

SUMMARY
The bacterial Mfd ATPase is increasingly recognized as a general transcription factor that participates in the resolution of transcription conflicts with other processes/roadblocks. This function stems from Mfd’s ability to preferentially act on stalled RNA polymerases (RNAPs). However, the mechanism underlying this preference and the subsequent coordination between Mfd and RNAP have remained elusive. Here, using a novel real-time translocase assay, we unexpectedly discovered that Mfd translocates autonomously on DNA. The speed and processivity of Mfd dictate a “release and catch-up” mechanism to efficiently patrol DNA for frequently stalled RNAPs. Furthermore, we showed that Mfd prevents RNAP backtracking or rescues a severely backtracked RNAP, allowing RNAP to overcome stronger obstacles. However, if an obstacle’s resistance is excessive, Mfd dissociates the RNAP, clearing the DNA for other processes. These findings demonstrate a remarkably delicate coordination between Mfd and RNAP, allowing efficient targeting and recycling of Mfd and expedient conflict resolution.
In Brief
A “release and catch-up” mechanism allows the bacterial protein Mfd to restart or remove stalled RNA polymerases
INTRODUCTION
Cellular processes often occur concurrently on chromosomes, and this crowded environment inevitably creates adverse conditions that must be timely resolved to ensure genome integrity and cellular viability (Garcia-Muse and Aguilera, 2016). In particular, fundamental processes that occur along DNA, such as transcription and replication, commonly encounter obstacles or “roadblocks” which may impede progress and ultimately result in mutations, DNA damage, or both. Thus, spatial and temporal coordination of cellular machineries can have far-reaching consequences.
In bacteria, the mutation frequency decline (Mfd) protein is widely recognized for contributing to transcriptional roadblock resolution. Mfd is best known for its recognition and displacement of a stalled RNA polymerase (RNAP) in transcription coupled repair (TCR) (Adebali et al., 2017; Selby and Sancar, 1993a). However, there is growing evidence that Mfd has broader roles in resolving roadblocks encountered by RNAP well beyond TCR (Deaconescu et al., 2012; Deaconescu and Suhanovsky, 2017; Selby, 2017). Many different types of roadblocks can stall RNAP, including DNA lesions that are not targets of TCR (Smith and Savery, 2008), protein or protein complexes bound in front of the RNAP (Belitsky and Sonenshein, 2011; Zalieckas et al., 1998), or factors that interact with the RNA transcript to arrest RNAP (Mustaev et al., 2016). Mfd helps to resolve these conflicts by facilitating transcription through these roadblocks or terminating transcription. Recent studies have illuminated the role of Mfd in transcription-replication collisions, which are a major intrinsic cause of genome instability (Dutta et al., 2011). These collisions are unavoidable because the transcription and replication machineries share the same DNA template. Mfd can facilitate the removal of RNAP ahead of a replication fork in vitro, allowing replication to continue unimpeded (Pomerantz and O’Donnell, 2010), and to reduce double stranded breaks that may result from collisions in vivo (Dutta et al., 2011).
Despite the importance of Mfd in transcription, it is unclear how Mfd targets a stalled transcription elongation complex (TEC) (Proshkin and Mironov, 2016). In each E. coli, there are a few thousand copies of RNAPs (Klumpp and Hwa, 2008; Shepherd et al., 2001), but only an estimated few hundred copies of Mfd per cell (Kad and Van Houten, 2012; Selby and Sancar, 1993b, 1994). The scarcity of Mfd calls for efficient appropriation of Mfd to stalled TECs. However, since a stalled TEC is expected to have minimal conformational changes from an elongating TEC (Brueckner et al., 2007), it is not known how Mfd differentiates between these two types of TECs. Even after an Mfd locates a stalled TEC, there remains little mechanistic understanding of how Mfd coordinates with RNAP and resolves obstacles.
Here, we unexpectedly discovered that Mfd can translocate on its own using novel real-time translocase tracking assays. However, Mfd translocates with a slower speed and shorter processivity than those of an elongating TEC. We further demonstrate that these intrinsic motor properties of Mfd lead to a remarkably delicate release and catch-up mechanism that allows Mfd to localize to stalled RNAPs and resolve conflicts of transcription at an obstacle.
RESULTS
Mfd translocates on its own
We developed a highly sensitive, real-time assay designed to track DNA translocases and applied it to Mfd. In the DNA “unzipping tracker” assay (Figures 1A, S1A, and S1B; Table S1; STAR Methods), a single dsDNA molecule is mechanically unzipped by an optical trap until the DNA fork reaches a bound translocase. Subsequently, the two unzipped DNA strands are held under a constant force (18 pN), which is high enough to keep the DNA unzipped (mean unzipping force ~ 15 pN) but also low enough to minimally perturb the translocase. If the translocase moves towards the fork, dsDNA is unzipped; whereas if the translocase moves away from the fork, dsDNA is rezipped. In contrast to many previous single molecule tracking methods, this method does not require tagging or anchoring of the translocase, which could affect the activity and function of the motor protein. Furthermore, when the unzipping force signature is used for sequence alignment (Hall et al., 2009; Shundrovsky et al., 2006), the translocase location may be pinpointed to near base pair accuracy and precision.
Figure 1. Mfd translocates along DNA on its own.

(A) The DNA “unzipping tracker” assay was used to monitor the translocation of Mfd away from the fork (top panel) and towards the fork (bottom panel) in real time under a constant unzipping force of 18 pN in 2 mM ATP. Multiple real-time tracking traces from different Mfd molecules are shown, with their time and location axes arbitrarily shifted. Each trace starts when the unzipping fork encountered an Mfd and ends when the Mfd dissociated from the DNA.
(B) A scatter plot of all traces for both translocation directions (N = 101). Each trace yields a single data point on this plot, with total distance of travel plotted against total time of travel. The means and standard errors of the means are shown for each direction. See also Figure S1 and Table S2.
Using the unzipping tracker, we discovered that Mfd alone translocates along DNA (Figure 1A). For a given measurement, Mfd moved processively either away from the fork or towards the fork and maintained its directionality during translocation, before eventual dissociation from the DNA. Analysis of multiple traces showed that translocation away from the fork had a mean speed of 7.2 ± 0.4 bp/s (mean ± SEM) and a processivity of 200 ± 37 bp, while translocation towards the fork had a mean speed of 5.8 ± 0.2 bp/s and a processivity of 247 ± 47 bp (Figure 1B; Table S2). Mfd moving towards the fork experienced a hindering force, whereas Mfd moving away from the fork experienced an assisting force. Thus the similar speeds and processivities for both directions suggest that the fork did not greatly influence Mfd activities. In addition, for each direction of movement, the distributions of the distance and time Mfd travelled before dissociation are well described by exponential functions (Figure S1C), suggesting that Mfd dissociation from DNA is stochastic.
These results directly demonstrate that Mfd is a bona fide autonomous translocase and its auto-inhibition function established in bulk experiments (Smith et al., 2007) does not prevent Mfd from translocating on naked DNA. The finding that Mfd can independently translocate also opens up the possibility that Mfd could locate a stalled RNAP by translocating on DNA via directed one-dimensional (1-D) motion, providing an alternative to a 3-D search mechanism. Mfd translocation on naked DNA allows it to broadly survey the bacterial genome for a TEC target, while the short processivity of Mfd ensures that it quickly gets off the DNA if a target is not promptly located. The 1-D and 3-D pathways together may allow Mfd to quickly target an arrested RNAP and could help expedite DNA repair or roadblock removal.
Mfd releases from TEC upon transcription resumption
The ability of Mfd to translocate independently raises new questions about its coordination with an elongating TEC. In comparison with a TEC, Mfd translocates with a lower speed and a shorter processivity. A TEC moves at an overall speed of ~ 14 bp/s under our conditions and can transcribe many thousands of base pairs without dissociation (Adelman et al., 2002). In contrast, Mfd moves slower at ~ 7 bp/s and can only cover approximately two hundred base pairs before dissociation. These differences impose significant constraints on how these two motor proteins coordinate their translocations. Consider a situation where Mfd interacts with a TEC while the TEC starts to actively elongate, which could occur when Mfd locates a stalled TEC and brings the TEC into active elongation (Park et al., 2002). What happens to the Mfd after the TEC starts to elongate? Does the Mfd separate from the elongating TEC?
We therefore investigated whether Mfd releases from the TEC upon transcription resumption. We directly mapped the locations of both Mfd and TEC using the “unzipping mapper” technique (Table S1; STAR Methods) (Brennan et al., 2016; Li et al., 2015). Unlike the “unzipping tracker”, which operates under a low force condition to minimize perturbation to a bound protein, the “unzipping mapper” exerts forces large enough to rapidly disrupt a bound protein and accurately locate the position of that bound protein on DNA (Hall et al., 2009; Koch and Wang, 2003; Shundrovsky et al., 2006). Here a force rise above the naked DNA baseline indicates tightening of DNA base-pairing and a force drop below the baseline indicates weakening of DNA base-pairing.
This experiment imposes stringent requirements on the measurement time window due to the short translocation time of Mfd (Figure 1). We therefore utilized a dual optical trap together with a multi-channel laminar flow cell which partitioned different buffers using flow, permitting rapid and controlled access to different buffer conditions (Figure S2A) (Forget and Kowalczykowski, 2012). Here, a TEC was stalled by nucleotide starvation at +20 on the trunk of an unzipping DNA fork template with its transcription direction away from the DNA fork (Table S4), and Mfd was either present or absent in the experiments (Figure S2B). In the presence of Mfd, Mfd was preloaded onto a TEC at ~ 36% efficiency (STAR Methods). The DNA molecule was rapidly (~ 2 s) transported to an adjacent flow channel containing all four NTPs and no Mfd, which permitted both transcription and Mfd translocation (Table S2). After a specified time (Δt), the DNA was then rapidly unzipped to map the locations of any bound proteins on the trunk (Figure 2A, left diagram).
Figure 2. Release and catch-up mechanism of Mfd.

(A) Mfd releases from TEC. The light pink and light green shaded regions indicate the ranges of expected locations of Mfd and TEC respectively at the time of the unzipping, with the width of each region determined by the molecule’s speed variations and measurement time uncertainties. The gray curve corresponds to unzipping naked DNA which serves as a baseline. The black and green traces were taken at Δt = 11 s. For the black trace, the two red dashed lines indicate the initial and final positions of Mfd, and the two green dashed lines indicate the initial and final positions of the TEC. See also Figure S2 and Table S2.
(B) Mfd catch-up experimental scheme.
(C) Representative unzipping traces for transcription time Δt = 10 s for experiments outlined in (B). The first trace (green curve) shows a TEC which had transcribed at ~ 14 bp/s; the second trace (red curve) shows an absence of TEC, indicating that Mfd had caught up with the TEC and removed it. The light green shaded areas indicate the possible range (standard deviation) of TEC location at Δt = 10 s (also see Figure S2C).
(D) Fraction of traces without a TEC as a function of the distance between stalls d. Each data point was determined from ~ 280 (with Mfd) or ~ 180 (without Mfd) traces, with error bars representing standard errors of the mean. The red dashed line is an exponential fit with the function f = a + b×exp(−d/d0) to the data taken in the presence of Mfd, with a baseline a determined from the fraction of RNAP dissociation in the absence of Mfd. The fitting yielded a characteristic distance d0 of 180 ± 30 bp. See also Figure S3C.
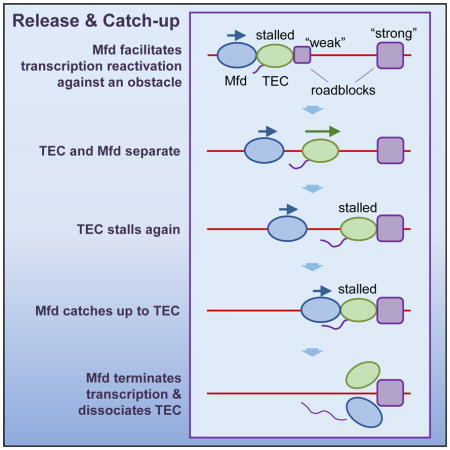
(E) The release and catch-up mechanism of Mfd. After Mfd separates from an elongating TEC, Mfd continues to translocate towards the TEC. If the TEC is stalled again within Mfd’s processivity, Mfd can catch up with the TEC.
Figure 2A shows example traces at Δt = 11 ± 2 s. In the absence of Mfd and presence of TEC translocation, traces with bound proteins only showed a single force peak above the naked DNA baseline, moving at a speed of 14 ± 4 bp/s (mean ± SD) (Figure S2C), consistent with TEC translocation alone. In the presence of both Mfd and TEC translocation, two types of traces with bound protein(s) were observed. One type showed a single force peak. These traces are consistent with TEC translocation alone (when Mfd failed to preload or Mfd was preloaded but dissociated from the DNA) or Mfd translocating with a TEC in close vicinity. More informatively, the second type of trace displayed two force peaks. The leading force peak was consistent with a TEC translocating independently at 14 ± 4 bp/s. The trailing force peak was indicative of Mfd releasing from TEC and then travelling independently in the same direction as the TEC at 7 ± 2 bp/s.
These data clearly demonstrate that upon transcription resumption, Mfd, which is initially bound to a TEC, releases from the TEC and continues to translocate behind an elongating TEC until dissociation from the DNA.
Release and catch-up mechanism of Mfd
The finding that upon transcription resumption, Mfd detaches from the TEC and the two motors translocate independently raises further questions about their coordination. Is the Mfd able to catch up with the TEC if the TEC stalls again? That is, can a single Mfd protein have multiple rounds of interactions with the same TEC when the TEC pauses frequently during transcription?
To answer these questions, we used an assay (Figure 2B) requiring only a single optical trap (Table S1; STAR Methods) and allowing for higher data throughput. In this assay, we first loaded Mfd onto a stalled TEC. The loading efficiency was ~ 55% (Figure S3C), corresponding to the fraction of the TEC that could be impacted by the Mfd. We then removed all free Mfd proteins in solution by replacing the buffer with one containing no Mfd but all four NTPs. This buffer condition permitted translocation of both Mfd (Table S2) and the TEC. Once Mfd separates from a TEC, Mfd may dissociate from DNA when the distance of its travel exceeds its processivity. Therefore, the effect of Mfd on subsequent transcription should depend on the time of exposure to all NTPs, Δt, which would correspond to a transcription distance d, the distance between two stall positions. After a specified time Δt, we replaced the buffer with one containing only ATP for 8 min to stall transcription but still permit Mfd to translocate. If Mfd was not able to catch up with the TEC, the 8 min ATP would ensure Mfd dissociation, and the TEC would remain stalled. On the other hand, if Mfd was able to catch up with the TEC, Mfd would disrupt the stalled TEC and remove it from the stalled location. To determine whether these outcomes could occur, we flushed out the ATP and used the “unzipping mapper” to determine the locations of any bound proteins on the DNA (Table S1) (Figures S3B and S3C).
For a given Δt, we detected two types of traces with their partitioning a function of Δt and thus the distance between stalls d (Figure 2C). In one type, a bound protein was located at a position consistent with a TEC having translocated and then stalled (Figure 2C, green curve; Figure S2C). This fraction could be due to an Mfd not being loaded onto the TEC initially, or an Mfd dissociating before catching up with the TEC. The other type showed naked DNA at the location where a stalled TEC would be expected (Figure 2C, red curve). The fraction of these traces above the no Mfd baseline was a result of the action of Mfd (Figure 2D), consistent with TEC disruption by Mfd. We found that this fraction follows an exponentially decaying dependence on transcription distance d, with a characteristic distance of 180 ± 30 bp (Figure 2D). Within measurement uncertainties, this characteristic distance is in agreement with Mfd’s processivity alone. The agreement between the two distances indicates that once a TEC resumes elongation, Mfd separates from the TEC and subsequently catches up to the TEC only if the distance is within Mfd’s processivity.
These findings suggest a highly coordinated dynamic interplay between an Mfd and a TEC. Mfd translocation leads to a “release and catch-up” mechanism, which has profound implications for transcription regulation (Figure 2E). As previously found, when an Mfd locates a stalled/paused TEC, it can bring the TEC into active elongation (Park et al., 2002). Here we show that the slower speed of Mfd compared with that of the TEC leads to separation of the Mfd from the TEC. If the TEC continues to elongate unimpeded for an extended distance, Mfd will dissociate from DNA and can target another stalled TEC. However, if the TEC pauses frequently (e.g., every 200 bp or less) and has difficulty escaping from a pause, Mfd can then catch up with the TEC and facilitate transcription through a pause site. In the absence of any interaction with a TEC, bound Mfd may be detrimental to the cell. Bound Mfd proteins may deplete available Mfd or also become roadblocks for other DNA-based processes. Thus the short processivity of Mfd ensures that Mfd does not stay on DNA when not needed, but Mfd’s association with DNA is effectively increased when Mfd is needed by transcription, making Mfd an efficient surveyor for a stalled TEC.
Mfd catches up to TEC at an ops pause
We further investigated how Mfd’s release and catch-up mechanism may impact TEC escape from a pause site. We focused on the rfaQ ops site, a well-characterized naturally occurring class II pause sequence that regulates bacterial gene expression (Artsimovitch and Landick, 2000). At an ops site, a TEC may pause due to backtracking, where RNAP reverse translocates along the DNA and the 3′ end of the RNA disengages from the active site, rendering the TEC inactive (Komissarova and Kashlev, 1997a; Nudler et al., 1997). Because Mfd has previously been shown to rescue a backtracked TEC (Park et al., 2002), we expect to observe this capability in a distance-dependent manner within the release and catch-up mechanism.
We conducted experiments with two different DNA templates each with an ops site located at a different distance d from the initially stalled TEC: d = 27 bp and 276 bp respectively (Table S4). The steps of these experiments were similar to those of Figures 2B–D, but with the replacement of the subsequent TEC stalling by nucleotide starvation with an ops pause site (Figure 3A; STAR Methods).
Figure 3. Mfd catching up to a TEC at an ops pause site.

(A) Experimental scheme. The experimental steps are similar to those of Figure 2B. Instead of a subsequent stall, RNAP could transiently pause at the ops site.
(B) and (C). Representative traces of transcription on two different templates, with the separation of the ops site from the initial +20 site being d = 27 bp and 276 bp, respectively. The black and red dashed lines indicate stall position at +20 and the expected ops pause position respectively.
(D) and (E). Probability density functions of TEC location. Plots were generated by pooling data from multiple traces of TEC location data: N = 205 and 255 for (−) and (+) Mfd respectively for d = 27 bp; N = 201 and 188 for (−) and (+) Mfd respectively for d = 276 bp. Only the TECs that had escaped from +20 were considered in these histograms. Arrows point to the peaks of TEC paused at the ops site. The vertical black and red dashed lines indicate the initial stall position at +20 and the expected ops pause position, respectively. See also Figure S3D.
(F) Pause reduction factor at the ops site, defined as the ratio of the ops peak area without Mfd preloading over that with Mfd preloading in (D) and (E). Error bars were calculated assuming a binomial distribution for the fraction of TEC paused at the ops site.
Without Mfd preloading, 10–15% of the TECs paused at the expected ops pause sites for both d = 27 bp and d = 276 bp after transcription resumption time Δt = 30 s and 70 s respectively (Figures 3B–E), while other TECs escaped from the ops site prior to detection (Figures 3D and 3E). More careful examination of the ops pause locations showed that the TEC backtracked by about ~ 5 bp (Figure S3D), consistent with previous biochemical findings of the backtracking distance at the ops pause (Artsimovitch and Landick, 2000).
With Mfd preloading, the fraction of the TEC paused at the ops site was reduced by approximately two fold on the d = 27 bp template (Figure 3D), but there were no detectable changes for the fraction at the ops site on the template with d = 276 bp (Figure 3E). This difference is further summarized in Figure 3F. This observation indicates that Mfd can reactivate a TEC paused at an ops site if this pause is located in close proximity to the Mfd, providing further evidence for the proposed release and catch-up mechanism of Mfd.
TEC becomes highly backtracked upon collision with a DNA fork junction
Before we examined Mfd’s impact on a TEC at an obstacle, we first considered how a TEC deals with an obstacle on its own. We carried out experiments where an actively elongating TEC translocated towards a DNA fork, a configuration resembling a simplified head-on collision of transcription with replication. We then followed transcription in real time upon collision of the TEC with the fork using an “unzipping staller” (Table S1; STAR Methods). In this case, after a TEC arrives at the unzipping fork, the two ends of the ssDNA strands are held fixed. As the TEC moves against the fork, it will rezip the DNA, resulting in an increase in the force. The continuous force build-up hinders the TEC’s forward translocation, leading to its stalling or dissociation. To allow for finer control of the start of the experiment, we used a dual optical trap together with a multi-channel laminar flow cell (Figure S4A).
As shown in Figure 4A, after a TEC encountered the DNA fork, it initially moved forward unidirectionally and processively though its motion was punctuated with transient pauses. The speed after pause removal (pause-free speed) was 15.5 ± 8.1 bp/s (mean ± SD) (Figure 4B) (STAR Method), consistent with earlier work (Adelman et al., 2002; Ma et al., 2013). As the TEC encountered a greater resistance of 19.5 ± 1.5 pN, corresponding to 4.5 ± 1.5 pN above baseline (15 pN) (Figure 4B), it became stalled (defined as a pause duration longer than 30 s) and subsequently experienced irreversible backtracking (Figure 4C). Intriguingly, we observed that ~ 50% of stalled TECs backtracked >20 bp, up to 100 bp or more within the measurement time window (Figure 4C; STAR Methods).
Figure 4. TEC working against a DNA fork.

(A) A representative trace of a TEC working against a DNA fork. See also Figure S4A.
(B) Histograms of the pause-free speed of TEC prior to stalling and the stall force. For each histogram, its mean and SD are indicated with the number of traces N = 16.
(C) TEC backtracking at a stall. In order to highlight backtracking, multiple traces, each with a different color, are shown. All traces are aligned with respect to the start of stall time and position. Bottom figure shows corresponding force.
In all measurements, backtracked RNAPs did not dissociate from DNA and remained bound over the entire duration of the measurements, demonstrating the exceptional stability of these complexes. They also appeared to undergo a biased random walk with the overall direction towards further backtracking (Figure 4C).
We thus demonstrate that when a TEC is working against an obstacle, it can undergo extensive backtracking. Our finding that backtracked complexes are exceptionally stable is in agreement with those from earlier studies (Cheung and Cramer, 2011; Komissarova and Kashlev, 1997b; Nudler et al., 1997). Because backtracked complexes may become major obstacles to replication if they remain on the DNA, effective mechanisms are crucial in mediating collision conflicts. Indeed, in addition to Mfd, a number of other factors have been identified to suppress backtracking: anti-backtracking factors (GreA and GreB) (Erie et al., 1993; Opalka et al., 2003; Tetone et al., 2017), rho-factor that travels with RNAP to terminate termination (Roberts et al., 2008), multiple RNAPs working in conjunction (Epshtein and Nudler, 2003; Jin et al., 2010), and ribosomes carrying out translation over the RNA transcript concurrent to transcription (Proshkin et al., 2010). The existence of these multiple, redundant mechanisms demonstrates critical cellular need for backtracking suppression.
Mfd facilitates and then terminates transcription at a DNA fork junction
To directly observe how Mfd impacts transcription against a DNA fork junction, we monitored real-time molecular events as in Figure 4 (STAR Methods) in the presence of Mfd. Three distinct types of real-time translocation traces were observed. In 40% of traces (N = 23 total) (Figure 5A, left panel), Mfd prevented extensive backtracking. The motor complex initially translocated steadily (pause-free speed of 15.1 ± 8.5 bp/s; Figure 5B) with its motion interrupted by frequent pausing. None of the pauses showed significant backtracking. The agreement of the pause-free speed with that of the TEC alone suggests that the observed motion involved an active TEC instead of Mfd alone. However, the TEC was able to continue to translocate against approximately 2–3 pN greater force than that of the TEC alone, indicating that Mfd assisted the TEC in forward translocation, in agreement with previous results from bulk assays (Park et al., 2002). As the force increased further (transition at 6.8 ± 1.5 pN above baseline; Figure 5C), the motor complex often underwent an abrupt decrease in pause-free speed to 7.1 ± 3.2 bp/s (Figure 5B) before stalling or dissociation. This decreased speed is consistent with that of Mfd alone or Mfd associated with a non-elongating RNAP (Figures 1, 2, S3A, and S6), indicating transcription termination. We therefore use this speed transition to partition regions of transcription facilitation and transcription termination.
Figure 5. TEC working against a DNA fork in the presence of Mfd.

(A) Two representative traces. The left trace is an example of prevention of extensive backtracking and the right an example of backtracking recovery. Blue dashed lines indicate transitions between Mfd facilitated transcription (green shaded areas) and Mfd terminated transcription (pink shaded areas). The start of a green shaded area was determined at when the force increased beyond that of the TEC alone as in the left panel, or at when a clear backtracking recovery was detected as in the right panel (also indicated by a red arrow). Within a green region, the instantaneous speed was consistent with that of a TEC (except during backtracking recovery). Within a pink region, the instantaneous speed was consistent with that of Mfd or Mfd with a non-elongating RNAP. See also Figure S6.
(B) Pause-free speed histograms before and after the transition from transcription elongation to transcription termination. Gaussian fits to these histograms are also shown along with the means and SDs of the fits. Number of traces used in both histograms: N = 23.
(C) Histograms of measured forces. The top histogram shows the stall force when no backtracking recovery was detected (TEC only). The middle histogram shows the force at the transition from transcription elongation to termination (TEC with the help of Mfd). The bottom histogram shows the stall force after the transition (Mfd with RNAP). The mean values and SDs are also indicated.
(D) An example trace showing multiple rounds of interactions of Mfd with a TEC. Each red arrow indicates a rescue event where a paused/stalled TEC was brought into active elongation by Mfd.
In a different 40% of traces (Figure 5A, right panel), Mfd rescued extensively backtracked TEC complexes. The TEC initially translocated forward and then became significantly backtracked (sometimes for more than 100 bp). However, backtracking was subsequently fully reversed via ~ 5–10 bp/s unidirectional forward motion which brought the complex entirely out of the backtracked state. Such a full recovery of a TEC from extensive backtracking was never observed in the absence of Mfd. This observation demonstrates Mfd-dependent recovery from extensive backtracking. We found that the TEC subsequently resumed elongation at a pause-free speed of 15.1 ± 8.5 bp/s and behaved in a manner similar to traces described in the previous paragraph.
Some traces even showed multiple backtracking rescue events (Figure 5D). Because of the long Mfd recruitment time (~ 50 s) (Figure S4B) as compared with the time interval (~ 10 s) between backtracking rescue events and/or TEC disruption events, these sequential events were consistent with a single Mfd motor having multiple rounds of interactions with a TEC that paused frequently during elongation via the release and catch-up mechanism. The short distance between these pauses (50 bp or less) are also well within the processivity of Mfd. Mfd may interact with a transiently (e.g., <3 s) paused TEC if the TEC is in close vicinity. Indeed we found that the presence of Mfd moderately reduced both the pause duration and pause density (Figure S5A). This suggests that Mfd may interact with TECs paused independently of backtracking, such as ubiquitous (Neuman et al., 2003) or “pre-translocation” pauses (Bai et al., 2004; Bai et al., 2009; Bai and Wang, 2010).
The remaining 20% of traces showed that the motor complex backtracked and did not recover within the observation time window, suggesting an absence of Mfd interaction. In addition, these traces stalled at a force similar to that of the TEC alone stall force (Figure 5C).
These results in Figure 5 show anti-pausing and termination dichotomy of Mfd on transcription. We then performed separate biochemical experiments using transcription gels to examine pausing in the presence and absence of Mfd (Figure S5B). We used nucleotide concentrations to modulate pausing and transcription speed relative to that of Mfd. We found that when transcription was carried out at a low nucleotide concentration, RNAP paused frequently and Mfd enhanced transcription termination/arrest. As the nucleotide concentration increased, Mfd facilitated transcription by reducing pause duration and frequency, demonstrating anti-pausing behavior. At a high nucleotide concentration, transcription occurred with minimal pausing and Mfd had little impact on either pausing or the overall transcription rate.
Mapping the structure of Mfd-TEC complex by unzipping DNA
Finally, to fully dissect the molecular details of Mfd interaction with a TEC, it is essential to establish a high-resolution interaction map of the Mfd-TEC complex with DNA. Although the structure of Mfd alone has been solved (Deaconescu et al., 2006), it is not known how Mfd changes its conformation upon interaction with a TEC and whether Mfd induces conformational changes in the TEC. As the first step in investigating this problem, we used the unzipping mapper technique, which disrupts each interaction sequentially within a complex along DNA, creating a detailed map of the locations and strengths of multiple interactions within a large protein-DNA complex to near base-pair resolution (Hall et al., 2009; Li et al., 2015; Li and Wang, 2012; Shundrovsky et al., 2006) (Table S1; STAR Methods).
As control experiments, we unzipped through a stalled TEC in both the forward (same direction as transcription) and the reverse (opposite to transcription) directions (Figures 6A and 6B; Table S4). Forward unzipping yielded a force drop (due to the transcription bubble), followed by a force rise immediately after the active site (due to RNAP clamping on the downstream DNA). Reverse unzipping yielded a force rise at +13 bp downstream of the active site. These results are consistent with previous findings (Inman et al., 2014; Jin et al., 2010).
Figure 6. Structural mapping of Mfd-TEC interactions.

(A) Example unzipping traces. A TEC was stalled at +20 position and each unzipping direction is indicated by an arrow. Vertical dashed lines indicate the positions of measured strong interactions. See also Figures S7A and S7B.
(B) Dwell-time histograms after pooling data from multiple traces. Total number of traces in each histogram (from top to bottom) are N = 132, 63, 73, 160, and 65. For dwell-time histograms in the presence of Mfd, only traces with an additional force rise upstream of the TEC, indicative of Mfd binding to TEC, were included for analysis. The pink arrow indicates a forward shift in the footprint of Mfd towards the TEC in the presence of ATPγS. (C) Dissociation constant Kd of the wild type Mfd as well as MfdR685A and MfdN817A for binding to TEC (STAR Methods). Error bars are SEMs.
(D) Dwell-time histograms of MfdR685A (green) and MfdN817A (red) in the absence (top) or presence (bottom) of 2 mM ATPγS. Number of traces for each histogram are 27 and 21 for MfdR685A and MfdN817A respectively in the absence of ATPγS, and 25 and 30 for MfdR685A and MfdN817A respectively in the presence of ATPγS.
(E) Proposed model of the cooperation of the two translocation domains of Mfd upon binding to nucleotide. See also Figure S7D.
In the presence of Mfd but with no nucleotides, unzipping revealed additional force signatures upstream of the active site (Figures 6A, 6B, and S7A), although this apo condition was previously thought not to support Mfd interaction with the TEC (Deaconescu et al., 2006; Selby and Sancar, 1995). In the forward direction, there were two distinct force peaks upstream of the active site of the TEC, at −43 bp and −16 bp, a clear signature for Mfd binding. By counting the fraction of TECs with a bound Mfd, we determined the dissociation equilibrium constant (Kd) of Mfd binding to be 20 ± 3 μM (Figure 6C). Although the binding of apo-Mfd to a TEC has a low affinity, it was detectable due to the exquisite sensitivity of the unzipping method. These data also provide a footprint of Mfd of approximately 27 bp, consistent with previous estimations (Deaconescu et al., 2006; Park et al., 2002). The −16 bp site is in partial overlap with the RNAP footprint, which extends to −18 bp upstream of the active site (Korzheva et al., 2000; Nudler, 1999). Therefore, this site could be a result of Mfd directly binding to the DNA by stepping into the footprint of the RNAP or Mfd-mediated RNAP interaction with DNA.
In the presence of ATPγS, Mfd’s footprint moved 12 bp towards the RNAP while the positions of the -16 bp site and the RNAP remained stationary (Figures 6A, 6B, and S7B), likely as a result of nucleotide-dependent conformational changes in Mfd.
To determine the orientation of Mfd relative to a TEC, we examined two Mfd mutants. Mfd’s translocase module is composed of two DNA translocation domains (TD1 and TD2), with a highly-conserved core for nucleotide binding and hydrolysis embedded near the TD1/TD2 interface (Deaconescu et al., 2006). While it is known that TD1 and TD2 represent the main DNA binding determinant (Selby and Sancar, 1993a), how these two domains orient relative to the TEC and how they coordinate their DNA binding activities have not been determined. Thus, we examined two Mfd variants, MfdR685A (mutation in TD1) and MfdN817A (mutation in TD2), which each showed severely impaired binding affinity to naked DNA in a bulk fluorescence anisotropy assay, but remained competent for ATP hydrolysis (Figure S7C).
In the apo state, MfdR685A still yielded a force peak at the −43 bp binding site, but had a diminished force peak at the −16 bp binding site (Figure 6D, top panel). Conversely, MfdN817A lacked a force peak at the −43 bp binding site, but retained the force peak at the −16 bp binding site. We therefore attribute the -43 binding site to TD2 and the −16 bp binding site to TD1. Thus TD1 binds to DNA at the front and is followed by TD2. In the presence of ATPγS, both variants produced a footprint on DNA similar to that of the wildtype Mfd, where Mfd’s footprint moved 12 bp towards the RNAP (Figure 6D, bottom panel).
These results show that upon nucleotide binding, TD2 moves towards TD1 while both the TD1 and RNAP remain stationary. Subsequent nucleotide hydrolysis and product release may allow TD1 to step into the RNAP. Thus our finding may be consistent with a model of Mfd translocation, where TD1 and TD2 alternate their stepping to translocate towards the RNAP (Figures 6E and S7D).
In addition, each mutant showed a significantly lower DNA binding affinity, as compared to wildtype Mfd, either in the presence or absence of ATPγS (Figure 6C). This finding implies that both TD1 and TD2 domains contribute to the overall DNA binding affinity observed for wild type (wt) Mfd. Interestingly, for either mutant, the decrease in affinity was more pronounced, by an order of magnitude, in the presence of ATPγS. This suggests that once Mfd binds ATP, TD1 and TD2 act cooperatively to interact with DNA and thus enhance the overall DNA affinity of Mfd (Figure 6E).
DISCUSSION
DNA translocases have traditionally been difficult to study because translocation often does not produce a readily measured product (Singleton et al., 2007). Our novel methodologies allow real-time tracking and measurement of the mechanical response of these translocation events at near base pair resolution. What emerges is a remarkably delicate coordination between Mfd and transcription (Figure 7). Contrary to previous biochemical findings (Smith et al., 2007), we found that Mfd can independently translocate at approximately 7 bp/s over approximately 200 bp. Intriguingly, these intrinsic motor properties of Mfd provide a simple explanation to how Mfd can localize to a paused/stalled TEC using a release and catch-up mechanism. Mfd may target a stalled or an elongating TEC without the need of discrimination based on their conformational differences. However, Mfd remains associated with a stalled TEC but detaches from an elongating TEC when its assistance is no longer beneficial. While independent translocation of Mfd allows it to continue to “patrol” for slowly moving TECs, the limited Mfd processivity restricts the search and ultimately ensures that Mfd dissociates from DNA, preventing it from becoming a roadblock for other processes and not depleting the pool of free Mfd.
Figure 7. Modes of Mfd regulation of transcription.

When a TEC encounters a major obstacle in the absence of Mfd and other anti-backtracking factors, the TEC may become stalled and experience extensive backtracking (A). Mfd is able to prevent TEC backtracking and/or rescue a backtracked complex into elongation. If a TEC elongates steadily after the initial rescue, Mfd detaches from the DNA (B). However, if a TEC stalls frequently, Mfd will use the release and catch-up mechanism to continue to facilitate elongation through multiple obstacles (C). When an obstacle becomes insurmountable, Mfd terminates transcription by disrupting the TEC, resulting in eventual dissociation of Mfd and RNAP from the DNA (D).
In some sense, Mfd can be regarded as a regulated co-factor for RNAP. Unlike other transcription factors that rely on recognizing protein conformations or chemical modifications for binding (Roberts et al., 2008), Mfd uses its ATP-dependent translocation to locate paused/stalled TECs. This “speed-gated” sensing affords kinetically regulated, tunable affinity of Mfd for a TEC. Because this type of coordination optimizes efficiency and mitigates conflicts and has minimal requirements for a specific DNA sequence, it may be more broadly employed in the cell.
The release and catch-up mechanism may provide insight into earlier ensemble results. An intriguing in vivo observation is that overexpression of Mfd, even by several hundred fold, does not grossly interfere with cell growth (Roberts and Park, 2004; Selby and Sancar, 1993a; Smith et al., 2012). This indicates that elevated Mfd concentration is not sufficient to offset the limited processivity of Mfd, preventing Mfd from becoming a major roadblock. Also Mfd’s persistent association with frequently paused TECs could efficiently bring backtracked TECs to elongation, facilitating cellular recovery from stress and DNA damage during which upregulation of UvrD during recovery may induce TEC backtracking (Epshtein et al., 2014). Consistent with this, an in vitro study showed that Mfd can stimulate the repair of a DNA lesion located downstream of an ops pause site (Haines et al., 2014). While Mfd can reach the lesion via association with a non-elongating RNAP, our work shows that Mfd can release from a TEC at the ops site and subsequently translocate to catch up to the TEC at the lesion or simply arrive at the lesion on its own. Thus, the novel release and catch-up mechanism proposed here may have broader implications in genome maintenance, both in terms of DNA repair and transcription-replication conflict resolution.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michelle D. Wang (mwang@physics.cornell.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains
RNAP was expressed at low levels in 5α-competent E. coli transformed with the plasmid pKA1 in Superbroth with 100 μg/mL ampicillin for 4 hours until A600nm reached 2.1. Cells were induced with IPTG to a final concentration of 1 mM for 4 hours.
Mfd protein was produced using a previously described expression system (Deaconescu and Darst, 2005). Overexpression plasmid pAD6 was transformed into BL21(DE3) or Rosetta(DE3)pLysS cells, which were then grown in LB media supplemented with the appropriate antibiotics at 37°C until the OD600nm reached 0.6–0.8. Induction was achieved with 1mM IPTG at 30°C for 4 hours. Mfd variants were prepared using standard PCR-based site-directed mutagenesis (Table S3), and then expressed as wild type Mfd.
METHOD DETAILS
DNA templates and protein preparations
The DNA unzipping segment containing a T7 A1 promoter was amplified from plasmid pRL574 (Schafer et al., 1991) using PCR with Phusion high-fidelity DNA polymerase and primers designed with SeqBuilder. The purified DNA was digested with DraIII (or AlwNI) restriction enzyme to produce a double-stranded DNA (dsDNA) fragment with a 3′-GAT ssDNA overhang. This DNA was then ligated to a pair of DNA Y-arms (Inman et al., 2014), with one arm labeled with biotin and the other arm labeled with dioxygenin through separate Klenow reactions with biotin-14-dATP and digoxigenin-11-dUTP, respectively. The labels on both arms allow DNA tethering between an anti-dig coated glass surface and a streptavidin-coated bead (489 nm in diameter). We prepared multiple DNA unzipping templates with unzipping directions that were either co-directional with, or head-on to, transcription. The co-directional DNA templates were used for experiments in Figures 1, 2, 3, 6, S1C, S2, S3, S7A, and S7B. The head-on templates were used for Figures 4, 5, 6, S4, S5A, and S6. A list of DNA primers used to generate these DNA templates is included in Table S3. Descriptions of the DNA unzipping templates used in this study are also provided in Table S4.
E. coli RNAP was purified using tagged purification (Adelman et al., 2002). In brief, the cells were lysed and sonicated on ice with a macro tip on a Branson Sonifier 250 with 60% duty cycle in small aliquots (<20 mL). Centrifugation was used to pellet cell debris and the supernatant containing DNA and DNA-bound proteins was saved. Cleared 5% (w/v) polyethyleneimine (PEI) pH 7.9 (made from 50% stock) was slowly added to the supernatant to a final concentration of 0.4% (w/v) in order to precipitate nucleic acids and their bound proteins out of solution. The DNA with bound RNAP was pelleted from the solution and after five washes in a 350 mM NaCl containing buffer, RNAP was eluteds from the PEI and DNA with a 1 M NaCl containing buffer. The eluted RNAP was purified to homogeneity by using chromatography on three columns: first on a HiPrep Heparin FF 16/10 column, followed by a HiPrep 26/60 Sephacryl S-300 HR column, and last on a QIAGEN Ni-NTA Superflow column. Fractions that contained holo-RNAP were pooled, concentrated, and dialyzed into RNAP storage buffer (50 mM Tris-HCl pH 8.0, 100 mM NaCl, 1 mM EDTA, 50% (v/v) glycerol, and 1 mM DTT) and stored at −20°C.
Mfd proteins were purified using small modifications of a published purification protocol (Deaconescu and Darst, 2005). After cell disruption and lysate clarification, protein was purified via Ni2+-affinity chromatography on a HisTrap column (GE Healthcare). Pooled fractions were then dialyzed against a low-salt buffer (75 mM NaCl, 20 mM Tris pH 8, 10% glycerol, 15 mM imidazole, 1 mM TCEP) and applied to a Heparin HiTrap column (GE Healthcare). Fractions eluted in this step were concentrated by centrifugation and applied to a Superdex200 10/300 size-exclusion chromatography column (GE Healthcare) equilibrated with a buffer consisting of 100mM NaCl, 20 mM Tris pH 8, 1mM TCEP. Variants were purified like wild-type with the exception of MfdR685A, for which the heparin affinity step was substituted with a ion-exchange chromatography step.
Single molecule transcription assays – Related to Figures 2–6
For single molecule experiments requiring transcription, TEC was formed in bulk on the trunk of the Y-structured DNA molecule and paused at +20 position via nucleotide depletion (Adelman et al., 2002; Jin et al., 2010). In brief, 3.5 nM DNA template with a T7A1 promoter was mixed with 17.5 nM E.coli RNAP holoenzyme in the presence of 250 μM ApU, 50 μM GTP, CTP, ATP, and 1 unit/μL SUPERase• RNAse inhibitor in a transcription buffer which contained 25 mM Tris-Cl pH 8.0, 100 mM KCl, 4 mM MgCl2, 1 mM DTT, 3% (v/v) glycerol, and 0.15 mg/mL acetylated BSA. The mixture was incubated at 37°C for 30 minutes and was then kept at 4°C. The DNA unzipping mapper showed that typically 90–95% of the DNA tethers contained a TEC assembled successfully at the expected stalled location (+20). For transcription chasing experiments that report the fraction of DNA tethers with a bound TEC after several rounds of buffer exchange (Figure 2), we measured the TEC assembling efficiency right before each chasing experiment and took account of this efficiency in the final fraction calculation.
Bulk transcription assays – Related to Figure S5B
The bulk transcription assays were performed under similar conditions as those of the single molecule experiments. TECs paused at +20 was prepared by incubation of 50 nM E. coli RNAP, 10 nM transcription DNA template attached to streptavidin coated magnetic beads, 250 μM ApU initiating dinucleotide, 50 μM ATP and CTP, and 5 μM GTP (containing 0.5 μCi/μL [α-32P]- GTP) in transcription buffer at 37°C for 30 min. The excess amounts of ApU, nucleotides and RNAPs were removed by washing the paused TECs with transcription buffer 3 times via magnetic bead pull down. Subsequently, Mfd was introduced and incubated for 5 min before transcription was resumed by addition of NTPs and 1 mM dATP, and then the reaction was quenched with 25 mM EDTA at a specified time point. All bulk transcription reactions were performed at room temperature (23.3°C), which is comparable to the single-molecule condition used in this study. The concentrations of Mfd and NTPs are indicated in Figure S5B. Transcripts were analyzed on 8% polyacrylamide sequencing gels and imaged with PhosphorImager (Typhoon).
Mfd ATPase assay – Related to Figure S7C
ATPase assays were carried out using an ATP/NADH-coupled ATPase assay at 37°C in a 100 μL reaction volume containing 40 nM wild-type or variant Mfd to measure ATP hydrolysis rates of Mfd. Assays were carried out in a buffer (40 mM HEPES pH 8.0, 100 mM NaCl, 5 mM KCl, 10 mM MgCl2, 4% glycerol (v/v), 2 mM DTT) containing 0.024 units/μL pyruvate kinase, 0.036 units/μL lactate dehydrogenase, 5.0 mM phosphoenolpyruvate, and 2.0 mM β-nicotinamide adenine dinucleotide (NADH). The reactions were started by the addition of ATP to a final concentration of 4.0 mM, and the absorbance at 340 nm was measured every 30 s for 1 h in a Cytation 3 cell-based multi-mode microplate reader (BioTek). Triplicate measurements were performed, and the linear decrease in absorbance was used to calculate the rate of NADH oxidation (which is equal to that of ATP hydrolysis) using the molar extinction coefficient for NADH of 6.22 mM−1 cm−1. Rates of phosphate release were corrected for spontaneous, non-enzymatic breakdown using a no Mfd control.
Mfd DNA binding assay – Related to Figure S7C
Mfd binding to dsDNA in the presence of Adenosine 5′-[γ-thio]triphosphate (ATPγS) was measured using a fluorescence anisotropy assay. A 40-base pair dsDNA fragment was generated by annealing two HPLC purified complimentary oligonucleotides, one of which contained a HEX fluorophore at the 5′ end (Table S3). 150 μL of fluorescently labeled DNA (10 nM) in buffer (20 mM HEPES pH 7.5, 50 mM NaCl, and 2 mM β-mercaptoethanol) was titrated with increasing concentrations of wild-type or variant Mfd in the presence of 2.0 mM ATPγS. After each addition of Mfd, the reaction was equilibrated for 5 min at 25°C before measurements were recorded. Fluorescence anisotropy was measured at 555 nm using a Fluoromax-4 spectrofluorometer (Horiba). Measurements were recorded in triplicate and Kd values were calculated using the following equation and nonlinear regression methods in the Graphpad Prism software package. ΔA is the change in anisotropy, ΔAT is the total anisotropy change, ET is the total Mfd concentration at each point in the titration, DT is the total DNA concentration, and Kd is the equilibrium dissociation constant.
Optical trapping techniques
This work requires the use of three different unzipping assays (Table S1): 1) unzipping tracker, 2) unzipping staller, and 3) unzipping mapper. These assays were implemented on two different optical trapping setups.
A surface-based optical trapping setup was used in experiments shown in Figures 1, 2B–D, 3, and 6. This optical trap was similar to that previously described (Brower-Toland et al., 2002) and was used to unzip a single DNA molecule using 489 nm diameter polystyrene beads by moving the microscope coverslip horizontally away from an optical trap (Figure S1A). These experiments started by tethering the two arms of a Y-structured DNA template between the surface of a microscope coverslip via a dioxygenin (dig) and anti-dig connection and a 489 nm bead via a biotin and streptavidin connection (Figure S1B). In order to ensure that each bead only tethered a single DNA molecule, we introduced a low concentration of DNA molecules (typically ~ 5 pM) into the sample chamber followed by incubation with 2 pM of beads. This yielded a surface tether density of one tether per 200–300 μm2.
A dual optical trap in combination with a multi-channel flow cell was used for experiments shown in Figures 2A, 4, and 5. The overall dual trap design was previously described (Inman et al., 2014) but with the addition of a custom multi-channel laminar flow cell. The channels of the flow cell were fed with syringes that were all driven by a single syringe pump set to 1.5 μL/min via blunt syringe tips in Tygon tubing. We estimate that this resulted in a fluid velocity of 200 μm/s at the trapping region. These experiments started by tethering the two arms of a Y-structured DNA molecules between two optically trapped 792 nm beads, via a dig and anti-dig connection for one bead and a biotin and streptavidin connection for the second bead (Figures S2A and S4). To prepare for the formation of such a DNA tether, we preincubated 30–40 pM DNA with 6 pM of streptavidin-coated beads for 30 min on ice and diluted this mixture by 250 times. To start the tether formation, we first trapped an anti-dioxygenin bead in the steered (moveable) trap, moved to a channel containing the streptavidin coated-beads (preincubated with DNA constructs), and then used the fixed (stationary) trap to trap a streptavidin coated-bead. Both beads were moved into a channel free of beads and then brought into close proximity repeatedly using an automated ‘fishing’ algorithm until a tether was formed between them (Landry et al., 2009). This configuration provided one order of magnitude improvement in trap stability (average drift rate ~ 0.02–0.03 bp/s) over time compared with surface based optical trapping techniques.
To determine if each DNA tether was a result of single DNA molecule, we performed control experiments to stretch the DNA molecules and confirmed that the mechanical properties for the DNA tethers were consistent with those of single DNA molecules.
DNA tethers suspended by an optical trap have finite lifetimes due to a combination of photo-damage induced by the trapping laser and the applied force (Landry et al., 2009). This places a time limit on the maximum duration of the experiments. Under the experimental conditions used, the mean tether lifetime was measured to be 75 s, with 30% of the tethers lasting over 100 s. These values are comparable to, or better than, those reported in the literature (Candelli et al., 2013; Landry et al., 2009). This lifetime was sufficiently long for experiments of Figures 1, 2, 3, and 6, but placed an upper limit on the overall measurement duration of the stalling experiments of Figures 4 and 5.
All optical trapping measurements were performed in a temperature-controlled room at 23.3°C. However, the temperature increased slightly to 25°C owing to local laser trap heating (Peterman et al., 2003). All reactions were also carried out at the room temperature of 23.3°C unless otherwise stated.
Single molecule experimental procedures
Below we provide detailed experimental procedures for the eight single-molecule experiments performed in this study.
Unzipping tracker for Mfd translocation – Related to Figures 1 and S1C
In the surface-based unzipping tracking experiment (Figures 1 and S1A; Table S1), after tethering the arms of a Y-structured DNA molecule containing a 4.4-kb DNA trunk to the surface of a coverslip in the sample chamber (Table S4), we introduced 1 μM wtMfd premixed with 2 mM ATP in a transcription buffer containing 25 mM Tris-Cl pH 8.0, 100 mM KCl, 4 mM MgCl2, 1 mM DTT, 3% (v/v) glycerol and 0.15 mg/mL acetylated BSA and sealed the channel entrance and exit with silicone high vacuum grease. We used an optical trap to unzip the DNA at a loading rate of 16 pN/s. If we did not detect any bound protein, we then moved to the next tether. If we detected a bound protein during unzipping (force rise above ~ 18 pN), we immediately switched the operation to a force clamp mode, where the laser intensity and bead position were held fixed while the coverslip was moved horizontally to maintain the unzipping force at ~ 18 pN. Thus Mfd translocation was tracked under a constant force until its final dissociation.
Direct evidence for Mfd release from a TEC – Related to Figures 2A, S2A, and S2B
The experiment was carried out with a dual optical trap that was combined with a multichannel laminar flow cell which partitioned different buffers using flow and allowed for fine control of the start of each experiment (Figure S2A). Since this experiment required continuous flow through of Mfd, to minimize Mfd consumption, we pre-loaded Mfd onto a stalled Mfd by mixing 0.4 μM Mfd with 5 μM ATPγS because Mfd has an increased affinity to TEC in the presence of ATPγS (Figure 6C). Using this method, we were able to obtain a preloading efficiency of 36 ± 7% with minimal non-specific binding of Mfd to DNA (N = 18 out of 50 traces, Figures S2B and S2C, see more information below). This preloading method, in comparison to that used for Figures 2B–D and Figure S6, required Mfd to be present at an order of magnitude lower concentration.
During an experiment, each of the two arms of a Y-structured DNA molecule was held in a separate optical trap. To prepare for this experiment, a TEC stalled by nucleotide starvation at +20 position was located on the 1.1-kb trunk with its transcription direction away from the DNA fork (also see “DNA templates and protein preparations” above and Table S4). 30–35 pM of these DNA-TEC complexes were then mixed with 6 pM streptavidin-coated 792-nm polystyrene beads to allow DNA attachment to the streptavidin bead. After ~30 minutes of incubation on ice, the mixture was diluted by ~ 250 times in the transcription buffer and 0.4 μM Mfd and 5 μM ATPγS were added to allow Mfd preloading onto TEC. This mixture was then introduced to channel 2. During a measurement, we started the experiment by trapping an antidig-coated 792-nm polystyrene bead in one trap in channel 1. This trapped bead was then moved to channel 2 where a second trap was turned on to capture a streptavidin-coated bead. The two beads were subsequently transported to an adjacent flow channel containing the transcription buffer supplemented with 1 mM NTPs but no Mfd to resume transcription and Mfd translocation (channel 3). In this channel, we quickly tethered the two arms of the DNA molecule between the two beads and unzipped the DNA at a speed of 200 nm/s. The NTP chasing time, Δt, was recorded from the moment the DNA entered the NTP channel to the detection of a bound protein.
To measure the preloading efficiency of Mfd to stalled TEC, we conducted experiments in a manner identical to the chasing experiment described above but replaced 1 mM NTPs in channel 3 by 1 mM ATPγS to ‘lock’ down Mfd to a TEC (Figure 6C). The preloading efficiency was calculated from the fraction of traces with additional force rise (indicative of Mfd binding) upstream of the TEC (Figure S2B; also see Figure 6A).
Mfd catching up to a stalled TEC – Related to Figures 2B–D and S3C
The two arms of a Y-structured DNA molecule with a 4.4-kb DNA trunk containing a TEC stalled via nucleotide depletion at +20 (see “DNA templates and protein preparations” and Table S4) were tethered between a glass coverslip of a microscope sample chamber and a bead to be held in an optical trap. Mfd was introduced into the sample chamber at 6 μM for 8 minutes in order to pre-load Mfd onto the stalled TEC, and we found the preloading efficiency to be ~ 55% (Figure S3C). The solution was replaced by 1 mM NTPs for a defined time ( Δt ) by flushing the sample chamber with a 5X chamber volume of 1 mM NTPs in the transcription buffer. This step removed free Mfd molecules, resumed the transcription, and allowed Mfd to translocate. We then flushed the sample chamber with a 20X chamber volume of 2 mM ATP in the transcription buffer and waited for 8 minutes. This buffer exchange stalled transcription but allowed Mfd to continue translocation. Finally, we flushed the sample chamber with a 20X chamber volume of 2 mM ATPγS in the transcription to stop Mfd activity before DNA was unzipped to check for any bound proteins.
In order to determine if the buffer replacement with 1 mM NTPs was able to effectively remove free Mfd from the sample chamber, we performed a control experiment which was identical to that described in Figure 2B for Δt = 0 except that no NTPs were included during the transcription resumption step. If there were residual free Mfd after this step, Mfd would be able to displace the stalled TEC at +20 during the subsequent step when 1 mM ATP was introduced, and TEC removal could be detected by the final unzipping mapping step. We found no detectable TEC removal, consistent with minimal presence of free Mfd in solution after the buffer exchange (Figure 2C). In addition, Figure 2C itself provides strong evidence for the lack of free Mfd after the NTP buffer exchange. The fraction of traces without a TEC strongly depends on the distance between the two stalls and approaches that of no-Mfd baseline at long distances. These features are not consistent any substantial role of free Mfd in the observed TEC removal. Taken together, we conclude that TEC removal observed in Figure 2C was a result of Mfd preloaded onto the TEC.
Mfd catching up to a TEC at an ops pause site – Related to Figures 3 and S3D
We modified the original pRL574 plasmid by site-directed mutagenesis to introduce the rfaQ ops sequence GGCGGTAGCGTG downstream of the +20 position. Two plasmids were generated with the conserved 3′ thymine residue T of the ops site, the major pause site (Artsimovitch and Landick, 2000), located 27 bp and 276 bp from +20 position respectively (Figure 3; Table S4). The plasmid sequences were verified by DNA sequencing.
Single-molecule experiments were conducted following the scheme described in Figure 3A. After the two arms of a Y-structured DNA molecule with a 4-kb DNA trunk containing a TEC stalled at +20 and an ops pause (see “DNA templates and protein preparations” and Table S4) were tethered between a glass coverslip and a 489 nm polystyrene bead, we introduced 6 μM Mfd into the sample chamber and incubated the protein for 8 minutes, allowing Mfd to preload onto TEC. We then transferred the sample chamber into a heat incubator with temperature preset to 37°C and quickly flushed the sample chamber with 15X chamber volume of 100 μM NTPs and 2 mM dATP in the transcription buffer pre-warmed to 37°C and incubated for Δt seconds. This flushing step removed free Mfd, resumed transcription, and allowed Mfd translocation. Mfd could potentially catch up with a TEC paused at the ops site and facilitate TEC pause escape. The chasing time Δt for each template was chosen to allow for the majority of the TECs to transcribe to or pass the ops pause (Figures 3D and 3E) so that TEC escape from the ops site might be more easily analyzed. Transcription was finally quenched by flushing the sample chamber with 30X chamber volume of a buffer without NTPs. The location of TEC after NTP chasing was determined by the DNA unzipping mapper using a loading rate of 16 pN/s.
TEC backtracking at the ops site was determined by measuring the distance between the peak location of TEC pausing at or near the ops site in the probability density function and the expected peak location of TEC at the ops site without backtracking (Figures S3D). The latter was estimated based on the previous finding that the force rise is located at ~2 bp downstream of the TEC’s active site (Jin et al., 2010).
Stalling a TEC – Related to Figures 4 and 5
Due to the fast rate of transcription and the single round of transcription in the experiment, the TEC stalling measurements placed a stringent requirement on the measurement time window. To fulfill this task, a dual optical trap was combined with a multi-channel flow cell which partitioned the flow cell into four different channels containing different buffers using laminar flows (Figure S4A). First, the first trap (trap 1) was used to capture an antidig-coated polystyrene bead (792 nm diameter) in channel 1. Trap 1 was then repositioned into channel 2, which contained streptavidin coated polystyrene beads (also 792 nm) attached to a Y-structure DNA molecule with a stalled TEC on a 2-kb Y-structure trunk with the direction of transcription towards the DNA fork (see “DNA templates and protein preparation” and Table S4). A second trap (trap 2) was turned on to capture such a bead. Both traps were moved to channel 3 to form a DNA dumbbell. The DNA trunk was then unzipped until the DNA fork was ~ 200 bp from the paused TEC. With the DNA extension held constant, the DNA dumbbell was repositioned into channel 4 which contained 1 mM NTPs in the transcription buffer and allowed transcription to resume. If a TEC progressed to the DNA fork and rewound the DNA, the unzipping force would increase.
For experiments with Mfd (Figures 5 and S5A), we included 1 μM Mfd and 1 mM NTPs in channel 4 (Figure S4A) and performed the experiment in a manner identical to that without Mfd. For information on the tether lifetime and optical trapping stability in our dual-trap configuration, please see the “Optical trapping technique” section.
Mfd’s recruitment rate to a stalled TEC – Related to Figures 5 and S4B
We measured the rate of Mfd recruitment to a stalled TEC. Here, we first stalled TEC at +20 via nucleotide depletion on a 2kb DNA trunk of Y-structured template and tethered the DNA arms between the surface of a sample chamber and a bead (Table S4). We then introduced a concentration of Mfd specified in Figure S4B and 2 mM ATP in the transcription buffer into the flow cell, and immediately proceeded to DNA unzipping mapper to check whether the TEC on a DNA tether was removed. The fraction of traces with TEC removal versus time was fit using an exponential function to obtain the characteristic time of TEC removal (Figure S4B, left). The TEC removal rate was determined based on this characteristic time at different Mfd concentrations (Figure S4B, right). We use the TEC removal rate as an estimate of the Mfd recruitment rate to a TEC.
Stalling a Mfd-RNAP complex – Related to Figure S6
Using the unzipping staller assay (Table S1), we measured the maximum force a translocating Mfd or an Mfd with a non-elongating RNAP can generate in a buffer condition identical to the tracking experiment of Mfd in Figure 1.
In the stalling experiment of Mfd on DNA, we first tethered a Y-structured DNA with a 2-kb DNA trunk between a coverslip and a bead in a typical sample chamber (Table S4). We then introduced into the sample chamber 1 μM Mfd and 2 mM ATP in the transcription buffer. The stalling experiment of Mfd translocating with a non-elongating RNAP uses a similar configuration except that the 2-kb DNA trunk contained a TEC stalled at +20 with the transcription direction towards the DNA fork (see “DNA templates and protein preparations” and Table S4). The TEC was incubated with 6 μM Mfd (without adding any nucleotides) for 8 minutes. We then washed the sample chamber with 15 sample volumes of the transcription buffer with 2 mM ATP but without any Mfd. This step should remove free Mfd from the sample chamber and allow Mfd to disrupt the TEC and further translocate downstream with the non-elongating RNAP.
In both experiments, the unzipping staller method (Table S1) was employed to measure the maximum force that Mfd or Mfd-RNAP complex can generate. In the first step, we unzipped DNA tethers with a loading rate of 16 pN/s to detect bound proteins. When the unzipping force increased above 20 pN, indicative of a bound protein, the unzipping staller mode was activated, holding the DNA extension constant. When the motor moved towards the DNA fork, the DNA fork was rezipped and the unzipping force increased until motor dissociation from DNA. The Mfd-RNAP stalling experiment was also carried out with a dual-trap within a laminar flow cell and yielded in similar results (data not shown).
Mfd-TEC mapping – Related to Figures 6, S7A, and S7B
Please refer to Table S1 for a general scheme of the DNA unzipping mapping experiment. Here, we tethered the two arms of a Y-structured DNA between a coverslip and a bead in a typical sample chamber. The trunk of the Y-structured template was 1.1-kb DNA and contained a TEC stalled at +20 position (see “DNA templates and protein preparations” and Table S4). We then flushed the sample chamber with a solution containing Mfd with and without ATPγS (see Main text and figure legends for concentrations of Mfd and ATPγS). To map the protein-DNA interactions at high resolution (Hall et al., 2009), the loading rate was set at 8 pN/s, which is lower than the typical rate (16 pN/s) used for quick mapping.
To determine the dissociation constant Kd of the wild type Mfd as well as MfdR685A and MfdN817A for binding to TEC (Figure 5C), measurements were made by unzipping from upstream of the TEC. Kd was calculated from the fraction of traces (pb) with detectable Mfd binding signatures: Kd = [Mfd] · (1/pb−1). Concentration of Mfd was 0.02 μM and 6 μM for wtMfd, 0.5 μM and 15 μM for MfdR685A, and 0.2 μM and 6 μM for MfdN817A, in the presence or in the absence of ATPγS, respectively. The means and the SEMs of Kd were determined assuming a binomial distribution in the fraction of bound protein. Total number of traces: 274, 364, and 648 for wtMfd, MfdR685A, and MfdN817A respectively in the absence of ATPγS; 222, 209, and 262 for wtMfd, MfdR685A, and MfdN817A respectively in 2 mM ATPγS.
QUANTIFICATION AND STATISTICAL ANALYSIS
All the data were obtained from at least three independent replicates. Statistical details of individual experiments, including number of traces and SD or SEM values, can be found in the manuscript text, Method Details sections, figure legends, and figures themselves. Additional details of data analysis are described below.
Data acquisition and data conversion
Data were acquired at 10 kHz, and converted into force and DNA extension as previously described (Johnson et al., 2007). Elasticity parameters for dsDNA and ssDNA, which are necessary for data conversion, were obtained from the force-extension curves of dsDNA and ssDNA (Hall et al., 2009).
DNA alignment algorithm
To improve the precision and accuracy in the unzipping data, we performed a cross-correlation optimization to align each experimental force versus number of base pairs unzipped curve with the corresponding theoretical curve using regions immediately preceding and following the protein disruption (Hall et al., 2009) using custom-software written in MATLAB.
Pause detection algorithm
To detect pauses in translocation of motor proteins, we employed a pause detection algorithm as previously described (Adelman et al., 2002; Bai et al., 2007; Shundrovsky et al., 2004). Data was low-pass filtered using a 2nd-order Savitzky-Golay filter with a time constant of 1 s. The instantaneous velocity was obtained by performing a linear fit to the number of base pairs unwound over time. A pause is defined if the unzipping fork dwell-time is at least 0.2 s at a given base pair location. The pause-free velocity was obtained by removing data points in the pausing regions from the translocation regions.
DATA AND SOFTWARE AVAILABILITY
For custom programs and scripts used in this study, please contact Dr. Michelle Wang (mwang@physics.cornell.edu).
Supplementary Material
(A) Schematic outlining steps involved in sample chamber preparation for a surface based optical trapping assay.
(B) Experimental configuration of unzipping tracker when used to track Mfd translocation.
(C) All experiments were carried out under a constant unzipping force of 18 pN. Histograms of distance and time traveled at 2 mM ATP are shown for traces presented in Figure 1B. Fitting parameters were deduced from a maximum likelihood method (MLM) for an exponential distribution. Smooth curves are best fits to exponential functions, with the fit parameters listed in Table S2.
(A) Related to Figure 2A – Experimental configuration. The experiment was carried it out with a dual optical trap that was combined with a multi-channel laminar flow cell which partitioned different buffers using flow and allowed for fine control of the start of each experimental step. See STAR Methods for more information.
(B) Related to Figure 2A – Control experiments to map the locations of Mfd and TEC before transcription resumption. We carried out a control experiment with and without preloading Mfd onto a TEC at + 20, prior to transcription resumption. Shown are two representative traces: TEC without and with a bound Mfd (also see Figure 6). Mfd binding to the TEC resulted in a force rise upstream of the TEC.
(C) Related to Figures 2A and 2B – Control experiment to determine the speed of TEC without Mfd. To determine the location of a TEC after at a given transcription time Δt, we carried out a control experiment in a manner identical to that of Figure 2B, except without Mfd. We found that ~ 90% of TECs were able to immediately resume transcription. A small number of TECs moved with lower speeds, possibly due to intrinsic pausing sites on the DNA template or late escape from the +20. For each time point Δt, the distribution of the elongating fraction was fit with a Gaussian function. The mean and the standard deviation of the Gaussian were used to determine the speed and its uncertainty (green shaded areas). N = 166, 185, 167 for Δt = 0, 10 s, and 70 s chasing times, respectively.
(A) Related to Figures 2B–D – Control experiment to determine the translocation speed of Mfd associated with a non-elongating RNAP. Here TECs stalled at +20 were incubated with 6 μM Mfd for 8 minutes and were subsequently washed with 2 mM ATP to remove free Mfd and allow Mfd translocation. After a specified time, 2 mM ATPγS was introduced to stop Mfd translocation and the DNA was unzipped to check for any bound proteins. The left panel shows example traces at different incubation times. The right panel is a summary of the data. Error bars indicate SEMs (N = 116, 20, 17, 45, 25 traces for 0, 2, 5, 8, and 10 minutes of ATP chasing, respectively). Data are linearly fit, yielding a speed of 2.8 ± 1.0 bp/s.
(B) Related to Figure 2C – An example trace consistent with no TEC but Mfd complexed with a non-elongating RNAP. In a fraction of traces, no bound protein was detected at or near the transcribed region (around the 2nd stall position, green vertical dashed line). However, a force peak was detected ~ 900 bp downstream of the second stalled site only when Mfd was present with TEC. The location of this peak is consistent with translocation from the second stalled site at ~ 2 bp/s in 8 minutes as would be expected for Mfd complexed with a non-elongating RNAP.
(C) Related to Figure 2D – A summary of different types of traces. Three types of traces were observed upon transcription resumption: TEC was located at the expected positions (TEC), TEC was removed from the DNA (no TEC), and TEC was disrupted and Mfd translocated with a non-elongating RNAP (no TEC, Mfd/RNAP). Error bars indicate SDs with the total number of traces indicated for each condition. For Δt = 0 s, ~ 55% of TECs were disrupted (the combined fractions of ‘no TEC’ and ‘no TEC, Mfd/RNAP’ in the top left panel), providing a measure of the efficiency Mfd loading onto the initially stalled TEC in Figure 2B and the first data point for (+) Mfd in Figure 2D. In addition, these data show the two types of no TEC traces were partitioned as a function of Δt (or d ): no TEC traces (RNAP dissociation) dominated as the transcript size increased.
(D) Related to Figure 3 – TEC backtracking at the ops site. Shown is a zoomed-in region of Figure 3E near the ops pause site without Mfd. The black dashed line indicates the expected location without backtracking (Inman et al., 2014; Jin et al., 2010). The green dashed line indicates the peak of the detected location, which was shifted about 5 bp upstream, providing an estimate of the distance of backtracking at the ops site.
(A) A dual optical trap was combined with a multi-channel flow cell which partitioned the flow cell into four different channels containing different buffers using laminar flows (see STAR Methods for more details). For experiments with Mfd, the same experimental procedure was employed except that channel 4 contained 1 μM Mfd in addition to NTPs.
(B) Control experiment to determine Mfd’s recruitment rate a stalled TEC. Mfd at a given concentration and 2 mM ATP were introduced to a sample chamber containing surface tethered DNA fork templates, each with a TEC stalled at +20 located on the trunk (STAR Methods). Each DNA tether was unzipped to determine if its TEC was removed by Mfd. The fraction of TECs removed was plotted versus reaction time and fit to an exponential function to obtain the removal rate. This TEC removal rate showed a linear relation as a function of Mfd concentration. This relation provides an estimate for the Mfd recruitment time of Figure 5: at 1 μM of Mfd, the recruitment time was around 50 s.
(A) Mfd slightly reduces transient pauses of an elongating TEC. Pause durations and pause densities were calculated for the portions of the traces with force below 18 pN, which is below the stall force of TEC alone, where TEC is fully active. Error bars are SEMs. The numbers of pause durations and distances between adjacent pauses identified in the TEC alone experiment are 75 and 61, respectively. The numbers of pause durations and distances between adjacent pauses identified in the TEC-Mfd experiment are 47 and 35, respectively.
(B) Mfd’s impact on transcription pausing and termination. TECs stalled at +20 via nucleotide depletion and were radioactively labelled by incorporation of [α-32P]-GTP. The paused TECs were incubated with 1 μM (top) or 0.1 μM (bottom) Mfd for 5 min. Transcription was resumed in the presence of either 20 μM NTPs, 1 mM dATP, and 1 μM Mfd (top) or 50 μM NTPs, 1 mM dATP, and 0.1 μM Mfd (bottom), and subsequently quenched after 30, 60, 120, and 240 s. Transcripts were assayed on 8% denaturing PAGE gels.
We separately investigated how Mfd or Mfd with a non-elongating RNAP may work against a DNA fork to provide further evidence for transcription termination at the end of traces in Figure 5. Using the unzipping staller assay, we measured the maximum force a translocating Mfd can generate in a condition identical to the tracking experiment of Mfd in Figure 1. For Mfd translocating with a non-transcribing RNAP, we first incubated TEC stalled at +20 with 6 μM Mfd for 8 minutes, and then washed the sample chamber with a copious amount of 2 mM ATP to remove unbound Mfd and to allow Mfd to disrupt the TEC and further translocate downstream with the non-transcribing RNAP (also see STAR Methods). The unzipping staller method (Table S1) was employed to measure the maximum force that the Mfd-RNAP complex can generate.
(A) Example traces of Mfd and Mfd-RNAP translocation during stalling at a DNA fork. Mfd moved at a faster overall speed with little pausing but had limited processivity and force generation capacity. Mfd with a non-elongating RNAP moved at a slower overall speed with frequent pausing/slipping but had a much greater processivity and force generation capacity.
(B) Pause-free speeds of Mfd and Mfd with a non-transcribing RNAP. The pause-free speeds, calculated using a pause-detecting algorithm (see STAR Methods), showed comparable values for Mfd alone and Mfd with a non-transcribing RNAP. Thus when Mfd is associated with a RNAP, it pauses much more often, resulting in slower overall speed. Total number of traces used in the two histograms: 41 and 37 for Mfd alone and Mfd with non-elongating RNAP, respectively.
(C) Maximum force generated by Mfd alone or Mfd with a non-transcribing RNAP. The Mfd-RNAP complex is capable of generating a much greater stall force than Mfd alone. In the context of TCR, such an enhanced motor capacity of Mfd ensures that transcription is terminated and RNAP is moved off the lesion site. In the context of a transcription-replication collision, this enhancement allows Mfd to bring a TEC out of a backtracked state and subsequently terminate transcription once the fork becomes insurmountable.
(A) Representative unzipping traces of TEC at +20 position in the presence of wtMfd without any nucleotide (N = 22). Arrow indicates the direction of unzipping. Cartoon on top with vertical dashed lines indicate overall structure of the complex with strong measured interactions.
(B) Same as (A) except 2 mM ATPγS was included in the unzipping buffer (N=20).
(C) Biochemical characterizations of wtMfd, MfdR685A, and MfdN817A. Left panel: ATP hydrolysis rates were measured using an ATP/NADH-coupled ATPase assay at 37°C with 40 nM wt or variant Mfd and 4 mM ATP. The results show that the two variants have ATPase activities comparable to wtMfd. Right panel: Mfd binding to dsDNA in the presence of ATPγS was measured using a fluorescence anisotropy assay. Mfd was titrated into a mixture of 10 nM terminally HEX-labeled 40mer dsDNA (Table S3) and 2 mM ATPγS. MfdR685A and MfdN817A show greatly reduced affinity to DNA, compared with wtMfd. Titrations were performed in triplicate and error bars are often smaller than symbols.
(D) Schematic of a possible model for Mfd translocation.
Highlights.
Mfd translocates independently at a speed of 7 bp/s with a processivity of 200 bp.
Mfd can locate a stalled/paused TEC via a release and catch-up mechanism.
A TEC on its own can backtrack for over 100 bp when elongating against a DNA fork.
Depending on load, Mfd either rescues a backtracked TEC or terminates transcription.
Acknowledgments
We thank members of the Wang laboratory for critical reading of the manuscript. We especially thank Robert Forties, Jie Ma, Jing Jin, Shanna Moore, Jim Baker, Jessica Killian, and Victoria Dorich for helpful discussion and technical assistance. This work was supported by the Howard Hughes Medical Institute, the National Science Foundation grant (MCB-0820293 to M.D.W), the National Institutes of Health (T32GM008267 to M.D.W; R01GM121975 and P20GM103430 to A.M.D), and an UTRA fellowship from Brown University (to S.P.). This research is based upon work conducted at the Rhode Island Genomics and the NSF/EPSCoR Proteomics Facility, supported in part by the National Science Foundation EPSCoR Grant No. 1004057.
Footnotes
AUTHOR CONTRIBUTIONS
T.T.L., Y.Y., C.T., J.T.I., M.L., and J.L. performed single molecule and bulk transcription experiments and analyzed the data. J.W.R. provided wtMfd for initial experiments. A.M.D, M.M.S. and S.P. purified and bulk characterized wtMfd and Mfd mutants. R.M.F. purified RNAP. T.T.L., Y.Y., J.T.I., and M.D.W. designed the single molecule assays. A.M.D. and M.M.S. designed the bulk Mfd assays. T.T.L. and M.D.W. drafted the manuscript. All authors contributed to revisions of the manuscript and intellectual discussions.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adebali O, Chiou YY, Hu JC, Sancar A, Selby CP. Genome-wide transcription-coupled repair in Escherichia coli is mediated by the Mfd translocase. P Natl Acad Sci USA. 2017;114:E2116–E2125. doi: 10.1073/pnas.1700230114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelman K, La Porta A, Santangelo TJ, Lis JT, Roberts JW, Wang MD. Single molecule analysis of RNA polymerase elongation reveals uniform kinetic behavior. P Natl Acad Sci USA. 2002;99:13538–13543. doi: 10.1073/pnas.212358999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artsimovitch I, Landick R. Pausing by bacterial RNA polymerase is mediated by mechanistically distinct classes of signals. P Natl Acad Sci USA. 2000;97:7090–7095. doi: 10.1073/pnas.97.13.7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L, Fulbright RM, Wang MD. Mechanochemical kinetics of transcription elongation. Phys Rev Lett. 2007:98. doi: 10.1103/PhysRevLett.98.068103. [DOI] [PubMed] [Google Scholar]
- Bai L, Shundrovsky A, Wang MD. Sequence-dependent kinetic model for transcription elongation by RNA polymerase. J Mol Biol. 2004;344:335–349. doi: 10.1016/j.jmb.2004.08.107. [DOI] [PubMed] [Google Scholar]
- Bai L, Shundrovsky A, Wang MD. Kinetic Modeling of Transcription Elongation. In: Henri Buc TS, editor. RNA Polymerases as Molecular Motors. Chapter 9. Royal Society of Chemistry; 2009. pp. 263–280. [Google Scholar]
- Bai L, Wang MD. Comparison of pause predictions of two sequence-dependent transcription models. J Stat Mech-Theory E. 2010 doi: 10.1088/1742-5468/2010/12/P12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belitsky BR, Sonenshein AL. Roadblock repression of transcription by Bacillus subtilis CodY. J Mol Biol. 2011;411:729–743. doi: 10.1016/j.jmb.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan LD, Forties RA, Patel SS, Wang MD. DNA looping mediates nucleosome transfer. Nat Commun. 2016:7. doi: 10.1038/ncomms13337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower-Toland BD, Smith CL, Yeh RC, Lis JT, Peterson CL, Wang MD. Mechanical disruption of individual nucleosomes reveals a reversible multistage release of DNA. P Natl Acad Sci USA. 2002;99:1960–1965. doi: 10.1073/pnas.022638399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueckner F, Hennecke U, Carell T, Cramer P. CPD damage recognition by transcribing RNA polymerase II. Science. 2007;315:859–862. doi: 10.1126/science.1135400. [DOI] [PubMed] [Google Scholar]
- Candelli A, Hoekstra TP, Farge G, Gross P, Peterman EJ, Wuite GJ. A toolbox for generating single-stranded DNA in optical tweezers experiments. Biopolymers. 2013;99:611–620. doi: 10.1002/bip.22225. [DOI] [PubMed] [Google Scholar]
- Cheung ACM, Cramer P. Structural basis of RNA polymerase II backtracking, arrest and reactivation. Nature. 2011;471:249–253. doi: 10.1038/nature09785. [DOI] [PubMed] [Google Scholar]
- Deaconescu AM, Artsimovitch I, Grigorieff N. Interplay of DNA repair with transcription: from structures to mechanisms. Trends in Biochemical Sciences. 2012;37:543–552. doi: 10.1016/j.tibs.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaconescu AM, Chambers AL, Smith AJ, Nickels BE, Hochschild A, Savery NJ, Darst SA. Structural basis for bacterial transcription-coupled DNA repair. Cell. 2006;124:507–520. doi: 10.1016/j.cell.2005.11.045. [DOI] [PubMed] [Google Scholar]
- Deaconescu AM, Darst SA. Crystallization and preliminary structure determination of Escherichia coli Mfd, the transcription-repair coupling factor. Acta Crystallogr F. 2005;61:1062–1064. doi: 10.1107/S1744309105035876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaconescu AM, Suhanovsky MM. From Mfd to TRCF and Back Again-A Perspective on Bacterial Transcription-coupled Nucleotide Excision Repair. Photochem Photobiol. 2017;93:268–279. doi: 10.1111/php.12661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, Shatalin K, Epshtein V, Gottesman ME, Nudler E. Linking RNA polymerase backtracking to genome instability in E. coli. Cell. 2011;146:533–543. doi: 10.1016/j.cell.2011.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epshtein V, Kamarthapu V, McGary K, Svetlov V, Ueberheide B, Proshkin S, Mironov A, Nudler E. UvrD facilitates DNA repair by pulling RNA polymerase backwards. Nature. 2014;505:372-+. doi: 10.1038/nature12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epshtein V, Nudler E. Cooperation between RNA polymerase molecules in transcription elongation. Science. 2003;300:801–805. doi: 10.1126/science.1083219. [DOI] [PubMed] [Google Scholar]
- Erie DA, Hajiseyedjavadi O, Young MC, von Hippel PH. Multiple RNA polymerase conformations and GreA: control of the fidelity of transcription. Science. 1993;262:867–873. doi: 10.1126/science.8235608. [DOI] [PubMed] [Google Scholar]
- Forget AL, Kowalczykowski SC. Single-molecule imaging of DNA pairing by RecA reveals a three-dimensional homology search. Nature. 2012;482:423–U178. doi: 10.1038/nature10782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Muse T, Aguilera A. Transcription-replication conflicts: how they occur and how they are resolved. Nat Rev Mol Cell Bio. 2016;17:553–563. doi: 10.1038/nrm.2016.88. [DOI] [PubMed] [Google Scholar]
- Haines NM, Kim YI, Smith AJ, Savery NJ. Stalled transcription complexes promote DNA repair at a distance. Proc Natl Acad Sci U S A. 2014;111:4037–4042. doi: 10.1073/pnas.1322350111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MA, Shundrovsky A, Bai L, Fulbright RM, Lis JT, Wang MD. High-resolution dynamic mapping of histone-DNA interactions in a nucleosome. Nat Struct Mol Biol. 2009;16:124–129. doi: 10.1038/nsmb.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman JT, Smith BY, Hall MA, Forties RA, Jin J, Sethna JP, Wang MD. DNA Y structure: a versatile, multidimensional single molecule assay. Nano Lett. 2014;14:6475–6480. doi: 10.1021/nl503009d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Bai L, Johnson DS, Fulbright RM, Kireeva ML, Kashlev M, Wang MD. Synergistic action of RNA polymerases in overcoming the nucleosomal barrier. Nat Struct Mol Biol. 2010;17:745–752. doi: 10.1038/nsmb.1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Bai L, Smith BY, Patel SS, Wang MD. Single-molecule studies reveal dynamics of DNA unwinding by the ring-shaped T7 helicase. Cell. 2007;129:1299–1309. doi: 10.1016/j.cell.2007.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kad NM, Van Houten B. Dynamics of lesion processing by bacterial nucleotide excision repair proteins. Prog Mol Biol Transl Sci. 2012;110:1–24. doi: 10.1016/B978-0-12-387665-2.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpp S, Hwa T. Growth-rate-dependent partitioning of RNA polymerases in bacteria. Proc Natl Acad Sci U S A. 2008;105:20245–20250. doi: 10.1073/pnas.0804953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch SJ, Wang MD. Dynamic force spectroscopy of protein-DNA interactions by unzipping DNA. Phys Rev Lett. 2003;91:028103. doi: 10.1103/PhysRevLett.91.028103. [DOI] [PubMed] [Google Scholar]
- Komissarova N, Kashlev M. RNA polymerase switches between inactivated and activated states by translocating back and forth along the DNA and the RNA. Journal of Biological Chemistry. 1997a;272:15329–15338. doi: 10.1074/jbc.272.24.15329. [DOI] [PubMed] [Google Scholar]
- Komissarova N, Kashlev M. Transcriptional arrest: Escherichia coli RNA polymerase translocates backward, leaving the 3′ end of the RNA intact and extruded. Proc Natl Acad Sci U S A. 1997b;94:1755–1760. doi: 10.1073/pnas.94.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzheva N, Mustaev A, Kozlov M, Malhotra A, Nikiforov V, Goldfarb A, Darst SA. A structural model of transcription elongation. Science. 2000;289:619–625. doi: 10.1126/science.289.5479.619. [DOI] [PubMed] [Google Scholar]
- Landry MP, McCall PM, Qi Z, Chemla YR. Characterization of photoactivated singlet oxygen damage in single-molecule optical trap experiments. Biophys J. 2009;97:2128–2136. doi: 10.1016/j.bpj.2009.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Hada A, Sen P, Olufemi L, Hall MA, Smith BY, Forth S, McKnight JN, Patel A, Bowman GD, et al. Dynamic regulation of transcription factors by nucleosome remodeling. Elife. 2015:4. doi: 10.7554/eLife.06249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Wang MD. Unzipping single DNA molecules to study nucleosome structure and dynamics. Methods Enzymol. 2012;513:29–58. doi: 10.1016/B978-0-12-391938-0.00002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Bai L, Wang MD. Transcription Under Torsion. Science. 2013;340:1580–1583. doi: 10.1126/science.1235441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustaev A, Vitiello CL, Gottesman ME. Probing the structure of Nun transcription arrest factor bound to RNA polymerase. Proc Natl Acad Sci U S A. 2016;113:8693–8698. doi: 10.1073/pnas.1601056113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman KC, Abbondanzieri EA, Landick R, Gelles J, Block SM. Ubiquitous transcriptional pausing is independent of RNA polymerase backtracking. Cell. 2003;115:437–447. doi: 10.1016/s0092-8674(03)00845-6. [DOI] [PubMed] [Google Scholar]
- Nudler E. Transcription elongation: Structural basis and mechanisms. Journal of Molecular Biology. 1999;288:1–12. doi: 10.1006/jmbi.1999.2641. [DOI] [PubMed] [Google Scholar]
- Nudler E, Mustaev A, Lukhtanov E, Goldfarb A. The RNA-DNA hybrid maintains the register of transcription by preventing backtracking of RNA polymerase. Cell. 1997;89:33–41. doi: 10.1016/s0092-8674(00)80180-4. [DOI] [PubMed] [Google Scholar]
- Opalka N, Chlenov M, Chacon P, Rice WJ, Wriggers W, Darst SA. Structure and function of the transcription elongation factor GreB bound to bacterial RNA polymerase. Cell. 2003;114:335–345. doi: 10.1016/s0092-8674(03)00600-7. [DOI] [PubMed] [Google Scholar]
- Park JS, Marr MT, Roberts JW. E. coli Transcription repair coupling factor (Mfd protein) rescues arrested complexes by promoting forward translocation. Cell. 2002;109:757–767. doi: 10.1016/s0092-8674(02)00769-9. [DOI] [PubMed] [Google Scholar]
- Peterman EJ, Gittes F, Schmidt CF. Laser-induced heating in optical traps. Biophys J. 2003;84:1308–1316. doi: 10.1016/S0006-3495(03)74946-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz RT, O’Donnell M. Direct restart of a replication fork stalled by a head-on RNA polymerase. Science. 2010;327:590–592. doi: 10.1126/science.1179595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proshkin S, Rahmouni AR, Mironov A, Nudler E. Cooperation Between Translating Ribosomes and RNA Polymerase in Transcription Elongation. Science. 2010;328:504–508. doi: 10.1126/science.1184939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proshkin SA, Mironov AS. Stalled RNA Polymerase Is a Target of the Mfd Factor. Mol Biol+ 2016;50:332–335. doi: 10.7868/S0026898416020191. [DOI] [PubMed] [Google Scholar]
- Roberts J, Park JS. Mfd, the bacterial transcription repair coupling factor: translocation, repair and termination. Curr Opin Microbiol. 2004;7:120–125. doi: 10.1016/j.mib.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Roberts JW, Shankar S, Filter JJ. RNA polymerase elongation factors. Annu Rev Microbiol. 2008;62:211–233. doi: 10.1146/annurev.micro.61.080706.093422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DA, Gelles J, Sheetz MP, Landick R. Transcription by Single Molecules of Rna- Polymerase Observed by Light-Microscopy. Nature. 1991;352:444–448. doi: 10.1038/352444a0. [DOI] [PubMed] [Google Scholar]
- Selby CP. Mfd Protein and Transcription-Repair Coupling in Escherichia coli. Photochem Photobiol. 2017;93:280–295. doi: 10.1111/php.12675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby CP, Sancar A. Molecular mechanism of transcription-repair coupling. Science. 1993a;260:53–58. doi: 10.1126/science.8465200. [DOI] [PubMed] [Google Scholar]
- Selby CP, Sancar A. Transcription-repair coupling and mutation frequency decline. J Bacteriol. 1993b;175:7509–7514. doi: 10.1128/jb.175.23.7509-7514.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby CP, Sancar A. Mechanisms of transcription-repair coupling and mutation frequency decline. Microbiol Rev. 1994;58:317–329. doi: 10.1128/mr.58.3.317-329.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby CP, Sancar A. Structure and function of transcription-repair coupling factor. I. Structural domains and binding properties. J Biol Chem. 1995;270:4882–4889. doi: 10.1074/jbc.270.9.4882. [DOI] [PubMed] [Google Scholar]
- Shepherd N, Dennis P, Bremer H. Cytoplasmic RNA polymerase in Escherichia coli. Journal of Bacteriology. 2001;183:2527–2534. doi: 10.1128/JB.183.8.2527-2534.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shundrovsky A, Santangelo TJ, Roberts JW, Wang MD. A single-molecule technique to study sequence-dependent transcription pausing. Biophysical Journal. 2004;87:3945–3953. doi: 10.1529/biophysj.104.044081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shundrovsky A, Smith CL, Lis JT, Peterson CL, Wang MD. Probing SWI/SNF remodeling of the nucleosome by unzipping single DNA molecules. Nat Struct Mol Biol. 2006;13:549–554. doi: 10.1038/nsmb1102. [DOI] [PubMed] [Google Scholar]
- Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- Smith AJ, Pernstich C, Savery NJ. Multipartite control of the DNA translocase, Mfd. Nucleic Acids Research. 2012;40:10408–10416. doi: 10.1093/nar/gks775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJ, Savery NJ. Effects of the bacterial transcription-repair coupling factor during transcription of DNA containing non-bulky lesions. DNA Repair. 2008;7:1670–1679. doi: 10.1016/j.dnarep.2008.06.020. [DOI] [PubMed] [Google Scholar]
- Smith AJ, Szczelkun MD, Savery NJ. Controlling the motor activity of a transcription-repair coupling factor: autoinhibition and the role of RNA polymerase. Nucleic Acids Res. 2007;35:1802–1811. doi: 10.1093/nar/gkm019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetone LE, Friedman LJ, Osborne ML, Ravi H, Kyzer S, Stumper SK, Mooney RA, Landick R, Gelles J. Dynamics of GreB-RNA polymerase interaction allow a proofreading accessory protein to patrol for transcription complexes needing rescue. P Natl Acad Sci USA. 2017;114:E1081–E1090. doi: 10.1073/pnas.1616525114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalieckas JM, Wray LV, Jr, Ferson AE, Fisher SH. Transcription-repair coupling factor is involved in carbon catabolite repression of the Bacillus subtilis hut and gnt operons. Mol Microbiol. 1998;27:1031–1038. doi: 10.1046/j.1365-2958.1998.00751.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Schematic outlining steps involved in sample chamber preparation for a surface based optical trapping assay.
(B) Experimental configuration of unzipping tracker when used to track Mfd translocation.
(C) All experiments were carried out under a constant unzipping force of 18 pN. Histograms of distance and time traveled at 2 mM ATP are shown for traces presented in Figure 1B. Fitting parameters were deduced from a maximum likelihood method (MLM) for an exponential distribution. Smooth curves are best fits to exponential functions, with the fit parameters listed in Table S2.
(A) Related to Figure 2A – Experimental configuration. The experiment was carried it out with a dual optical trap that was combined with a multi-channel laminar flow cell which partitioned different buffers using flow and allowed for fine control of the start of each experimental step. See STAR Methods for more information.
(B) Related to Figure 2A – Control experiments to map the locations of Mfd and TEC before transcription resumption. We carried out a control experiment with and without preloading Mfd onto a TEC at + 20, prior to transcription resumption. Shown are two representative traces: TEC without and with a bound Mfd (also see Figure 6). Mfd binding to the TEC resulted in a force rise upstream of the TEC.
(C) Related to Figures 2A and 2B – Control experiment to determine the speed of TEC without Mfd. To determine the location of a TEC after at a given transcription time Δt, we carried out a control experiment in a manner identical to that of Figure 2B, except without Mfd. We found that ~ 90% of TECs were able to immediately resume transcription. A small number of TECs moved with lower speeds, possibly due to intrinsic pausing sites on the DNA template or late escape from the +20. For each time point Δt, the distribution of the elongating fraction was fit with a Gaussian function. The mean and the standard deviation of the Gaussian were used to determine the speed and its uncertainty (green shaded areas). N = 166, 185, 167 for Δt = 0, 10 s, and 70 s chasing times, respectively.
(A) Related to Figures 2B–D – Control experiment to determine the translocation speed of Mfd associated with a non-elongating RNAP. Here TECs stalled at +20 were incubated with 6 μM Mfd for 8 minutes and were subsequently washed with 2 mM ATP to remove free Mfd and allow Mfd translocation. After a specified time, 2 mM ATPγS was introduced to stop Mfd translocation and the DNA was unzipped to check for any bound proteins. The left panel shows example traces at different incubation times. The right panel is a summary of the data. Error bars indicate SEMs (N = 116, 20, 17, 45, 25 traces for 0, 2, 5, 8, and 10 minutes of ATP chasing, respectively). Data are linearly fit, yielding a speed of 2.8 ± 1.0 bp/s.
(B) Related to Figure 2C – An example trace consistent with no TEC but Mfd complexed with a non-elongating RNAP. In a fraction of traces, no bound protein was detected at or near the transcribed region (around the 2nd stall position, green vertical dashed line). However, a force peak was detected ~ 900 bp downstream of the second stalled site only when Mfd was present with TEC. The location of this peak is consistent with translocation from the second stalled site at ~ 2 bp/s in 8 minutes as would be expected for Mfd complexed with a non-elongating RNAP.
(C) Related to Figure 2D – A summary of different types of traces. Three types of traces were observed upon transcription resumption: TEC was located at the expected positions (TEC), TEC was removed from the DNA (no TEC), and TEC was disrupted and Mfd translocated with a non-elongating RNAP (no TEC, Mfd/RNAP). Error bars indicate SDs with the total number of traces indicated for each condition. For Δt = 0 s, ~ 55% of TECs were disrupted (the combined fractions of ‘no TEC’ and ‘no TEC, Mfd/RNAP’ in the top left panel), providing a measure of the efficiency Mfd loading onto the initially stalled TEC in Figure 2B and the first data point for (+) Mfd in Figure 2D. In addition, these data show the two types of no TEC traces were partitioned as a function of Δt (or d ): no TEC traces (RNAP dissociation) dominated as the transcript size increased.
(D) Related to Figure 3 – TEC backtracking at the ops site. Shown is a zoomed-in region of Figure 3E near the ops pause site without Mfd. The black dashed line indicates the expected location without backtracking (Inman et al., 2014; Jin et al., 2010). The green dashed line indicates the peak of the detected location, which was shifted about 5 bp upstream, providing an estimate of the distance of backtracking at the ops site.
(A) A dual optical trap was combined with a multi-channel flow cell which partitioned the flow cell into four different channels containing different buffers using laminar flows (see STAR Methods for more details). For experiments with Mfd, the same experimental procedure was employed except that channel 4 contained 1 μM Mfd in addition to NTPs.
(B) Control experiment to determine Mfd’s recruitment rate a stalled TEC. Mfd at a given concentration and 2 mM ATP were introduced to a sample chamber containing surface tethered DNA fork templates, each with a TEC stalled at +20 located on the trunk (STAR Methods). Each DNA tether was unzipped to determine if its TEC was removed by Mfd. The fraction of TECs removed was plotted versus reaction time and fit to an exponential function to obtain the removal rate. This TEC removal rate showed a linear relation as a function of Mfd concentration. This relation provides an estimate for the Mfd recruitment time of Figure 5: at 1 μM of Mfd, the recruitment time was around 50 s.
(A) Mfd slightly reduces transient pauses of an elongating TEC. Pause durations and pause densities were calculated for the portions of the traces with force below 18 pN, which is below the stall force of TEC alone, where TEC is fully active. Error bars are SEMs. The numbers of pause durations and distances between adjacent pauses identified in the TEC alone experiment are 75 and 61, respectively. The numbers of pause durations and distances between adjacent pauses identified in the TEC-Mfd experiment are 47 and 35, respectively.
(B) Mfd’s impact on transcription pausing and termination. TECs stalled at +20 via nucleotide depletion and were radioactively labelled by incorporation of [α-32P]-GTP. The paused TECs were incubated with 1 μM (top) or 0.1 μM (bottom) Mfd for 5 min. Transcription was resumed in the presence of either 20 μM NTPs, 1 mM dATP, and 1 μM Mfd (top) or 50 μM NTPs, 1 mM dATP, and 0.1 μM Mfd (bottom), and subsequently quenched after 30, 60, 120, and 240 s. Transcripts were assayed on 8% denaturing PAGE gels.
We separately investigated how Mfd or Mfd with a non-elongating RNAP may work against a DNA fork to provide further evidence for transcription termination at the end of traces in Figure 5. Using the unzipping staller assay, we measured the maximum force a translocating Mfd can generate in a condition identical to the tracking experiment of Mfd in Figure 1. For Mfd translocating with a non-transcribing RNAP, we first incubated TEC stalled at +20 with 6 μM Mfd for 8 minutes, and then washed the sample chamber with a copious amount of 2 mM ATP to remove unbound Mfd and to allow Mfd to disrupt the TEC and further translocate downstream with the non-transcribing RNAP (also see STAR Methods). The unzipping staller method (Table S1) was employed to measure the maximum force that the Mfd-RNAP complex can generate.
(A) Example traces of Mfd and Mfd-RNAP translocation during stalling at a DNA fork. Mfd moved at a faster overall speed with little pausing but had limited processivity and force generation capacity. Mfd with a non-elongating RNAP moved at a slower overall speed with frequent pausing/slipping but had a much greater processivity and force generation capacity.
(B) Pause-free speeds of Mfd and Mfd with a non-transcribing RNAP. The pause-free speeds, calculated using a pause-detecting algorithm (see STAR Methods), showed comparable values for Mfd alone and Mfd with a non-transcribing RNAP. Thus when Mfd is associated with a RNAP, it pauses much more often, resulting in slower overall speed. Total number of traces used in the two histograms: 41 and 37 for Mfd alone and Mfd with non-elongating RNAP, respectively.
(C) Maximum force generated by Mfd alone or Mfd with a non-transcribing RNAP. The Mfd-RNAP complex is capable of generating a much greater stall force than Mfd alone. In the context of TCR, such an enhanced motor capacity of Mfd ensures that transcription is terminated and RNAP is moved off the lesion site. In the context of a transcription-replication collision, this enhancement allows Mfd to bring a TEC out of a backtracked state and subsequently terminate transcription once the fork becomes insurmountable.
(A) Representative unzipping traces of TEC at +20 position in the presence of wtMfd without any nucleotide (N = 22). Arrow indicates the direction of unzipping. Cartoon on top with vertical dashed lines indicate overall structure of the complex with strong measured interactions.
(B) Same as (A) except 2 mM ATPγS was included in the unzipping buffer (N=20).
(C) Biochemical characterizations of wtMfd, MfdR685A, and MfdN817A. Left panel: ATP hydrolysis rates were measured using an ATP/NADH-coupled ATPase assay at 37°C with 40 nM wt or variant Mfd and 4 mM ATP. The results show that the two variants have ATPase activities comparable to wtMfd. Right panel: Mfd binding to dsDNA in the presence of ATPγS was measured using a fluorescence anisotropy assay. Mfd was titrated into a mixture of 10 nM terminally HEX-labeled 40mer dsDNA (Table S3) and 2 mM ATPγS. MfdR685A and MfdN817A show greatly reduced affinity to DNA, compared with wtMfd. Titrations were performed in triplicate and error bars are often smaller than symbols.
(D) Schematic of a possible model for Mfd translocation.