crystallography, it is clear that this signal transfer must be supported by dynamic plasticity of the receptors, which needs to be investigated by other methods. NMR spectroscopy in solution complements GPCR crystal structure determination by its unique ability of detecting multiple, simultaneously populated conformational states, as has been reported, for example, in studies of the β2–adrenergic receptor (β2AR) (Bokoch et al., 2010; Eddy et al., 2016; Horst et al., 2013; Kim et al., 2013; Kofuku et al., 2012; Kofuku et al., 2014; Liu et al., 2012a; Manglik et al., 2015; Nygaard et al., 2013), the μ-opioid receptor (MOR) (Okude et al., 2015; Sounier et al., 2015), the β1–adrenergic receptor (Isogai et al., 2016), and the human A2A adenosine receptor (A2AAR) (Ye et al., 2016). Thereby, drugs bound in the orthosteric pocket can alter the relative populations of two or multiple locally different conformational states on the intracellular surface, which signal to partner proteins (Didenko et al., 2013; Manglik and Kobilka, 2014). Here, these potentialities of NMR in solution were used to investigate the structural basis of the functional role of Asp522.50 (superscripts indicate the Ballesteros-Weinstein nomenclature; (Ballesteros and Weinstein, 1995), which has been reported to be a key residue in an allosteric switch along the 30-Ångstrom signaling pathway from the drug binding pocket of A2AAR to its intracellular surface (Figure 1A).
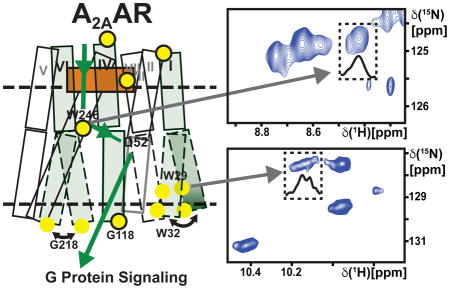
Figure 1. A2AAR Signaling via an Allosteric Center at Asp522.50 Probed by an Extensive Network of Assigned NMR Signals.

(A) Scheme of agonist-induced signaling in A2AAR and A2AAR[D52N], i.e., receptors with and without an active allosteric switch at position 522.50, respectively. The gray shape represents A2AAR, the green shape the signal-inducing drug, and the upper and lower thin horizontal lines indicate the extracellular and intracellular membrane surfaces, respectively. (B) Pie chart showing the frequency of amino acid types occurring at position 522.50 in all class A GPCRs: Asp 84%, Glu 10%, Arg and others 6% (Munk et al., 2016). (C) Ribbon representation of the crystal structure of the antagonist ZM241385 (green) complex of A2AAR expressed in Pichia pastoris (PDB 6AQF). The location of the fusion protein BRIL in the intracellular loop 3 (ICL3), where G218 was eliminated in the fusion protein, is indicated by a thin broken line; G218 was left intact in the A2AAR sequence used for NMR studies (see Figure S1 for the location of G218 at the intracellular tip of trans-membrane helix VI in a structure without fusion protein). NMR-assigned tryptophan residues and their sequence positions are highlighted in blue, assigned glycine residues are orange, and the residue Asp522.50 is shown in red. (D) 2D [15N,1H]-TROSY correlation spectrum of A2AAR in complex with ZM241385. Dashed boxes highlight the Trp indole 15N–1H and Gly backbone 15N–1H regions, which are shown on expanded scales in (E) and (F), respectively, with sequence-specific assignments indicated next to the signals. The peaks numbered 1 to 30 were used to monitor the global folds of the A2AAR variants used here for resonance assignments and function-related studies.
The key role of Asp522.50 in regulating signaling to intracellular partner proteins has been experimentally documented for over 30 different class A GPCRs (Katritch et al., 2014). Asp522.50 is also one of the most highly conserved residues, with aspartic acid at this position in about 85% of the approximately 700 class A GPCRs in the human proteome (Figure 1B) (Katritch et al., 2014). For some GPCRs, replacement of Asp522.50 with an uncharged amino acid resulted in marked decrease of G protein signaling; examples include the cannabinoid receptors CB1 and CB2 (Tao and Abood, 1998) and the type 1 neurotensin receptor (NTSR1) (Martin et al., 1999). For other receptors, including the angiotensin II receptor (Bihoreau et al., 1993), MOR (Xu et al., 1999) and A2AAR (Massink et al., 2014), replacement of Asp522.50 has been reported to completely abolish G protein signaling. In addition to its role in regulating G protein signaling, Asp522.50 was seen in high-resolution crystal structures of A2AAR and DOR to form coordinative bonds with a sodium ion (Liu et al., 2012b), which provided an initial rationale for independently observed physiological effects of sodium ions on opiate ligand binding and receptor signaling (Cooper et al., 1982; Pert et al., 2009).
A2AAR belongs to the purinergic receptor family that binds endogenous adenosine and regulates vasodilation and inflammation (Ohta and Sitkovsky, 2001), and affects the central nervous system (Dunwiddie and Masino, 2001). A2AAR is a validated target for the treatment of Parkinson’s disease (Bara-Jimenez et al., 2003), and more recently it has been identified as a potential therapeutic target for cancer immunotherapies (Young et al., 2016). Relating to this biomedical interest, A2AAR has been subject to intense studies, including crystal structure determinations of complexes with antagonists (Doré et al., 2011; Jaakola et al., 2008) and agonists (Lebon et al., 2011; Xu et al., 2011), and for a ternary complex of A2AAR with an agonist and a “mini-GS” G protein mimetic (Carpenter et al., 2016).
A recent crystal structure determination of the variant receptor A2AAR[D52N] in complex with the full agonist UK432097 (PDB 5W5F) showed that this structure is nearly identical to that of the A2AAR complex with the same agonist (PDB 3QAK) (Xu et al., 2011), including nearly identical conformations of the intracellular signaling surfaces. In this paper we further explore the structural basis of the Asp522.50-related allosteric effects on the receptor function, showing that NMR in solution can provide novel insights. Based on sequence-specific assignment of numerous amino acid residues distributed throughout the receptor (Figures 1C and S1), we report observations of a strong interplay between Asp522.50 and the “toggle switch” tryptophan at position 2466.48 (Rosenbaum et al., 2009; Schwartz et al., 2006; Shi et al., 2002), and of local structural polymorphisms on the A2AAR intracellular surface which can be directly related to the functionality of the Asp522.50 allosteric switch.
Results
We obtained sequence-specific resonance assignments for well-resolved resonances in the NMR spectra of A2AAR (Figure 1D), including the indole 15N–1H-signals of all six tryptophans (Figure 1E) and the backbone 15N–1H signals of eight glycines (Figure 1F). The assigned signals provided NMR probes of conformation in the hydrophobic core, at the extracellular surface with the entry to the orthosteric ligand binding cavity, and at the intracellular surface (Figure 1C). The labeling also included helix VI, which has been observed in crystal structures to undergo a large structural rearrangement, relative to an antagonist complex, upon formation of a complex with an agonist (Xu et al., 2011) and in a ternary complex with an agonist and a G protein or G protein mimetic (Carpenter et al., 2016). We then investigated the response of the assigned NMR signals to variable efficacy of bound drugs. To obtain information on the allosteric switch at Asp522.50, we compared corresponding NMR data of A2AAR and A2AAR[D52N], where the allosteric center is known to be inactive (Massink et al., 2014). This experimental approach was based on the use of a novel expression system for GPCRs, which allowed us to obtain sequence-specific NMR assignments by single-residue amino acid replacements.
Expression of Stable-Isotope-Labeled A2AAR in Pichia pastoris
A2AAR was expressed in Pichia pastoris, which enabled uniform labeling with the stable isotopes 2H,13C and 15N. The use of extensive deuteration provided the expected improvement of the A2AAR NMR spectra (Figure S2), which made the present project feasible. Optimization of each step of the expression and purification process resulted in the capability to routinely produce milligram-per-liter quantities of homogeneous isotope-labeled A2AAR (see STAR Methods). A2AAR reconstituted into LMNG/CHS mixed micelles for NMR studies was highly homogeneous, as observed by analytical SEC, which showed that samples were monodisperse and did not contain detectable amounts of aggregated protein (Figure S3B).
To ensure that the present solution NMR studies were performed with A2AAR that was pharmacologically and structurally identical to A2AAR produced in the widely used Sf9 cells (Carpenter et al., 2016; Jaakola et al., 2008; Xu et al., 2011), we performed extensive characterization of A2AAR produced from Pichia. Ligand binding experiments performed with A2AAR which was either embedded in membranes isolated from Pichia preparations (Figure S3, C and D) or reconstituted in LMNG/CHS micelles (Figure S3A) showed nearly identical activity to A2AAR produced in Sf9 cells. A crystal structure of Pichia-produced A2AAR in complex with the antagonist ZM241385 was found to be identical to an earlier corresponding structure of A2AAR produced in Sf9 (Figure S3, E and F).
Sequence-Specific Resonance Assignments in A2AAR
Uniform stable-isotope labeling with 15N and 2H enabled the recording of A2AAR NMR spectra where all tryptophan indole 15N–1H and most glycine backbone 15N–1H NMR signals were well separated from the other signals (Figures 1, E and F). In addition, the signals numbered 1 to 30 in the 2D [15N,1H]-TROSY correlation spectrum (Figure 1D) were also well separated, so that chemical shift changes or variations of the line shapes could readily be detected. Primary assignments were obtained for the A2AAR complex with the antagonist ZM2411385, based on comparisons of the spectra of A2AAR and A2AAR variants containing single-residue amino acid replacements. Throughout, we checked that the native globular fold was preserved in the variant proteins, by monitoring the chemical shifts of the signals 1 to 30 (Figure 1D) as illustrated for the tryptophan assignments with the Figures S4 to S6 and Table S2.
The Trp indole 15N–1H signals were assigned from the missing peak for the variant A2AAR in which the corresponding tryptophan was replaced (Figure 2, A and B). The signal for Trp1294.50 was weaker than the other signals, but was reproducibly observed in multiple sample preparations and was present in the spectra of all variants with a different tryptophan replacement. Replacement of Trp2466.48 with Phe resulted in a missing signal outside of the tryptophan indole 15N–1H spectral region (Figures 1E and 2B); this outlying chemical shift is due to large ring current fields in the chemical environment of Trp2466.48, as confirmed by ring current calculations based on corresponding A2AAR crystal structures (Table S1) (Johnson Jr and Bovey, 1958; Koradi et al., 1996; Liu and Wüthrich, 2016; MacDonald and Phillips, 1967; Perkins and Wüthrich, 1979; Wüthrich, 1969; Wüthrich, 1986).
Figure 2. Sequence-specific Assignment of the Trp Indole 15N–1H and Eight Gly Backbone 15N–1H NMR Lines.

Panels A and B document the assignments for the tryptophans, and the panels C to I those of eight glycines. Each panel displays a region of the 2D [15N,1H]-TROSY correlation spectrum of A2AAR in complex with ZM241385. On the left are contour plots and on the right are 1D cross sections taken at the 15N chemical shifts indicated by dashed lines in the contour plots. The spectrum of A2AAR is shown in blue, and the spectra of the variant proteins used for the assignment of the residues indicated in the individual panels are shown in red; locations in the 3D A2AAR structure are also indicated, where “TM” stands for “trans-membrane helix”, and “ICL” and “ECL”, respectively, for intracellular and extracellular loop. Comparison of the two spectra resulted in sequence-specific assignment of the signal identified by an asterisk, by observation of the absence in the variant protein of the signal to be assigned. In the right panel of (C), the most intense signals have been truncated.
Backbone 15N–1H signals of eight glycines were assigned by single-residue replacements of Gly with Ala (Figure 2, C–I). Overall, assignment of the Gly signals was straightforward, since, with the sole exception of Gly 51.31, they were strong and well resolved.
Extensive deuteration has first been achieved with soluble proteins in the late 1960s (Markley et al., 1968), and it became mandatory with the introduction of transverse relaxation-optimized spectroscopy (TROSY) for studies of large structures (Pervushin et al., 1997). A limitation arising from the use of 2H2O as the solvent during protein expression is that back-protonation of the amide groups may be incomplete. While all six Trp indole 15N–1H signals expected in A2AAR were observed, albeit with low intensity for Trp129 (Figures 1 and 2), we could therefore not so far establish whether or not the TROSY spectrum of Figure 1D includes signals from all residues in the polypeptide chain. In this situation, we selected 30 well-resolved polypeptide backbone signals, which all represent large conformation-dependent 1H chemical shifts and are therefore sensitive probes of conformational rearrangements (Wüthrich, 1986), to monitor the preservation of the overall three-dimensional fold in the variant A2AARs used for the NMR assignments (Figures S4–S6; Table S2).
Interplay of the “Toggle Switch” Tryptophan 2466.48 with the Allosteric Center at Asp522.50
Tryptophan 2466.48 is referred to in the GPCR literature as the “toggle switch” or “rotamer toggle switch”, since early studies predicted large changes in the rotamer state of Trp2466.48 between GPCR complexes with agonists and antagonists (Nygaard et al., 2013; Shi et al., 2002). Trp2466.48 is located in helix VI at the bottom of the ligand-binding cavity (Figure 1). Variations in the conformation of Trp6.48 between antagonist and agonist complexes have been widely observed in crystal structures of class A human GPCRs (Rosenbaum et al., 2009), including A2AAR (Jaakola et al., 2008; Xu et al., 2011) and very recently the human CB1 receptor (Hua et al., 2017).
As was done for the other tryptophan indole 15N–1H signals, a sequence-specific assignment for Trp2466.48 was obtained for the A2AAR complex with the antagonist ZM241385 (Figures 2B and 3A). This ring current-shifted signal outside of the characteristic Trp indole 15N–1H spectral region can be expected to be highly sensitive to local conformational rearrangements, since these may cause large chemical shift changes (Johnson Jr and Bovey, 1958; MacDonald and Phillips, 1967; Wüthrich, 1969; Wüthrich, 1986). The NMR signal of Trp2466.48 in A2AAR complexes with agonists was therefore de novo identified by amino acid replacement (Figure 3B), revealing a large chemical shift change relative to complexes with antagonists (Figure 3A). Table S1 shows that the observed chemical shift change is in good qualitative agreement with the difference between the ring current shifts calculated from the crystal structures. Within the expected accuracy of ring current shift analyses (Perkins and Wüthrich, 1979; Wüthrich, 1986), this observation established direct correlations for conformational changes near Trp2466.48 between inactive and active-like states of A2AAR seen in solution and in crystal structures (Figure 4A).
Figure 3. NMR Assignment of the Toggle Switch Trp2466.48 in A2AAR and A2AAR[D52N] and Chemical Structures of Ligands Used in this Study.

(A and B) Assignment of the Trp2466.48 indole 15N–1H signal of A2AAR complexes with different ligands. (A) Antagonist ligand ZM241385. (B) Agonist ligand NECA. (C and D) Corresponding assignments for Trp2466.48 in the A2AAR[D52N] complexes with ZM241385 (C) and NECA (D). On the left are contour plots of 2D [15N,1H]-TROSY correlation spectra and on the right are cross sections taken at the 15N or 1H chemical shifts indicated in the contour plots by dashed vertical or horizontal lines, respectively. The spectra of A2AAR and A2AAR[D52N] are shown in blue, and the spectra for the variant proteins used for the resonance assignments are shown in red. Comparison of the two spectra resulted in sequence-specific assignments for the signals identified by asterisks, by observation of the absence in the variant protein of the signal to be assigned. (E) Chemical structures of the ligands used in the current study, where the adenine moiety found in almost all A2AAR antagonists and agonists is highlighted in green, and the ribose moiety found only in A2AAR agonists is highlighted in orange.
Figure 4. Local Environments of Selected Trp Residues in Crystal Structures of A2AAR and Response of Trp Indole 15N–1H NMR Lines to Variable Efficacy of Bound Drugs.

(A) Global superposition of the environment of Trp2466.48 in the crystal structures of A2AAR complexes with ZM241385 (red; PDB 3EML) and UK432097 (silver-blue; PDB 3QAK), and the ternary complex with NECA and a “mini-Gs” protein (grey; PDB 5G53). Trp2466.48 and the nearby residues F2426.44, I923.40 and P1895.50 are labeled and shown in stick representation. (B and C) Environment of two Trp residues in the A2AAR complexes with six different ligands (Figure 3E), i.e., ZM241385 (red; PDB 3EML), CGS21680 (orange; PDB 4UHR), UK432097 (silver-blue, PDB 3QAK), NECA (yellow; PDB 2YDV), caffeine (green; PDB 3PWH) and XAC (blue; PDB 3REY). Global superpositions of the crystal structures are shown, with ribbon representations of the backbone and stick representations of aromatic residues of interest. (B) Trp143. (C) Trp291.55. These structures were used to calculate the ring current shifts in Table S1. (D) Survey of the response of NMR probes in A2AAR to drug efficacy. Assigned Gly and Trp residues are shown as spheres positioned in the structure of the A2AAR-ZM241385 complex (PDB 3PWH). Red spheres indicate that a response was observed, either by different chemical shifts, variation of the signal line shapes, or both. Black spheres indicate that there was no response. The curved dashed line at the top of the receptor indicates the position of ECL2, which was not observed in the crystal structure. (E) – (G) Contour plots of the tryptophan indole 15N–1H region of 2D [15N,1H]-TROSY correlation spectra of [u-15N,~70% 2H]-A2AAR bound to ligands with different efficacies, as identified in each panel. Complexes with antagonists are in the left column and those with agonists on the right. Colors of the spectra correspond to the colors of the corresponding crystal structures of A2AAR–ligand complexes in panels (B) and (C). (H) – (K) Projections of the contents of the boxes in the corresponding contour plots along the 15N chemical shift axis onto the lower boundary of the boxes. The same colors are used as for the contour plots and peak assignments are indicated.
In view of the high sensitivity of the indole 15N–1H chemical shift of Trp2466.48 to local conformational rearrangements, we established de novo resonance assignments also for the variant protein A2AAR[D52N]. Two-residue variants were thus prepared, where in addition to Asp522.50, Trp2466.48 was replaced by phenylalanine. This showed that there are large chemical shifts between complexes of the two proteins with antagonists (Figure 3, A and C) and agonists (Figure 3, B and D). Furthermore, there are large chemical shift differences of the Trp2466.48 NMR signal between the complexes with agonists and antagonists for A2AAR (Figure 3, A and B), and A2AAR[D52N] (Figure 3, C and D). Response to variable efficacy of bound drugs is thus preserved in A2AAR[D52N], where the allosteric switch is known to be inactivated (Massink et al., 2014).
Considering the close proximity of Asp522.50 to the orthosteric ligand binding site (Figure 1C), we further checked the response to bound drugs with similar efficacy but different chemical structures (Figure 3E). Closely similar chemical shift differences were observed between complexes with different combinations of agonists and antagonists (Figure S7), indicating that the observed NMR spectral changes were indeed a response to different drug efficacies, rather than being due to direct contacts with the different ligand chemical structures (Figure 3E).
Overall, the data in Figure 3 reveal a tight interplay between the toggle switch Trp2466.48 and the allosteric center at Asp522.50, as manifested by the large chemical shift differences between the spectra of A2AAR and A2AAR[D52N] with corresponding ligands (Figure 3). We further see that inactivation of the allosteric center does not abolish the response of the toggle switch Trip2466.48 indole 15N–1H signal to variable efficacy of drugs bound to the orthosteric site.
Assigned NMR Signals Manifest Specific Response on the Intracellular A2AAR Surface to Variable Efficacy of Bound Drugs
In contrast to the large ring current shifts for the Trp2466.48 indole 15N–1H signal, ring current shifts calculated for the remaining Trp 15N–1H indole groups were much smaller for all complexes studied (Table S1), and the ring current shift calculations predicted measureable chemical shift changes between antagonist and agonist complexes only for Trp291.55 (Table S1). For glycine residues in A2AAR, at most very small ring current shifts of the backbone 15N–1H signals were calculated. The results shown in Figures 4, E–G, and 5 thus appear to be in line with recent observations on 19F-NMR probes in GPCRs, which indicated that large chemical shift changes between different functional states of GPCRs can be expected only in locations where the observed NMR signals originate from atom groups experiencing sizeable ring current fields (Liu and Wüthrich, 2016).
Figure 5. Probing Drug Efficacy-Dependent Signaling in A2AAR by Gly Backbone 15N–1H Resonances.

Contour plots are shown of 2D [15N,1H]-TROSY correlation spectra of [u-15N,~70% 2H]-A2AAR complexes with ligands of different efficacies, as identified by the color code in (A) (same colors as in Figure 4B, C, E–G): (A) Gly1184.39; (B) Gly1424.63; (C and D) Gly158, Gly160 and Gly218; (E) G114. In (A), (B) and (E) each panel shows a superposition of the spectra of complexes with two different ligands, whereas in (C) and (D) each panel displays the spectrum of one complex.
To investigate the response to variable efficacy of bound drugs, we recorded 2D [15N,1H]-TROSY correlation spectra of [u-15N,~70% 2H]-A2AAR complexes with six ligands of different pharmacological efficacies (Figure 3E) for which crystal structures are available (Carpenter et al., 2016; Doré et al., 2011; Jaakola et al., 2008; Lebon et al., 2011; Xu et al., 2011). Figure 4D affords a survey of the response to drugs of different efficacies seen at the assigned tryptophan indole 15N–1H and glycine backbone 15N–1H signals. Between agonist and antagonist complexes, significant changes were observed for 11 of the 14 assigned residues, i.e., either 1H or 15N chemical shift differences greater than 0.05 ppm and 0.20 ppm, respectively, or changes in signal fine structure.
Details of the response to variable drug efficacy indicated in Figure 4D are shown in Figure 4, E–K, for Trp indole 15N–1H resonances and in Figure 5 for Gly backbone 15N–1H resonances. For Trp291.55 we observe multiple components in the NMR signal for complexes with antagonists, and a single component for the agonist complex. There are small chemical shift changes which result in partial overlap of the signals for Trp291.55 and Trp143 in the agonist spectra, whereas two clearly separated signals are seen in the spectra of antagonist complexes. Trp2687.33 shows a single resonance component throughout. Its chemical shift is invariant among the three antagonist complexes, and we observe very small 1H chemical shift variations among the three agonist complexes. For Trp1294.50 the data of Figure 2A shows that the resonance is very weak in the complex with the antagonist ZM241385. We conclude that this is due to incomplete back-protonation of the indole 15N-1H group, since there is no evidence of excessive line broadening. In the other complexes, the Trp1294.50 NMR line was not detected, indicating that the back exchange was less efficient than in the complex with ZM241385.
Gly1184.39 shows a relatively large response to ligand efficacy, with an approximately 1 ppm shift in the 15N dimension between complexes with agonists and antagonists that indicates a change in local backbone conformation at the intracellular end of helix IV. Gly114 in ICL2 presents small chemical shift changes in the 15N and 1H dimensions between complexes with ligands of different efficacies. Gly1424.63 also shows a clear response to ligand efficacy, with chemical shift changes in both the 15N and 1H dimensions between complexes with different ligands. For Gly218 we observe two components of unequal intensities only in spectra of complexes with antagonists. In addition, there is a chemical shift change for Gly218 in the 15N and 1H dimensions between complexes with antagonists and agonists.
Inactivation of the Allosteric Center at Asp522.50 Modulates the Response to Variable Drug Efficacy at the A2AAR Intracellular Surface
To investigate effects from inactivation of the allosteric center at Asp522.50 by replacement with Asn522.50, we recorded 2D [15N,1H]-TROSY correlation spectra of [u-15N,~70% 2H]-A2AAR[D52N] complexes with the antagonist ZM241385 and the agonist NECA, and compared these data to spectra of the corresponding complexes with A2AAR. A survey of the effects from this single-amino acid replacement observed by this comparison in Figure 6A shows that only the regions of the three-dimensional structure of A2AAR from the toggle switch Trp2466.48 to the intracellular surface are affected, whereas there is no response on the extracellular surface and at the orthosteric drug binding site.
Figure 6. NMR Response on the Extracellular and Intracellular Surfaces of A2AAR to Perturbation of the Allosteric Center at Asp522.50.

(A) Survey of the response of NMR Signals in A2AAR to replacement of Asp522.50 in the allosteric center with Asn522.50. Assigned Trp and Gly residues are shown as spheres positioned in the structure of A2AAR-ZM241385 (PDB 3PWH). Cyan coloring indicates that changes were observed either in chemical shifts or in NMR line shapes. Red coloring indicates that there was no response. The curved dashed line at the top of the receptor indicates the position of ECL2, which was not observed in the crystal structure. (B)–(E) 2D [15N,1H]-TROSY correlation spectra documenting the results surveyed in (A). On the left and in the center are 2D contour plots, and on the right are projections along the 15N dimension onto the lower boundary of the regions indicated by the dashed boxes in the contour plots. (B) and (C) Gly218. (D) and (E) Trp291.55, Trp321.58 and Trp143. The proteins and the ligands are identified in the figure; the panels on the right show superpositions of the projections for the two proteins.
Most signals located on the intracellular surface showed either differences in chemical shifts, NMR signal intensities, or fine structures between A2AAR and A2AAR[D52N]. For Gly218 we observed two resonance components in NMR spectra of A2AAR-antagonist complexes, whereas in the A2AAR[D52N]-antagonist complex only a single resonance component is seen. The chemical shift of this single resonance is closely similar to the chemical shift of the upfield component in A2AAR, and the resonance of Gly218 also shows an approximately 5-fold increase in intensity (Figure 6B). In the A2AAR[D52N]-agonist complex, G218 also showed increased NMR signal intensity compared to the same A2AAR complex (Figure 6C). Trp291.55 showed a broad, relatively weak signal in A2AAR complexes with antagonists and agonists, and in spectra of A2AAR-antagonist complexes two components are observed (Figures 4, H and I; 6D). In contrast, Trp291.55 in A2AAR[D52N] shows only a single component in spectra of both the antagonist and agonist complexes (Figure 6, D and I). Also, the intensity of the Trp291.55 signal is about 5-fold more intense in A2AAR[D52N]. For Trp321.58 in A2AAR, we observed two resonance components in the agonist complexes (Figure 4, J and K), whereas for A2AAR[D52N] only a single component was observed in the agonist complex (Figure 6E). As an illustration of the inert behavior of residues near the extracellular surface (Figure 6A), Figure 6, D and E, shows that Trp143 has the same chemical shift and signal intensity in spectra of A2AAR and A2AAR[D52N] for both agonist and antagonist complexes.
Discussion
The result visualized in Figure 6A is highly intriguing in the context of literature data on allosteric coupling from G protein binding toward the drug binding sites in human GPCRs. Thus, studies of β2AR led to the conclusion that complex formation with an intracellular partner protein allosterically modulated the conformation of the orthosteric drug binding cavity at the extracellular receptor surface (DeVree et al., 2016; Staus et al., 2016). Analogous observations were based on NMR spectroscopic studies of the β1-adrenergic receptor (β1AR) (Isogai et al., 2016). In the present study, inactivation of the allosteric center changes the conformational dynamics on the intracellular surface and does not affect the conformation or dynamics of the extracellular receptor surface (Figures 6). This suggests that the view of GPCRs consisting of two semi-independent subdomains, an orthosteric domain that reacts to varied molecular “triggers” and an intracellular domain where signaling pathways converge (Katritch et al., 2012; Venkatakrishnan et al., 2016), may also apply to A2AAR, where the allosteric center links together the two regions through residue Asp2.50.
In A2AAR, as previously observed for other GPCRs (Eddy et al., 2016; Horst et al., 2013; Kim et al., 2013; Kofuku et al., 2014; Liu et al., 2012a; Manglik et al., 2015; Nygaard et al., 2013; Okude et al., 2015; Sounier et al., 2015), observations of local conformational polymorphisms by NMR spectroscopy in solution complement the available A2AAR crystal structures (Carpenter et al., 2016; Cheng et al., 2017; Doré et al., 2011; Jaakola et al., 2008; Lebon et al., 2011; Segala et al., 2016; Sun et al., 2017; Xu et al., 2011) with key insights into dynamic signaling-related processes. Comparison of agonist-bound crystal structures of A2AAR (PDB 3QAK) and A2AAR[D52N] (PDB 5W5F) bound to the same agonist showed no structural differences at the intracellular surface, even though the two proteins demonstrated striking differences in G protein signaling. Thus, the presently discovered relations between the presence of local polymorphisms at the intracellular tips of helices I and VI and signaling at the intracellular surface (Figure 7) would have gone unnoticed without the use of solution NMR techniques.
Figure 7. Visualization of Correlations Between Structural and Functional Response to Drug Efficacy and Activity of the Allosteric Center at Asp522.50 in A2AAR.

(A) and (B) Schematic side-views of A2AAR and A2AAR[D52N]. The transmembrane helices are represented by two adjoining rectangles, each indicating the extracellular and intracellular subdomains of the receptor (see text). The three helices carrying NMR reporter groups near the intracellular surface are shaded, and the residues with assigned NMR lines are indicated by yellow spheres, where framed spheres indicate that a single NMR line was observed, and unframed spheres correspond to multiple-component resonances. The helices drawn with broken lines indicate the local polymorphisms seen in the NMR spectra of the residues with unframed yellow spheres. The broken horizontal lines indicate the extracellular and intracellular membrane surfaces. The black arrow indicates the signaling pathway from the orthosteric drug binding site to the intracellular surface. In A2AAR, signaling has been correlated in the present work with local polymorphisms at the intracellular tips of the helices I and VI. In A2AAR[D52N], signaling to the intracellular surface is quenched (Massink et al., 2014); the broken arrow indicates that an NMR-detectable structural response to variable efficacy of the bound drug was observed. The connection between the toggle switch Trp2466.48 and the allosteric switch at Asp522.50 is supported by the observation of a strong interplay between these two centers in the present work.
A mechanism of how inactivation of the allosteric center could result in a loss of signaling may involve an interplay between Asp522.50 and Trp2466.48, as the loss of signaling-related dynamics at the A2AAR[D52N] intracellular surface (Figure 7) is correlated with changes in the local environment of Trp2466.48 in A2AAR[D52N] (Figure 3). The chemical shift of Trp2466.48 is highly sensitive to the proximity and relative orientation of the nearby ring of Phe2426.44 (Table S1); thus differences in the chemical shift of Trp2466.48 between A2AAR and A2AAR[D52N] reflect changes in the relative orientation of these two residues between the native and variant proteins. Comparisons of inactive and active-like GPCRs have documented coupling of structural reorientation of Phe2426.44 to structural rearrangements of helices associated with receptor activation (Elling et al., 2006; Rasmussen et al., 2011; Schwartz et al., 2006; Wacker et al., 2013). Thus, the present experimental evidence for reorientation of Phe2426.44 in A2AAR[D52N] may partly explain differences in signaling between A2AAR and A2AAR[D52N].
NMR studies of rhodopsin also provided support for an involvement of Trp6.48 reorientation in driving changes at the intracellular surface. Specifically, studies of rhodopsin in the solid state revealed a reorientation of Trp6.48 relative to the covalently bound retinal upon activation with light (Crocker et al., 2006), which is coupled with an outward reorientation of the intracellular end of helix VI as part of a proposed two-step activation mechanism (Kimata et al., 2016). Trp6.48 has also been implicated in studies of so called “efficacy switches” whereby a single point mutation can modulate the efficacy of a bound ligand. In the chemokine receptor CCR5, the amino acid replacement G286F7.42 converted some antagonists into full agonists (Steen et al., 2013). Based on molecular modeling, a proposed mechanism for this effect involved reorientation of Trp6.48 (Steen et al., 2013). In the δ–opioid receptor (DOR), an efficacy switch was observed for the variant D2.50N, which converted some antagonists into biased agonists (Fenalti et al., 2014). Based on the present results that Trp2466.48 and Phe2426.48 are reoriented in the D522.50N variant (Figure 3), we hypothesize that the efficacy switch in the DOR D2.50N variant may involve a similar reorientation of Trp6.48. As Trp6.48 is thus clearly important for receptor activation, and because of the high sequence conservation of both Trp2466.48 and Phe2426.44 among class A GPCRs (Katritch et al., 2012), the indole 15N-1H resonance of Trp2466.48 provides a highly sensitive probe for future NMR studies of class A GPCR activation mechanisms.
The current study provides a means for assessing the extent to which concepts of GPCR “microswitches” and “microdomains”, first described in early literature of rhodopsin photoactivation, can be extended to other class A receptors. GPCR microswitches are local clusters of highly conserved amino acids proximately located in the receptor, which undergo coordinated structural changes between inactive and active states. The concept of microswitches or “functional microdomains” emerged from earlier studies that applied optical spectroscopy and site-directed mutagensis to delineate contributions of specific amino acids to rhodopsin photoactivation (Fahmy et al., 1995; Franke et al., 1990; Lin and Sakmar, 1996; Sakmar and Fahmy, 1995). Of particular relevance to the current study is literature data that reported changes in UV-absorbance of tryptophan residues upon rhodopsin photoactivation (Lin and Sakmar, 1996). Trp1263.41 and Trp2656.48 in rhodopsin were observed to transition from more hydrophobic to more hydrophilic environments upon activation; in particular, Trp2656.48 experienced the largest change in local environment among all the rhodopsin tryptophans (Lin and Sakmar, 1996). Additional literature data correlated changes in interactions among rhodopsin transmembrane residues with varied functional responses (Beck et al., 1998; Han et al., 1998; Shieh et al., 1997), giving rise to the idea of “functional microdomains” (Lin et al., 2000). The present study corroborates the main ideas of functional microswitches and microdomains in the adenosine A2A receptor and provides a more solid structural foundation with tools that were not available at the time of these earlier rhodopsin studies.
The observations presented in this study were made possible by a multi-parameter NMR characterization of the conformation and dynamics of a human GPCR in solution. Most earlier NMR studies of human GPCRs have utilized a smaller number of reporter groups introduced by chemical conjugation after protein production, including 19F-NMR probes introduced by labeling of surface-exposed cysteine residues (Chung et al., 2012; Horst et al., 2013; Kim et al., 2013; Klein-Seetharaman et al., 1999; Liu et al., 2012a; Manglik et al., 2015), and 13CH3-probes introduced by reductive methylation of lysine residues (Bokoch et al., 2010; Kofuku et al., 2012; Kofuku et al., 2014; Nygaard et al., 2013; Okude et al., 2015; Sounier et al., 2015). While these have been highly successful in providing information at targeted locations, the stable-isotope-labeling strategy used here provides a more comprehensive view of GPCR activation. For future studies, this approach provides an avenue for obtaining ever denser networks of assigned NMR signals for monitoring function-related conformational rearrangements in GPCRs at ever higher resolution.
With regard to the physiological relevance, it is of key importance that the present study used a construct of the human A2AAR that was closely similar to the native receptor, i.e., it did not contain either thermostabilizing point mutations or fusion proteins inserted into receptor loops, as is extensively used to make GPCRs amenable for crystallographic studies and also applied to other experimental techniques. Eliminating thermostabilizing modifications enables one to obtain information of the global dynamics and plasticity of GPCRs. Thus, the present study provides a framework whereby dynamic processes at the intracellular surface can be observed in parallel to monitoring structural changes in other regions of the receptor.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Please direct any requests for further information or reagents to the Lead Contact Kurt Wüthrich (wuthrich@scripps.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbes
E. coli cells were cultured in LB medium. P. pastoris cells were cultured in BMGY and BMMY media (see Protein Expression below).
METHODS DETAILS
Mutagenesis
PCR-based site-directed mutagenesis (QuickChange II, Strategene, CA), was used to generate variant A2AARs with single amino acid replacements. Primers for mutagenesis are listed in the Table S4.
Protein Expression
For production of A2AAR for NMR studies, the gene encoding human A2AAR(1–316) containing a point mutation to remove the only glycosylation site (N154Q), an N-terminal FLAG tag, and a 10 X C-terminal His tag was cloned into a pPIC9K vector (Invitrogen) at the BamHI and NotI restriction sites. The pharmacological response of this construct to binding of orthosteric ligands has been demonstrated to be unchanged from the WT sequence (Palmer and Stiles, 1997). We expressed all A2AAR NMR samples in Pichia pastoris and demonstrated that the protein was functionally and structurally identical to A2AAR expressed in insect cells by validating the pharmacological response of A2AAR to orthosteric ligands and by crystal structure comparison of the antagonist ZM241385 complexes of A2AAR expressed, respectively, in Sf9 (PDB 4EIY) and in P. pastoris (PDB 6QAF), both with the protein BRIL fused into ICL3 (Figure S3). The construct was introduced by electroporation into the BG12 strain of Pichia pastoris (Biogrammatics, Carlsbad, CA).
For production of A2AAR for crystal structure determination, the employed construct was nearly identical to a previously published construct (Chun et al., 2012; Liu et al., 2012b) which contained a fusion with residues 1 to 106 of apo-cytochrome b562, in the place of the residues 209 to 218 in the ICL3, and was thermostabilized by the amino acid replacements M7W, H102I and K106L. The only differences to this earlier construct were the removal of an N-terminal HA leader sequence and the amino acid exchange N154Q.
Expression was screened by small-scale protein production and Western Blots, i.e. about fifteen clones per construct were grown in 4 mL cultures and protein production was assessed by anti-FLAG Western Blots. From these clones, the highest expressing clone was selected for future experiments. Glycerol stocks of the high expressing clone were used to inoculate a 5 mL starter culture in buffered minimal glycerol (BMGY) media, which was grown at 30 °C to an O.D. of 7–10 in a 15 mL culture tube. Cells were collected by centrifugation at 3000 × g for 10 minutes and used to inoculate 50 mL BMGY medium in a 250 mL baffled flask, which was grown at 30 °C to an O.D. of 15–20. This entire culture was then used to inoculate 500 mL BMGY media and allowed to grow at 30 °C to reach an O.D. of 15–20. Cells were collected by centrifugation at 3000 × g for 15 minutes and resuspended in buffered minimal methanol (BMMY) medium without methanol and the temperature was lowered to 27 °C. The cultures were allowed to shake for 3–4 hours to ensure complete metabolisation of glycerol before methanol was added to a final concentration of 0.5% w/v to induce protein expression. Two additional 0.5% w/v aliquots of methanol were added 12 and 24 hours after induction. Expression proceeded for a total of 36 hours until cells were harvested by centrifugation at 3500 × g for 15 minutes. The isolated cell pellets were frozen in liquid nitrogen and stored at −80 °C for future processing.
To generate deuterated, 15N isotopically labeled A2AAR samples, cells were adapted to increasing amounts of deuterium oxide in BMGY media at 30 °C over a period of about 9 days. Adapted cells were grown in BMGY media containing 99.8% D2O and 15N ammonium sulfate and protein expression was induced in BMMY media containing 99.8% D2O and 15N ammonium sulfate.
NMR Sample Preparation
A2AAR-containing cell pellets were resuspended in lysis buffer (50 mM sodium phosphate pH 7.0, 100 mM NaCl, 5% glycerol (w/v), and protease inhibitor cocktail solution prepared in-house) and broken by two passes through a cell disruptor (Constant Systems) operating at 40,000 PSI. Membranes were isolated by ultracentrifugation at 200,000 × g.
Isolated membranes were resuspended in buffer (10 mM HEPES pH 7.0, 10 mM KCl, 20 mM MgCl2, 1M NaCl) and treated with 1 mM theophylline, protease inhibitor cocktail solution (prepared in-house), and 2 mg/mL iodoacetamide 1 hour prior to solubilization. Protein was extracted from the membrane in buffer containing 0.5% (w/v) 2,2-didecylpropane-1,3-bis-β-D-maltopyranoside (LMNG), 0.025% cholesteryl hemisuccinate (CHS), 50 mM HEPES pH 7.0, and 500 mM NaCl for 5–6 hours followed by ultracentrifugation at 200,000 × g for 30 minutes to remove unsolubilized material. Co2+-charged affinity resin (Talon, Clontech) and imidazole (30 mM final concentration), were added to the supernatant and incubated overnight at 4 °C. Resin was washed with 20 column volumes wash buffer 1 (25 mM HEPES pH 7.0, 500 mM NaCl, 10 mM MgCl2, 0.1% LMNG, 0.005% CHS, 8 mM ATP, 30 mM imidazole), 2 times 20 column volumes wash buffer 2 (25 mM HEPES pH 7.0, 250 mM NaCl, 0.05% LMNG, 0.0025% CHS, 5% glycerol, 30 mM imidazole and ligand), and eluted with buffer 3 (25 mM HEPES pH 7.0, 250 mM NaCl, 0.05% LMNG, 0.0025% CHS, 5% glycerol, 300 mM imidazole and ligand). After elution, samples were exchanged into NMR buffer (20 mM HEPES pH 7.0, 75 mM NaCl, 0.025% LMNG, 0.00125% CHS, and ligand), using a PD-10 desalting column (G.E. Healthcare). All buffers were prepared with ligand at saturating concentration. Samples were concentrated to 280 μL in a Vivaspin-6 concentrator with a 30 kDa MWCO (Sartorius) and 20 μL D2O was added prior to transferring to a 5 mm Shigemi NMR tube. All samples were purified in protonated buffers containing the antagonist theophylline. Theophylline was exchanged with other ligands during the protein purification.
NMR Spectroscopy
2-dimensional [15N,1H]-transverse relaxation-optimized spectroscopy (TROSY) (Pervushin et al., 1997) correlation spectra were measured on a Bruker Avance 800 MHz spectrometer equipped with a 5-mm TXI-HCN probe running Topspin 3.1 (Bruker Biospin). Experiments were measured at 307 Kelvin. The sample temperature was calibrated using a standard sample (4% methanol in methanol-d4). Chemical shifts were referenced to an internal DSS standard. TROSY spectra were typically recorded with acquisition periods of 98 ms in 1H and 22.5 ms in 15N, with a 1 s recycle delay for a total experimental time of about 18 hours per experiment. NMR data were processed and analyzed in Topspin 3.5pl2 (Bruker Biospin). All data were processed identically; prior to Fourier transformation, the data matrices were zero filled to 1024 (t1) × 4096 (t2) complex points and multiplied by a Gaussian window function in the acquisition dimension and a 75°-shifted sine bell window function in the indirect dimension.
Radioligand Binding Assays in Membranes
Radioligand binding assays were performed using membranes prepared from Pichia pastoris expressing WT human A2AAR or one of the variant proteins. For saturation binding experiments, increasing concentrations (ranging from 2 nM to 80 nM) of [3H]2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine ([3H]CGS21680, 35.2 Ci/mmol) or increasing concentrations of [3H]4-[2-[7-amino-2-(2-furyl)-1,2,4-triazolo[1,5-a][1,3,5]triazin-5-yl-amino]ethylphenol ([3H]ZM241385, 0.2 nM to 12 nM) were incubated with membranes (5 μg protein) at 25°C for 60 min in a total of 200 μl Tris–HCl buffer (50 mM, pH 7.5) containing 10 mM MgCl2. Adenosine-5′-N-ethyluronamide (NECA, 100 μM) or 8-[4-[[[[(2-aminoethyl)amino]carbonyl]methyl)oxy]phenyl]-1,3-dipropylxanthine (XAC, 10 μM) was used to determine the non-specific binding. For displacement binding experiments, increasing concentrations of ligands were incubated with [3H]CGS21680 (5 nM) and membrane preparations at 25 °C for 60 min. Binding reactions were terminated by filtration through Whatman GF/B filters under reduced pressure using an MT-24 cell harvester (Brandell, Gaithersburg, MD, USA) and followed by washing twice with 5 ml cold Tris-HCl buffer. Radioactivity was measured using a scintillation counter (Tri-Carb 2810TR).
Radioligand Binding Assays for A2AAR Reconstituted in LMNG/CHS Micelles
Radioligand binding assays were performed using a scintillation proximity assay previously described (Bocquet et al., 2015). Ligand binding was measured using purified A2AAR reconstituted in LMNG/CHS micelles. Binding assays were carried out in a total volume of 175 μl per well in 96-well plates with binding buffer (150 mM NaCl, 0.05% LMNG, 0.0025% CHS, 25 mM HEPES, pH 7.0) containing approximately 0.05 μg A2AAR, 1nM [3H] ZM241385 (American Radiolabelled Chemicals, Inc.), a dose range of competing compound (10−12 – 10−5, M), and 250 μg of YSi copper His-tag SPA beads (Perkin Elmer), which were incubated for 60 min at 4 °C on a shaker. [ 3H] ZM241385 (25 μL) and competing compound (25 μL) were added to the 96-well plates first followed by A2AAR (25 μL) and SPA beads (100 μL). Binding was detected in a MicroBeta2 TriLux plate scintillation counter (Perkin Elmer) using SPA mode. ZM241385 binding affinity (KD) was determined using homologous competition binding, and competition binding assays were done to determine Ki values for NECA, XAC, and CGS21680. Controls were done to optimize the ratio of SPA beads to protein to minimize non-specific binding and ensure no more than 10% of free ligand was bound.
A2AAR Crystallization, Data Collection, Structure Calculation and Refinement
A2AAR-BRIL in complex with ZM241385 was reconstituted into lipidic cubic phase (LCP) by previously reported methods (Caffrey and Cherezov, 2009). Diffraction quality crystals were obtained with the same conditions as used for the crystallization of Sf9-produced A2AAR-BRIL(Chun et al., 2012; Liu et al., 2012b).
X-ray diffraction data were collected on the 23ID-D beamline (GM/CA CAT) at the Advanced Photon Source, Argonne, IL using a Pilatus3 6M detector (X-ray wavelength 1.033 Å). The crystals were exposed with a 10 μm mini-beam for 1 second and 1.0° oscillation per frame. HKL2000 (Otwinowski and Minor, 1997) was used for integrating, scaling and merging data from 18 best-diffracting crystals of the A2AAR–BRIL from Pichia pastoris in complex with ZM241385.
Initial phase information of A2AAR–BRIL from Pichia pastoris was obtained by molecular replacement (MR) with PHASER (McCoy et al., 2007) using A2AAR–BRIL (PDB: 4EIY) as a search model. Refinement was performed with REFMAC5 (Murshudov et al., 1997; Otwinowski and Minor, 1997) and autoBUSTER (Bricogne G., 2016) followed by manual examination and rebuilding of the refined coordinates in the program COOT (Emsley and Cowtan, 2004), using both |2Fo|–-|Fc| and |Fo| – |Fc| maps. TLS refinement strategy with two TLS groups (GPCR and BRIL domains) was incorporated in the refinement.
The final model of the A2AAR–BRIL complex contains residues −2 to −1 from the expression tag, 1–208, 219–306 of A2AAR, 1001–1042 and 1058–1106 of BRIL, and the ligand ZM241385 (ZMA as defined in the coordinate file), 3 cholesterols, 7 oleic acids, 5 OLC (or OLB), and 15 waters. The remaining C-terminal residues beyond 306 were disordered and not visible in the electron density maps and were therefore not modelled. The final model has good stereochemistry with no Ramachandran outliers (98.7% in favored and 1.3% in allowed regions), as determined by Molprobity (Chen et al., 2010). Data collection and refinement statistics are summarized in Table S3.
Quantification and Statistical Analysis
Quantification and analysis of ligand binding for A2AAR in Pichia membranes
Binding parameters were calculated using Prism 6 software (GraphPad). IC50 values obtained from displacement curves were converted to Ki values using the Cheng–Prusoff equation (Yung-Chi and Prusoff, 1973). All data are expressed as the mean ± standard error from three independent experiments performed in triplicate.
Quantification of ligand binding for A2AAR in LMNG/CHS performed by the scintillation proximity assay
The data were analyzed by Prism 6.05 (GraphPad) to give KD and Ki values and reported as the mean ± S.D. and done three times or more in triplicate.
Data and Software Availability
NMR assignments have been deposited in the Biological Magnetic Resonance Data Bank (BMRB) with Entry ID … The structure of A2AAR-BRIL in complex with ZM241385 expressed in P. pastoris has been deposited in the Protein Data Bank (PDB) with the ID PDB 6QAF.
Supplementary Material
Gly218 is shown in the crystal structure of thermostabilized A2AAR in complex with the antagonist ZM241385 (PDB 3PWH) (Doré et al., 2011) at the intracellular end of helix VI.
2D [15N,1H]-TROSY correlation spectra are shown. (A) [u-15N]-A2AAR. (B) [u-15N, ~70% 2H]-A2AAR. The proteins are in complex with the antagonist ZM241385. Both experiments were recorded at 800 MHz and 35 °C. The concentration of [u-15N]-A2AAR was ~500 μM, the concentration of [u-15N, ~70% 2H]-A2AAR was ~250μM. Measurement times were 24 hours for [u-15N]-A2AAR and 16 hours for [u-15N, ~70% 2H]-A2AAR. Both spectra were identically processed, as described in STAR Methods, and plotted at the same contour levels.
(A) Pharmacological ligand-binding activity of A2AAR reconstituted in LMNG/CHS mixed micelles: ZM241385 (top left), XAC (top right), CGS21680 (bottom left), NECA (bottom right). The measured KD or Ki values are listed in each panel. Radioligand competition binding used the scintillation proximity assay (Bocquet et al., 2015), as described in STAR Methods. (B) Analytical size exclusion chromatogram of detergent-solubilized [u-15N,~70% 2H]-A2AAR. The blue trace shows purified A2AAR as a single symmetric peak eluted at 8.4 minutes, consistent with a monodispersed and highly pure sample. A chromatogram of Bio-Rad molecular weight standards is superimposed in grey, with five major peaks at 6.6, 7.9, 8.8, 9.8, and 12.0 minutes; the molecular weights provided by the manufacturer are indicated. A2AAR elutes between the molecular weight standard peaks of 44 kDa (8.8 minutes) and 158 kDa (7.9 minutes). (C and D) Pharmacological activity of A2AAR in Pichia membranes determined using radioligand saturation binding. (C) 3H-labeled ZM241385. (D) 3H-labeled CGS21680. The KD values of 2.14 ± 0.75 nM for ZM241385 and 27.7 ± 5.3 nM for CGS21680 are consistent with previously published values determined for A2AAR in Sf9 membranes (Jaakola et al., 2008), and nearly identical to the data for A2AAR in LMNG/CHS micelles (panel A). (E and F) The crystal structure of the Pichia-expressed fusion protein of A2AAR with apo-cytochrome b562RIL (A2AAR-BRIL) (cyan; PDB 6AQF) is superimposed onto A2AAR-BRIL produced in Sf9 cells (red; PDB 3EML) (Liu et al., 2012b). The ligand ZM241385 in A2AAR from Pichia is colored green and in A2AAR from Sf9 cells it is colored yellow. (E) Overall comparison of the two structures. (F) Expansion of the ligand binding pocket, where the ECL3 was omitted for improved clarity (its position is indicated by a broken line; see also panel E).
2D [15N,1H]-TROSY correlation spectra are shown. The peaks numbered 1 to 30 were used to monitor the fold of the variant proteins.
2D [15N,1H]-TROSY correlation spectra are shown. The peaks numbered 1 to 30 were used to monitor the fold of the variant proteins.
2D [15N,1H]-TROSY correlation spectra are shown. The peaks numbered 1 to 30 were used to monitor the fold of the variant proteins.
The blue contour plots in Figure 3, A and B, are reproduced in the two upper panels. The two lower panels show corresponding spectral regions for complexes of A2AAR with ligands of similar efficacy but different chemical structures (Figure 3E), as indicated in the Figure.
Highlights.
Comprehensive view of GPCR signaling pathways using NMR probes
Allosteric signaling monitored by NMR probes distributed throughout A2AAR
Function-related conformational polymorphisms at intracellular A2AAR surface
Strong coupling between allosteric switch and signaling-activation motif
Acknowledgments
The authors acknowledge support from NIH/NIGMS PSI: Biology grant U54 GM094618 and R01GM115825, and from the NIDDK Intramural Research Program. M.T.E. acknowledges funding from an American Cancer Society postdoctoral fellowship. K.W. is the Cecil H. and Ida M. Green Professor of Structural Biology at The Scripps Research Institute. The authors thank Sophie Nguyen for help with the production of A2AAR biomass, Jeffrey Velasquez for help in cloning of A2AAR constructs, Yekaterina Kadyshevskaya for help with the preparation of figures, Dr. Vsevold Katritch for input on the manuscript, and Angela Walker for careful checking of the manuscript.
Footnotes
Author Contributions
M.T.E. carried out protein production and sample purification and recorded the NMR spectra; M.T.E. and K.W. analyzed the NMR data with input from T.D., P.S. and R.H.; Z.G., K.A.J., and K.L.W. performed the radioligand binding assays; M.T.E. and K.M.M. produced A2AAR-BRIL for crystal structure determination; M.T.E., M-Y.L., and M.A. crystallized A2AAR-BRIL; G.W.H. and M-Y.L. solved the A2AAR-BRIL crystal structure; M.T.E., R.C.S and K.W. designed the project; M.T.E. and K.W. prepared the manuscript with input from all the authors.
Supplemental Information includes STAR methods, seven figures, and three tables.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Audet M, Bouvier M. Restructuring G-Protein-Coupled Receptor Activation. Cell. 2012;151:14–23. doi: 10.1016/j.cell.2012.09.003. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosi. 1995;25:366–428. [Google Scholar]
- Bara-Jimenez W, Sherzai A, Dimitrova T, Favit A, Bibbiani F, Gillespie M, Morris M, Mouradian M, Chase T. Adenosine A2A receptor antagonist treatment of Parkinson’s disease. Neurology. 2003;61:293–296. doi: 10.1212/01.wnl.0000073136.00548.d4. [DOI] [PubMed] [Google Scholar]
- Beck M, Sakmar TP, Siebert F. Spectroscopic evidence for interaction between transmembrane helices 3 and 5 in rhodopsin. Biochemistry. 1998;37:7630–7639. doi: 10.1021/bi9801560. [DOI] [PubMed] [Google Scholar]
- Bihoreau C, Monnot C, Davies E, Teutsch B, Bernstein KE, Corvol P, Clauser E. Mutation of Asp74 of the rat angiotensin II receptor confers changes in antagonist affinities and abolishes G-protein coupling. Proc Natl Acad Sci USA. 1993;90:5133–5137. doi: 10.1073/pnas.90.11.5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocquet N, Kohler J, Hug MN, Kusznir EA, Rufer AC, Dawson RJ, Hennig M, Ruf A, Huber W, Huber S. Real-time monitoring of binding events on a thermostabilized human A2A receptor embedded in a lipid bilayer by surface plasmon resonance. BBA - Biomembranes. 2015;1848:1224–1233. doi: 10.1016/j.bbamem.2015.02.014. [DOI] [PubMed] [Google Scholar]
- Bokoch MP, Zou Y, Rasmussen SGF, Liu CW, Nygaard R, Rosenbaum DM, Fung JJ, Choi HJ, Thian FS, Kobilka TS, et al. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature. 2010;463:108–112. doi: 10.1038/nature08650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricogne G, BE, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C, Womack TO. BUSTER version 2.10.1. Cambridge, United Kingdom: Global Phasing Ltd; 2016. [Google Scholar]
- Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter B, Nehmé R, Warne T, Leslie AGW, Tate CG. Structure of the adenosine A(2A) receptor bound to an engineered G protein. Nature. 2016;536:104–107. doi: 10.1038/nature18966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D: Biological Crystallography. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng RKY, Segala E, Robertson N, Deflorian F, Doré AS, Errey JC, Fiez-Vandal C, Marshall FH, Cooke RM. Structures of Human A1 and A2A Adenosine Receptors with Xanthines Reveal Determinants of Selectivity. Structure. 2017;25:1275–1285. e1274. doi: 10.1016/j.str.2017.06.012. [DOI] [PubMed] [Google Scholar]
- Chun E, Thompson AA, Liu W, Roth CB, Griffith MT, Katritch V, Kunken J, Xu F, Cherezov V, Hanson MA, et al. Fusion Partner Toolchest for the Stabilization and Crystallization of G Protein-Coupled Receptors. Structure/Folding and Design. 2012;20:967–976. doi: 10.1016/j.str.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KY, Kim TH, Manglik A, Alvares R, Kobilka BK, Prosser RS. Role of Detergents in Conformational Exchange of a G Protein-coupled Receptor. J Biol Chem. 2012;287:36305–36311. doi: 10.1074/jbc.M112.406371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DM, Londos C, Gill DL, Rodbell M. Opiate Receptor-Mediated Inhibition of Adenylate Cyclase in Rat Striatal Plasma Membranes. J Neurochem. 1982;38:1164–1167. doi: 10.1111/j.1471-4159.1982.tb05365.x. [DOI] [PubMed] [Google Scholar]
- Crocker E, Eilers M, Ahuja S, Hornak V, Hirshfeld A, Sheves M, Smith SO. Location of Trp265 in metarhodopsin II: implications for the activation mechanism of the visual receptor rhodopsin. J Mol Biol. 2006;357:163–172. doi: 10.1016/j.jmb.2005.12.046. [DOI] [PubMed] [Google Scholar]
- DeVree BT, Mahoney JP, Vélez-Ruiz GA, Rasmussen SGF, Kuszak AJ, Edwald E, Fung JJ, Manglik A, Masureel M, Du Y, et al. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature. 2016;535:182–186. doi: 10.1038/nature18324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didenko T, Liu JJ, Horst R, Stevens RC, Wüthrich K. Fluorine-19 NMR of integral membrane proteins illustrated with studies of GPCRs. Curr Opin Struct Biol. 2013;23:740–747. doi: 10.1016/j.sbi.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doré AS, Robertson N, Errey JC, Ng I, Hollenstein K, Tehan B, Hurrell E, Bennett K, Congreve M, Magnani F, et al. Structure of the Adenosine A2A Receptor in Complex with ZM241385 and the Xanthines XAC and Caffeine. Structure. 2011;19:1283–1293. doi: 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- Eddy MT, Didenko T, Stevens RC, Wüthrich K. β2-Adrenergic Receptor Conformational Response to Fusion Protein in the Third Intracellular Loop. Structure. 2016;24:2190–2197. doi: 10.1016/j.str.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elling CE, Frimurer TM, Gerlach L-O, Jorgensen R, Holst B, Schwartz TW. Metal ion site engineering indicates a global toggle switch model for seven-transmembrane receptor activation. J Biol Chem. 2006;281:17337–17346. doi: 10.1074/jbc.M512510200. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica Section D: Biological Crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Fahmy K, Siebert F, Sakmar TP. Photoactivated state of rhodopsin and how it can form. Biophys Chem. 1995;56:171–181. doi: 10.1016/0301-4622(95)00030-2. [DOI] [PubMed] [Google Scholar]
- Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V, Roth BL, Stevens RC. Molecular control of d-opioid receptor signalling. Nature. 2014;506:191–196. doi: 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke RR, König B, Sakmar TP, Khorana HG, Hofmann KP. Rhodopsin mutants that bind but fail to activate transducin. Science. 1990;250:123–125. doi: 10.1126/science.2218504. [DOI] [PubMed] [Google Scholar]
- Han M, Smith SO, Sakmar TP. Constitutive Activation of Opsin by Mutation of Methionine 257 on Transmembrane Helix 6. Biochemistry. 1998;37:8253–8261. doi: 10.1021/bi980147r. [DOI] [PubMed] [Google Scholar]
- Horst R, Liu JJ, Stevens RC, Wüthrich K. β2-adrenergic receptor activation by agonists studied with 19F NMR spectroscopy. Angew Chem Int Ed Engl. 2013;52:10762–10765. doi: 10.1002/anie.201305286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T, Vemuri K, Nikas SP, Laprairie RB, Wu Y, Qu L, Pu M, Korde A, Jiang S, Ho JH, et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature. 2017;547:468–471. doi: 10.1038/nature23272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai S, Deupi X, Opitz C, Heydenreich FM, Tsai CJ, Brueckner F, Schertler GF, Veprintsev DB, Grzesiek S. Backbone NMR reveals allosteric signal transduction networks in the β1-adrenergic receptor. Nature. 2016;530:237–241. doi: 10.1038/nature16577. [DOI] [PubMed] [Google Scholar]
- Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, IJzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C, Jr, Bovey F. Calculation of nuclear magnetic resonance spectra of aromatic hydrocarbons. J Chem Phys. 1958;29:1012–1014. [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol Sci. 2012;33:17–27. doi: 10.1016/j.tips.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, Stevens RC. Allosteric sodium in class A GPCR signaling. Trends Biochem Sci. 2014;39:233–244. doi: 10.1016/j.tibs.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Chung KY, Manglik A, Hansen AL, Dror RO, Mildorf TJ, Shaw DE, Kobilka BK, Prosser RS. The role of ligands on the equilibria between functional states of a G protein-coupled receptor. J Am Chem Soc. 2013;135:9465–9474. doi: 10.1021/ja404305k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata N, Pope A, Eilers M, Opefi CA, Ziliox M, Hirshfeld A, Zaitseva E, Vogel R, Sheves M, Reeves PJ. Retinal orientation and interactions in rhodopsin reveal a two-stage trigger mechanism for activation. Nature Comm. 2016;7:12683. doi: 10.1038/ncomms12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein-Seetharaman J, Getmanova EV, Loewen MC, Reeves PJ, Khorana HG. NMR spectroscopy in studies of light-induced structural changes in mammalian rhodopsin: Applicability of solution 19F NMR. Proc Natl Acad Sci USA. 1999;96:13744–13749. doi: 10.1073/pnas.96.24.13744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuku Y, Ueda T, Okude J, Shiraishi Y, Kondo K, Maeda M, Tsujishita H, Shimada I. Efficacy of the β2-adrenergic receptor is determined by conformational equilibrium in the transmembrane region. Nature Comm. 2012;3:1045. doi: 10.1038/ncomms2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuku Y, Ueda T, Okude J, Shiraishi Y, Kondo K, Mizumura T, Suzuki S, Shimada I. Functional dynamics of deuterated β2-adrenergic receptor in lipid bilayers revealed by NMR spectroscopy. Angew Chem Int Ed Engl. 2014;53:13376–13379. doi: 10.1002/anie.201406603. [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wüthrich K. MOLMOL: A program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AGW, Tate CG. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SW, Han M, Sakmar TP. Analysis of functional microdomains of rhodopsin. Methods Enzymol. 2000;315:116–130. doi: 10.1016/s0076-6879(00)15839-2. [DOI] [PubMed] [Google Scholar]
- Lin SW, Sakmar TP. Specific Tryptophan UV-Absorbance Changes Are Probes of the Transition of Rhodopsin to Its Active State. Biochemistry. 1996;35:11149–11159. doi: 10.1021/bi960858u. [DOI] [PubMed] [Google Scholar]
- Liu D, Wüthrich K. Ring current shifts in 19F-NMR of membrane proteins. J Biomol NMR. 2016;65:1–5. doi: 10.1007/s10858-016-0022-4. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Horst R, Katritch V, Stevens RC, Wüthrich K. Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science. 2012a;335:1106–1110. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, Han GW, Roth CB, Heitman LH, IJzerman AP, et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012b;337:232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald C, Phillips W. Manifestations of the Tertiary Structures of Proteins in High-Frequency NMR. J Am Chem Soc. 1967;89:6332. doi: 10.1021/ja01000a061. [DOI] [PubMed] [Google Scholar]
- Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, Lerch MT, Kobilka TS, Thian FS, Hubbell WL, et al. Structural Insights into the Dynamic Process of β2-Adrenergic Receptor Signaling. Cell. 2015;161:1101–1111. doi: 10.1016/j.cell.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kobilka B. The role of protein dynamics in GPCR function: insights from the β2AR and rhodopsin. Curr Opin Cell Biol. 2014;27:136–143. doi: 10.1016/j.ceb.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markley JL, Putter I, Jardetzky O. High-resolution nuclear magnetic resonance spectra of selectively deuterated staphylococcal nuclease. Science. 1968;161:1249–1251. doi: 10.1126/science.161.3847.1249. [DOI] [PubMed] [Google Scholar]
- Martin S, Botto JM, Vincent JP, Mazella J. Pivotal role of an aspartate residue in sodium sensitivity and coupling to G proteins of neurotensin receptors. Mol Pharmacol. 1999;55:210–215. doi: 10.1124/mol.55.2.210. [DOI] [PubMed] [Google Scholar]
- Massink A, Gutierrez-de-Teran H, Lenselink EB, Ortiz Zacarias NV, Xia L, Heitman LH, Katritch V, Stevens RC, Jzerman IAP. Sodium Ion Binding Pocket Mutations and Adenosine A2A Receptor Function. Mol Pharmacol. 2014;87:305–313. doi: 10.1124/mol.114.095737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of applied crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munk C, Isberg V, Mordalski S, Harpsøe K, Rataj K, Hauser A, Kolb P, Bojarski A, Vriend G, Gloriam D. GPCRdb: the G protein-coupled receptor database–an introduction. Br J Pharmacol. 2016 doi: 10.1111/bph.13509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallographica Section D: Biological Crystallography. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP, et al. The dynamic process of β2-adrenergic receptor activation. Cell. 2013;152:532–542. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- Okude J, Ueda T, Kofuku Y, Sato M, Nobuyama N, Kondo K, Shiraishi Y, Mizumura T, Onishi K, Natsume M, et al. Identification of a Conformational Equilibrium That Determines the Efficacy and Functional Selectivity of the μ-Opioid Receptor. Angew Chem Int Ed Engl. 2015;54:15771–15776. doi: 10.1002/anie.201508794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Palmer TM, Stiles GL. Identification of an A2a adenosine receptor domain specifically responsible for mediating short-term desensitization. Biochemistry. 1997;36:832–838. doi: 10.1021/bi962290v. [DOI] [PubMed] [Google Scholar]
- Perkins SJ, Wüthrich K. Ring current effects in the conformation dependent NMR chemical shifts of aliphatic protons in the basic pancreatic trypsin inhibitor. Biochim Biophys Acta. 1979;576:409–423. doi: 10.1016/0005-2795(79)90416-1. [DOI] [PubMed] [Google Scholar]
- Pert CB, Pasternak G, Snyder SH. Opiate agonists and antagonists discriminated by receptor binding in brain. Science and Psychiatry: Groundbreaking Discoveries in Molecular Neuroscience. 2009:19. doi: 10.1126/science.182.4119.1359. [DOI] [PubMed] [Google Scholar]
- Pervushin K, Riek R, Wider G, Wüthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Rasmussen SGF, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakmar TP, Fahmy K. Properties and Photoactivity of Rhodopsin Mutants. Isr J Chem. 1995;35:325–337. [Google Scholar]
- Schwartz TW, Frimurer TM, Holst B, Rosenkilde MM, Elling CE. Molecular Mechanism of 7TM Receptor Activation—A Global Toggle Switch Model. Annu Rev Pharmacol Toxicol. 2006;46:481–519. doi: 10.1146/annurev.pharmtox.46.120604.141218. [DOI] [PubMed] [Google Scholar]
- Segala E, Guo D, Cheng RKY, Bortolato A, Deflorian F, Doré AS, Errey JC, Heitman LH, Jzerman IAP, Marshall FH, et al. Controlling the Dissociation of Ligands from the Adenosine A 2AReceptor through Modulation of Salt Bridge Strength. J Med Chem. 2016;59:6470–6479. doi: 10.1021/acs.jmedchem.6b00653. [DOI] [PubMed] [Google Scholar]
- Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, Javitch JA. Beta2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J Biol Chem. 2002;277:40989–40996. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- Shieh T, Han M, Sakmar TP, Smith SO. The steric trigger in rhodopsin activation. J Mol Biol. 1997;269:373–384. doi: 10.1006/jmbi.1997.1035. [DOI] [PubMed] [Google Scholar]
- Sounier R, Mas C, Steyaert J, Laeremans T, Manglik A, Huang W, Kobilka BK, Déméné H, Granier S. Propagation of conformational changes during μ-opioid receptor activation. Nature. 2015;524:375–378. doi: 10.1038/nature14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus DP, Strachan RT, Manglik A, Pani B, Kahsai AW, Kim TH, Wingler LM, Ahn S, Chatterjee A, Masoudi A, et al. Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature. 2016;535:448–452. doi: 10.1038/nature18636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen A, Thiele S, Guo D, Hansen LS, Frimurer TM, Rosenkilde MM. Biased and constitutive signaling in the CC-chemokine receptor CCR5 by manipulating the interface between transmembrane helices 6 and 7. J Biol Chem. 2013;288:12511–12521. doi: 10.1074/jbc.M112.449587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Bachhawat P, Chu MLH, Wood M, Ceska T, Sands ZA, Mercier J, Lebon F, Kobilka TS, Kobilka BK. Crystal structure of the adenosine A 2Areceptor bound to an antagonist reveals a potential allosteric pocket. Proc Natl Acad Sci USA. 2017;114:2066–2071. doi: 10.1073/pnas.1621423114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Q, Abood ME. Mutation of a highly conserved aspartate residue in the second transmembrane domain of the cannabinoid receptors, CB1 and CB2, disrupts G-protein coupling. J Pharmacol Exp Ther. 1998;285:651–658. [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Heydenreich FM, Flock T, Miljus T, Balaji S, Bouvier M, Veprintsev DB, Tate CG, et al. Diverse activation pathways in class A GPCRs converge near the G-protein-coupling region. Nature. 2016;536:484–487. doi: 10.1038/nature19107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, et al. Structural features for functional selectivity at serotonin receptors. Science. 2013;340:615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wüthrich K. High-resolution proton nuclear magnetic resonance spectroscopy of cytochrome c. Proc Natl Acad Sci USA. 1969;63:1071–1078. doi: 10.1073/pnas.63.4.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wüthrich K. NMR of Proteins and Nucleic Acids. John Wiley & Sons; 1986. [Google Scholar]
- Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Van Eps N, Zimmer M, Ernst OP, Prosser RS. Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature. 2016;533:265–268. doi: 10.1038/nature17668. [DOI] [PubMed] [Google Scholar]
- Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, Huang Q, Liu J, Takeda K, Teng MWL, et al. Co-inhibition of CD73 and A2AR Adenosine Signaling Improves Anti-tumor Immune Responses. Cancer Cell. 2016;30:391–403. doi: 10.1016/j.ccell.2016.06.025. [DOI] [PubMed] [Google Scholar]
- Yung-Chi C, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gly218 is shown in the crystal structure of thermostabilized A2AAR in complex with the antagonist ZM241385 (PDB 3PWH) (Doré et al., 2011) at the intracellular end of helix VI.
2D [15N,1H]-TROSY correlation spectra are shown. (A) [u-15N]-A2AAR. (B) [u-15N, ~70% 2H]-A2AAR. The proteins are in complex with the antagonist ZM241385. Both experiments were recorded at 800 MHz and 35 °C. The concentration of [u-15N]-A2AAR was ~500 μM, the concentration of [u-15N, ~70% 2H]-A2AAR was ~250μM. Measurement times were 24 hours for [u-15N]-A2AAR and 16 hours for [u-15N, ~70% 2H]-A2AAR. Both spectra were identically processed, as described in STAR Methods, and plotted at the same contour levels.
(A) Pharmacological ligand-binding activity of A2AAR reconstituted in LMNG/CHS mixed micelles: ZM241385 (top left), XAC (top right), CGS21680 (bottom left), NECA (bottom right). The measured KD or Ki values are listed in each panel. Radioligand competition binding used the scintillation proximity assay (Bocquet et al., 2015), as described in STAR Methods. (B) Analytical size exclusion chromatogram of detergent-solubilized [u-15N,~70% 2H]-A2AAR. The blue trace shows purified A2AAR as a single symmetric peak eluted at 8.4 minutes, consistent with a monodispersed and highly pure sample. A chromatogram of Bio-Rad molecular weight standards is superimposed in grey, with five major peaks at 6.6, 7.9, 8.8, 9.8, and 12.0 minutes; the molecular weights provided by the manufacturer are indicated. A2AAR elutes between the molecular weight standard peaks of 44 kDa (8.8 minutes) and 158 kDa (7.9 minutes). (C and D) Pharmacological activity of A2AAR in Pichia membranes determined using radioligand saturation binding. (C) 3H-labeled ZM241385. (D) 3H-labeled CGS21680. The KD values of 2.14 ± 0.75 nM for ZM241385 and 27.7 ± 5.3 nM for CGS21680 are consistent with previously published values determined for A2AAR in Sf9 membranes (Jaakola et al., 2008), and nearly identical to the data for A2AAR in LMNG/CHS micelles (panel A). (E and F) The crystal structure of the Pichia-expressed fusion protein of A2AAR with apo-cytochrome b562RIL (A2AAR-BRIL) (cyan; PDB 6AQF) is superimposed onto A2AAR-BRIL produced in Sf9 cells (red; PDB 3EML) (Liu et al., 2012b). The ligand ZM241385 in A2AAR from Pichia is colored green and in A2AAR from Sf9 cells it is colored yellow. (E) Overall comparison of the two structures. (F) Expansion of the ligand binding pocket, where the ECL3 was omitted for improved clarity (its position is indicated by a broken line; see also panel E).
2D [15N,1H]-TROSY correlation spectra are shown. The peaks numbered 1 to 30 were used to monitor the fold of the variant proteins.
2D [15N,1H]-TROSY correlation spectra are shown. The peaks numbered 1 to 30 were used to monitor the fold of the variant proteins.
2D [15N,1H]-TROSY correlation spectra are shown. The peaks numbered 1 to 30 were used to monitor the fold of the variant proteins.
The blue contour plots in Figure 3, A and B, are reproduced in the two upper panels. The two lower panels show corresponding spectral regions for complexes of A2AAR with ligands of similar efficacy but different chemical structures (Figure 3E), as indicated in the Figure.