

Abstract
Autophagy and inflammasome complex assembly are physiological processes that control homeostasis, inflammation, and immunity. Autophagy is a ubiquitous pathway that degrades cytosolic macromolecules or organelles, as well as intracellular pathogens. Inflammasomes are multi-protein complexes that assemble in the cytosol of cells upon detection of pathogen- or danger-associated molecular patterns. A critical outcome of inflammasome assembly is the activation of the serine protease caspase-1, which activates the pro-inflammatory cytokine precursors pro-IL-1β and pro-IL-18. Studies on chronic inflammatory diseases, heart diseases, Alzheimer's disease, and multiple sclerosis revealed that autophagy and inflammasomes intersect and regulate each other. In the context of infectious diseases, however, less is known about the interplay between autophagy and inflammasome assembly, although it is becoming evident that pathogens have evolved multiple strategies to inhibit and/or subvert these pathways and to take advantage of their intricate crosstalk. An improved appreciation of these pathways and their subversion by diverse pathogens is expected to help in the design of anti-infective therapeutic interventions.
Keywords: inflammasome, autophagy, bacterial and viral infection, inflammatory disease, therapeutic target
Introduction
Autophagy and inflammasome complex activation are physiological pathways that control tissue homeostasis and immunity. Like any other biological processes, if dysregulated, they can exert detrimental effects as exemplified by numerous inflammatory diseases and cancer [1,2]. It is now appreciated that autophagy and the inflammasomes intersect and regulate each other to maintain tissue homeostasis [3]. The goal of this review is to summarize current knowledge about the inflammasome complex and autophagy and their crosstalk in the context of infection.
Inflammasomes
Inflammasomes are multi-protein complexes that assemble in the cytosol of infected or damaged cells (recently reviewed in Refs. [4,5]). Assembly of these molecular platforms can be initiated by various signals, but they all converge toward the activation of the cysteine protease caspase-1, which ultimately orchestrates the elimination of pathogens and damaged cells. The assembly of an inflammasome is initiated upon engagement of sensor proteins (pattern recognition receptors, or PRRs) with pathogen-associated molecular patterns (PAMPs), or with damage-associated molecular patterns (DAMPs). These sensors are classified based on structural characteristics and include cytosolic nucleotide-binding oligomerization domain (NOD or NBD or NACHT) and leucine-rich repeat (LRR) receptors (NLRs), absent in melanoma 2 (AIM2)-like receptors (ALRs), and pyrin (Fig. 1). NLRs are further subdivided into NLRP and NLRC depending on the presence of a pyrin (PYD) or a caspase activation and recruitment (CARD) domains. The sensor recruits and nucleates an adaptor protein, the apoptosis-associated speck-like protein (ASC) containing CARD and PYD domains. Consequently, the association of pro-caspase-1 with the sensor-ASC complex promotes its auto-activation into a cysteine protease [6]. The assembly of an inflammasome relies on homotypic protein–protein interactions CARD–CARD or PYD–PYD domains located on sensors, the ASC adaptor, and pro-caspase-1. The adaptor ASC amplifies the signal through oligomerization into PYD filaments and recruitment of multiple pro-caspase-1 molecules via interaction with CARD. Different microbial products may engage distinct receptors giving rise to the activation of one or several canonical inflammasomes, among which the most studied are NLRP1, NLRP3, NLRC4, AIM 2, and pyrin. The NLRP3 inflammasome is also a master sensor of DAMPs, such as reactive oxygen species, adenosine 5′-triphosphate (ATP), mitochondrial DNA (mtDNA), and K+ efflux. DAMPS form during tissue damage due to infection, or to non-infectious conditions such as disorders of autoimmunity, although the modalities of NLRP3 activation remain unsettled. The canonical inflammasomes may differ by their sensing receptors but converge toward the activation of caspase-1. Murine caspase-11 (caspase-4 and -5 in humans) forms a non-canonical inflammasome consisting of the lipid A portion of LPS interacting with the CARD domain of pro-caspase-11. Activated caspase-11 then controls caspase-1 activation via promoting the NLRP3 inflammasome and also exerts caspase-1-independent activities such as regulation of vesicular trafficking [7,8]. Once activated, caspase-1 processes several substrates, regulating numerous pathways. Caspase-1 major substrates include the pro-inflammatory cytokine precursors pro-IL-1β and pro-IL-18, which initial transcription and translation generally involve activation of Toll-like receptors (TLRs) by a process described as “priming.” Additional substrates include the pro-pyroptotic cell death molecule gasdermin, and molecules promoting fusion of bacteria-containing vesicles with lysosomes, tissue repair, membrane biogenesis, and metabolism [9–12]. Since the discovery that the common mouse used in caspase-1 research was indeed a double capase-1−/−/caspase-11−/− knockout, the community is revisiting claimed downstream effectors of these caspases [13].
Fig. 1.
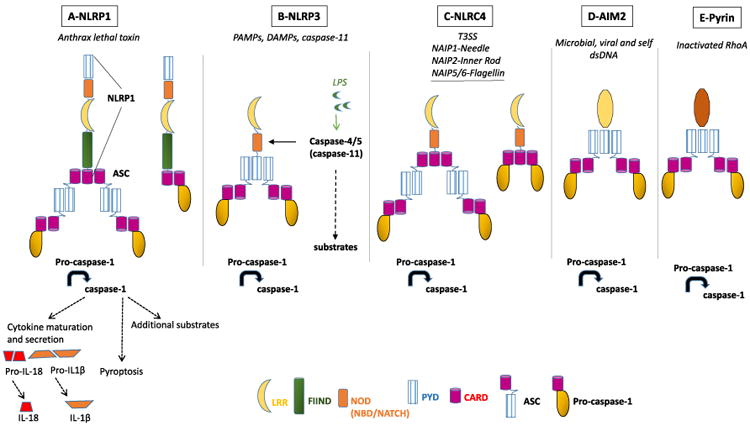
Canonical and non-canonical inflammasomes. (A) NLRP1 is activated by proteolysis consecutive to cell intoxication by the anthrax lethal toxin leading to the recruitment of the adaptor ASC and pro-caspase-1. Alternatively, activated NLRP1 can directly recruit pro-caspase-1 through its CARD domain. The drawing represents human NLRP1, and murine NLRP1 lacks the CARD domain. Note that NLRP1 carries both PYD and CARD domains and therefore can also recruit ASC via its PYD, as presented in panel B. (B) NLRP3 is activated in response to cell stress and infection, leading to the recruitment of ASC and pro-caspase-1. The non-canonical inflammasome consists of LPS-activated caspase-4/5 (in human), or caspase-11 (in mice), which then promotes NLRP3 activation. (C) NLRC4 inflammasome is activated by T3SS or by flagellin produced by Gram-negative bacteria. Activated NLRC4 can successively recruit ASC and pro-caspase-1, or can recruit directly pro-caspase-1. (D) AIM2 inflammasome is activated upon binding to double-stranded DNA. (E) Pyrin inflammasome is activated by bacterial toxins that inactivate the RhoA GTPase. Note: direct recruitment of pro-caspase-1 is not represented in panels D and E, but can proceed via CARD domain as presented in panels A and C.
Excessive activation and dysregulation of inflammasomes are associated with inflammatory and autoimmune diseases such as colitis, diabetes, multiple sclerosis, Alzheimer's disease, sepsis, and cancer. Therefore, tight regulation of inflammasome activation by the autophagy machinery is critical for tissue homeostasis and health.
Autophagy
Autophagy is a ubiquitous and versatile homeostatic pathway that disposes of intracellular macromolecules or organelles as well as infectious agents [14, 15]. Autophagy may be nonselective when cytosolic components are randomly captured to generate nutrients under starvation conditions, or can be selective when damaged organelles or intracellular pathogens must be targeted for their autophagic elimination. Autophagy involves over 30 proteins encoded by autophagy-related genes, or Atgs, including the highly studied class III phosphatidylinositol 3-kinase VPS34, the ubiquitin-like LC3 (Atg8 in yeast), and beclin-1 (Atg6 in yeast) proteins. This machinery sequentially recruits membranes to sequester a cargo into a double-membrane vesicle called the phagophore, which matures into an autophagosome (Fig. 2). Maturation of the autophagosome into an autophagolysosome then allows for the digestion and recycling of the cargo. Various forms of selective autophagy have been described based on the nature of the intracellular cargo to be degraded. For example, mitophagy clears damaged or senescent mitochondria and xenophagy eliminates intracellular pathogens whether they are cytosolic or reside within in a vacuole. Cargos can be targeted for selective autophagic degradation by both ubiquitin-dependent and -independent mechanisms [16]. The ubiquitinated cargo is recognized by adaptor proteins [p62/SQSTM1, NBR1, optineurin, nuclear domain 10 protein 52 (NDP52), TAX1BP1], which associate them with the forming phagophore via interaction with the autophagy protein LC3. Cytosolic receptors such as galectin-8 can also target some cargos for autophagy independently of prior ubiquiti-nation. In contrast to the assembly of inflammasomes, which occurs only when provoked, autophagy always proceeds at a constant low level in every eukaryotic cell to maintain a steady supply of nutrients. Basal autophagy also plays a housekeeping role by continuously removing damaged organelles, protein aggregates, and long-lived proteins. The resulting nutrients are recycled to participate in de novo macromolecule synthesis or energy production. Autophagy can be dramatically induced in response to various environmental stresses, such as starvation, hypoxia, oxidative stress, radiation, and infection [17,18]. The induction of autophagy is accompanied by increased synthesis of the autophagy proteins, increased autophagic degradation, or increased autophagic flux (defined as a measure of autophagic degradation activity [19]).
Fig. 2.
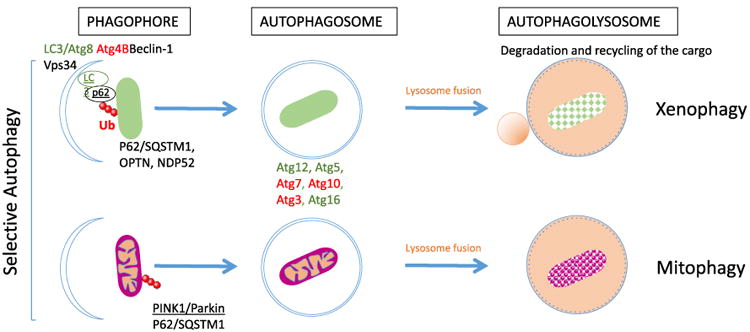
Selective autophagy. In selective autophagy, some molecular determinants or ubiquitin chains on the cargo are recognized by several autophagy receptors including p62 (or SQSTM1), which link the cargo to the autophagy machinery. PINK1/Parkin is a mitochondria-specific ubiquitination system, which can be recognized by autophagy receptors. Some Atgs are important to initiate formation of the phagophore as, for example, Beclin-1, Vps34, and LC3, whereas, additional Atgs are sequentially recruited for elongation and closure of the autophagosome including Atg3, Atg5, Atg7, Atg10, Atg12, and Atg16 (only a few molecules are presented here, reviewed in Ref. [15]). The autophagosome fuses with lysosomes to generate the autophagolysosome for degradation and recycling of the cargo.
Autophagy is at the crossroad of multiple homeostatic pathways and is therefore critical for a diversity of physiological and pathological processes. Relevant to this review, autophagy is an innate immune mechanism that kills pathogens and controls inflammation. Xenophagy decreases pathogen burden and participates in the processing of antigens for activation of the adaptive immunity. On the other hand, mitophagy not only maintains normal production of energy and metabolism but also prevents inflammation by limiting both the release of mtDNA and the production of reactive oxygen species by damaged mitochondria, which would otherwise be potent activators of NLRP3 [20,21]. Therefore, by eliminating damaged mitochondria and other organelles, autophagy limits the release of pro-inflammatory cytokines.
Crosstalk between autophagy and the inflammasome machineries
Autophagy and inflammasomes are functionally interconnected; they both control cell homeostatic processes such as metabolism, energy production, maintenance of organelles, and critically control inflammation and the clearance of pathogens. It is therefore important to understand how autophagy and inflammasome machineries intersect and regulate each other.
TLRs, NLRs, and retinoic acid-inducible gene I (RIG-I)-like receptors have all been shown to activate autophagy by directly inducing posttranslational modifications of autophagy proteins, by increasing their synthesis, or by directing the localization of autophagy proteins [22]. In particular, recent studies have identified that NOD2-mediated induction of autophagy in intestinal epithelial cells was necessary to hold bacterial burden, thereby limiting inflammation [23]. NOD2 was identified as one of the inflammatory bowel disease susceptibility genes involved in autophagy [24,25]. NOD2 stimulation in dendritic cells also induces autophagy to promote bacterial clearance and antigen presentation [26]. In this process, NOD2 in conjunction with NOD1 recruits ATG16L1 to the site of bacterial entry to target the forming phagosome to the autophagy machinery [27]. Interestingly, p62 forms a complex with NOD2 and increases NOD2 signaling by stabilizing NOD2 oligomerization [28]. In the intestines, the NLRP6 inflammasome is important for mucus secretion by globlet cells. In NLRP6- or capase-1-deficient mice, mucus secretion and autophagy are impaired and mice are more susceptible to enteric infection. Notably, mucus secretion requires autophagy, thus linking inflammasome activity to autophagy in the intestines [29]. Activation of PRRs is also critical for cell priming for inflammasome activation. This priming involves the synthesis of inflammasome components and substrates. LPS is commonly used as a tool for cell priming via TLR4 and MyD88 signaling, increasing the synthesis of pro-IL-1β and other components necessary for the inflammasome pathways.
On the other hand, a number of PRRs and inflammasomes can inhibit autophagy. For example, NLRC4 and NLRP4 negatively regulate autophagy via association with and sequestration of the autophagy protein beclin1 [30]. Another mechanism of inhibition of autophagy involves caspase-1-mediated proteolytic cleavage of autophagy proteins and cytosolic transducers that are involved in autophagy activation. In particular, caspase-1 can degrade beclin 1 and the mitophagy-specific protein parkin [31].
Autophagy can promote inflammasome activity. The autophagosome has been proposed to promote the release of cytokines activated by caspase-1, thereby positively cooperating with the inflammasome. For example, recruitment of the autophagy machinery by the activated AIM2 inflammasome has been proposed to mediate the release of IL-1β via exocytosis of the autophagosome [32]. Thus, IL-1β can be released in an autophagy-dependent manner that requires SNAREs and syntaxins [33]. Additional studies showed that autophagy is required for the release of active IL-1β or IL-18 downstream of other inflammasomes [34]. Autophagy is also involved in the release of HMGB1 and secretory lysosomes [35,36]. However, it is not clear how these substrates are selected for this secretion instead of degradation by autophagy. On the contrary, autophagy can suppress the activation of several inflammasomes by multiple mechanisms [37]. Autophagy can indirectly limit the activation of inflammasomes via removal of old/damaged organelles or pathogens, which decreases the release of PAMPs or DAMPs. In particular, mitochondria play a central role in the autophagy–inflammasome interplay. The capture and degradation of damaged mitochondria limits the accumulation mtDNA and reactive oxygen species. Reciprocally, autophagy genetic deficiencies, or autophagy inhibition by pharmacological compounds increases the activation of inflamma-somes via accumulation of PAMPs, DAMPs, cytokine precursors, and their downstream signaling pathways. The autophagosome may directly sequester and degrade activators, components, or products of inflammasomes. For example, autophagy controls IL-1β production by degrading the cytokine precursor pro-IL-1β [38]. More recently, basal autophagy was also shown to remove monomeric inactive MyD88 and degrade TRIF to limit pro-inflammatory signals mediated by TLR and IL-1R that produce pro-IL-1β and pro-IL-18 [39]. Autophagy can target and destroy AIM2 and NLRP3 in human macrophages, a process thought to limit inflammation [40]. In this process, the autophagy adaptor molecule p62 binds to ubiquitinated ASC or AIM2 and induces their degradation by selective autophagy (Fig. 3). In conclusion, autophagy and inflammasome are parts of complex regulatory networks and can inhibit or activate one another in multiple fashions.
Fig. 3.
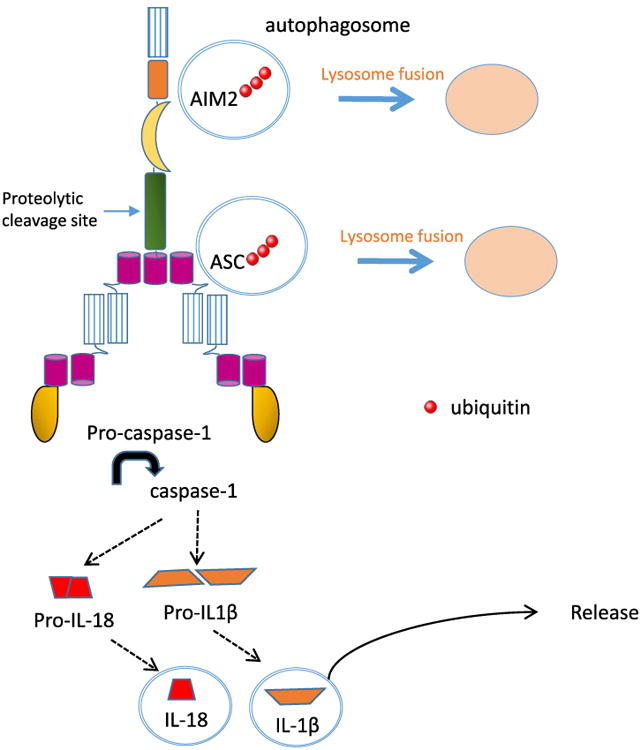
The mechanism by which autophagy controls activation of the inflammasome. Inflammasome sensors such as AIM2 and adaptor molecule ASC are ubiquitinated then targeted for degradation by autophagy. Similarly, the production of pro-IL-1β is regulated by autophagy. On the other hand, active IL-1β and IL-18 may exploit the autophagy machinery for packaging and release to extracellular milieu.
Pathogens differentially interfere with the inflammasomes and autophagy machineries
Here we discuss how several intracellular pathogens including Listeria monocytogenes, Salmonella enterica serovar Typhimurium, Francisella tularencis, Burkholderia cenocepacia, Mycobacterium tuberculosis (M.tb), non-tuberculous mycobacteria, and Influenza virus can modulate the activation of inflammasomes or autophagy machineries. It appears that each pathogen affects and is affected by inflammasomes and autophagy in a very unique mode.
Listeria monocytogenes
L. monocytogenes is a Gram-positive pathogen responsible for the foodborne disease listeriosis [41]. This facultative intracellular pathogen replicates in professional phagocytes and in normally non-phagocytic cells such as epithelial cells [42,43]. Its intracellular life cycle involves the secreted pore-forming toxin listeriolysin O (LLO) and two phospho-lipases C (PI-PLC and PC-PLC) that disrupt the membrane of the phagosome to allow bacterial replication in the cytosol [44]. In the cytosol, the bacterial surface protein ActA promotes the growth of F-actin at one bacterial pole, propelling the bacterium until it forms an extracellular protrusion [43,45]. The bacterium-containing protrusion is then taken up by a neighboring cell into a double-membrane vacuole that is disrupted by the concerted activities of LLO, PI-PLC and PC-PLC to initiate a new cycle of infection [46].
L. monocytogenes infection leads to the activation of caspase-1 as observed using in vitro and in vivo experimental models. In mice, this activation is necessary for early control of L. monocytogenes via production of IL-18 and interferon (IFN)γ. However, caspase-1 appears dispensable for acquisition of sterilizing adaptive immunity and no role for caspase-11 was identified in C57BL/6 mice [47–49]. Using cell culture models, several studies showed that L. monocytogenes activates NLRP3 and AIM2 inflammasomes. Activation of NLRP3 was reported to require LLO-mediated bacterial escape from the phagosome [47,50]. In the cytosol, L. monocytogenes components such as RNA and the secreted protein p60 activate NLRP3 [51,52]. Extracellular L. monocytogenes can also activate NLRP3 independently of host-cell invasion through secretion of LLO. Indeed, the efflux of K+ subsequent to plasma membrane perforation by LLO leads to NLRP3 activation [53]. In addition, bacterial DNA released upon cytosolic lysis of L. monocytogenes activates the AIM2 inflammasome [54,55]. More recently, L. monocytogenes was proposed to activate the NLRP1B inflammasome, via causing energy stress in fibroblasts and macrophages [56]. It is thought that L. monocytogenes maintains a low-level of inflammasome activation in order to establish a successful infection. Hence, an L. monocytogenes strain engineered to activate the NLRC4 inflammasome was substantially attenuated in the murine model of infection [57,58].
Several studies report that L. monocytogenes can be targeted by the autophagy machinery and that autophagy limits its intracellular growth in vivo and in vitro [59–62]. Mice harboring Atg5−/−-deficient monocyte/macrophages and granulocytes had increased L. monocytogenes burden and decreased survival [63]. Also, C57BL/6 irf8−/− mice displayed increased L. monocytogenes burden due to lower levels of autophagy, and it was proposed that the transcription factor IRF8 downstream from IFNγ controls the expression of seven of the autophagy genes during Listeria infection [64]. The autophagy adaptor optineurin is phosphorylated by the TANK binding kinase 1, which enhances its affinity for LC3 and L. monocytogenes degradation by autophagy in HeLa cells in an LLO-dependent fashion [65]. Macro-phage infection models suggested that autophagy can capture L. monocytogenes located in the phagosome, but after bacteria had started to replicate in the cytosol [62]. Furthermore, L. monocytogenes can activate autophagy at an early stage of infection in an LLO-dependent fashion and LLO is sufficient to activate autophagy in macrophages, which is consistent with the idea that the autophagy machinery recognizes damaged phagosomes [66]. However, an absence or low levels of autophagy during L. monocytogenes infection has been reported, unless the bacterial metabolic activity was affected by the antibiotic chloramphenicol [61]. Therefore, in order to survive within host cells, L. monocytogenes actively limits its recognition and destruction by the autophagy machinery. A strategy to avoid autophagy of the L. monocytogenes-containing phagosome is the secretion of phosphatidylinositol-specific phospholipase C (PI-PLC) that counteracts the synthesis of phosphatidylinositol 3-phosphate [59,67]. PI-PLC prevents LC3 lipidation, which is a key step in the formation of the autophagosome [66]. To limit selective autophagy in the cytosol, L. monocytogenes recruits host proteins that prevent ubiquitination of its surface and subsequent recruitment of p62 or NDP52 [68]. ActA recruits the host protein Ena/VASP and the Arp2/3 complex masking the bacterial surface, rather than via inducing bacterial motility, as it was initially thought [69]. This disguise strategy is exploited by a second virulence factor, the surface protein InlK, which recruits a conserved mammalian ribonucleo-protein, the major vault protein (MVP) [70].
Taken together, L. monocytogenes has intrinsic features that can activate autophagy and multiple inflammasomes; however, this pathogen limits activation of these machineries to ensure its intracellular survival. To date, it is unknown if L. monocytogenes exploits or subvert the crosstalk between these two pathways. It would also be of interest to determine if treatments that potentiate autophagy or the inflammasomes facilitate elimination of L. monocytogenes and if this would involve beneficial co-regulation between both pathways.
Salmonella enterica serovar Typhimurium
S. enterica serovar Typhimurium is one of the most studied Gram-negative bacterium that causes gastroenteritis, typhoid fever, and enteric fever. This bacterium resides in the Salmonella containing vacuole within host cells. Two Type Three Secretion Systems (T3SS) play an essential role in mediating uptake and vacuole maturation for formation of the intracellular niche.
Salmonella has proved to be a valuable model system to dissect the different molecular pathways that contribute to inflammasome formation and activation. The necessity for inflammasome activation to control and clear Salmonella has been demonstrated through infection of caspase-1−/−, IL-18−/−, or IL-1β−/− mice, which are much more susceptible to oral infection than wild-type mice and succumb to Salmonella more rapidly with increased bacterial burden in the spleen and mesenteric lymph nodes [71,72]. Only NALP3−/− NLRC4−/− mice, but not NLRC4−/− or NLRP3−/− single knockout mice, were susceptible to infection similar to caspase-1−/−, indicating that NLRP3 and NLRC4 are redundant or overlapping pathways for caspase-1 activation by Salmonella [73]. Critical to the host response to Salmonella is the recognition of ligands that activate these inflammasomes. Flagellin is a ligand for NAIP5 and 6 NLRC4 [9,72,74], the needle and inner rod of the T3SS are NAIP1 and NAIP2 ligands, respectively [55,75–77]. As an inflammasome evasion strategy, Salmonella can repress expression of PAMPs through the PhoP–PhoQ regulatory system. Interestingly, the consequences of inflammasome activation depend on the nature of the activated inflammasome. Activation of NLRP3 by LPS results in the production of IL-1β and IL-18. In contrast, activation of NLRC4 in response to Salmonella T3SS and/or LPS leads to rapid pyroptosis in a gasdermin D-dependent fashion. In the absence of gasdermin D, cells undergo mainly apoptosis instead of pyroptosis [78], which leads to increased infection benefiting Salmonella. In contrast to murine cells, human cells generate IL-1β in a caspase-1-dependent manner but also use caspase-4 for IL-1β release and the induction of cell death [79]. The process of pyroptosis is thought to bypass the beneficial induction of apoptosis by Salmonella [80]. Cytokine production and cell death by pyroptosis both contribute to the control or clearance of Salmonella, yet other mechanisms also exist. It is likely that during Salmonella infection, multiple receptors are engaged in tandem, generating a mixed response that is evident in the in vivo mouse models. In addition, Salmonella that lacks aconitase has attenuated virulence that is dependent on rapid NLRP3 inflammasome activation [81]. Neutrophils can also activate the NLRC4 inflammasome in response to Salmonella infection and make IL-1β but do not undergo pyroptosis (in contrast to macrophages) leading to sustained IL-1β production [82]. The GBP cytosolic proteins are host defense molecules whose expression is activated by the IFNs [83]. GBPs can target the Salmonella-containing vacuole causing its rupture and the release of PAMPs such as LPS leading to activation of the non-canonical NLRP3 [84]. It is important to note that GBPs also activate inflammasomes via multiple mechanisms [85].
Upon infection of epithelial cells, most Salmonella reside in a vacuole. However, due to damage of the vacuole by the T3SS or by host proteins, a significant fraction (around 15%–20%) of cytosolic bacteria or bacteria-containing damaged vacuoles are coated by ubiquitin and associate with LC3-positive nascent autophagosomes [86]. Depletion of a single autophagy receptor leads to hyperproliferation of Salmonella [87]. Three different autophagy receptors, p62 [88], NDP52 [87], and optineurin [89], recognize ubiquitinated Salmonella and target them to autophagosomes. In particular, the autophagy receptor optineurin is phosphorylated by protein kinase TANK binding kinase 1 (TBK1) to promote autophagy of Salmonella [90]. Linking inflammasome to autophagy during Salmonella infection, both processes can be activated by common pathways such as the damage of the Salmonella-containing vacuole by GBPs [22,91].
Francisella tularensis
F. tularensis is a highly infectious, facultative, intracellular, gram-negative bacterium, and is the causative agent of tularemia [92]. F. tularensis has been classified into four subspecies: F. tularensis subsp. tularensis (F. tularensis; Type A; includes Schu S4 strain), F. tularensis subsp. holarctica (F. holarctica; Type B; includes live vaccine strain), F. mediasiatica, and F. tularensis subsp. novicida (F. novicida). Francisella is characterized by its low infectious dose and ability to cause severe illness and death if untreated. The most life-threatening forms of tularemia are particularly associated with Type A infections (Schu S4) regardless of the host species [93], whereas F. holarctica is more virulent for mice than for humans. In contrast, F. novicida is highly virulent for mice but is almost completely avirulent for humans [94]. Of note, avirulent F. novicida can infect human monocytes and induce a more robust inflammatory response as compared to virulent F. tularensis and F. holarctica [95–97]. Macrophage infection by Francisella is characterized by a multifaceted life cycle that is essential for pathogenesis. It begins with recognition of the bacterium at the cell surface via the TLR2 receptor [98,99], followed by uptake by an asymmetrical exuberant pseudopod loop (looping phagocytosis) resulting in bacterium enclosure within a spacious vacuole [100]. Although the Francisella-containing phagosome acquires early endosomal markers, the bacterium arrests phagosome–lysosome fusion and rapidly escapes into the cytosol for extensive replication [101–103].
In the cytosol, intracellular Francisella subspecies can be recognized by intracellular PRRs including AIM2 [104,105], NLRP3 [106,107], and pyrin [108,109], depending on murine or human hosts. It is well established that AIM2 senses Francisella DNA [110], NLRP3 detects a broad range of signals, whereas no specific PAMP is identified for pyrin activation. Pyrin's role is controversial; originally, it was proposed as an anti-inflammatory molecule, but over the last several years, a growing body of evidence has demonstrated that the pyrin inflammasome is pro-inflammatory [111–115]. This leads to controlled active caspase-1 and IL-1β/IL-18 release followed by pyroptosis. At later stages of infection, some intracellular Francisella (in murine but not in human macro-phages) are found in autophagosomes but they are still intact and not degraded [116].
To survive within an intracellular environment, bacteria should either avoid or suppress recognition by inflammasome or autophagy machineries. Schu S4, the most pathogenic strain of Francisella, can actively dampen and subvert the inflammatory response [96], reduce the expression of autophagy-related genes, and compromise relevant signaling pathways in human hosts. In particular, F. tularensis Schu S4, significantly downregulates the expression of autophagy related genes such as BEC1, ATG5, ATG12, ATG16L2, ATG7, ATG4A, and PIK3R1 but increases expression of IFNG and IFNB1 [117,118]. The observed downregulation of autophagy-related genes correlates with Francisella's ability to avoid degradation by autophagy during active cytosolic proliferation. In addition to the suppression of autophagy genes, F. tularensis SchuS4 may actively evade ubiquitination and subsequent recognition by the autophagy adaptors p62/SQSTM1 and NBR1 [119,120]. Interestingly, Francisella's O-antigen plays a protective role against uptake by the autophagy machinery [121]. Pyrin, which senses Francisella, directly interacts with essential autophagy molecules such as beclin1, ULK1, and ATL16L1 [122]. Autophagy proteins bind to pyrin's B-box and coiled-coil domain accompanied by the assembly of an autophagosome [122]. Homotypic interaction between PYD of pyrin, ASC, and other PYD-containing molecules (NLRP3) may bring the inflammasome's components into the autophagosome for degradation. It seems that Francisella modulates both pathways to establish a convenient niche for intracellular survival.
Burkholderia cenocepacia
Burkholderia pseudomallei is a facultative intracellular pathogen and the causative agent of melioidosis [123]. B. cenocepacia is a member of the B. cepacia complex and is frequently isolated from cystic fibrosis patients [124]. Acquisition of B. cenocepacia by cystic fibrosis patients leads to colonization and inflammation with two possible outcomes. B. cenocepacia can lead to moderate infection without severe change in pulmonary status, or severe infection with accelerated pulmonary deterioration accompanied by bacteremia, necrotizing pneumonia, and death. B. cenocepacia survives and may replicate within free-living amoebae [125] as well as in murine and human macrophages [126–131]. B. cenocepacia also survives in epithelial cells [132]. B. cenocepacia survives within a vacuole (phagosome) incystic fibrosis macrophages where the B. cenocepacia-containing vacuole acquires the early endosomal marker EEA1 and Rab5, and then slowly acquires LAMP-1 [126] while acquiring CD63 and Rab7 [133]. B. cenocepacia expresses one T3SS, two type 4 (T4SS-1 and T4SS-2), and one type 6 (T6SS) secretion systems [134–136]. B. cenocepacia employs its secretion systems, especially T6SS, to survive in macrophages. T6SS effectors are delivered into the cytosol where they affect the function of Rho-family GTPases. This disrupts the trafficking of the phagosome and causes a defect in the recruitment of soluble components of the NADPH oxidase to the membrane of the phagosome [128–130,137]. These functional defects, by a yet unidentified mechanism, lead to the activation of the NLRP3 and pyrin inflammasomes and subsequent cell death by pyroptosis and sustained inflammation [137]. B. cenocepacia infection of human mononuclear cells also leads to the production of IL-1β and IL-18 through NLRC4. In mouse macrophages, however, B. cenocepacia employs the NLRP3 inflammasome to induce IL-1β secretion and pyroptosis [138]. LPS is essential and sufficient for the production of IL-1β in macrophages [131].
Autophagy has been implicated in the restriction of B. pseudomallei in macrophages [139]. Electron microscopy analyses of infected cells demonstrated that B. pseudomallei is sequestered in LC3-positive single-membrane phagosomes rather than double-membrane autophagosomes. On the other hand, engulfed B. cenocepacia resides in a vacuole that elicits the characteristic double membranes of typical autophagosomes [128,140]. The B. cenocepacia-containing phagosome also acquires LC3 [129]. However, even in wild-type murine macrophages, the bacteria-containing vacuoles remain in contact with newly formed endosomes and delays fusion with lysosomes [126]. Delay in acidification correlates with delayed recruitment of the phagosomal vacuolar ATPase [137,138]. Recent results have shown that in macrophages derived from cystic fibrosis (F508del CFTR) patients, and in a corresponding mouse model, B. cenocepacia persists in single-membrane vacuoles that lack LC3 and rarely fuse with lysosomes. These infected cells produce higher levels of IL-1β than infected CFTR-normal macrophages do [128]. Interestingly, B. cenocepacia infection leads to the downregulation of the expression of several autophagy-related genes [140]. This effect occurred in both human and mouse macrophages and was mediated through a specific microRNa cluster (Mir17-92) [140].
Mycobacterium tuberculosis
M.tb is the main causative agent of tuberculosis [141]. The main host cell harboring M.tb is the alveolar macrophage) [142]. Several mechanisms can be used by alveolar macrophage to control M.tb including bacterial degradation in the phagolysosomes, the oxidative response, TNF-induced apoptosis, and autophagy (reviewed in Ref. [143]). Studies have also shown that the inflammasome and autophagy contribute to the clearance of M.tb infection. Under some circumstances, M.tb is capable of bypassing all these killing mechanisms by using specific host-cell receptors and trafficking networks to create a safe intracellular niche [120–124]. M.tb has developed strategies to bypass the inflammatory process mediated by the assembly of the inflammasome. Initial reports indicated that M.tb infection does not activate IL-1β production [144]. Mechanistic in vitro and in vivo studies using a mutant strain lacking Zmp1 (Rv0198c), a putative Zn2+ metalloprotease, indicated that this mycobacterial protein is directly implicated in inhibiting caspase-1-dependent activation and secretion of IL-1β [144]. As is the case for non-flagellated Shigella [145], the production of IL-1β was observed via NLRC4-dependent- and ASC-inflammasome activation in macrophages only when using an M.tb zmp1-KO strain, resulting in a better control of the infection through increased acidification of M.tb-containing phagosomes [144]. In this model, the possibility of an alternate succession of NLRC4- and ASC-dependent inflammasomes was also suggested [144] as described in Salmonella infections [6]. Other reports indicate that M.tb is in fact capable of inflammasome activation and release of IL-1β employing the M.tb region of differentiation locus (RD1) encoding ESX-1 type VII secretion system [146–148]. M.tb inflammasome activation depends on the presence of bacterial DNA in the host-cell cytosol engaging AIM2 [149,150]. Further deletion of AIM2 and the observation of IL-1β and IL-18 production uncovered the role of NLRP3-dependent inflammasome system in M.tb infection in myeloid cells [151]. In this case, protein tyrosine kinase Syk is thought to participate in the M.tb-NLRP3-inflammasome activation [152], akin to what is observed for fungal pathogens [153]. The oxidative response generated by M.tb infection also plays a role, where radical oxygen intermediate-dependent NLRP-3 inflammasome activation depends on the generation of cardiolipin at the mitochondrial membrane [154], and generation of nitric oxide negatively regulates NLRP-3-inflammasome activation by directed S-nitrosylation of NLRP-3 [155]. The NLRP-3-mediated activation of the inflammasome involves potassium efflux [151]. However, M.tb is also capable of generating IL-1β independently of NLRP-3 and caspase-1 [151]. Subsequent studies indicate that M.tb is able to differentially engage the inflammasome/IL-1β pathway or the cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS)/cyclic dinucleotide sensor STING/type-I IFN pathway during infection, and confirmed that this was directly dependent on the bacterium levels of EsXA and/or other EsxA/ESX-1 type VII secretion system-associated factors [151, 156–158], where less EsXA drives selective protective production of IL-1β [156]. Furthermore, as nitric oxide, IFN-β has being reported as a host factor induced by M.tb during infection to negatively regulate the NLRP3 inflammasome to bypass inflammation [159,160]. As IFN-β is reported to increase activity of the AIM2 inflammasome [161], induction of IFN-β at lower levels may suppress the NLRP3 inflammasome without activating the AIM2 inflammasome (concept extensively reviewed in Ref. [162]). Nonetheless, mechanisms behind regulation of the NLRP-3 inflammasome during M.tb infection need to be further explored, especially in the context of developing novel host mediated therapies looking at its link with the autophagy cellular process.
M.tb survives in infected host cells by blocking phagosome maturation or escaping from phagosomes to the cytosol [163]. Studies indicate that autophagy could also be subverted by M.tb to reside into an autophagosome that is not acidified [164]. Overall, the autophagy process depends on the activation status of Ser/Thr kinase Tor [165]. When Tor is active, autophagy is suppressed, but when Tor is inactivated, autophagy is promoted. Importantly, phosphatidylinositol 3-phosphate and hVPS34 are involved in phagosome and autophagosome maturation, only with a different autophagy-specific subunit termed Atg6 (or beclin 1) that is essential for autophagy [166]. Induction of autophagy can overcome phagosome maturation block imposed by M.tb [167]. Thus, physiological (nutrient starvation), pharmacological (rapamycin, sodium valproate, vitamin D), or immunological agonists of autophagy (TNF, IFN-α) stimulate mycobacterial phagosome maturation, allowing the host cell to control M.tb infection [163]. Specifically, IFN-γ acts on promoting autophagy via the induction of TNF and the immunity-related GTPase protein IRGM1 [49–51,167,168]. The role of IRGM1 in induction of autophagy has been well characterized in several human polymorphism studies, as well as the potential role of other closely related to autophagy genes such as P2X7, VDR, NOD2, and TLR8 (summarized in Ref. [168]). Other cytokines such as IL-4, IL-10, and IL-13 have been shown to inhibit autophagy via the protein kinase B (Akt), signal transducer and activator of transcription (STAT)-3 and STAT-6 signaling pathways [169].
Recent in vivo studies, using a mouse strain lacking Atg5 expression only in myeloid cells, showed that Atg5 is required to control of M.tb growth [170,171], thus indicating that this protein is important for restriction. However, subsequent in vivo studies looking at other autophagy proteins reported that except for Atg5, other proteins are not implicated in M.tb control. Thus, the role of autophagy in M.tb infection is uncertain. In this instance, several hypotheses can be put forward [172]. It is possible that atg5 could be involved in a yet unrevealed autophagy-independent trafficking network contributing to the control of M.tb intracellular growth like other TAG proteins [173,174]. For instance, Atg5 is thought to be involved in controlling host-cell death via interactions with Bcl-xL and FADD [175,176]. An alternative explaination is the existence of an only atg5-dependent autophagy program [172,177]. Studies have demonstrated that treatment of M tb-infected human macrophages with ATP enhances intracellular killing of mycobacteria [178]. These anti-microbial responses appear to be dependent on both apoptosis and autophagy. Apoptosis of infected macrophages reduces intracellular viability of M.tb [179]. ATP is also an inducer of inflammasome activation and IL-1βsecretion in LPS-stimulated macrophages. The interplay between autophagy and the inflammasome during M.tb infection still needs extensive studies to discern their reciprocal interactions.
Nontuberculous mycobacteria
In contrast to M tb, few studies have examined inflammasome activation in cells infected with non-tuberculous mycobacteria (NTM). Nonetheless, Mycobacterium marinum has been shown to activate the NLRP3 inflammasome via the Esx-1 secretion system. Using both parental and Esx-1 mutant bacteria and NRLP3- and ASC-deficient mice, Esx-1 was found to be necessary for increased caspase-1-dependent NRLP3 activation and increased IL-1β release. NLRP3 activation, however, did not restrict M. marinum growth, suggesting that inflammasome activation was not beneficial for the host [147]. In contrast, inflammasome activation and IL-1β secretion restricted growth by Mycobacterium kansasii in human macrophages [180]. Mycobacterium abscessus also activates NRLP3 in human macrophages via Syk kinase and the C-type lectin, Dectin-1 [181]. NLRP3 or ACS knockdown by RNA interference resulted in a significant reduction of IL-1β and a significant increase of viable M. abscessus in macrophages, suggesting that inflammasome activation is important in host defense with this pathogenic NTM. The diversity of NTM likely contributes to pathogen-specific effects and defining the mechanisms involved in regulating NTM-inflammasome activation requires further exploration.
A recent study showed that approximately 5% of M. marinum were found to be in autophagosomes in the zebrafish model [182]. Ubiquitinated cytosolic M. marinum is sequestered in LC3-negative double-membrane compartments that do not require ATG5 to assemble [183]. In one study, azithromycin appeared to impair autophagic and phagosomal degradation resulting in decreased mycobacterial clearance (including M. abscessus) by preventing lysosomal acidification and intracellular killing [184]. The smooth morphotype of M. abscessus was also recently shown to restrict intraphagosomal acidification resulting in reduced apoptosis and autophagy [185]. However, a detailed understanding of autophagy and/or inflammasome pathways as drivers of intracellular persistence and pathogenesis in NTM is still undefined and crosstalk between the two pathways is unknown. Given the recalcitrance of some NTM to antibiotic therapy, it would be of particular interest to determine if autophagy and/or inflammasome-potentiating treatments aid in the elimination of NTM and the involvement of possible co-regulation between these pathways.
Influenza virus
Influenza viruses are respiratory pathogens in humans that can cause both seasonal epidemics and global pandemics [186]. Distinguishing influenza virus from the bacterial pathogens discussed above, these viruses replicate their genomes in the cell nucleus and rely on host machinery to produce and organize the proteins needed for virus assembly and propagation. Likewise, the molecules detected by the innate immune system in influenza virus and bacterial infections are largely distinct, although similar pathways are activated, including the type I IFN system, inflammasomes, and autophagy, that determine the severity and ultimate outcome of infections.
Type I IFNs are a particularly critical component of the innate immune system for the early control of influenza virus infections, as mice deficient in the receptor for these antiviral cytokines succumb to virus doses that are sub-lethal in wild-type mice [187]. To induce IFNs, influenza virus RNAs possessing 5′-tri-phosphate ends are detected by RIG-I in most cell types [188–190]. This allows RIG-I interaction with mitochondrial antiviral signaling protein (MAVS) at the mitochondria and subsequent signaling that activates specific transcription factors required for induction of IFNs [191–194]. In addition, a minor role for the RNA sensor Melanoma Differentiation-Associated protein 5 (MDA5), which also signals through MAVS, has been reported recently [195], and TLR7 is the primary sensor for influenza virus leading to IFN production specifically in plasmacytoid DCs [190,196]. Double knockout of MAVS and TLR7 thus results in increased mortality during infection due to loss of type I IFN production [197]. In addition to IFNs, DCs and macrophages also secrete IL-1β when infected with influenza virus in vitro, indicating that inflammasome activation also occurs during infection [198,199]. Confirming the importance of this cytokine in vivo, the IL-1R is required for effective development of adaptive immunity during infection in the mouse model [199,200]. NLRP3 and caspase-1 were each found to be required for maximal IL-1β secretion by infected macrophages and DCs [199], and knockout of these molecules in mice results in enhanced lethality from influenza virus infection [201,202]. Linking the type I IFN induction system to inflammasome activation, RIG-I contributes to upregulation of NLRP3 inflammasome components through type I IFN signaling, and remarkably, RIG-I can also form a non-canonical inflammasome complex with ASC and caspase-1 [203]. Further highlighting the importance and connectivity of these pathways, the influenza virus NS1 protein directly binds to RIG-I and antagonizes cellular production of both IFNs and IL-1β [189,204–206]. Thus, the viral sensing pathways that lead to type I IFN and IL-1β secretion are interrelated and are both critically involved in anti-influenza virus immunity.
NLRP3 inflammasome activation during influenza virus infections occurs through at least four distinct and potentially redundant mechanisms that are largely virus specific. First, NLRP3 inflammasomes can be activated by viral ssRNA or dsRNA that are produced in abundance during infection [201]. Second, the matrix protein 2 (M2) of influenza virus, a proton-selective ion channel, can activate the NLRP3 inflammasome in a manner that is dependent on its ion channel activity and localization to the Golgi apparatus, indicating that aberrant intracellular proton gradients can serve to activate this inflammasome [207]. Third, the influenza virus PB1-F2 protein is able to activate the NLRP3 inflammasome, dependent on the formation of high-molecular-weight aggregates that disrupt mitochondria [208,209]. Fourth, the cellular protein RNase L, which is upregulated and activated by IFNs during influenza virus infection, produces RNA fragments possessing unusual termini that are capable of triggering activation of the NLRP3 inflammasome [210]. The relative contributions of each of these inflammasome activators during infection are unclear, although the varied mechanisms of inflammasome triggering further support the significance of this pathway in anti-influenza virus immunity.
Type I IFN production and inflammasome activation are also intricately intertwined with cellular autophagy in controlling the outcome of influenza virus infection. Interestingly, the production of specific influenza virus proteins (hemagglutinin and M2) stimulates autophagy [211], and the virus requires the autophagy pathway for optimal replication [212,213]. However, M2 concurrently limits autophagosome fusion with lysosomes [214,215]. Taken together, this suggests that inducing the formation of autophagosome membranes while dampening the degradative potential of these compartments is beneficial to the virus. Importantly, cells also respond to influenza virus infection by upregulating mitophagy through activation of the NOD2–RIPK2 signaling axis, which leads to phosphorylation and activation of the critical autophagy factor ULK1 [216]. In the absence of RIPK2 or ULK1, accumulation of damaged mitochondria during infection leads to further activation of the NLRP3 inflammasome during infection [216]. In addition to limiting NLRP3 activation, mitophagy can also decrease the number of mitochondrial signaling platforms for RIG-I, thus also reducing production of IFNs. The increased mortality of RIPK2 KO mice during influenza virus infection suggests that preventing excessive activation of type I IFN and inflammasome pathways is an essential role for autophagy in limiting virus-induced pathology [216]. Overall, the results of the studies discussed here demonstrate that complex interactions between type I IFN pathways, inflammasomes, and autophagy must be properly balanced to limit pathology while effectively clearing infection.
Conclusions
This review summarizes recent findings on the activation and subversion of the autophagy and inflammasome surveillance pathways during infection by a collection of pathogens. It is clear that each pathogen interacts with these pathways in a very unique fashion. While the autophagosome and the inflammasome implement checks and balances on each other, infectious agents likely employ their virulence factors to alter these checks and tip the balance toward a precise environment that favors their survival. As highlighted in this review, knowledge about the autophagy/inflammasome interconnection and their subversion is still lacking for many pathogens. We also realize that the way autophagy and inflammasomes interact is more sophisticated than previously expected. Therefore, each infectious disease still requires comprehensive studies to tease out the relationships between of autophagy and inflammasomes, which may lead to identification of therapeutic targets.
Acknowledgments
Studies in the Amer laboratory are supported by Ohio State University Bridge funds, the Public Health Preparedness for Infectious Diseases, the Center for Clinical and Translational Science), R01HL127651, R01AI124121, R21AI113477, R21AI120013, and a Cystic Fibrosis Foundation research grant. Studies in the Seveau's lab are supported by the National Institute of Health under awards R01AI107250 and R21AI105588. Studies in the Yount's lab are supported by the National Institute of Health under award R01AI130110. Joanne Turner and Jordi B. Torrelles laboratories are supported by a National Institute on Aging grant number [AG051428] Jordi B. Torrelles by a National Institute of Allergy and Infectious Diseases grant number [AI093570] Joanne Turner by a National Institute of Allergy and Infectious Diseases grant number [R01AI116917]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- PRRs
pattern recognition receptors
- PAMPs
pathogen-associated molecular patterns
- DAMPs
damage-associated molecular patterns
- NLRs
Nucleotide-binding domain, leucine-rich repeat containing family
- AIM2
absent in melanoma 2
- PYD
pyrin domain
- CARD
caspase activation and recruitment domain
- ASC
apoptosis-associated specklike protein
- mtDNA
mitochondrial DNA
- TLRs
Toll-like receptors
- LLO
listeriolysin O
- PI-PLC and PC-PLC
phospholipases C
- NDP52
nuclear domain 10 protein 52
- T3SS
Type three secretion system
- M.tb
Mycobacterium tuberculosis
- NTM
nontuberculous mycobacteria
- IFN
interferon
- RIG-I
retinoic acid-inducible gene I
- MAVS
mitochondrial antiviral signaling protein
- M2
matrix protein 2
References
- 1.Abdelaziz DH, Khalil H, Cormet-Boyaka E, Amer AO. The cooperation between the autophagy machinery and the inflammasome to implement an appropriate innate immune response: do they regulate each other? Immunol Rev. 2015;265:194–204. doi: 10.1111/imr.12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fesus L, Demeny MA, Petrovski G. Autophagy shapes inflammation. Antioxid Redox Signal. 2011;14:2233–2243. doi: 10.1089/ars.2010.3485. [DOI] [PubMed] [Google Scholar]
- 3.Netea-Maier RT, Plantinga TS, van de Veerdonk FL, Smit JW, Netea MG. Modulation of inflammation by autophagy: consequences for human disease. Autophagy. 2016;12:245–260. doi: 10.1080/15548627.2015.1071759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kesavardhana S, Kanneganti TD. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int Immunol. 2017;29:201–210. doi: 10.1093/intimm/dxx018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma D, Kanneganti TD. The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol. 2016;213:617–629. doi: 10.1083/jcb.201602089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 7.Akhter A, Caution K, Abu Khweek A, Tazi M, Abdulrahman BA, Abdelaziz DH, Voss OH, Doseff AI, Hassan H, Azad AK, Schlesinger LS, Wewers MD, Gavrilin MA, Amer AO. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity. 2012;37:35–47. doi: 10.1016/j.immuni.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caution K, Gavrilin MA, Tazi M, Kanneganti A, Layman D, Hoque S, Krause K, Amer AO. Caspase-11 and caspase-1 differentially modulate actin polymerization via RhoA and Slingshot proteins to promote bacterial clearance. Sci Rep. 2015;5:18479. doi: 10.1038/srep18479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamkanfi M, Kanneganti TD, Franchi L, Nunez G. Caspase-1 inflammasomes in infection and inflammation. J Leukoc Biol. 2007;82:220–225. doi: 10.1189/jlb.1206756. [DOI] [PubMed] [Google Scholar]
- 10.Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. 2017;27:673–684. doi: 10.1016/j.tcb.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 12.Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 13.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 14.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 16.Khaminets A, Behl C, Dikic I. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol. 2016;26:6–16. doi: 10.1016/j.tcb.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 17.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 18.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loos B, du Toit A, Hofmeyr JH. Defining and measuring autophagosome flux—concept and reality. Autophagy. 2014;10:2087–2096. doi: 10.4161/15548627.2014.973338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saitoh T, Akira S. Regulation of inflammasomes by autophagy. J Allergy Clin Immunol. 2016;138:28–36. doi: 10.1016/j.jaci.2016.05.009. [DOI] [PubMed] [Google Scholar]
- 21.Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, NLRP3 inflammasome and auto-inflammatory/immune diseases. Clin Exp Rheumatol. 2016;34:12–16. [PubMed] [Google Scholar]
- 22.Deretic V. Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Curr Opin Immunol. 2012;24:21–31. doi: 10.1016/j.coi.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Negroni A, Colantoni E, Vitali R, Palone F, Pierdomenico M, Costanzo M, Cesi V, Cucchiara S, Stronati L. NOD2 induces autophagy to control AIEC bacteria infectiveness in intestinal epithelial cells. Inflamm Res. 2016;65:803–813. doi: 10.1007/s00011-016-0964-8. [DOI] [PubMed] [Google Scholar]
- 24.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O'Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel JF, Sahbatou M, Thomas G. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 25.Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol. 2008;8:458–466. doi: 10.1038/nri2340. [DOI] [PubMed] [Google Scholar]
- 26.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 27.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, Boneca IG, Allaoui A, Jones NL, Nunez G, Girardin SE, Philpott DJ. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 28.Park S, Ha SD, Coleman M, Meshkibaf S, Kim SO. p62/SQSTM1 enhances NOD2-mediated signaling and cytokine production through stabilizing NOD2 oligomerization. PLoS One. 2013;8:e57138. doi: 10.1371/journal.pone.0057138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM, Frankel G, Levy M, Katz MN, Philbrick WM, Elinav E, Finlay BB, Flavell RA. NLRP6 inflammasome orchestrates the colonic host–microbial interface by regulating goblet cell mucus secretion. Cell. 2014;156:1045–1059. doi: 10.1016/j.cell.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jounai N, Kobiyama K, Shiina M, Ogata K, Ishii KJ, Takeshita F. NLRP4 negatively regulates autophagic processes through an association with beclin1. J Immunol. 2011;186:1646–1655. doi: 10.4049/jimmunol.1001654. [DOI] [PubMed] [Google Scholar]
- 31.Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, Horng T. Inflammasome activation leads to caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci U S A. 2014;111:15514–15519. doi: 10.1073/pnas.1414859111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang LJ, Huang HY, Huang MP, Liou W, Chang YT, Wu CC, Ojcius DM, Chang YS. The microtubule-associated protein EB1 links AIM2 inflammasomes with autophagy-dependent secretion. J Biol Chem. 2014;289:29322–29333. doi: 10.1074/jbc.M114.559153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kimura T, Jia J, Kumar S, Choi SW, Gu Y, Mudd M, Dupont N, Jiang S, Peters R, Farzam F, Jain A, Lidke KA, Adams CM, Johansen T, Deretic V. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 2017;36:42–60. doi: 10.15252/embj.201695081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011;30:4701–4711. doi: 10.1038/emboj.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thorburn J, Frankel AE, Thorburn A. Regulation of HMGB1 release by autophagy. Autophagy. 2009;5:247–249. doi: 10.4161/auto.5.2.7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeSelm CJ, Miller BC, Zou W, Beatty WL, van Meel E, Takahata Y, Klumperman J, Tooze SA, Teitelbaum SL, Virgin HW. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell. 2011;21:966–974. doi: 10.1016/j.devcel.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 38.Joosten LA, Netea MG, Dinarello CA. Interleukin-1beta in innate inflammation, autophagy and immunity. Semin Immunol. 2013;25:416–424. doi: 10.1016/j.smim.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 39.Into T, Horie T, Inomata M, Gohda J, Inoue JI, Murakami Y, Niida S. Basal autophagy prevents autoactivation or enhancement of inflammatory signals by targeting monomeric MyD88. Sci Rep. 2017;7:1009. doi: 10.1038/s41598-017-01246-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlech WF, III, Lavigne PM, Bortolussi RA, Allen AC, Haldane EV, Wort AJ, Hightower AW, Johnson SE, King SH, Nicholls ES, Broome CV. Epidemic listeriosis—evidence for transmission by food. N Engl J Med. 1983;308:203–206. doi: 10.1056/NEJM198301273080407. [DOI] [PubMed] [Google Scholar]
- 42.Cossart P. Illuminating the landscape of host–pathogen interactions with the bacterium Listeria monocytogenes. Proc Natl Acad Sci U S A. 2011;108:19484–19491. doi: 10.1073/pnas.1112371108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tilney LG, Portnoy DA. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J Cell Biol. 1989;109:1597–1608. doi: 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaughnessy LM, Hoppe AD, Christensen KA, Swanson JA. Membrane perforations inhibit lysosome fusion by altering pH and calcium in Listeria monocytogenes vacuoles. Cell Microbiol. 2006;8:781–792. doi: 10.1111/j.1462-5822.2005.00665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P. L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell. 1992;68:521–531. doi: 10.1016/0092-8674(92)90188-i. [DOI] [PubMed] [Google Scholar]
- 46.Gedde MM, Higgins DE, Tilney LG, Portnoy DA. Role of listeriolysin O incell-to-cell spread of Listeria monocytogenes. Infect Immun. 2000;68:999–1003. doi: 10.1128/iai.68.2.999-1003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meixenberger K, Pache F, Eitel J, Schmeck B, Hippenstiel S, Slevogt H, N'Guessan P, Witzenrath M, Netea MG, Chakraborty T, Suttorp N, Opitz B. Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1beta, depending on listeriolysin O and NLRP3. J Immunol. 2010;184:922–930. doi: 10.4049/jimmunol.0901346. [DOI] [PubMed] [Google Scholar]
- 48.Tsuji NM, Tsutsui H, Seki E, Kuida K, Okamura H, Nakanishi K, Flavell RA. Roles of caspase-1 in Listeria infection in mice. Int Immunol. 2004;16:335–343. doi: 10.1093/intimm/dxh041. [DOI] [PubMed] [Google Scholar]
- 49.Man SM, Karki R, Briard B, Burton A, Gingras S, Pelletier S, Kanneganti TD. Differential roles of caspase-1 and caspase-11 in infection and inflammation. Sci Rep. 2017;7:45126. doi: 10.1038/srep45126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Warren SE, Mao DP, Rodriguez AE, Miao EA, Aderem A. Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J Immunol. 2008;180:7558–7564. doi: 10.4049/jimmunol.180.11.7558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aaltonen T, Abazov VM, Abbott B, Acharya BS, Adams M, et al. Evidence for a particle produced in association with weak bosons and decaying to a bottom-antibottom quark pair in higgs boson searches at the tevatron. Phys Rev Lett. 2012;109:071804. doi: 10.1103/PhysRevLett.109.071804. [DOI] [PubMed] [Google Scholar]
- 52.Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, Bertin J, Coyle A, Grant EP, Akira S, Nunez G. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440:233–236. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- 53.Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 54.Sauer JD, Witte CE, Zemansky J, Hanson B, Lauer P, Portnoy DA. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe. 2010;7:412–419. doi: 10.1016/j.chom.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Neiman-Zenevich J, Stuart S, Abdel-Nour M, Girardin SE, Mogridge J. Listeria monocytogenes and Shigella flexneri activate the NLRP1B inflammasome. Infect Immun. 2017;85 doi: 10.1128/IAI.00338-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sauer JD, Pereyre S, Archer KA, Burke TP, Hanson B, Lauer P, Portnoy DA. Listeria monocytogenes engineered to activate the Nlrc4 inflammasome are severely attenuated and are poor inducers of protective immunity. Proc Natl Acad Sci U S A. 2011;108:12419–12424. doi: 10.1073/pnas.1019041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Theisen E, Sauer JD. Listeria monocytogenes and the inflammasome: from cytosolic bacteriolysis to tumor immunotherapy. Curr Top Microbiol Immunol. 2016;397:133–160. doi: 10.1007/978-3-319-41171-2_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Birmingham CL, Canadien V, Gouin E, Troy EB, Yoshimori T, Cossart P, Higgins DE, Brumell JH. Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy. 2007;3:442–451. doi: 10.4161/auto.4450. [DOI] [PubMed] [Google Scholar]
- 60.Meyer-Morse N, Robbins JR, Rae CS, Mochegova SN, Swanson MS, Zhao Z, Virgin HW, Portnoy D. Listeriolysin O is necessary and sufficient to induce autophagy during Listeria monocytogenes infection. PLoS One. 2010;5:e8610. doi: 10.1371/journal.pone.0008610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rich KA, Burkett C, Webster P. Cytoplasmic bacteria can be targets for autophagy. Cell Microbiol. 2003;5:455–468. doi: 10.1046/j.1462-5822.2003.00292.x. [DOI] [PubMed] [Google Scholar]
- 62.Py BF, Lipinski MM, Yuan J. Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy. 2007;3:117–125. doi: 10.4161/auto.3618. [DOI] [PubMed] [Google Scholar]
- 63.Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, Cadwell K, Delgado MA, Ponpuak M, Green KG, Schmidt RE, Mizushima N, Deretic V, Sibley LD, Virgin HW. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4:458–469. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gupta M, Shin DM, Ramakrishna L, Goussetis DJ, Platanias LC, Xiong H, Morse HC, III, Ozato K. IRF8 directs stress-induced autophagy in macrophages and promotes clearance of Listeria monocytogenes. Nat Commun. 2015;6:6379. doi: 10.1038/ncomms7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Puri M, La Pietra L, Mraheil MA, Lucas R, Chakraborty T, Pillich H. Listeriolysin O regulates the expression of optineurin, an autophagy adaptor that inhibits the growth of Listeria monocytogenes. Toxins (Basel) 2017;9 doi: 10.3390/toxins9090273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mitchell G, Ge L, Huang Q, Chen C, Kianian S, Roberts MF, Schekman R, Portnoy DA. Avoidance of autophagy mediated by PlcA or ActA is required for Listeria monocytogenes growth in macrophages. Infect Immun. 2015;83:2175–2184. doi: 10.1128/IAI.00110-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tattoli I, Sorbara MT, Yang C, Tooze SA, Philpott DJ, Girardin SE. Listeria phospholipases subvert host autophagic defenses by stalling pre-autophagosomal structures. EMBO J. 2013;32:3066–3078. doi: 10.1038/emboj.2013.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mostowy S, Sancho-Shimizu V, Hamon MA, Simeone R, Brosch R, Johansen T, Cossart P. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J Biol Chem. 2011;286:26987–26995. doi: 10.1074/jbc.M111.223610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, Mimuro H, Nakagawa I, Yanagawa T, Ishii T, Kakizuka A, Sztul E, Chakraborty T, Sasakawa C. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol. 2009;11:1233–1240. doi: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- 70.Dortet L, Mostowy S, Samba-Louaka A, Gouin E, Nahori MA, Wiemer EA, Dussurget O, Cossart P. Recruitment of the major vault protein by InlK: a Listeria monocytogenes strategy to avoid autophagy. PLoS Pathog. 2011;7:e1002168. doi: 10.1371/journal.ppat.1002168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lara-Tejero M, Sutterwala FS, Ogura Y, Grant EP, Bertin J, Coyle AJ, Flavell RA, Galan JE. Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J Exp Med. 2006;203:1407–1412. doi: 10.1084/jem.20060206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Abdelaziz DH, Amr K, Amer AO. Nlrc4/Ipaf/CLAN/CARD12: more than a flagellin sensor. Int J Biochem Cell Biol. 2010;42:789–791. doi: 10.1016/j.biocel.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med. 2010;207:1745–1755. doi: 10.1084/jem.20100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Matusiak M, Van Opdenbosch N, Vande Walle L, Sirard JC, Kanneganti TD, Lamkanfi M. Flagellin-induced NLRC4 phosphorylation primes the inflammasome for activation by NAIP5. Proc Natl Acad Sci U S A. 2015;112:1541–1546. doi: 10.1073/pnas.1417945112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci U S A. 2013;110:14408–14413. doi: 10.1073/pnas.1306376110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao Y, Shi J, Shi X, Wang Y, Wang F, Shao F. Genetic functions of the NAIP family of inflammasome receptors for bacterial ligands in mice. J Exp Med. 2016;213:647–656. doi: 10.1084/jem.20160006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hoffmann C, Galle M, Dilling S, Kappeli R, Muller AJ, Songhet P, Beyaert R, Hardt WD. In macrophages, caspase-1 activation by SopE and the type III secretion system-1 of S. typhimurium can proceed in the absence of flagellin. PLoS One. 2010;5:e12477. doi: 10.1371/journal.pone.0012477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fink SL, Cookson BT. Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol. 2007;9:2562–2570. doi: 10.1111/j.1462-5822.2007.01036.x. [DOI] [PubMed] [Google Scholar]
- 80.Puri AW, Broz P, Shen A, Monack DM, Bogyo M. Caspase-1 activity is required to bypass macrophage apoptosis upon Salmonella infection. Nat Chem Biol. 2012;8:745–747. doi: 10.1038/nchembio.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wynosky-Dolfi MA, Snyder AG, Philip NH, Doonan PJ, Poffenberger MC, Avizonis D, Zwack EE, Riblett AM, Hu B, Strowig T, Flavell RA, Jones RG, Freedman BD, Brodsky IE. Oxidative metabolism enables Salmonella evasion of the NLRP3 inflammasome. J Exp Med. 2014;211:653–668. doi: 10.1084/jem.20130627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen KW, Gross CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, Schroder K. The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge. Cell Rep. 2014;8:570–582. doi: 10.1016/j.celrep.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 83.Gupta SL, Rubin BY, Holmes SL. Interferon action: induction of specific proteins in mouse and human cells by homologous interferons. Proc Natl Acad Sci U S A. 1979;76:4817–4821. doi: 10.1073/pnas.76.10.4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S, Roose-Girma M, Bumann D, Kayagaki N, Takeda K, Yamamoto M, Broz P. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature. 2014;509:366–370. doi: 10.1038/nature13157. [DOI] [PubMed] [Google Scholar]
- 85.Kim BH, Chee JD, Bradfield CJ, Park ES, Kumar P, MacMicking JD. Interferon-induced guanylate-binding proteins in inflammasome activation and host defense. Nat Immunol. 2016;17:481–489. doi: 10.1038/ni.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Birmingham CL, Brumell JH. Autophagy recognizes intracellular Salmonella enterica serovar Typhimurium in damaged vacuoles. Autophagy. 2006;2:156–158. doi: 10.4161/auto.2825. [DOI] [PubMed] [Google Scholar]
- 87.Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–1221. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 88.Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183:5909–5916. doi: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
- 89.Rogov VV, Suzuki H, Fiskin E, Wild P, Kniss A, Rozenknop A, Kato R, Kawasaki M, McEwan DG, Lohr F, Guntert P, Dikic I, Wakatsuki S, Dotsch V. Structural basis for phosphorylation-triggered autophagic clearance of Salmonella. Biochem J. 2013;454:459–466. doi: 10.1042/BJ20121907. [DOI] [PubMed] [Google Scholar]
- 90.Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dotsch V, Bumann D, Dikic I. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–233. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cadwell K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat Rev Immunol. 2016;16:661–675. doi: 10.1038/nri.2016.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sjostedt A. Tularemia: history, epidemiology, pathogen physiology, and clinical manifestations. Ann N Y Acad Sci. 2007;1105:1–29. doi: 10.1196/annals.1409.009. [DOI] [PubMed] [Google Scholar]
- 93.Mohapatra NP, Soni S, Rajaram MV, Strandberg KL, Gunn JS. Type A Francisella tularensis acid phosphatases contribute to pathogenesis. PLoS One. 2013;8:e56834. doi: 10.1371/journal.pone.0056834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ellis J, Oyston PC, Green M, Titball RW. Tularemia. Clin Microbiol Rev. 2002;15:631–646. doi: 10.1128/CMR.15.4.631-646.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Telepnev M, Golovliov I, Sjostedt A. Francisella tularensis LVS initially activates but subsequently down-regulates intracellular signaling and cytokine secretion in mouse monocytic and human peripheral blood mononuclear cells. Microb Pathog. 2005;38:239–247. doi: 10.1016/j.micpath.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 96.Gillette DD, Tridandapani S, Butchar JP. Monocyte/macrophage inflammatory response pathways to combat Francisella infection: possible therapeutic targets? Front Cell Infect Microbiol. 2014;4:18. doi: 10.3389/fcimb.2014.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bolger CE, Forestal CA, Italo JK, Benach JL, Furie MB. The live vaccine strain of Francisella tularensis replicates in human and murine macrophages but induces only the human cells to secrete proinflammatory cytokines. J Leukoc Biol. 2005;77:893–897. doi: 10.1189/jlb.1104637. [DOI] [PubMed] [Google Scholar]
- 98.Jones JW, Broz P, Monack DM. Innate immune recognition of Francisella tularensis: activation of type-I interferons and the inflammasome. Front Microbiol. 2011;2:16. doi: 10.3389/fmicb.2011.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ghonime MG, Mitra S, Eldomany RA, Wewers MD, Gavrilin MA. Inflammasome priming is similar for Francisella species that differentially induce inflammasome activation. PLoS One. 2015;10:e0127278. doi: 10.1371/journal.pone.0127278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Clemens DL, Horwitz MA. Uptake and intracellular fate of Francisella tularensis in human macrophages. Ann N Y Acad Sci. 2007;1105:160–186. doi: 10.1196/annals.1409.001. [DOI] [PubMed] [Google Scholar]
- 101.Golovliov I, Baranov V, Krocova Z, Kovarova H, Sjostedt A. An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect Immun. 2003;71:5940–5950. doi: 10.1128/IAI.71.10.5940-5950.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mariathasan S, Weiss DS, Dixit VM, Monack DM. Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J Exp Med. 2005;202:1043–1049. doi: 10.1084/jem.20050977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gavrilin MA, Bouakl IJ, Knatz NL, Duncan MD, Hall MW, Gunn JS, Wewers MD. Internalization and phago-some escape required for Francisella to induce human monocyte IL-1beta processing and release. Proc Natl Acad Sci U S A. 2006;103:141–146. doi: 10.1073/pnas.0504271103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, Datta P, Mc Cormick M, Huang L, Mc Dermott E, Eisenlohr L, Landel CP, Alnemri ES. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol. 2010;11:385–393. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O'Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, Dixit VM, Monack DM. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci U S A. 2010;107:9771–9776. doi: 10.1073/pnas.1003738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Atianand MK, Duffy EB, Shah A, Kar S, Malik M, Harton JA. Francisella tularensis reveals a disparity between human and mouse NLRP3 inflammasome activation. J Biol Chem. 2011;286:39033–39042. doi: 10.1074/jbc.M111.244079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Duffy EB, Periasamy S, Hunt D, Drake JR, Harton JA. FcgammaR mediates TLR2- and Syk-dependent NLRP3 inflammasome activation by inactivated Francisella tularensis LVS immune complexes. J Leukoc Biol. 2016;100:1335–1347. doi: 10.1189/jlb.2A1215-555RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gavrilin MA, Mitra S, Seshadri S, Nateri J, Berhe F, Hall MW, Wewers MD. Pyrin critical to macrophage IL-1beta response to Francisella challenge. J Immunol. 2009;182:7982–7989. doi: 10.4049/jimmunol.0803073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gavrilin MA, Wewers MD. Francisella recognition by inflammasomes: differences between mice and men. Front Microbiol. 2011;2:11. doi: 10.3389/fmicb.2011.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chae JJ, Cho YH, Lee GS, Cheng J, Liu PP, Feigenbaum L, Katz SI, Kastner DL. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity. 2011;34:755–768. doi: 10.1016/j.immuni.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ratner D, Orning MP, Proulx MK, Wang D, Gavrilin MA, Wewers MD, Alnemri ES, Johnson PF, Lee B, Mecsas J, Kayagaki N, Goguen JD, Lien E. The Yersinia pestis effector YopM inhibits pyrin inflammasome activation. PLoS Pathog. 2016;12:e1006035. doi: 10.1371/journal.ppat.1006035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ratner D, Orning MP, Lien E. Bacterial secretion systems and regulation of inflammasome activation. J Leukoc Biol. 2017;101:165–181. doi: 10.1189/jlb.4MR0716-330R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Park YH, Wood G, Kastner DL, Chae JJ. Pyrin inflammasome activation and RhoA signaling in the auto-inflammatory diseases FMF and HIDS. Nat Immunol. 2016;17:914–921. doi: 10.1038/ni.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chung H, Vilaysane A, Lau A, Stahl M, Morampudi V, Bondzi-Simpson A, Platnich JM, Bracey NA, French MC, Beck PL, Chun J, Vallance BA, Muruve DA. NLRP3 regulates a non-canonical platform for caspase-8 activation during epithelial cell apoptosis. Cell Death Differ. 2016;23:1331–1346. doi: 10.1038/cdd.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Checroun C, Wehrly TD, Fischer ER, Hayes SF, Celli J. Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc Natl Acad Sci U S A. 2006;103:14578–14583. doi: 10.1073/pnas.0601838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Butchar JP, Cremer TJ, Clay CD, Gavrilin MA, Wewers MD, Marsh CB, Schlesinger LS, Tridandapani S. Microarray analysis of human monocytes infected with Francisella tularensis identifies new targets of host response subversion. PLoS One. 2008;3:e2924. doi: 10.1371/journal.pone.0002924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cremer TJ, Amer A, Tridandapani S, Butchar JP. Francisella tularensis regulates autophagy-related host cell signaling pathways. Autophagy. 2009;5:125–128. doi: 10.4161/auto.5.1.7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chong A, Wehrly TD, Child R, Hansen B, Hwang S, Virgin HW, Celli J. Cytosolic clearance of replication-deficient mutants reveals Francisella tularensis interactions with the autophagic pathway. Autophagy. 2012;8:1342–1356. doi: 10.4161/auto.20808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Case ED, Chong A, Wehrly TD, Hansen B, Child R, Hwang S, Virgin HW, Celli J. The Francisella O-antigen mediates survival in the macrophage cytosol via autophagy avoidance. Cell Microbiol. 2014;16:862–877. doi: 10.1111/cmi.12246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kimura T, Jain A, Choi SW, Mandell MA, Johansen T, Deretic V. TRIM-directed selective autophagy regulates immune activation. Autophagy. 2016;0 doi: 10.1080/15548627.2016.1154254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lazar Adler NR, Govan B, Cullinane M, Harper M, Adler B, Boyce JD. The molecular and cellular basis of pathogenesis in melioidosis: how does Burkholderia pseudomallei cause disease? FEMS. Microbiol Rev. 2009;33:1079–1099. doi: 10.1111/j.1574-6976.2009.00189.x. [DOI] [PubMed] [Google Scholar]
- 124.Drevinek P, Mahenthiralingam E. Burkholderia cenocepacia in cystic fibrosis: epidemiology and molecular mechanisms of virulence. Clin Microbiol Infect. 2010;16:821–830. doi: 10.1111/j.1469-0691.2010.03237.x. [DOI] [PubMed] [Google Scholar]
- 125.Marolda CL, Hauroder B, John MA, Michel R, Valvano MA. Intracellular survival and saprophytic growth of isolates from the Burkholderia cepacia complex in free-living amoebae. Microbiology. 1999;145(Pt 7):1509–1517. doi: 10.1099/13500872-145-7-1509. [DOI] [PubMed] [Google Scholar]
- 126.Lamothe J, Thyssen S, Valvano MA. Burkholderia cepacia complex isolates survive intracellularly without replication within acidic vacuoles of Acanthamoeba polyphaga. Cell Microbiol. 2004;6:1127–1138. doi: 10.1111/j.1462-5822.2004.00424.x. [DOI] [PubMed] [Google Scholar]
- 127.Saini LS, Galsworthy SB, John MA, Valvano MA. Intracellular survival of Burkholderia cepacia complex isolates in the presence of macrophage cell activation. Microbiology. 1999;145(Pt 12):3465–3475. doi: 10.1099/00221287-145-12-3465. [DOI] [PubMed] [Google Scholar]
- 128.Abdulrahman BA, Khweek AA, Akhter A, Caution K, Kotrange S, Abdelaziz DH, Newland C, Rosales-Reyes R, Kopp B, McCoy K, Montione R, Schlesinger LS, Gavrilin MA, Wewers MD, Valvano MA, Amer AO. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy. 2011;7:1359–1370. doi: 10.4161/auto.7.11.17660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Abdulrahman BA, Khweek AA, Akhter A, Caution K, Tazi M, Hassan H, Zhang Y, Rowland PD, Malhotra S, Aeffner F, Davis IC, Valvano MA, Amer AO. Depletion of the ubiquitin-binding adaptor molecule SQSTM1/p62 from macrophages harboring cftr DeltaF508 mutation improves the delivery of Burkholderia cenocepacia to the autophagic machinery. J Biol Chem. 2013;288:2049–2058. doi: 10.1074/jbc.M112.411728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kopp BT, Abdulrahman BA, Khweek AA, Kumar SB, Akhter A, Montione R, Tazi MF, Caution K, McCoy K, Amer AO. Exaggerated inflammatory responses mediated by Burkholderia cenocepacia in human macrophages derived from cystic fibrosis patients. Biochem Biophys Res Commun. 2012;424:221–227. doi: 10.1016/j.bbrc.2012.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kotrange S, Kopp B, Akhter A, Abdelaziz D, Abu Khweek A, Caution K, Abdulrahman B, Wewers MD, Mc Coy K, Marsh C, Loutet SA, Ortega X, Valvano MA, Amer AO. Burkholderia cenocepacia O polysaccharide chain contributes to caspase-1-dependent IL-1beta production in macrophages. J Leukoc Biol. 2011;89:481–488. doi: 10.1189/jlb.0910513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Burns JL, Jonas M, Chi EY, Clark DK, Berger A, Griffith A. Invasion of respiratory epithelial cells by Burkholderia (Pseudomonas) cepacia. Infect Immun. 1996;64:4054–4059. doi: 10.1128/iai.64.10.4054-4059.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Huynh KK, Plumb JD, Downey GP, Valvano MA, Grinstein S. Inactivation of macrophage Rab7 by Burkholderia cenocepacia. J Innate Immun. 2010;2:522–533. doi: 10.1159/000319864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sajjan SU, Carmody LA, Gonzalez CF, Li Puma JJ. A type IV secretion system contributes to intracellular survival and replication of Burkholderia cenocepacia. Infect Immun. 2008;76:5447–5455. doi: 10.1128/IAI.00451-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tegos GP, Haynes MK, Schweizer HP. Dissecting novel virulent determinants in the Burkholderia cepacia complex. Virulence. 2012;3:234–237. doi: 10.4161/viru.19844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang R, Li Puma JJ, Gonzalez CF. Two type IV secretion systems with different functions in Burkholderia cenocepacia K56-2. Microbiology. 2009;155:4005–4013. doi: 10.1099/mic.0.033043-0. [DOI] [PubMed] [Google Scholar]
- 137.Valvano MA. Intracellular survival of Burkholderia cepacia complex in phagocytic cells. Can J Microbiol. 2015;61:607–615. doi: 10.1139/cjm-2015-0316. [DOI] [PubMed] [Google Scholar]
- 138.Rosales-Reyes R, Aubert DF, Tolman JS, Amer AO, Valvano MA. Burkholderia cenocepacia type VI secretion system mediates escape of type II secreted proteins into the cytoplasm of infected macrophages. PLoS One. 2012;7:e41726. doi: 10.1371/journal.pone.0041726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cullinane M, Gong L, Li X, Lazar-Adler N, Tra T, Wolvetang E, Prescott M, Boyce JD, Devenish RJ, Adler B. Stimulation of autophagy suppresses the intracellular survival of Burkholderia pseudomallei in mammalian cell lines. Autophagy. 2008;4:744–753. doi: 10.4161/auto.6246. [DOI] [PubMed] [Google Scholar]
- 140.Tazi MF, Dakhlallah DA, Caution K, Gerber MM, Chang SW, Khalil H, Kopp BT, Ahmed AE, Krause K, Davis I, Marsh C, Lovett-Racke AE, Schlesinger LS, Cormet-Boyaka E, Amer AO. Elevated Mirc1/Mir17-92 cluster expression negatively regulates autophagy and CFTR (cystic fibrosis transmembrane conductance regulator) function in CF macrophages. Autophagy. 2016;12:2026–2037. doi: 10.1080/15548627.2016.1217370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.WHO. Global Tuberculosis Report 2016. 2016 [Google Scholar]
- 142.Arnett E, Krishnan N, Roberston BD, Schlesinger L. Host pathogen biology for airborne Mycobacterium tuberculosis. In: Hickey A, Misra A, Fourie P, editors. Drug Delivery Systems for Tuberculosis Preveniton and Treatment. John Wiley & Sons, Ltd; Chichester, UK: 2016. pp. 11–47. [Google Scholar]
- 143.Guirado E, Schlesinger LS, Kaplan G. Macrophages in tuberculosis: friend or foe, Semin. Immunopathol. 2013;35:563–583. doi: 10.1007/s00281-013-0388-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Master SS, Rampini SK, Davis AS, Keller C, Ehlers S, Springer B, Timmins GS, Sander P, Deretic V. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe. 2008;3:224–232. doi: 10.1016/j.chom.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, Mimuro H, Inohara N, Sasakawa C, Nunez G. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 2007;3:e111. doi: 10.1371/journal.ppat.0030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Koo IC, Wang C, Raghavan S, Morisaki JH, Cox JS, Brown EJ. ESX-1-dependent cytolysis in lysosome secretion and inflammasome activation during mycobacterial infection. Cell Microbiol. 2008;10:1866–1878. doi: 10.1111/j.1462-5822.2008.01177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Carlsson F, Kim J, Dumitru C, Barck KH, Carano RA, Sun M, Diehl L, Brown EJ. Host-detrimental role of Esx-1-mediated inflammasome activation in mycobacterial infection. PLoS Pathog. 2010;6:e1000895. doi: 10.1371/journal.ppat.1000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Simeone R, Bottai D, Brosch R. ESX/type VII secretion systems and their role in host–pathogen interaction. Curr Opin Microbiol. 2009;12:4–10. doi: 10.1016/j.mib.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 149.Saiga H, Kitada S, Shimada Y, Kamiyama N, Okuyama M, Makino M, Yamamoto M, Takeda K. Critical role of AIM2 in Mycobacterium tuberculosis infection. Int Immunol. 2012;24:637–644. doi: 10.1093/intimm/dxs062. [DOI] [PubMed] [Google Scholar]
- 150.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dorhoi A, Nouailles G, Jorg S, Hagens K, Heinemann E, Pradl L, Oberbeck-Muller D, Duque-Correa MA, Reece ST, Ruland J, Brosch R, Tschopp J, Gross O, Kaufmann SH. Activation of the NLRP3 inflammasome by Mycobacterium tuberculosis is uncoupled from susceptibility to active tuberculosis. Eur J Immunol. 2012;42:374–384. doi: 10.1002/eji.201141548. [DOI] [PubMed] [Google Scholar]
- 152.Wong KW, Jacobs WR., Jr Critical role for NLRP3 in necrotic death triggered by Mycobacterium tuberculosis. Cell Microbiol. 2011;13:1371–1384. doi: 10.1111/j.1462-5822.2011.01625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gross O, Poeck H, Bscheider M, Dostert C, Hannesschlager N, Endres S, Hartmann G, Tardivel A, Schweighoffer E, Tybulewicz V, Mocsai A, Tschopp J, Ruland J. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. 2009;459:433–436. doi: 10.1038/nature07965. [DOI] [PubMed] [Google Scholar]
- 154.Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, Eisenbarth SC, Nauseef WM, Cassel SL, Sutterwala FS. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39:311–323. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, Sassetti CM. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nat Immunol. 2013;14:52–60. doi: 10.1038/ni.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, Schmid-Burgk JL, Schmidt T, Hornung V, Cole ST, Ablasser A. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe. 2015;17:799–810. doi: 10.1016/j.chom.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 157.Stoop EJ, Bitter W, van der Sar AM. Tubercle bacilli rely on a type VII army for pathogenicity. Trends Microbiol. 2012;20:477–484. doi: 10.1016/j.tim.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 158.Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, Anes E. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol. 2010;12:1046–1063. doi: 10.1111/j.1462-5822.2010.01450.x. [DOI] [PubMed] [Google Scholar]
- 159.Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, Oland S, Gordon S, Sher A. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity. 2011;35:1023–1034. doi: 10.1016/j.immuni.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Novikov A, Cardone M, Thompson R, Shenderov K, Kirschman KD, Mayer-Barber KD, Myers TG, Rabin RL, Trinchieri G, Sher A, Feng CG. Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1beta production in human macrophages. J Immunol. 2011;187:2540–2547. doi: 10.4049/jimmunol.1100926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Briken V, Ahlbrand SE, Shah S. Mycobacterium tuberculosis and the host cell inflammasome: a complex relationship. Front Cell Infect Microbiol. 2013;3:62. doi: 10.3389/fcimb.2013.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–448. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 163.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 164.Arcos J, Sasindran SJ, Moliva JI, Scordo JM, Sidiki S, Guo H, Venigalla P, Kelley HV, Lin G, Diangelo L, Silwani SN, Zhang J, Turner J, Torrelles JB. Mycobacterium tuberculosis cell wall released fragments by the action of the human lung mucosa modulate macrophages to control infection in an IL-10-dependent manner. Mucosal Immunol. 2016;8(307):1–12. doi: 10.1038/mi.2016.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 166.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 167.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 168.Songane M, Kleinnijenhuis J, Netea MG, van Crevel R. The role of autophagy in host defence against Mycobacterium tuberculosis infection. Tuberculosis (Edinb) 2012;92:388–396. doi: 10.1016/j.tube.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 169.Turner J, D'Souza CD, Pearl JE, Marietta P, Noel M, Frank AA, Appelberg R, Orme IM, Cooper AM. CD8-and CD95/95L-dependent mechanisms of resistance in mice with chronic pulmonary tuberculosis. Am J Respir Cell Mol Biol. 2001;24:203–209. doi: 10.1165/ajrcmb.24.2.4370. [DOI] [PubMed] [Google Scholar]
- 170.Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, Delgado-Vargas M, Timmins GS, Bhattacharya D, Yang H, Hutt J, Lyons CR, Dobos KM, Deretic V. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A. 2012;109:E3168–76. doi: 10.1073/pnas.1210500109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. 2012;150:803–815. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Behar SM, Baehrecke EH. Tuberculosis: autophagy is not the answer. Nature. 2015;528:482–483. doi: 10.1038/nature16324. [DOI] [PubMed] [Google Scholar]
- 173.Deretic V, Jiang S, Dupont N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol. 2012;22:397–406. doi: 10.1016/j.tcb.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Shravage BV, Hill JH, Powers CM, Wu L, Baehrecke EH. Atg6 is required for multiple vesicle trafficking pathways and hematopoiesis in Drosophila. Development. 2013;140:1321–1329. doi: 10.1242/dev.089490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–1132. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 176.Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, Sharma AK, Amin S, Hu CD, Zhang J, Kester M, Wang HG. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem. 2012;287:12455–12468. doi: 10.1074/jbc.M111.309104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bento CF, Empadinhas N, Mendes V. Autophagy in the fight against tuberculosis. DNA Cell Biol. 2015;34:228–242. doi: 10.1089/dna.2014.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lammas DA, Stober C, Harvey CJ, Kendrick N, Panchalingam S, Kumararatne DS. ATP-induced killing of mycobacteria by human macrophages is mediated by purinergic P2Z(P2X7) receptors. Immunity. 1997;7:433–444. doi: 10.1016/s1074-7613(00)80364-7. [DOI] [PubMed] [Google Scholar]
- 179.Molloy S. Host response: double trouble for TB. Nat Rev Microbiol. 2012;10:730. doi: 10.1038/nrmicro2908. [DOI] [PubMed] [Google Scholar]
- 180.Chen CC, Tsai SH, Lu CC, Hu ST, Wu TS, Huang TT, Said-Sadier N, Ojcius DM, Lai HC. Activation of an NLRP3 inflammasome restricts Mycobacterium kansasii infection. PLoS One. 2012;7:e36292. doi: 10.1371/journal.pone.0036292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lee HM, Yuk JM, Kim KH, Jang J, Kang G, Park JB, Son JW, Jo EK. Mycobacterium abscessus activates the NLRP3 inflammasome via Dectin-1–Syk and p62/SQSTM1. Immunol Cell Biol. 2012;90:601–610. doi: 10.1038/icb.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hosseini R, Lamers GE, Hodzic Z, Meijer AH, Schaaf MJ, Spaink HP. Correlative light and electron microscopy imaging of autophagy in a zebrafish infection model. Autophagy. 2014;10:1844–1857. doi: 10.4161/auto.29992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Collins CA, De Maziere A, van Dijk S, Carlsson F, Klumperman J, Brown EJ. Atg5-independent sequestration of ubiquitinated mycobacteria. PLoS Pathog. 2009;5:e1000430. doi: 10.1371/journal.ppat.1000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Renna M, Schaffner C, Brown K, Shang S, Tamayo MH, Hegyi K, Grimsey NJ, Cusens D, Coulter S, Cooper J, Bowden AR, Newton SM, Kampmann B, Helm J, Jones A, Haworth CS, Basaraba RJ, DeGroote MA, Ordway DJ, Rubinsztein DC, Floto RA. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J Clin Invest. 2011;121:3554–3563. doi: 10.1172/JCI46095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Roux AL, Viljoen A, Bah A, Simeone R, Bernut A, Laencina L, Deramaudt T, Rottman M, Gaillard JL, Majlessi L, Brosch R, Girard-Misguich F, Vergne I, de Chastellier C, Kremer L, Herrmann JL. The distinct fate of smooth and rough Mycobacterium abscessus variants inside macrophages. Open Biol. 2016;6 doi: 10.1098/rsob.160185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Taubenberger JK, Kash JC. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe. 2010;7:440–451. doi: 10.1016/j.chom.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Arimori Y, Nakamura R, Yamada H, Shibata K, Maeda N, Kase T, Yoshikai Y. Type I interferon limits influenza virus-induced acute lung injury by regulation of excessive inflammation in mice. Antivir Res. 2013;99:230–237. doi: 10.1016/j.antiviral.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 188.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reise Sousa C, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 189.Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F. Reis e Sousa C., RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 190.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 191.McWhirter SM, Tenoever BR, Maniatis T. Connecting mitochondria and innate immunity. Cell. 2005;122:645–647. doi: 10.1016/j.cell.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 192.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 193.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 194.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 195.Benitez AA, Panis M, Xue J, Varble A, Shim JV, Frick AL, Lopez CB, Sachs D, tenOever BR. In vivo RNAi screening identifies MDA5 as a significant contributor to the cellular defense against influenza A virus. Cell Rep. 2015;11:1714–1726. doi: 10.1016/j.celrep.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 197.Pillai PS, Molony RD, Martinod K, Dong H, Pang IK, Tal MC, Solis AG, Bielecki P, Mohanty S, Trentalange M, Homer RJ, Flavell RA, Wagner DD, Montgomery RR, Shaw AC, Staeheli P, Iwasaki A. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science. 2016;352:463–466. doi: 10.1126/science.aaf3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lopez CB, Garcia-Sastre A, Williams BR, Moran TM. Type I interferon induction pathway, but not released interferon, participates in the maturation of dendritic cells induced by negative-strand RNA viruses. J Infect Dis. 2003;187:1126–1136. doi: 10.1086/368381. [DOI] [PubMed] [Google Scholar]
- 199.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Pang IK, Ichinohe T, Iwasaki A. IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8(+) T cell responses to influenza A virus. Nat Immunol. 2013;14:246–253. doi: 10.1038/ni.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Allen IC, Scull MA, Moore CB, Holl EK, Mc Elvania-Te Kippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Thomas PG, Dash P, Aldridge JR, Jr, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL, Doherty PC, Kanneganti TD. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 2009;30:566–575. doi: 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Pothlichet J, Meunier I, Davis BK, Ting JP, Skamene E, von Messling V, Vidal SM. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog. 2013;9:e1003256. doi: 10.1371/journal.ppat.1003256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Mibayashi M, Martinez-Sobrido L, Loo YM, Cardenas WB, Gale M, Jr, Garcia-Sastre A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol. 2007;81:514–524. doi: 10.1128/JVI.01265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Moriyama M, Chen IY, Kawaguchi A, Koshiba T, Nagata K, Takeyama H, Hasegawa H, Ichinohe T. The RNA-and TRIM25-binding domains of influenza virus NS1 protein are essential for suppression of NLRP3 inflammasome-mediated interleukin-1beta secretion. J Virol. 2016;90:4105–4114. doi: 10.1128/JVI.00120-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Cheong WC, Kang HR, Yoon H, Kang SJ, Ting JP, Song MJ. Influenza A virus NS1 protein inhibits the NLRP3 inflammasome. PLoS One. 2015;10:e0126456. doi: 10.1371/journal.pone.0126456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ichinohe T. Respective roles of TLR, RIG-I and NLRP3 in influenza virus infection and immunity: impact on vaccine design. Expert Rev Vaccines. 2010;9:1315–1324. doi: 10.1586/erv.10.118. [DOI] [PubMed] [Google Scholar]
- 208.McAuley JL, Tate MD, MacKenzie-Kludas CJ, Pinar A, Zeng W, Stutz A, Latz E, Brown LE, Mansell A. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 2013;9:e1003392. doi: 10.1371/journal.ppat.1003392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Yoshizumi T, Ichinohe T, Sasaki O, Otera H, Kawabata S, Mihara K, Koshiba T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat Commun. 2014;5:4713. doi: 10.1038/ncomms5713. [DOI] [PubMed] [Google Scholar]
- 210.Aaltonen T, Abazov VM, Abbott B, Acharya BS, Adams M, et al. Tevatron constraints on models of the Higgs boson with exotic spin and parity using decays to bottom–antibottom quark pairs. Phys Rev Lett. 2015;114:151802. doi: 10.1103/PhysRevLett.114.151802. [DOI] [PubMed] [Google Scholar]
- 211.Zhirnov OP, Klenk HD. Influenza A virus proteins NS1 and hemagglutinin along with M2 are involved in stimulation of autophagy in infected cells. J Virol. 2013;87:13107–13114. doi: 10.1128/JVI.02148-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zhou Z, Jiang X, Liu D, Fan Z, Hu X, Yan J, Wang M, Gao GF. Autophagy is involved in influenza A virus replication. Autophagy. 2009;5:321–328. doi: 10.4161/auto.5.3.7406. [DOI] [PubMed] [Google Scholar]
- 213.Hahn DR, Na CL, Weaver TE. Reserve autophagic capacity in alveolar epithelia provides a replicative niche for influenza A virus. Am J Respir Cell Mol Biol. 2014;51:400–412. doi: 10.1165/rcmb.2013-0437OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Gannage M, Dormann D, Albrecht R, Dengjel J, Torossi T, Ramer PC, Lee M, Strowig T, Arrey F, Conenello G, Pypaert M, Andersen J, Garcia-Sastre A, Munz C. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe. 2009;6:367–380. doi: 10.1016/j.chom.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Beale R, Wise H, Stuart A, Ravenhill BJ, Digard P, Randow F. A LC3-interacting motif in the influenza A virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe. 2014;15:239–247. doi: 10.1016/j.chom.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, Huang G, Green M, Kundu M, Chi H, Xavier RJ, Green DR, Lamkanfi M, Dinarello CA, Doherty PC, Kanneganti TD. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol. 2013;14:480–488. doi: 10.1038/ni.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]