

Abstract
Developmental ethanol exposure is a well known cause of lifelong cognitive deficits, behavioral hyperactivity, emotional dysregulation, and more. In healthy adults, sleep is thought to have a critical involvement in each of these processes. Our previous work has demonstrated that some aspects of cognitive impairment in adult mice exposed at postnatal day 7 (P7) to ethanol (EtOH) correlate with slow-wave sleep (SWS) fragmentation (Wilson, et al., 2016). We and others have also previously demonstrated that co-treatment with LiCl on the day of EtOH exposure prevents many of the anatomical and physiological impairments observed in adults. Here we explored cognitive function, diurnal rhythms (activity, temperature), SWS, and parvalbumin (PV) and perineuronal net (PNN)-positive cell densities in adult mice that had received a single day of EtOH exposure on P7 and saline treated littermate controls. Half of the animals also received a LiCl injection on P7. The results suggest that developmental EtOH resulted in adult behavioral hyperactivity, cognitive impairment, and reduced SWS compared to saline controls. Both of these effects were reduced by LiCl treatment on the day of EtOH exposure. Finally, developmental EtOH resulted in decreased PV/PNN-expressing cells in retrosplenial (RS) cortex and dorsal CA3 hippocampus at P90. As with sleep and behavioral activity, LiCl treatment reduced this decrease in PV expression. Together, these results further clarify the long-lasting effects of developmental EtOH on adult behavior, physiology, and anatomy. Furthermore, they demonstrate the neuroprotective effects of LiCl co-treatment on this wide range of developmental EtOH’s long-lasting consequences.
Keywords: Fetal alcohol syndrome, sleep fragmentation, slow-wave sleep, lithium chloride, insomnia, diurnal rhythm, parvalbumin, perineuronal nets
Introduction
Sleep plays a vital role in memory, perception, cognition, emotional regulation, as well as a variety of physiological processes (Stickgold, et al., 2001; Yoo, et al., 2007; Diekelmann, et al., 2010; Killgore, 2010; Harvey, 2011; Abel, et al., 2013; Talamini, et al., 2013). Disruptions in sleep can impact any or all of these processes. These sleep effects are bidirectional; for example, reduced sleep can impair memory consolidation (Killgore, 2010; Havekes, et al., 2017; Krause, et al., 2017), while enhanced sleep duration or quality can facilitate memory consolidation (Huber, et al., 2004; Marshall, et al., 2006; Barnes, et al., 2014). Impaired or fragmented sleep (i.e., short sleep bouts, frequent sleep/wake state transitions) is associated with a variety of disorders (Wulff, et al., 2010; Krause, et al., 2017), and is increasingly seen as contributing factor in some psychopathologies, rather than just a consequence or side-effect of the psychopathology. For example, treatment of insomnia that is co-morbid with depression can reduce depressive symptoms (Manber, et al., 2008).
Among a variety of other consequences (Abel, et al., 1986; Riley, et al., 2005; Mattson, et al., 2010), developmental exposure to ethanol disrupts subsequent sleep structure in maturing humans (D’Angiulli, et al., 2006; Pesonen, et al., 2009; Jan, et al., 2010; Wengel, et al., 2011; Chen, et al., 2012) and other animals (Stone, et al., 1996; Veatch, 2006; Criado, et al., 2008; Ehlers, et al., 2010; Wilson, et al., 2016), resulting in severe sleep fragmentation. Importantly, our recent work in a mouse model of developmental ethanol exposure suggests that the extent of sleep impairment in adulthood predicts cognitive function as assessed by contextual fear conditioning (Wilson, et al., 2016). This leads to the hypothesis that repair or prevention of developmental ethanol exposure effects on sleep could be a potential treatment for cognitive and/or emotional outcomes.
The effects of developmental ethanol on adult sleep could be related to the hyper-excitability of cortical/limbic circuits (D’Angiulli, et al., 2006; Criado, et al., 2008; Wilson, et al., 2011) and/or the loss of GABAergic interneurons (Coleman, et al., 2012; Sadrian, et al., 2014; Smiley, et al., 2015), given the role of GABAergic circuits in sleep-wake cycles (Manfridi, et al., 2001; Saper, et al., 2010; Xu, et al., 2015; Zucca, et al., 2017), and other sleep-dependent processes. In particular, parvalbumin (PV) expressing GABAergic interneurons in hippocampus are also important for sleep-dependent memory consolidation of contextual fear memory (Ognjanovski, et al., 2017). Thus, the deficits in contextual fear memory consolidation produced by EtOH may be related to its effects on the GABAergic interneuron populations involved in this and other sleep-related functions, if not sleep structure itself.
Lithium, a common treatment for bipolar disorder, has been shown to have neuroprotective effects in several neuropathological conditions including traumatic brain injury (Yu, et al., 2012), intracerebral hemorrhage (Kang, et al., 2012), and stroke (Doeppner, et al., 2017). Lithium has been demonstrated to affect a variety of molecular cascades related to neural plasticity, neurogenesis, neural migration, and cell survival (Chuang, 2004; Luo, 2009; Luo, 2010; Yu, et al., 2012; Doeppner, et al., 2017). Lithium treatment near the time of developmental ethanol exposure in mouse models has also been demonstrated to ameliorate many of ethanol’s immediate and long-lasting consequences (Zhong, et al., 2006; Chakraborty, et al., 2008; Young, et al., 2008; Luo, 2010; Sadrian, et al., 2012), though lithium itself can be a teratogen (Sharma, et al., 1986).
Here, as a beginning to our investigation of sleep as a target for treatment of developmental ethanol’s behavioral effects, we explored whether lithium chloride (LiCl) co-treatment with postnatal day 7 ethanol could prevent adult sleep fragmentation that co-occurs with diverse other behavioral and neuroanatomical outcomes. These results significantly extend our previous work by assessing whether the neuroprotective effects of LiCl extend to the sleep, behavioral, and neuroanatomical effects of developmental EtOH. It additionally addresses more detailed neuroanatomical questions by assessing the effects of developmental EtOH on PV cell number and perineuronal nets (PNN) which frequently surround PV neurons, in specific areas of hippocampus and cortex, and the ability of LiCl to repair these effects. Although LiCl might itself be a teratogen (Sharma, et al., 1986), this work begins investigation into whether preventative treatments which may involve the mechanisms of EtOH’s developmental action may be a viable target for future investigation.
Experimental Procedures
Subjects
A total of 124 C57BL/6By mice, bred and housed at the Nathan Kline Institute animal facility, were maintained on ad lib food and water at all times. All procedures involving animals were approved by the Nathan Kline Institute IACUC and were in accordance with NIH regulations for the proper treatment of animals. Dams and litters were housed in standard mouse cages. Subcutaneous (s.c.) injection of ethanol into postnatal day 7 (P7) mice is a well-established model of developmental ethanol neuropathology (Olney, et al., 2002b; Wozniak, et al., 2004; Izumi, et al., 2005; Gil-Mohapel, et al., 2010). While the effects of the frequency of alcohol consumption, potency consumed, and developmental timing of alcohol exposure are factors which produce a range of fetal alcohol induced developmental deficits (May, et al., 2011), this model focuses insult during the rodent brain growth spurt period that is developmentally equivalent to third trimester of human gestation (Schlessinger, et al., 1975). P7 pups were injected with ethanol (2.5 g/kg; s.c.) twice at 0 hr and 2 hrs as originally described for C57BL/6 mice (Olney, et al., 2002a; Olney, et al., 2002b). This model induces a peak truncal blood alcohol level of ~0.5g/dL at 0.5, 1, 3, and 6 hrs following the second ethanol injection, as assessed with the Alcohol Reagent Set (Pointe Scientific, Canton, MI)(Saito, et al., 2007). This alcohol level is similar to previous reports by others (Wozniak, et al., 2004; Young, et al., 2006; Saito, et al., 2007). Lithium chloride (0.6M LiCl in saline, 10μl/g, 6 mEq/kg body weight) or saline was injected intraperitoneally 15 min after the first ethanol injection as described in (Zhong, et al., 2006; Chakraborty, et al., 2008; Sadrian, et al., 2012). Pups were returned to the litter after treatment, and typically gain weight normally in the following days (Coleman, et al., 2012), though this was not assessed in the current study to limit postnatal handling. Weaning occurred at P25–30 and mice were tested as young adults at 3 months old. Sex differences in the effects of P7 EtOH have not been previously observed (Wilson, et al., 2011; Sadrian, et al., 2012; Sadrian, et al., 2014), nor were any significant differences observed between sexes here (with the exception of one neuroanatomical analysis described below), thus, data from males and females were combined.
Telemetry recordings and slow-wave analyses
Mice (postnatal age 85–100) were anesthetized with isoflurane and surgically implanted with a single stainless steel (125μ diameter) electrode in the frontal cortex. The electrode and reference were connected to a telemetry transmitter (DSI, model ETA-F10), which was implanted subcutaneously under the back. The telemetric device also transmitted body temperature and movement data. Given that these transmitters did not allow EMG measures, REM sleep was not monitored. Following surgery, animals were allowed to recover in their home cage for 3–7 days before onset of 24hr recordings. Basal sleep/wake and diurnal activity were recorded continuously for 2–3 days in the home cage. Animals were housed individually during all recordings, with animals in different conditions recorded simultaneously. Data from no more than one mouse/condition/litter are included here.
Local field potentials (LFPs) were acquired, digitized at 1000Hz and analyzed using Spike2 software (CED, Inc). Slow-wave activity was identified by analysis of delta frequency (0.1–5Hz) oscillations. LFPs were low-pass filtered and r.m.s. delta amplitude extracted. As previously described (Wilson, et al., 2016), epochs (14s) of high delta power were identified as exceeding the mean power over a given 24hr period by at least 1 standard deviation. Recording artifacts were removed before calculating mean and standard deviation. Analyses of slow-wave bouts included: mean percent time in slow-wave over a 24 hour period, and mean number of slow-wave/fast-wave bout transitions. For diurnal homecage activity analysis, data were blocked into 1hr bins, and mean time-dependent activity levels were calculated over 2–3 consecutive days.
Contextual fear conditioning
A separate group of animals were subjected to a contextual fear conditioning task. Conditioning procedures, selected to be consistent with our extensive previous work, are described as follows. Conditioning occurred during the light phase. Animals were placed in a conditioning chamber (9 x 22 x 20cm; W x L x H) with a shock grid floor, and a visual and peppermint scented context. We note that odor is an ecologically important component of the multi-sensory context for fear conditioning in nocturnal rodents (e.g.,(Huckleberry, et al., 2016)) and inclusion of odor as a part of the context does not reduce hippocampal involvement (Wang, et al., 2013). Animals were allowed to habituate to the chamber for 5min before receiving four 0.5mA, 1s foot shocks, with an average inter-shock interval of 2min. The following day, animals were returned to the peppermint scented conditioning chamber for a 5min test of contextual freezing. Testing was videotaped for blind analysis via hand scoring, where freezing was defined as a complete absence of movement other than breathing. Time spent freezing during the 5 minutes of testing was quantified for analysis.
Immunohistochemistry and cell counting
Ten to fifteen mice (male and female combined, derived from at least 5 litters) were treated with saline, EtOH, and LiCl at P7 as described above. At 3 months old, these mice were perfusion-fixed with 4% paraformaldehyde and 4% sucrose in cacodylate buffer solution (pH 7.2). The heads were removed and immersed in the perfusion solution overnight to fix further. Brains were then extracted, transferred to phosphate buffered saline (PBS), and kept at 4°C for 2–5 days until cut with a vibratome into 50 μm thick coronal sections. For dual labeling of PV and PNN, free-floating sections were rinsed in PBS, permeabilized in methanol for 10 min, and incubated for 30 min in blocking solution (PBS containing 0.1% Triton X-100 and 5% BSA), followed by incubation overnight with anti-parvalbumin antibody (PV25, Swant, Marly, Switzerland) in PBS containing 3% BSA and 0.1% Triton X-100. Sections were then rinsed in 0.1% Triton X-100 in PBS three times and further incubated with biotin-conjugated wisteria floribunda agglutinin (WFA) (Sigma-Aldrich, St. Louis, MO) in 0.1% Triton X-100 in PBS containing 1% BSA for 2hr at r.t., followed by incubation with a mixture of Alexa Fluor 488 goat anti-rabbit IgG (Life Technologies, Grand Island, NY) and streptavidin-conjugated Alexa Fluor 594 (Fisher Scientific, Pittsburgh, PA) in 0.1% Triton X-100 in PBS containing 1% BSA for 1hr at r.t.. Finally, sections were rinsed in PBS three times, mounted, and coverslipped using ProLong Gold Antifade Reagent (Life Technologies). All photomicrographs were taken through a 4X or a 20X objective with a Nikon Eclipse TE2000 inverted microscope attached to a digital camera DXM1200F. Because our previous studies (Smiley et al., 2015) showed that both 2-dimensional and stereological 3-dimensional counting methods gave similar significant reduction in PV-positive (PV+) cell densities in the cortex by P7 ethanol treatment, the two-dimensional counting method was used in the present studies. The number of PV+ and WFA+ cells in each area of interest (AOI) and total dimensions of each AOI were measured using the Image-Pro software version 6.0 (Media Cybernetics, Silver Spring, MD). AOIs for cell counting were RS cortex, dorsal CA1, and CA3 hippocampus. These AOIs were defined according to the Atlas of Mouse Brain (Paxinos, et al., 2004). The cell density of each AOI was calculated as the mean cell number per square millimeter using 3 to 6 sections per brain around bregma −0.94 to −2.3 mm for RS cortex and bregma −1.22 to −2.3 mm for dorsal CA1 and CA3 hippocampus.
Statistical analyses
Statistical analyses were conducted using Statview or Prism software. As is standard for this type of analysis, we selected to use ANOVAs; Bonferroni post-hoc tests were selected as appropriate for our sample size and to correct for multiple comparisons. SWS%, Sleep fragmentation, contextual freezing, and neuroanatomical analyses were conducted using 2 way ANOVA (EtOH/Saline Treatment X Saline/LiCl Drug). Post-hoc determinations comparing all groups (total of 6 determinations/analysis) were conducted where main effects or interactions were found. For the diurnal activity analysis, a Group x Time repeated measures ANOVA was conducted, with Fisher LSD post-hoc tests for main effects comparing all groups (6 determinations/analysis).
RESULTS
Growth and development
At three months old, the mean body weights for all groups were similar (Sal-Sal: M = 24.41, SEM = 0.93; EtOH-Sal: M = 24.06, SEM = 0.93; Sal-LiCl: M = 23.29, SEM = 0.76; EtOH-LiCl: M = 21.53, SEM = 0.74), though there was a main effect of Lithium on body weight (2 way ANOVA, Treatment x Drug, main effect of Drug, F (1, 57) = 4.627, p < 0.05). However, Bonferroni post-hoc tests (p < 0.05) did not reveal any significant differences between groups.
Behavior and sleep
P7 ethanol impaired adult contextual fear conditioning. As shown in Fig. 1, time spent freezing in the fear conditioning context was significantly reduced in P7 EtOH-Sal treated mice, trained and tested as adults, compared to P7 saline-treated adult mice. LiCl co-treatment with EtOH on P7 reduced this adult cognitive impairment (2 way ANOVA, Treatment x Drug, main effect of treatment, F(1,32) = 6.50, p < 0.05). Post-hoc tests (p < 0.05, 6 total determinations) comparing all groups revealed that only EtOH-Sal was significantly different from Sal-Sal controls. In contrast, EtOH-LiCl was not significantly different from Sal-Sal or Sal-LiCl treatment groups. It also failed to reach significance from EtOH-Sal however, thus the LiCl treatment provided partial blockade of EtOH effects on this task, but not complete.
Figure 1.
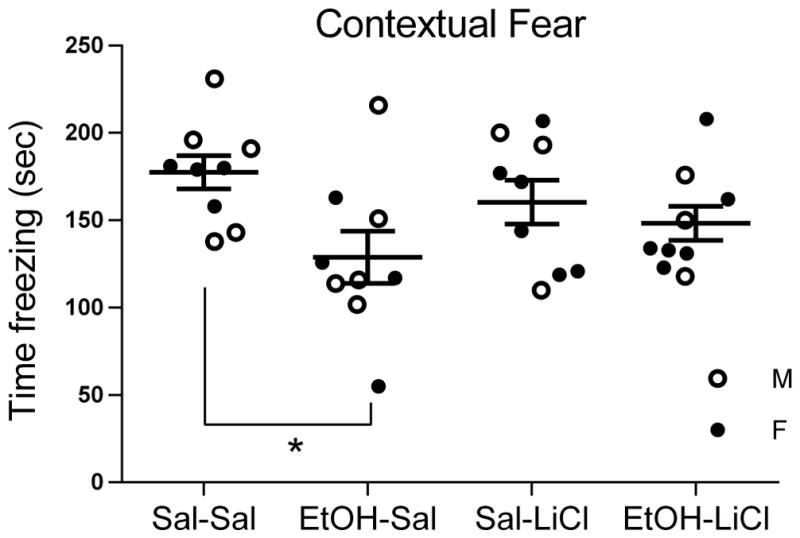
The impairment in adult contextual fear conditioning induced by P7 EtOH was prevented by co-treatment with LiCl. Mean time freezing +/− SEM is indicated on the y-axis. EtOH-Sal is the only group that is significantly different from Sal-Sal (* = p <.05, 6 total post-hoc comparisons).
Our previous work has demonstrated that adult cognitive impairment following developmental EtOH exposure is correlated with slow-wave sleep (SWS) fragmentation (Wilson, et al., 2016). Here we assessed SWS in separate animals as shown in Fig. 2. Delta frequency band amplitude was monitored in the frontal cortex with telemetry (Fig. 2A). Recording was performed continuously for 2–3 days in mice singly housed in their home cage, and periods of elevated delta were identified as SWS periods. As shown in Fig. 2B, P7 EtOH-Sal-treated adult mice displayed significantly reduced time in SWS over a 24 hr period. LiCl co-treatment with EtOH on P7 completely prevented this adult SWS reduction (Fig. 2B; 2 way ANOVA, Treatment x Drug, main effect of treatment F(1,35) = 11.20 p < 0.01; main effect of drug, F(1,35) = 8.05, p < 0.01). Bonferroni post-hoc tests (p < 0.05, 6 total determinations) revealed that as expected, EtOH-Sal mice spent significantly less time in SWS than Sal-Sal treated controls, and likewise, significantly less than EtOH exposed mice co-treated with LiCl. There was no difference between Sal-Sal and EtOH-LiCl groups. Additionally, post-hoc tests revealed an apparent benefit of LiCl-only treatment over the EtOH-LiCl co-treated group (p < 0.001). Similarly, LiCl co-treatment with EtOH prevented the SWS fragmentation seen in EtOH-Sal adults (Fig. 2C; significant treatment X drug interaction, F(1,35 = 6.44, p < 0.05). Post-hoc tests (p < 0.05, 6 total determinations) revealed EtOH-Sal groups transitioned from SWS more than either Sal-Sal, Sal-LiCl or EtOH-LiCl groups. Sal-Sal, Sal-LiCl and EtOH-LiCl groups were not significantly different.
Figure 2.

A) Slow-wave sleep (SWS) was monitored with an electrode in the frontal cortex attached to a telemetry transmitter for wireless recording. Delta band r.m.s amplitude was extracted and used to quantify SWS bouts. B) The reduction in percent time in SWS in adults induced by P7 ethanol was prevented by co-treatment with LiCl (post-hoc Bonferroni tests, 6 total post-hoc comparisons). Mean % time in SWS +/− SEM is indicated on the y-axis. Males are represented by open circles and females by filled circles here and in C. C) The increase in SWS-waking transitions (sleep fragmentation) in adults induced by P7 ethanol was prevented by co-treatment with LiCl (post-hoc Bonferroni tests, 6 total post-hoc comparisons). Mean number of transitions from SWS +/− SEM is indicated on the y-axis. * = p < 0.05; ** = p < .01; *** = p < .001.
A final assay was behavioral hyperactivity in the home cage assessed with the telemetry system. As shown in Fig. 3, and previously reported (Wilson, et al., 2016), P7 EtOH-Sal-treated adult mice displayed significant hyperactivity compared to Sal-Sal in their home cage, with maximal hyperactivity during the dark phase of the diurnal cycle. As with our other behavioral assays, co-treatment with LiCl blocked this adult hyperactivity (repeated measures ANOVA, Group x Time; main effect of Group, F(3,1104) = 7.18, p < 0.001; main effect of Time, F(23,1104) = 22.04, p < 0.001; Group x Time interaction, F(69,1104) = 1.33, p < 0.05).
Figure 3.
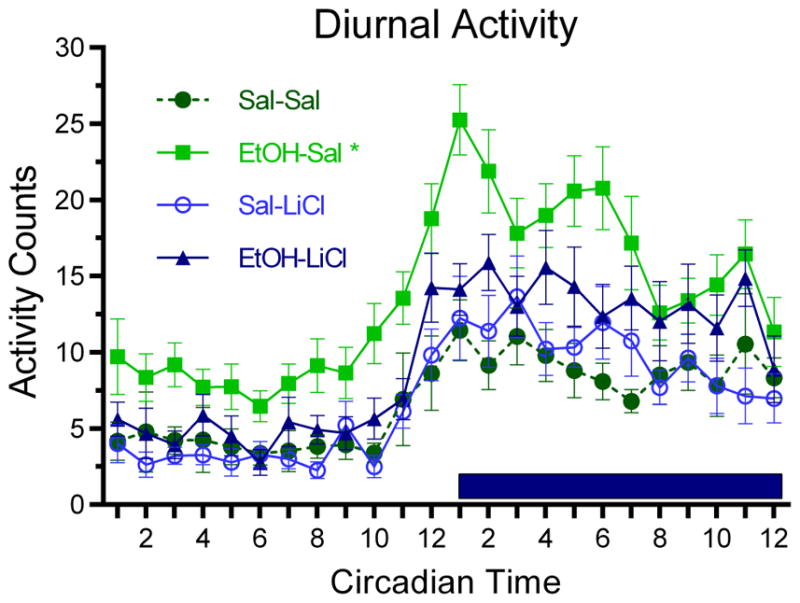
P7 ethanol induced behavioral hyperactivity in the home cage compared to all other groups. Activity is quantified in arbitrary units by the DSI telemetry implant; mean hourly activity +/− SEM is indicated on the y-axis. This hyperactivity was present during both the light and dark phases of the diurnal cycle; dark phase is indicated by the dark bar. Co-treatment with LiCl prevented this behavioral hyperactivity. Asterisk signifies a main group effect with EtOH-Sal being significantly different from all other groups.
Together these results demonstrate that co-treatment with LiCl on the day of developmental EtOH treatment reduces impairments in cognition, sleep, and hyperactivity expressed in adulthood.
Neuroanatomy
Our previous work has demonstrated that P7 EtOH exposure decreases PV+ neuron densities in the cortex and hippocampus in adult mice (Sadrian, et al., 2014; Smiley, et al., 2015). In the present study, the effects of EtOH and LiCl on PV neurons were examined in more specific regions—RS cortex (Todd, et al., 2015) and dorsal CA1/CA3 hippocampus (Gewirtz, et al., 2000; Maren, et al., 2013; Wang, et al., 2013) which have been implicated in the consolidation of contextual fear memory. Because PNNs (lattice-like structures detected by WFA lectin binding) selectively surround a subset of PV neurons (Brauer et al., 1993; Hartig et al., 1994) and regulate the development and plasticity of those neurons (Celio et al., 1998; Ye and Miao, 2013; Balmer, 2016; Lensjo et al., 2017), WFA+ cell densities were also measured in the RS cortex and dorsal CA1/CA3 hippocampal regions. As shown in Fig. 4A, which is a representative image of a control brain section dual-labeled with PV antibody and WFA lectin, PNNs were abundantly expressed in RS cortex, while they were low in CA3 hippocampus. Fig. 4B shows enlarged images of RS and CA3 regions from different treatment group of animals. As expected, WFA+ cells were generally positive for PV, although some cells were only PV or WFA+ (Fig. 4B). Fig. 5 shows quantitative results of the effects of EtOH/LiCl on PV+ and WFA+ cell densities. In RS cortex, EtOH treatment (EtOH-Sal) significantly reduced PV+ cell densities, while LiCl co-treatment with EtOH (EtOH-LiCl) prevented the reduction (2 way ANOVA, Treatment x Drug, main effect of treatment F(1,52) = 9.37, p < 0.01; main effect of drug, F (1, 52) = 12.44, p < .001). Post-hoc tests (p < 0.05, 6 total comparisons) revealed EtOH-Sal was different from all other groups). The main effect of sex and the interaction between sex and treatment groups were not significant. Similarly, EtOH-Sal group significantly reduced WFA+ cell densities and lithium co-treatment with EtOH restored the reduction (2 way ANOVA, Treatment x Drug, main effect of treatment F(1,36) = 18.02, p < 0.001; main effect of drug, F(1,36) = 16.02, p < 0.001; treatment X drug interaction, F(1,36) = 18.06, p < 0.001). Post-hoc tests (p < 0.05, 6 total determinations) revealed EtOH-Sal was different from all other groups. However, there was a significant sex difference, and males showed ~10% higher WFA+ cell densities (ANOVA F(1,34)=7.22, p=0.011), with no significant interaction between sex and treatment groups. In CA3, similar to RS, the EtOH-Sal group significantly reduced PV+ cell densities, while LiCl co-treatment prevented the reduction (2 way ANOVA, Treatment x Drug, main effect of treatment F(1,48) = 13.88, p < 0.001; treatment X drug interaction, F(1,48) = 6.204, p < .05, EtOH-Sal diff from all other groups based on post-hoc tests, p < 0.05), and there were no significant effects of sex and no significant interaction between sex and treatment groups. In addition, there was a tendency that EtOH reduced WFA-positive cell densities and LiCl co-treatment attenuated the loss, but the data did not reach statistical significance. In contrast to RS and CA3, there were no significant main effects or significant interaction in either PV-positive cell or WFA-positive cell densities in CA1 hippocampal region.
Figure 4.

A) A control (Sal-Sal) brain section was dual-labeled using anti-PV antibody and WFA lectin. The image of PV+ cells (red), WFA+ cells (green), and the merged image are shown. The bar indicates 300 μm. B) Brain sections from Sal-Sal, EtOH-Sal, Sal-LiCl, and EtOH-LiCl groups were dual-labeled using anti-PV antibody and WFA lectin. The merged images of PV (red)+ cells and WFA (green)+ cells were shown here. The bar indicates 50 μm.
Figure 5.

P7 EtOH-induced PV+ cell density reduction in RS cortex (A) and CA3 hippocampus (B) was prevented by LiCl co-treatment with EtOH. PV+ cell density and WFA+ cell density were measured in RS cortex and CA3 hippocampus in the brain sections derived from Sal-Sal, EtOH-Sal, Sal-LiCl, and EtOH-LiCl groups. Legend in A indicates PV+ and WFA+ cell counts in panels A and B. Asterisks indicate that the EtOH-Sal group was significantly different from other groups by Bonferroni post-hoc test after ANOVA (p < .05, 6 total post-hoc comparisons) C) WFA cell density was about 10% higher in the retrosplenial cortex of males (ANOVA F(1,34)=7.22, * = p < .05), although there was no significant interaction between sex and treatment groups.
DISCUSSION
The present results demonstrate that LiCl co-treatment with developmental EtOH exposure reduces adult sleep fragmentation and associated behavioral abnormalities. Furthermore, this co-treatment blocks EtOH-induced reduction in PV neuron density in the dorsal CA3 hippocampus and the RS cortex. Loss of GABAergic neurons could contribute to disruption in sleep-dependent contextual fear memory consolidation, and/or to sleep-related local circuit dysregulation (Saper, et al., 2010; Xu, et al., 2015; Zucca, et al., 2017), thus LiCl’s protective effect on both sleep and behavior could be related to this GABAergic sparing, though future work is required to confirm this link. It is also important to note that these data do not allow us to conclude that preventing sleep fragmentation with LiCl co-treatment underlies the improvement in adult cognition and behavioral hyperactivity in developmental EtOH exposed mice. Nonetheless, the data further strengthen the association between adult sleep fragmentation and cognitive and behavioral impairments following developmental EtOH (Wilson, et al., 2016). Ongoing work is exploring whether direct manipulations of sleep can influence cognitive and behavioral function in adults exposed to developmental EtOH.
In addition to blocking adult sleep fragmentation, LiCl co-treatment reduced both cognitive (contextual fear conditioning) and general behavioral (homecage activity levels) impairments in EtOH treated mice. This extends our previous findings showing that the impairment in object placement memory, which is impaired in adult mice following P7 ethanol, is also spared by LiCl co-treatment (Sadrian, et al., 2012). Although we cannot rule out a potential contribution of behavioral hyperactivity to the reduction in freezing shown by EtOH treated mice, these results nonetheless demonstrate that developmental EtOH exposed mice had impaired performance on this cognitive-behavioral task, and that LiCl co-treatment restored performance to normal levels.
Both contextual fear memory and object placement memory have been linked at least in part to hippocampal function (Gewirtz, et al., 2000; Maren, et al., 2013; Hunt, et al., 2016) though see (Gewirtz, et al., 2000; Wiltgen, et al., 2006), and sleep plays an important role in consolidation of those memories (Graves, et al., 2003; Marshall, et al., 2007). Importantly, recent studies suggest critical roles of hippocampal PV neurons in this sleep-dependent memory consolidation (Caliskan, et al., 2016; Ognjanovski, et al., 2017), potentially linking developmental EtOH’s impacts on both sleep and hippocampal PV to contextual fear memory impairment seen here, although PV+ cell density was only reduced in dorsal CA3, but not in dorsal CA1 region (Fig. 4 and 5). The RS cortex also has been implicated in contextual fear memory (Todd, et al., 2015), although roles of PV neurons in this function have not been elucidated. Circuits underlying hyperactivity are less clear, though multiple previous studies have demonstrated developmental ethanol-induced behavioral hyperactivity in both open arenas (Wozniak, et al., 2004) and in the home cage (Wilson, et al., 2016), and again, sleep and sleep deprivation impacts hyperactivity (Yoon, et al., 2012).
LiCl co-treatment, which blocks the wave of cell death in the hours following P7 ethanol exposure (Zhong, et al., 2006; Chakraborty, et al., 2008; Young, et al., 2008; Luo, 2010; Sadrian, et al., 2012) also prevented the reduction in PV+ neurons in RS cortex and dorsal CA3 regions (Fig. 4 and 5). It has been previously hypothesized that this ethanol-induced loss of GABAergic interneurons may induce a shift in the excitation/inhibition balance (Sadrian, et al., 2013) – a shift evidenced by hyperexcitability and hyper-functional connectivity in corticolimbic circuits (D’Angiulli, et al., 2006; Wilson, et al., 2011), and increased susceptibility to seizures (Bonthius, et al., 2001; Bell, et al., 2010) following developmental EtOH exposure. Sleep-wake cycle disruption may similarly be an indicant of dysfunction in GABAergic inhibition. Sleep-wake cycles are dependent on circadian rhythms, sensory input (e.g., light levels), and recent experience (e.g., stress levels), and brainstem, hypothalamic, and basal forebrain circuits collectively regulate switching between sleep-wake states and state maintenance (Saper, et al., 2010). GABAergic circuits are a critical component in both sleep-wake transitions and state stability (Buzsaki, 2006; Saper, et al., 2010; Zucca, et al., 2017). Thus, a decrease in this cell population caused by P7 EtOH could contribute the observed sleep disruption, and sparing these cells with LiCl co-treatment may contribute to the observed sleep restoration. Further studies including the effects of P7 EtOH on PV or other GABAergic neurons in sleep-related brain regions, such as hypothalamus and basal forebrain, may be important to assess this hypothesis. The long-lasting reduction in PV+ cell density observed in the cortex and hippocampus (Fig. 5) may be caused by P7 EtOH-induced acute cell death or by P7 EtOH-induced disturbance in maturation of PV+ cells. The RS cortex was one of the regions most affected by P7 EtOH-induced acute cell death (Olney, et al., 2002a; Saito, et al., 2007), and PV+ cell density reduction in this area in the adult brain was accompanied by a decrease in the density of WFA+ cells, which were mostly PV+ cells (Fig. 5), suggesting cell loss rather than reduction in PV expression by P7 EtOH. However, further studies are necessary.
In summary, LiCl can serve as a preventative co-treatment with developmental ethanol exposure to block the emergence of adult sleep fragmentation, cognitive impairment, behavioral hyperactivity, and PV cell loss. These results support previous work showing that the extent of adult sleep fragmentation following P7 EtOH predicts cognitive impairment (Wilson, et al., 2016). The loss of PV neurons in these animals could directly contribute to deficits in contextual fear memory (Caliskan, et al., 2016; Ognjanovski, et al., 2017), indirectly impact memory via disruption of sleep-dependent memory consolidation (Stickgold, et al., 2007; Abel, et al., 2013), or be unrelated to either of these major effects. Current work is aimed at determining whether more selective manipulations of sleep can ameliorate the long-lasting cognitive and behavioral consequences of developmental ethanol exposure.
HIGHLIGHTS.
Developmental EtOH exposure induces sleep fragmentation and hyperactivity in adults
Developmental EtOH reduces PV/PNN-expressing cells in RS cortex and CA3 hippocampus
Co-treatment with LiCl prevents adult impairment in sleep, activity, cognition, and PV/PNN cell loss
Acknowledgments
This work was supported by a grant from NIAAA (R01- AA023181) to M.S. and D.A.W.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abel EL, Sokol RJ. Fetal alcohol syndrome is now leading cause of mental retardation. Lancet. 1986;2:1222. doi: 10.1016/s0140-6736(86)92234-8. [DOI] [PubMed] [Google Scholar]
- Abel T, Havekes R, Saletin JM, Walker MP. Sleep, plasticity and memory from molecules to whole-brain networks. Curr Biol. 2013;23:R774–788. doi: 10.1016/j.cub.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DC, Wilson DA. Slow-wave sleep-imposed replay modulates both strength and precision of memory. J Neurosci. 2014;34:5134–5142. doi: 10.1523/JNEUROSCI.5274-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SH, Stade B, Reynolds JN, Rasmussen C, Andrew G, Hwang PA, Carlen PL. The remarkably high prevalence of epilepsy and seizure history in fetal alcohol spectrum disorders. Alcohol Clin Exp Res. 2010;34:1084–1089. doi: 10.1111/j.1530-0277.2010.01184.x. [DOI] [PubMed] [Google Scholar]
- Bonthius DJ, Pantazis NJ, Karacay B, Bonthius NE, Taggard Da, Lothman EW. Alcohol exposure during the brain growth spurt promotes hippocampal seizures, rapid kindling, and spreading depression. Alcohol Clin Exp Res. 2001;25:734–745. [PubMed] [Google Scholar]
- Buzsaki G. Rhythms of the brain. Oxford University Press; New York: 2006. [Google Scholar]
- Caliskan G, Muller I, Semtner M, Winkelmann A, Raza AS, Hollnagel JO, Rosler A, Heinemann U, Stork O, Meier JC. Identification of Parvalbumin Interneurons as Cellular Substrate of Fear Memory Persistence. Cereb Cortex. 2016;26:2325–2340. doi: 10.1093/cercor/bhw001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty G, Saito M, Mao RF, Wang R, Vadasz C, Saito M. Lithium blocks ethanol-induced modulation of protein kinases in the developing brain. Biochem Biophys Res Commun. 2008;367:597–602. doi: 10.1016/j.bbrc.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ML, Olson HC, Picciano JF, Starr JR, Owens J. Sleep problems in children with fetal alcohol spectrum disorders. J Clin Sleep Med. 2012;8:421–429. doi: 10.5664/jcsm.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang DM. Neuroprotective and neurotrophic actions of the mood stabilizer lithium: can it be used to treat neurodegenerative diseases? Crit Rev Neurobiol. 2004;16:83–90. doi: 10.1615/critrevneurobiol.v16.i12.90. [DOI] [PubMed] [Google Scholar]
- Coleman LG, Jr, Oguz I, Lee J, Styner M, Crews FT. Postnatal day 7 ethanol treatment causes persistent reductions in adult mouse brain volume and cortical neurons with sex specific effects on neurogenesis. Alcohol. 2012;46:603–612. doi: 10.1016/j.alcohol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criado JR, Wills DN, Walker BM, Ehlers CL. Effects of adolescent ethanol exposure on sleep in adult rats. Alcohol. 2008;42:631–639. doi: 10.1016/j.alcohol.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angiulli A, Grunau P, Maggi S, Herdman A. Electroencephalographic correlates of prenatal exposure to alcohol in infants and children: a review of findings and implications for neurocognitive development. Alcohol. 2006;40:127–133. doi: 10.1016/j.alcohol.2006.09.031. [DOI] [PubMed] [Google Scholar]
- Diekelmann S, Born J. The memory function of sleep. Nat Rev Neurosci. 2010;11:114–126. doi: 10.1038/nrn2762. [DOI] [PubMed] [Google Scholar]
- Doeppner TR, Kaltwasser B, Sanchez-Mendoza EH, Caglayan AB, Bahr M, Hermann DM. Lithium-induced neuroprotection in stroke involves increased miR-124 expression, reduced RE1-silencing transcription factor abundance and decreased protein deubiquitination by GSK3beta inhibition-independent pathways. J Cereb Blood Flow Metab. 2017;37:914–926. doi: 10.1177/0271678X16647738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers CL, Criado JR. Adolescent ethanol exposure: does it produce long-lasting electrophysiological effects? Alcohol. 2010;44:27–37. doi: 10.1016/j.alcohol.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewirtz JC, McNish KA, Davis M. Is the hippocampus necessary for contextual fear conditioning? Behav Brain Res. 2000;110:83–95. doi: 10.1016/s0166-4328(99)00187-4. [DOI] [PubMed] [Google Scholar]
- Gil-Mohapel J, Boehme F, Kainer L, Christie BR. Hippocampal cell loss and neurogenesis after fetal alcohol exposure: insights from different rodent models. Brain Res Rev. 2010;64:283–303. doi: 10.1016/j.brainresrev.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Graves LA, Heller EA, Pack AI, Abel T. Sleep deprivation selectively impairs memory consolidation for contextual fear conditioning. Learn Mem. 2003;10:168–176. doi: 10.1101/lm.48803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey AG. Sleep and circadian functioning: critical mechanisms in the mood disorders? Annu Rev Clin Psychol. 2011;7:297–319. doi: 10.1146/annurev-clinpsy-032210-104550. [DOI] [PubMed] [Google Scholar]
- Havekes R, Abel T. The tired hippocampus: the molecular impact of sleep deprivation on hippocampal function. Curr Opin Neurobiol. 2017;44:13–19. doi: 10.1016/j.conb.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber R, Ghilardi MF, Massimini M, Tononi G. Local sleep and learning. Nature. 2004;430:78–81. doi: 10.1038/nature02663. [DOI] [PubMed] [Google Scholar]
- Huckleberry KA, Ferguson LB, Drew MR. Behavioral mechanisms of context fear generalization in mice. Learn Mem. 2016;23:703–709. doi: 10.1101/lm.042374.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt PS, Barnet RC. Adolescent and adult rats differ in the amnesic effects of acute ethanol in two hippocampus-dependent tasks: Trace and contextual fear conditioning. Behav Brain Res. 2016;298:78–87. doi: 10.1016/j.bbr.2015.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Kitabayashi R, Funatsu M, Izumi M, Yuede C, Hartman RE, Wozniak DF, Zorumski CF. A single day of ethanol exposure during development has persistent effects on bi-directional plasticity, N-methyl-D-aspartate receptor function and ethanol sensitivity. Neuroscience. 2005;136:269–279. doi: 10.1016/j.neuroscience.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Jan JE, Asante KO, Conry JL, Fast DK, Bax MC, Ipsiroglu OS, Bredberg E, Loock CA, Wasdell MB. Sleep Health Issues for Children with FASD: Clinical Considerations. Int J Pediatr. 2010;2010 doi: 10.1155/2010/639048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang K, Kim YJ, Kim YH, Roh JN, Nam JM, Kim PY, Ryu WS, Lee SH, Yoon BW. Lithium pretreatment reduces brain injury after intracerebral hemorrhage in rats. Neurol Res. 2012;34:447–454. doi: 10.1179/1743132812Y.0000000015. [DOI] [PubMed] [Google Scholar]
- Killgore WD. Effects of sleep deprivation on cognition. Prog Brain Res. 2010;185:105–129. doi: 10.1016/B978-0-444-53702-7.00007-5. [DOI] [PubMed] [Google Scholar]
- Krause AJ, Simon EB, Mander BA, Greer SM, Saletin JM, Goldstein-Piekarski AN, Walker MP. The sleep-deprived human brain. Nat Rev Neurosci. 2017;18:404–418. doi: 10.1038/nrn.2017.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J. GSK3beta in ethanol neurotoxicity. Mol Neurobiol. 2009;40:108–121. doi: 10.1007/s12035-009-8075-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J. Lithium-mediated protection against ethanol neurotoxicity. Front Neurosci. 2010;4:41. doi: 10.3389/fnins.2010.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manber R, Edinger JD, Gress JL, San Pedro-Salcedo MG, Kuo TF, Kalista T. Cognitive behavioral therapy for insomnia enhances depression outcome in patients with comorbid major depressive disorder and insomnia. Sleep. 2008;31:489–495. doi: 10.1093/sleep/31.4.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfridi A, Brambilla D, Mancia M. Sleep is differently modulated by basal forebrain GABA(A) and GABA(B) receptors. Am J Physiol Regul Integr Comp Physiol. 2001;281:R170–175. doi: 10.1152/ajpregu.2001.281.1.R170. [DOI] [PubMed] [Google Scholar]
- Maren S, Phan KL, Liberzon I. The contextual brain: implications for fear conditioning, extinction and psychopathology. Nat Rev Neurosci. 2013;14:417–428. doi: 10.1038/nrn3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall L, Born J. The contribution of sleep to hippocampus-dependent memory consolidation. Trends Cogn Sci. 2007;11:442–450. doi: 10.1016/j.tics.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Marshall L, Helgadottir H, Molle M, Born J. Boosting slow oscillations during sleep potentiates memory. Nature. 2006;444:610–613. doi: 10.1038/nature05278. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Roesch SC, Fagerlund A, Autti-Ramo I, Jones KL, May PA, Adnams CM, Konovalova V, Riley EP Collaborative Initiative on Fetal Alcohol Spectrum D. Toward a neurobehavioral profile of fetal alcohol spectrum disorders. Alcohol Clin Exp Res. 2010;34:1640–1650. doi: 10.1111/j.1530-0277.2010.01250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PA, Gossage JP. Maternal risk factors for fetal alcohol spectrum disorders: not as simple as it might seem. Alcohol Res Health. 2011;34:15–26. [PMC free article] [PubMed] [Google Scholar]
- Ognjanovski N, Schaeffer S, Wu J, Mofakham S, Maruyama D, Zochowski M, Aton SJ. Parvalbumin-expressing interneurons coordinate hippocampal network dynamics required for memory consolidation. Nat Commun. 2017;8:15039. doi: 10.1038/ncomms15039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW, Tenkova T, Dikranian K, Muglia LJ, Jermakowicz WJ, D’Sa C, Roth KA. Ethanol-induced caspase-3 activation in the in vivo developing mouse brain. Neurobiol Dis. 2002a;9:205–219. doi: 10.1006/nbdi.2001.0475. [DOI] [PubMed] [Google Scholar]
- Olney JW, Tenkova T, Dikranian K, Qin YQ, Labruyere J, Ikonomidou C. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Brain Res Dev Brain Res. 2002b;133:115–126. doi: 10.1016/s0165-3806(02)00279-1. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse atlas in stereotaxic coordinates. Academic Press; London, U.K: 2004. [Google Scholar]
- Pesonen AK, Raikkonen K, Matthews K, Heinonen K, Paavonen JE, Lahti J, Komsi N, Lemola S, Jarvenpaa AL, Kajantie E, Strandberg T. Prenatal origins of poor sleep in children. Sleep. 2009;32:1086–1092. doi: 10.1093/sleep/32.8.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley EP, McGee CL. Fetal alcohol spectrum disorders: an overview with emphasis on changes in brain and behavior. Exp Biol Med (Maywood) 2005;230:357–365. doi: 10.1177/15353702-0323006-03. [DOI] [PubMed] [Google Scholar]
- Sadrian B, Lopez-Guzman M, Wilson DA, Saito M. Distinct neurobehavioral dysfunction based on the timing of developmental binge-like alcohol exposure. Neuroscience. 2014;280:204–219. doi: 10.1016/j.neuroscience.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadrian B, Subbanna S, Wilson DA, Basavarajappa BS, Saito M. Lithium prevents long-term neural and behavioral pathology induced by early alcohol exposure. Neuroscience. 2012;206:122–135. doi: 10.1016/j.neuroscience.2011.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadrian B, Wilson DA, Saito M. Long-lasting neural circuit dysfunction following developmental ethanol exposure. Brain Sci. 2013;3:704–727. doi: 10.3390/brainsci3020704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Mao RF, Wang R, Vadasz C, Saito M. Effects of gangliosides on ethanol-induced neurodegeneration in the developing mouse brain. Alcohol Clin Exp Res. 2007;31:665–674. doi: 10.1111/j.1530-0277.2007.00351.x. [DOI] [PubMed] [Google Scholar]
- Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron. 2010;68:1023–1042. doi: 10.1016/j.neuron.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger AR, Cowan WM, Gottlieb DI. An autoradiographic study of the time of origin and the pattern of granule cell migration in the dentate gyrus of the rat. J Comp Neurol. 1975;159:149–175. doi: 10.1002/cne.901590202. [DOI] [PubMed] [Google Scholar]
- Sharma A, Rawat AK. Teratogenic effects of lithium and ethanol in the developing fetus. Alcohol. 1986;3:101–106. doi: 10.1016/0741-8329(86)90019-4. [DOI] [PubMed] [Google Scholar]
- Smiley JF, Saito M, Bleiwas C, Masiello K, Ardekani B, Guilfoyle DN, Gerum S, Wilson DA, Vadasz C. Selective reduction of cerebral cortex GABA neurons in a late gestation model of fetal alcohol spectrum disorder. Alcohol. 2015;49:571–580. doi: 10.1016/j.alcohol.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickgold R, Hobson JA, Fosse R, Fosse M. Sleep, learning, and dreams: off-line memory reprocessing. Science. 2001;294:1052–1057. doi: 10.1126/science.1063530. [DOI] [PubMed] [Google Scholar]
- Stickgold R, Walker MP. Sleep-dependent memory consolidation and reconsolidation. Sleep Med. 2007;8:331–343. doi: 10.1016/j.sleep.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone WS, Altman HJ, Hall J, Arankowsky-Sandoval G, Parekh P, Gold PE. Prenatal exposure to alcohol in adult rats: relationships between sleep and memory deficits, and effects of glucose administration on memory. Brain Res. 1996;742:98–106. doi: 10.1016/s0006-8993(96)00976-6. [DOI] [PubMed] [Google Scholar]
- Talamini LM, Bringmann LF, de Boer M, Hofman WF. Sleeping worries away or worrying away sleep? Physiological evidence on sleep-emotion interactions. PLoS One. 2013;8:e62480. doi: 10.1371/journal.pone.0062480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd TP, Bucci DJ. Retrosplenial Cortex and Long-Term Memory: Molecules to Behavior. Neural Plast. 2015;2015:414173. doi: 10.1155/2015/414173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch LM. Disruptions in sleep time and sleep architecture in a mouse model of repeated ethanol withdrawal. Alcohol Clin Exp Res. 2006;30:1214–1222. doi: 10.1111/j.1530-0277.2006.00134.x. [DOI] [PubMed] [Google Scholar]
- Wang ME, Fraize NP, Yin L, Yuan RK, Petsagourakis D, Wann EG, Muzzio IA. Differential roles of the dorsal and ventral hippocampus in predator odor contextual fear conditioning. Hippocampus. 2013;23:451–466. doi: 10.1002/hipo.22105. [DOI] [PubMed] [Google Scholar]
- Wengel T, Hanlon-Dearman AC, Fjeldsted B. Sleep and sensory characteristics in young children with fetal alcohol spectrum disorder. J Dev Behav Pediatr. 2011;32:384–392. doi: 10.1097/DBP.0b013e3182199694. [DOI] [PubMed] [Google Scholar]
- Wilson DA, Masiello K, Lewin MP, Hui M, Smiley JF, Saito M. Developmental ethanol exposure-induced sleep fragmentation predicts adult cognitive impairment. Neuroscience. 2016;322:18–27. doi: 10.1016/j.neuroscience.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DA, Peterson J, Basavaraj BS, Saito M. Local and regional network function in behaviorally relevant cortical circuits of adult mice following postnatal alcohol exposure. Alcohol Clin Exp Res. 2011;35:1974–1984. doi: 10.1111/j.1530-0277.2011.01549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltgen BJ, Sanders MJ, Anagnostaras SG, Sage JR, Fanselow MS. Context fear learning in the absence of the hippocampus. J Neurosci. 2006;26:5484–5491. doi: 10.1523/JNEUROSCI.2685-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, Young C, Olney JW, Muglia LJ. Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol Dis. 2004;17:403–414. doi: 10.1016/j.nbd.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Wulff K, Gatti S, Wettstein JG, Foster RG. Sleep and circadian rhythm disruption in psychiatric and neurodegenerative disease. Nat Rev Neurosci. 2010;11:589–599. doi: 10.1038/nrn2868. [DOI] [PubMed] [Google Scholar]
- Xu M, Chung S, Zhang S, Zhong P, Ma C, Chang WC, Weissbourd B, Sakai N, Luo L, Nishino S, Dan Y. Basal forebrain circuit for sleep-wake control. Nat Neurosci. 2015;18:1641–1647. doi: 10.1038/nn.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SS, Gujar N, Hu P, Jolesz FA, Walker MP. The human emotional brain without sleep--a prefrontal amygdala disconnect. Curr Biol. 2007;17:R877–878. doi: 10.1016/j.cub.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Yoon SY, Jain U, Shapiro C. Sleep in attention-deficit/hyperactivity disorder in children and adults: past, present, and future. Sleep Med Rev. 2012;16:371–388. doi: 10.1016/j.smrv.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Young C, Olney JW. Neuroapoptosis in the infant mouse brain triggered by a transient small increase in blood alcohol concentration. Neurobiol Dis. 2006;22:548–554. doi: 10.1016/j.nbd.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Young C, Straiko MM, Johnson SA, Creeley C, Olney JW. Ethanol causes and lithium prevents neuroapoptosis and suppression of pERK in the infant mouse brain. Neurobiol Dis. 2008;31:355–360. doi: 10.1016/j.nbd.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Wang Z, Tchantchou F, Chiu CT, Zhang Y, Chuang DM. Lithium ameliorates neurodegeneration, suppresses neuroinflammation, and improves behavioral performance in a mouse model of traumatic brain injury. J Neurotrauma. 2012;29:362–374. doi: 10.1089/neu.2011.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Yang X, Yao W, Lee W. Lithium protects ethanol-induced neuronal apoptosis. Biochem Biophys Res Commun. 2006;350:905–910. doi: 10.1016/j.bbrc.2006.09.138. [DOI] [PubMed] [Google Scholar]
- Zucca S, D’Urso G, Pasquale V, Vecchia D, Pica G, Bovetti S, Moretti C, Varani S, Molano-Mazon M, Chiappalone M, Panzeri S, Fellin T. An inhibitory gate for state transition in cortex. Elife. 2017:6. doi: 10.7554/eLife.26177. [DOI] [PMC free article] [PubMed] [Google Scholar]