

Abstract
Vaccinia virus (VACV) envelope protein D8 is one of three glycosaminoglycan adhesion molecules and binds to the linear polysaccharide chondroitin sulfate (CS). D8 is also a target for neutralizing antibody responses that are elicited by the smallpox vaccine, which has enabled the first eradication of a human viral pathogen and is a useful model for studying antibody responses. However, to date, VACV epitopes targeted by human antibodies have not been characterized at atomic resolution. Here, we characterized the binding properties of several human anti-D8 antibodies and determined the crystal structures of three VACV-mAb variants, VACV-66, VACV-138, and VACV-304, separately bound to D8. Although all these antibodies bound D8 with high affinity and were moderately neutralizing in the presence of complement, VACV-138 and VACV-304 also fully blocked D8 binding to CS-A, the low affinity ligand for D8. VACV-138 also abrogated D8 binding to the high-affinity ligand CS-E, but we observed residual CS-E binding was observed in the presence of VACV-304. Analysis of the VACV-138– and VACV-304–binding sites along the CS-binding crevice of D8, combined with different efficiencies of blocking D8 adhesion to CS-A and CS-E allowed us to propose that D8 has a high- and low-affinity CS-binding region within its central crevice. The crevice is amenable to protein engineering to further enhance both specificity and affinity of binding to CS-E. Finally, a wild-type D8 tetramer specifically bound to structures within the developing glomeruli of the kidney, which express CS-E. We propose that through structure-based protein engineering, an improved D8 tetramer could be used as a potential diagnostic tool to detect expression of CS-E, which is a possible biomarker for ovarian cancer.
Keywords: antibody, glycosaminoglycan, protein crystallization, protein structure, vaccine, viral protein, X-ray crystallography
Introduction
Smallpox is caused by infection with variola virus and was a major health threat until successful global vaccination efforts led to its complete eradication from the general human population (1). This eradication of an infectious agent was achieved by immunizing with vaccinia virus (VACV),2 a related orthopoxvirus with low virulence and the active ingredient in the smallpox vaccine (2, 3). Immunization with VACV leads to the robust production of highly neutralizing antibodies targeting several VACV envelope proteins, including A27, A33, B5, D8, H3, L1, and others, which are expressed on different viral envelopes (3, 4). A27, D8, H3, and L1 are expressed on the outer membrane of the intracellular mature virion (IMV), whereas A33 and B5 are found in the more fragile extracellular enveloped virion (EEV), which has an additional host cell-derived envelope. Both the IMV and EEV particles are infectious virions, with IMV being more abundant and mainly responsible for spread between hosts, and EEV being involved in cell-to-cell spread within the host after infection with the IMV (5). As a result, a potent antibody-mediated immune response against VACV targets both infectious virions.
Among the targeted envelope proteins, A27, D8, and H3 are glycosaminoglycan (GAG) adhesion molecules. GAGs are linear, mostly sulfated polysaccharides with repeating disaccharide units. The precise sulfation of each GAG is regulated by specific sulfotransferases, leading to the production of different isoforms. GAGs are covalently attached to various core proteins to form the proteoglycan (PG). They are ubiquitously expressed on cell surfaces and in extracellular matrices (6). In addition to providing structural integrity, GAGs participate in various physiological processes such as cell adhesion, cell proliferation, migration, neurite outgrowth, microbial adhesion and infection, inflammation, and angiogenesis (7–11). Many pathogens encode GAG-binding proteins as a major route of entry (12). Although A27 and H3 (13–15) bind to heparan sulfate (HS), D8 binds to chondroitin sulfate (CS) (16). Chondroitin sulfate (β-GlcA-β-(1–3)GalNAc-sulfate) is the most abundant GAG and has 4 major isotypes (chondroitin-4-sulfate, CS-A; chondroitin-6-sulfate, CS-C; chondroitin-2,6-sulfate, CS-D; chondroitin-4,6-disulfate, CS-E) in mammals (17). We have determined previously the structure of the VACV D8 protein, which revealed an N terminally located carbonic anhydrase (CAH) domain (residues 1–235), followed by a disordered stalk region (residues 236–267) that is required for higher oligomeric assembly of the D8 hexamer in solution, and for embedding D8 in the virial envelope (18, 19). We have further identified CS-E as the optimal ligand for D8 and identified a positively charged crevice as the CS-binding site (19). Computational docking suggested that the crevice is capable of binding to at least an octasaccharide of CS-E (18).
We also previously characterized the binding pattern for a panel of D8-reactive murine antibodies, all of which targeted the CAH domain rather than the stalk region, and can be grouped into 4 different specificity groups based on cross-blocking studies (18, 20, 21). One of these antibodies, LA5, is moderately neutralizing in the presence of complement and prevents D8 binding to CS-E, because LA5 binds above the CS-E binding crevice (18, 19).
However, to date, VACV epitopes that are targeted by human antibodies have not been characterized to atomic resolution using X-ray crystallography, and in total only the structures of eight murine antibodies bound to their VACV antigens A27, A33, D8, and L1 have been determined (19, 22–25).
Previously, we reported isolation of a panel of 21 anti-D8 human monoclonal antibodies (mAb) and assessed their neutralizing and protective capabilities (26). In this study, we have characterized the epitopes for some of these human D8 antibodies and determined the first crystal structures of human antibodies bound to D8, using three clones designated VACV-66, VACV-138, and VACV-304. Similarly to the murine antibodies, human antibodies bound to the CAH domain of D8 and were moderately neutralizing in the presence of complement. Two antibodies, VACV-138 and VACV-304, also fully blocked D8 binding to CS-A, whereas VACV-304 only partially blocked D8 binding to the high-affinity ligand CS-E, revealing a high- and low-affinity CS-binding region within the central D8 crevice.
Results
Human anti-D8 mAbs identify a novel D8 epitope
We analyzed four human anti-D8 mAbs VACV-66, VACV-138, VACV-249, and VACV-304 for their ability to block each other's binding to recombinant D8 using a real-time binding assay biolayer interferometry. VACV-66 was the only antibody that did not compete with any other mAb for D8 binding, suggesting it bound a distinct epitope on D8 (Fig. 1A). VACV-138, VACV-249, and VACV-304 all competed with each other for binding to D8, suggesting that their epitopes on D8 were overlapping. However, a previous study suggested that cross-blocking of VACV-249 with VACV-304 depends on the order of antibodies used in the assay, suggesting the overlap is only partial (26). Next, we used ELISA to analyze the ability of VACV-66 and VACV-304, as representatives of the two major human mAb groups, to block binding of murine anti-D8 mAbs in a previously characterized antibody panel (21). We included representative murine mAbs from specificity groups I, II, and IV to ask whether the human mAbs recognized common or unique epitopes on D8 (Fig. 1B). Although VACV-304 blocked binding of mouse CC7.1 (group II), human VACV-66 appeared to recognize a unique epitope on D8, notwithstanding that there was slight cross-blocking with the murine group IV antibodies HG12 and LA5 (Fig. 1). LA5 had been shown previously to block D8 binding to its ligand CS-E (18, 19), raising the possibility that some human mAbs may also prevent D8 binding to CS-E.
Figure 1.

Cross-blocking of human and mouse D8 Abs reveal a novel epitope. A, human antibody cross-blocking reveals two specificity groups (columns labeled 1 and 2). B, overlap between mouse and human mAbs identify a separate epitope for VACV-66. Black cells correspond to Abs that fully block D8 binding of each other and are thus assumed to share overlapping epitopes. Empty cells correspond to Abs that did not affect the D8 binding of each other. Relative strength of cross-blocking is indicated in shades of gray, because epitopes may overlap partially and allow simultaneous binding of both antibodies. Columns 1–4 indicate the four different specificity groups.
Human and murine anti-D8 mAbs possess moderate neutralizing potency
We next tested the ability of anti-D8 mAbs to neutralize IMV in a flow cytometry-based assay. Although the murine control anti-L1 mAb M12B9 neutralized IMV in the absence of complement, most anti-D8 mAbs required the presence of complement to neutralize IMV, regardless of whether they were of human or murine origin (Fig. 2). Murine mAb JF11 was the only anti-D8 mAb that did not neutralize, whereas neutralization mediated by mAb JE11 also was weak. Compared with L1, D8 appeared to be a target only for antibodies with a moderate level of neutralizing potency, likely because the GAG adhesion molecules A27 and H3 can compensate for the function of D8.
Figure 2.

Human anti-D8 mAbs are moderately neutralizing. Shown is VACV IMV neutralization activity of purified human (VACV-249, VACV-304, and VACV-66) and mouse (JE11, JF11, and LA5) anti-D8 mAbs in the absence (black bars) or presence (red bars) of complement. Murine anti-L1 mAb (M12B9) was used as positive control and anti-L1 mAb 39D4 as control for a weakly neutralizing mAb.
Human anti-D8 mAbs bind with high affinity
Next, we determined the binding affinity of human anti-D8 mAbs for recombinant D8. Although VACV-66, VACV-138, and VACV-249 bound D8 with relatively low affinity (KD = 0.35 to 1.9 nm), VACV-304 bound with much higher affinity (KD = 16 pm, Fig. 3). mAb VACV-249 had a comparably low association rate (kon = 2 × 105 1/ms), whereas VACV-66, VACV-138, and VACV-304 bound similarly fast (kon = 6.5 to 7.2 × 105 1/ms). Surprisingly, the high affinity of VACV-304 was a result of extremely slow dissociation, which was roughly 100 times slower compared with VACV-66 (koff = 1 × 10−5 versus 1 × 10−3 1/s). This finding suggested that VACV-304 formed a stable complex with D8 that was characterized by a long half-life in solution. As the kinetic measurements were performed with immobilized intact IgG, only the 1:1 binding interaction between a single Fab and D8 was measured. Increases in avidity due to antibody bivalency were not addressed in this experimental design. As a result, binding of the antibody to D8 embedded in the virion membrane during natural infection is likely even higher.
Figure 3.

Real-time binding kinetics using biolayer interferometry. mAbs were immobilized on anti-human Fc capture antibody sensors and the 1:1 binding of each Fab to monomeric D8 was measured by dipping into increasing concentrations (colored curves) of D8. Kinetic measurements were unaffected by antibody avidity. All antibodies bind with high affinity.
Human antibodies can interfere with D8 adhesion to CS
We next asked if any of the antibodies could prevent binding of D8 to its host ligand chondroitin sulfate-E (CS-E), similarly to the murine antibody LA5 (18), and whether the ability to block CS binding correlated with their neutralization capacity. mAbs were pre-bound to oligomeric D8, because monomeric D8 does not bind to CS-A. The D8–mAb complexes were assessed for their binding ability to CS-A and CS-E using GAG microarrays (Fig. 4). Although VACV-66 did not impair D8 binding to CS-A or CS-E, both VACV-138 and VACV-304 fully abrogated D8 binding to CS-A. Although binding to CS-E was greatly reduced when VACV-304 bound to D8, a low level of binding (25% of maximum binding) was consistently observed. As CS-E is the high-affinity D8 ligand, and CS-A is only bound by oligomeric D8, VACV-304 likely binds at one of the extremities of the crevice. This has been confirmed by structural studies discussed below. VACV-304 would then leave the remainder of the D8 crevice uncovered and available to bind CS-E. CS-A, on the other hand, would not be able to bind to only a partially accessible D8 crevice due to its lower binding affinity compared with CS-E. Although the mAbs do not show great differences in extent of IMV neutralization (∼20–50%), all of the mAbs that prevented CS adhesion (VACV-138, VACV-304, and LA5) neutralize to slightly higher levels compared with mAbs VACV-66, JE11, and JF11 that did not block CS adhesion (Figs. 2 and 3) (18). VACV-249 was not included in the CS microarray but since its binding site on D8 partially overlaps with that of VACV-138 and VACV-304, it is likely that VACV-249 also could impair D8 binding to CS-E.
Figure 4.

Interference of D8 binding to CS-A or CS-E by human mAbs. Hexameric D8 was incubated with mAbs VACV-66, VACV-138, VACV-304, or alone prior to binding to GAG microarrays. Antibody blocking was assessed by comparing the amount of D8 and D8–mAb complex bound to the microarray. Blocking experiments were performed in triplicate (± S.E., error bars). Although VACV-66 does not block D8 binding to CS-A or CS-E, VACV-138 blocks binding to both. VACV-304 fully blocks binding to CS-A, whereas slight binding to CS-E was observed (∼25% of maximum binding).
Structural characterization of the D8 epitopes targeted by human mAbs
To identify and characterize the D8 epitopes that are targeted by the human mAbs, we determined the crystal structures of VACV-66, VACV-138, and VACV-304 bound to the CAH domain of D8, to resolutions of 2.23, 2.9, or 2.9 Å, respectively (Table 1).
Table 1.
Data collection and refinement statistics
| Data collection | |||
| Antibody complex | D8/VACV-66 | D8/VACV-138 | D8/VACV-304 |
| PDB ID | 5USH | 6B9J | 5USL |
| Space group | P212121 | C2221 | P4 |
| Cell dimension | |||
| a, b, c, (Å) | 55.9, 61.5, 444.4 | 234.3 253.8, 73.8 | 118.0, 118.0, 104.6 |
| ɑ, β, ɣ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution range (Å) (outer shell) | 38.8–2.23 (2.35–2.23) | 117.1–2.9 (3.06–2.90) | 47.8–2.9 (3.06–2.90) |
| No. of unique reflections | 75,859 (10,617) | 48,731 (7,089) | 30,196 (4,480) |
| Rpim (%) | 3.5 (40.5) | 7.1 (60.7) | 11.2 (57.2) |
| Multiplicity | 6.2 (4.9) | 6.2 (6.4) | 3.2 (3.3) |
| Average I/σI | 15.7 (1.9) | 8.3 (1.4) | 6.5 (1.5) |
| Completeness (%) | 99.6 (97.5) | 99.4 (99.8) | 94.8 (97.1) |
| Refinement statistics | |||
| No. atoms | 10,202 | 9,990 | 10,022 |
| Protein | 10,028 | 9,923 | 10,022 |
| Waters | 174 | 48 | 0 |
| Ramachandran plot (%) | |||
| Favored | 97.0 | 94.7 | 95.2 |
| Allowed | 99.6 | 99.8 | 99.8 |
| Root mean square deviations | |||
| Bonds (Å) | 0.007 | 0.007 | 0.004 |
| Angles (°) | 1.23 | 1.19 | 0.85 |
| B-factors (Å2) | |||
| Protein | 52.0 | 50.5 | 56.6 |
| Waters | 44.7 | 63.3 | / |
| R factor (%) | 21.8 | 24.1 | 24.7 |
| Rfree (%) | 25.7 | 26.8 | 29.7 |
VACV-66–D8 complex
VACV-66 binds on the side of D8, away from the CS-binding crevice, explaining the inability of VACV-66 to interfere with D8 attachment to CS-E (Fig. 5). The VACV-66-binding site on D8 is rotated 90° compared with the CS-E–binding crevice and is adjacent to that of the murine group II, III, and IV antibodies, correlating with a low degree of cross-blocking with representative antibodies from these groups (Fig. 1).
Figure 5.

Structural overview of human mAb–D8 complexes. A, binding of VACV-66, VACV-138, or VACV-304 to D8. B, mAb footprint on D8 is colored by chain on the D8 protein otherwise shown in gray. Heavy-chain footprint is shown in orange and light-chain in green. The CS-binding site is indicated with a black dot (A) and a yellow line (B). Both VACV-138 and VACV-304 bind above the CS-binding crevice, whereas VACV-66 binds on the side of D8 away from the crevice.
The Fab binds to a total of 21 discontinuous D8 residues. The heavy chain (HC) buries a total of 1683 Å2, whereas the light chain (LC) buries a total of 313 Å2 (Table S1). The HC dominates the overall interactions and binds to 20 D8 residues (Gln-3, Gln-4, Asn-18, Arg-20, Leu-21, Phe-56, Pro-58, Tyr-61, Lys-109, His-110, Asp-111, Asp-112, Ile-115, Leu-170, Asp-179, Val-181, Asp-228, Thr-229, Glu-230, and Tyr-232), whereas the LC contacts three D8 residues (Asn-18, Ala-19, and Arg-20). All three HC CDRs are involved in the interaction, forming an intricate H-bond network of 21 H-bonds and one salt bridge, whereas the LC forms a cation–π interaction between Tyr-33L and Arg-20 of D8 (Fig. 6 and Table S2). VACV-66 exhibited a relatively fast dissociation from D8, possibly as a result of the lack of strong electrostatic, or hydrophobic interactions with D8. VACV-66 is the only one of the three antibodies studied that did not engage in salt bridges with the antigen (Table S2). The VACV-66-binding site on D8 also is rather shallow and broad, with only minimal participation of the LC in antigen binding.
Figure 6.

Interactions between mAbs and D8. A–C, binding VACV-66 (A), VACV-138 (B), or VACV-304 (C) to D8. D8 is shown in gray and the mAb CDRs are colored as follows: L1 (green), L2 (light green), L3 (dark green), H1 (yellow), H2 (cyan), and H3 (orange). Potential H-bonds are indicated with blue dotted lines.
VACV-138–D8 complex
VACV-138 blocks D8 adhesion to CS and binds directly over the CS-binding crevice (Fig. 5). VACV-138 was not included in the murine antibody cross-blocking study, but shares several contact residues with murine LA5, which also interferes with CS-E adhesion. Compared with LA5, however, the footprint of VACV-138 is not located centrally above the binding crevice but rather is shifted along the crevice. VACV-138 engages D8 in a slightly tilted manner compared with the more centrally placed murine LA5 (19). The VACV-138 footprint is delineated by Lys-48 and Glu-105 at one end, and Thr-39 and Asn-175 at the other end, thereby sitting over both sides of the crevice.
The Fab binds to a total of 21 discontinuous D8 residues and the HC buries a total of 1465 Å2, whereas the LC buries a total of 435 Å2. As a result, the LC is slightly more involved in D8 binding compared with mAb VACV-66. Nevertheless, the HC dominates the overall interactions and binds to 17 D8 residues, whereas the LC contacts five D8 residues (Table S1). All three HC CDRs bind D8 with a total of 15 H-bonds and three salt bridges, whereas the LC only forms one polar interaction using L3 (Fig. 6 and Table S2). The additional electrostatic interactions compared with VACV-66 are consistent with the reduced dissociation from D8 and an overall higher binding affinity.
VACV-304–D8 complex
Similar to VACV-138, VACV-304 also blocks D8 adhesion to CS and binds over the CS-binding crevice (Fig. 5). However, in contrast to VACV-138, VACV-304 binds closer to the end of the crevice, delineated by Lys-48 and Lys-98 on one end and Asn-145 and Glu-105 on the other end. Likely due to the slightly altered footprint compared with VACV-138 and the loss of interaction with D8 residue Asn-175, the CS-binding crevice is more accessible upon VACV-304 binding and thus residual binding to the high-affinity ligand CS-E was observed (Fig. 4). In addition, slight cross-blocking with LA5 was observed, because VACV-304 and LA5 share the D8 contact residues Thr-39, Arg-44, and Asn-145, which are the peripheral binding residues of LA5 (19).
The Fab binds to a total of 21 discontinuous D8 residues, and the HC buries a total of 1010 Å2, whereas the LC buries a total of 977 Å2. The HC binds to 14 D8 residues, whereas the LC contacts 10 D8 residues (Table S1). All three HC CDRs bind D8 with a total of 8 H-bonds, whereas the LC forms 5 H-bonds and 1 salt bridge using L2 and L3 (Fig. 6 and Table S2). In contrast to VACV-66 and VACV-138, the LC of VACV-304 is far more involved in D8 binding, as judged by the buried surface area and the number of contact residues. This finding is consistent with the highest observed binding affinity among the tested human mAbs.
In summary, the Fab binding affinity correlates well with the number of electrostatic interactions, as well as the involvement of the LC in D8 binding. VACV-66, which has the lowest affinity, has the least involvement of the LC in binding and lacks salt bridges, whereas VACV-304 has the highest affinity, establishes several salt bridges and also the most contacts using the LC. The affinity differences are driven mostly by the off-rate, suggesting a locking mechanism when both LC and HC bind to D8, leading to a reduced off-rate. VACV-304 has a 100-fold reduced off-rate compared with VACV-66.
Antibody binding to D8 mutants
We generated a panel of 10 D8 mutants in which either one (Gln-3, Tyr-85, Tyr-101, Glu-105, Glu-106, Asp-112, Asn-1675, and Arg-220) or two (Arg-44/Arg-220 and Lys-48/Arg-220) residues were replaced with alanine to assess the effect of a particular amino acid side chain in antibody binding (Fig. S1). The D8 double mutants Lys-48/Arg-220 and Arg-44/Arg-220 had been shown previously to abrogate D8 binding to CS-E (18).
mAb VACV-66
None of the mutations tested significantly affected VACV-66 binding. Of the 10 mutants, Gln-3 and Asp-112 are contact residues but did not appear to be critical for binding. In the structure, Gln-3 forms a H-bond with Tyr-55H of VACV-66, but because the carboxylate of Asp-179 also contacts Tyr-55H, the interaction with Gln-3 appears to be dispensable. Asp-112 forms two potential H-bonds with Y60H but because the mutation has no effect, loss of these interactions is likely compensated for by the other contact residues. Several D8 mutants appear to increase antibody dissociation slightly, but because they are located outside the binding interface they likely have an allosteric rather than a direct effect on the antibody binding.
mAb VACV-138
Binding of VACV-138 to the D8 mutants R44A/R220A was abrogated, because Arg-44 is a centrally located contact residue that binds to D52H through two salt bridges. The K48A/R220A mutant only showed slightly increased dissociation, suggesting contact of VACV-138 with Lys-48, which lies at the periphery of the epitope. This finding was somewhat surprising, given that Lys-48 engages both Thr-28H with an H-bond and Asp-31H using a salt bridge. Arg-220, which is present in both mutants, is not part of the epitope, and as such the R220A single-position mutant did not exhibit reduced binding to VACV-138. The E105A mutation also demonstrated no effect on binding because it is located at the periphery of the epitope as well (Fig. 5).
mAb VACV-304
Because VACV-304 binds D8 slightly offset along the CS-binding crevice compared with VACV-138, VACV-304 binding to the mutant R44A/R220A was not impaired, in contrast to the binding of VACV-138. Arg-44 is not a contact residue for VACV-304, although VACV-304 and VACV-138 bind at a similar D8 site. This finding is in contrast to that for VACV-138, for which the binding to the two aforementioned mutants was reversed. Lys-48 is a LC contact and appeared important for D8 binding, because it engaged both Asp-50L with a salt bridge and also forms extensive non-polar VdW interactions with Phe-31L.
The D8 crevice has a low- and high-affinity binding region for CS-E
As VACV-304 binds to one extremity of the CS-E–binding crevice, residual binding to the high-affinity ligand CS-E was observed consistently (Fig. 4). At least half of the binding crevice was not covered upon VACV-304 binding (Fig. 7). As CS-A binding was not observed in the presence of VACV-304, we postulate that there is an affinity threshold necessary for binding, which is not reached by CS-A but is by CS-E, correlating with the finding that CS-A is a lower affinity ligand for D8. It should be noted that monomeric D8 does not bind to CS-A but only to CS-E (18). In addition, LA5 binds to the opposite extremity of the binding crevice and fully blocks CS-E binding. Therefore, we speculate that the binding crevice in D8 has a high- and low-affinity CS-binding region. A detailed view at the D8/CS-E computationally docked model correlates well with a high-affinity CS-binding region located at the C-terminal end of the D8 crevice, which starts with residues Arg-220 and His-176 and ends in the middle of the crevice around Lys-108 and Arg-44 (Fig. S2). This region is covered by the antibody LA5 but is accessible when VACV-304 is bound. In this model, more polar interactions with CS-E are predicted in the high-affinity region compared with the low-affinity region, where only a few interactions are predicted. In conclusion, the low-affinity CS-binding region is amenable to protein engineering to increase the specificity and affinity toward CS-E. Such a protein would be useful in generating a highly specific and high-affinity CS-E-binding reagent for detecting changes in CS-E expression in various tissues, which would correlate with cancer progression, because CS-E has been identified as a potential biomarker for ovarian cancer (27).
Figure 7.
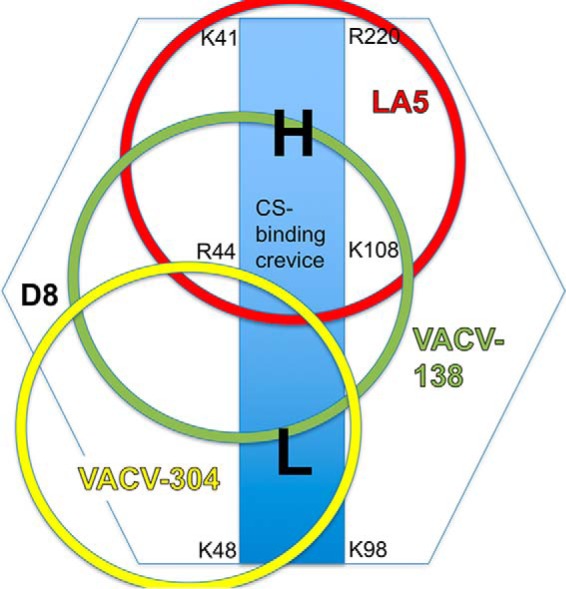
Footprint of CS-blocking mAbs on D8. Shown is a schematic representation of mAb binding sites (circles) and illustration of a high- (H) and low- (L) affinity binding region for CS within the central binding crevice (blue rectangle) of D8.
Immunofluorescence microscopy using D8 tetramers
As monomeric D8 specifically binds to the glycosaminoglycan CS-E, we asked whether we could use D8 in microscopy to identify CS-E expression in tissues and generated a D8 tetramer conjugated to Alexa Fluor 488. Because the CS-E-specific antibody GD3G7 was reported to bind to developing glomeruli of the kidney (27), we assessed the staining of D8 on kidney sections isolated from 1-week-old mice. Glomeruli were stained in green using phalloidin, a toxin with high affinity for F-actin enriched in these structures and D8 control tetramer, which cannot bind to CS-E, in red (Fig. 8). Although D8 does indeed bind to specific structures in glomeruli, it only stains localized regions, suggesting a highly specific binding pattern. The D8 control tetramer showed no binding to the glomerulus, in contrast to wild-type D8. However, there was a low-grade immunoreactivity of this reagent to adjacent regions. This altered binding behavior likely is the result of the four introduced mutations (K41A, R44A, K48A, R220A) that greatly reduce the positive charge of D8 and may favor hydrophobic interactions to unknown structures outside the glomerulus.
Figure 8.

D8 tetramers specifically detect chondroitin sulfate E in tissues. Kidney tissue samples were flash-frozen, sectioned, fixed in 4% paraformaldehyde and reacted with 156 ng/ml of either D8 or D8 control tetramers coupled to Alexa Fluor 488. Samples were counterstained with phalloidin to reveal dense F-actin in the glomerulus and with the DNA stain Hoechst. The D8 tetramer immunoreactivity was localized within the glomeruli. Negative control with the mutated D8 tetramer revealed the absence of binding in the glomerulus but low-grade immunoreactivity in adjacent regions. Bar = 20 μm.
Discussion
Many microbes exist that express GAG-binding proteins to adhere to their host cells for subsequent cell entry and infection (28, 29). These adhesion molecules have evolved to acquire unique binding specificities. VACV expresses three envelope proteins, H3, A27, and D8, that are glycosaminoglycan adhesion molecules. Although H3 and A27 bind to heparin and HS, D8 binds chondroitin sulfate (13–16). Antibodies that target A27, H3, and D8 can block viral adhesion to the host cell and protect from infection, especially if multiple viral envelope proteins are targeted simultaneously (26, 30). It is difficult to correlate mAb-binding affinity and the ability of a single anti-D8 mAb to block viral cell adhesion with neutralization potency, because VACV expresses several antigens that can functionally compensate for each other. This is especially true for the three GAG adhesion molecules A27, H3, and D8. Blocking viral adhesion via one antigen is likely not sufficient to prevent adhesion and subsequent infection. However, many antibodies targeting several different antigens are elicited during the cause of viral infection, and together these antibodies confer protection. As a result, the smallpox vaccine is considered the gold standard of vaccines (3).
Because GAGs are the most abundant heteropolysaccharides in the body and virtually expressed on every cell, they are ideal receptors for viral adhesion. They consist of long unbranched chains with repeating dissacharide units. Most GAGs, including CS are sulfated. The precise sulfation of CS is regulated by specific sulfotransferases, leading to the production of different isoforms. All GAGs are attached to specific serine residues of a core-protein to form the PG (32). They are ubiquitously expressed on cell surfaces and in extracellular matrices. In addition to providing structural integrity, GAGs participates in various physiological processes such as cell adhesion, cell proliferation, migration, neurite outgrowth, microbial adhesion and infection, inflammation and angiogenesis (7–11). The up-regulation of individual GAG-specific sulfotransferases during cellular stress results in the expression of different GAG isoforms and can be a biomarker for cellular changes leading to disease or a method to monitor disease progression. Chondroitin sulfate (GlcAβ1–3GalNAcβ1–4)n is the most abundant GAG and has 3 major isotypes (chondroitin-4-sulfate, CS-A; chondroitin-6-sulfate, CS-C, chondroitin-4,6-O-disulfate, CS-E) (17). The CS sulfotransferase GalNAc4S-6ST, which converts CS-A into CS-E is up-regulated in ovarian cancer and results in an enrichment of CS-E on the cancer cell surface, whereas CS-C and CS-E are enriched in colon carcinomas (33, 34). CS-E has been proposed to be involved in ovarian cancer tumorigenesis as it mediates binding of the vascular endothelial growth factor (27, 33). More recently, the malaria protein VAR2CSA was shown to bind a form of CS-A that is both expressed in the placenta and various cancers, especially on cancer-associated proteoglycans including CD44 and CSPG but not on healthy tissues (35). These findings suggest that detection of defined isoforms of CS can be a useful diagnostic in monitoring cancer progression.
Several anti-CS-specific antibodies exist that target CS-A. Antibodies against different CS isoforms (CS-A, CS-C, and CS-E) exist but often their specificity toward the individual sulfation patterns is limited, or their epitope is rather small (1–2 disaccharides), leading to antibody cross-reactivity with other similar sulfated isoforms (36–39). The CS-E-specific antibody GD3G7 also binds moderately to oversulfated dermatan sulfate (DS) from hagfish (IdoA-GalNac4S6S) and shark skin DS (40). Therefore, the antibody also recognizes IdoA, a component of dermatan sulfate and is not exclusively CS specific. A new CS-E-specific antibody has recently been reported that can distinguish between iduronic and glucuronic acid and is, similar to D8, specific for CS (and not DS) (39). However, as the GAGs were mostly obtained commercially and purified from natural source, heterogeneity in the CS sulfation patterns is to be expected, i.e. squid cartilage from Sigma is labeled as containing ∼61% CS-E, among other CS structures. An additional limitation is that no structural information of any antibody bound to any CS exists so the antibody recognition of and interaction with a particular GAG molecular species is unknown. Some antibodies, such as the CS-E-specific antibody from Ref. 36 are very specific but also moderately binds to the CS-E disaccharide. This could lead to off-target binding, e.g. the CS-E-specific antibody could stain tissues or cellular structures that are enriched for CS-A or CS-C but contain one or two CS-E disaccharides to which it binds, falsely suggesting that this cell expresses largely CS-E. In summary, the different CS-binding antibodies have different limitations and a more specific CS-binding reagent that recognizes longer stretches, such as minimally a hexamer is needed. This specific binding reagent would allow a more careful distinction of cell surface-expressed CS isoforms and their role in disease onset or progression, ultimately leading to better diagnostics needed for biomarker discovery.
Because D8 specifically binds to CS-E using a longitudinal binding crevice capable of binding at least 4 disaccharide units (18), we propose that the D8 protein is a suitable scaffold for the design of a highly specific CS-E–binding protein as a potential diagnostic tool to monitor cancer progression that is linked to CS-E up-regulation, such as ovarian cancer.
Experimental procedures
Human antibody generation
Human anti-D8 mAbs VACV-66, VACV-138, VACV-249, and VACV-304 were generated as reported previously (26). Hybridoma cells secreting VACV-specific mAbs were generated previously (26) and grown in serum-free medium (Gibco). mAbs were purified from culture supernatants using a HiTrap MabSelect Sure column (GE Healthcare). Nucleotide sequences of variable gene segments were determined by Sanger sequencing from cloned cDNA generated by reverse transcription PCR of mRNA, using variable gene-specific primers, as reported previously (41).
Murine antibody generation
Murine anti-D8 mAbs JE11, JF11, and LA5, as well as anti-L1 mAbs M12B9 and 39D4, were reported previously (19, 20, 23).
Competition-binding ELISA
The ELISA was performed as previously reported (21). Briefly, recombinant D8 (residues 1–263) was coated on 96-well flat-bottom plates, washed with PBS + 0.05% Tween 20, and blocked by adding 10% fetal bovine serum. The antibody of interest was added to the D8-bound wells, washed off, and the ability of a second antibody to bind simultaneously to D8 was assessed. For this, the second antibody was HRP-conjugated, and its binding to D8 was assessed by measuring optical density at 490 nm. Total cross-blocking results in no signal, whereas a distinct binding site on D8 would result in maximum signal of the second antibody.
Antibody-mediated neutralization of virus
In vitro antibody neutralization was performed using a flow cytometry-based assay, as reported previously (23, 26, 42). Briefly, Vero E6 cells (ATCC, CRL-1586) were seeded in 96-well plates (1 × 105 cells/well) and allowed to adhere for 5 h. Cells were then infected with 1.25 × 104 PFU of purified VACV-GFP and mAbs (final mAbs concentration of 20 μg/ml) for 12 h at 37 °C and 5% CO2 in 50 μl in the presence (2% final concentration) or absence of sterile baby rabbit complement (CEDARLANE).
D8 constructs and tetramer generation
The VACV (Acam2000) D8 ectodomain (residues 1–263), as well as its CAH (residues 1–235), were cloned separately into the pET22b vector to generate hexameric (residues 1–263) or monomeric (residues 1–235) D8 protein forms. For D8 tetramer generation, we added a C-terminal 15-amino acid glycine-serine linker (GGGGS)3 followed by a biotin protein ligase (birA) tag and a His6 tag to the CAH construct. A CS-E binding-deficient D8 quadruple mutant (K41A,R44A,K48A,R220A) was generated as a specificity control for immunofluorescence experiments. The resulting expression plasmids were transformed into CodonPlus BL21 cells (Agilent), and D8 expression and purification was performed as reported (19). Briefly, expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside at 37 °C for 4 h at a cell density of 0.6 (A600). Cells were pelleted by centrifugation (10 min at 4000 × g), resuspended in 50 mm Tris, pH 8.0, and sheared with a microfluidizer (3 rounds at 1400 bars, Microfluidics). Crude lysate was clarified by centrifugation (1 h at 50,000 × g), and D8 was purified using ion metal affinity chromatography on 1-ml HisTrap cartridges (GE Healthcare). After washing the column with 20 cv of 50 mm Tris, pH 8.0, 300 mm NaCl, and 20 mm imidazole, followed by an additional wash step with 20 cv of 50 mm imidazole containing wash buffer, D8 was eluted with 50 mm Tris, pH 8.0, 300 mm NaCl, and 250 mm imidazole. D8 was dialyzed against 10 mm Tris, pH 8.0, 200 mm NaCl prior to size exclusion chromatography (SEC). For tetramer preparation, birA-tagged D8 was expressed and purified in a similar manner and biotinylated using the biotin protein ligase, according to the manufacturer's suggestions (Avidity). Free biotin was removed using 10-kDa MWCO filtration devices, and D8 was tetramerized by incubation with Alexa Fluor-conjugated streptavidin using a molar excess of D8 (5 D8 monomers per 1 streptavidin tetramer). D8 tetramer was purified from excess D8 monomer using SEC.
Glycosaminoglycan microarray assay
CS GAGs enriched in CS-A, CS-C, CS-D, and CS-E (Seikagaku Corp., Tokyo, Japan), DS (Sigma), hyaluronic acid (HA; Sigma), heparin (Hep; Neoparin, Alameda, CA), HS (Sigma), or CS (Sigma) were printed on poly-dl-lysine-coated glass surfaces, as described previously (43, 44). Antibodies were preincubated at a 10-fold molar excess with hexameric D8 before assessing D8 binding to CS-A and CS-E. Experiments were conducted as previously published (18).
Real-time binding studies using biolayer interferometry
Intact IgGs were loaded onto anti-human Fc sensor tips (20 nm concentration) and binding to increasing concentrations of monomeric D8 (0.625 to 40 nm) was measured, as previously reported (18). For qualitative assessment of antibody binding to variant D8 protein, a single antibody was loaded on the sensor tip and binding to wild-type D8 or seven variant D8 proteins was measured simultaneously using a saturating concentration of monomeric D8 (5 μm). Only a complete abrogation of binding was considered as significant, because overall binding amplitudes differed between individual D8 variant proteins.
Fab preparation
Purified IgGs for mAbs VACV-66, VACV-138, and VACV-304 (1 mg/ml) were digested with 2% (w/w) activated papain (Sigma, number P3125) for 4 h at 37 °C in in 100 mm Tris-HCl, pH 7.5, 1 mm EDTA, 10 mm cysteine. The papain digestion was stopped by adding 20 mm iodoacetamide. Digestion mixtures were dialyzed against PBS for subsequent protein A purification to remove undigested IgG and Fc. The protein A flow-through containing Fabs was concentrated and purified by SEC on a Superdex S200 GL10/300 column (GE Healthcare), using 50 mm Hepes, pH 7.5, 150 mm NaCl as running buffer.
Fab–D8 complex preparation
20% molar excess of monomeric D8 protein was used to prepare the D8–Fab complex with Fab made from mAbs VACV-66, VACV-138, or VACV-304. Complexes were prepared at a low concentration (∼0.2 mg/ml) and incubated on ice for 5 min, concentrated, and separated from unbound D8 using SEC on a Superdex 200 100/300 GL column (GE Healthcare). Fractions corresponding to the D8–Fab complexes (molecular mass ∼82 kDa) were pooled and concentrated to ∼10 mg/ml for subsequent crystallization.
Crystallization and structure determination
Initially, suitable crystallization conditions were determined using a liquid handling robot (Phenix, Art Robbins Ltd.) and commercially available screens. Quality diffracting crystals then were grown manually using the sitting drop vapor diffusion method by mixing 0.5 μl of protein with 0.5 μl of precipitate; 20% PEG 3350, 200 mm sodium malonate, pH 5.0 (D8/VACV-66 and D8/VACV-138), or 20% PEG 400, 200 mm sodium citrate tribasic (D8/VACV-304). Crystals were flash-cooled at 100 K in mother liquor containing 20% glycerol. Data collection for the D8/VACV-66 crystal included modification from a regular φ scan (SSRL BL11–1, κ = 30°; 0.25 oscillations), due to a very long axis (c = 444 Å). Diffraction data were integrated using XDS (45). All diffraction data were scaled with Scala and phase determination by molecular replacement (MR) was carried out using Phaser (46). MR template for the Fab VACV-66 was PDB code 4FQQ (highest sequence similarity), separated into variable and constant domains, whereas PDB 4E9O (D8) was used as a template for D8. MR search models for VACV-138 and VACV-304 were PDB codes 1RZ7 and 3LMJ, respectively. MR solutions first were refined in ccp4 using rigid body refinement, followed by iterative rounds of model building in COOT (47) and restrained refinement using TLS refinement in REFMAC5, as part of the CCPP4 suite (48–50). Refinement was carried out to a final Rcryst and Rfree of 21.8 and 25.7% (D8/VACV-66), 25.0 and 26.9% (D8/VACV-138), or 24.7 and 29.7% (D8/VACV-304). Data collection and refinement statistics are presented in Table 1.
Structural analyses and presentation
The quality of the models was examined with the program Molprobity (51). Buried surface areas and atomic contacts were calculated using PDBePISA (www.ebi.ac.uk/msd-srv/prot_int/pistart.html).3 Figures were prepared using PyMOL.
Accession numbers
The coordinates and structure factors of the D8–Fab complexes have been deposited in the Protein Data Bank (www.rcsb.org) with codes 5USH, 6B9J, and 5USL. The genetic features of the human anti-D8 antibodies were reported previously (26).
Immunofluorescence
One-week-old mice were sacrificed by CO2 inhalation, followed by immediate decapitation. Kidneys were removed and frozen in Tissue-Tek optimum cutting temperature medium (Sakura Finetek). Eight-μm cryostat sections were fixed with methanol-free 4% formaldehyde (EMS Diasum) for 10 min at room temperature. Following 2 washes with PBS (Gibco), slides were blocked with 5% normal donkey serum, 0.3% Triton X-100 in PBS for 1 h and incubated with 156 ng/ml of D8 or control tetramer that is unable to bind to CS-E (D8 coupled to Alexa Fluor 488 streptavidin diluted in 1% BSA, 0.3% Triton X-100 in PBS. Following an overnight incubation at 4 ºC slides were washed with PBS and counterstained with Alexa Fluor 647 phalloidin (Life Technologies, 1:100) and Hoechst (Life Technologies, 10 μg/ml). Slides were washed and mounted with Prolong Gold using number 1.5 coverslips. Microscopy was performed using a Zeiss LSM780 confocal laser scanning microscope using 63 ×1.4NA objective. Imaging was done using 405, 488, and 633 nm laser lines in a sequential mode to eliminate cross-talk between channels. Acquired Z-stacks were projected to a single plane and contrast was adjusted using Fiji (31). All images were acquired and processed in an identical way. Experiments were repeated 3 times with 2 different batches of tetramers, yielding similar results.
Author contributions
M. H. M. conducted most of the biochemical and structural experiments, analyzed the results, and drafted the paper. A. S. assisted with biochemical and structural experiment. I. M. G. generated human antibodies. G. M. performed the GAG microarray. Z. M. performed immunofluorescence microscopy using the D8 tetramers. M. H. assisted with crystallization experiments, J. W. generated the D8 tetramers, X. M. and Y. X. generated the murine antibodies, T. K. performed antibody cross-blocking ELISA and antibody neutralization experiments. T. D. assisted with data collection, K. L. conceived and supervised the immunofluorescence microscopy experiment and edited the manuscript. S. C. supervised and conceived antibody neutralization experiments and edited the manuscript. B. P. conceived and supervised the antibody cross-blocking experiment and edited the manuscript. L. C. H. W. conceived and supervised the GAG microarray experiment and edited the paper. J. E. C. conceived and supervised human antibody generation and edited the manuscript. D. M. Z. conceived the biochemical and structural experiments, supervised the overall project and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Angela Lamberth for technical assistance in preparing and staining the tissue sections for immunofluorescence microscopy. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the United States Dept. of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences Grant P41GM103393.
This work was supported by NIAID, National Institutes of Health, Department of Health and Human Services Contracts HHSN272200900048C (BP), HHSN272200900047C, and HHSN272201400024C (to J. E. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5USH, 6B9J, and 5USL) have been deposited in the Protein Data Bank (http://wwpdb.org/).
This article contains Tables S1 and S2 and Figs. S1 and S2.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- VACV
- vaccinia virus
- CS
- chondroitin sulfate
- GAG
- glycosaminoglycan
- HC
- heavy chain
- LC
- light chain
- IMV
- intracellular mature virion
- EEV
- extracellular enveloped virion
- PG
- proteoglycan
- HS
- heparan sulfate
- CAH
- carbonic anhydrase
- DS
- dermatan sulfate
- SEC
- size exclusion chromatography
- HA
- hyaluronic acid
- MR
- molecular replacement
- PDB
- Protein Data Bank.
References
- 1. Henderson D. A. (2011) The eradication of smallpox: an overview of the past, present, and future. Vaccine 29, D7–D9 10.1016/j.vaccine.2011.06.080 [DOI] [PubMed] [Google Scholar]
- 2. Amanna I. J., Slifka M. K., and Crotty S. (2006) Immunity and immunological memory following smallpox vaccination. Immunol. Rev. 211, 320–337 10.1111/j.0105-2896.2006.00392.x [DOI] [PubMed] [Google Scholar]
- 3. Moss B. (2011) Smallpox vaccines: targets of protective immunity. Immunol. Rev. 239, 8–26 10.1111/j.1600-065X.2010.00975.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Edghill-Smith Y., Golding H., Manischewitz J., King L. R., Scott D., Bray M., Nalca A., Hooper J. W., Whitehouse C. A., Schmitz J. E., Reimann K. A., and Franchini G. (2005) Smallpox vaccine-induced antibodies are necessary and sufficient for protection against monkeypox virus. Nat. Med. 11, 740–747 10.1038/nm1261 [DOI] [PubMed] [Google Scholar]
- 5. Roper R. L., Wolffe E. J., Weisberg A., and Moss B. (1998) The envelope protein encoded by the A33R gene is required for formation of actin-containing microvilli and efficient cell-to-cell spread of vaccinia virus. J. Virol. 72, 4192–4204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Esko J. D., Kimata K., and Lindahl U. (2009) Proteoglycans and Sulfated Glycosaminoglycans. in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., and Etzler M. E., eds) 2nd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 7. Sugahara K., and Mikami T. (2007) Chondroitin/dermatan sulfate in the central nervous system. Curr. Opin. Struct. Biol. 17, 536–545 10.1016/j.sbi.2007.08.015 [DOI] [PubMed] [Google Scholar]
- 8. Sugahara K., Mikami T., Uyama T., Mizuguchi S., Nomura K., and Kitagawa H. (2003) Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr. Opin. Struct. Biol. 13, 612–620 10.1016/j.sbi.2003.09.011 [DOI] [PubMed] [Google Scholar]
- 9. Bishop J. R., Schuksz M., and Esko J. D. (2007) Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446, 1030–1037 10.1038/nature05817 [DOI] [PubMed] [Google Scholar]
- 10. Fuster M. M., and Esko J. D. (2005) The sweet and sour of cancer: glycans as novel therapeutic targets. Nat. Rev. Cancer 5, 526–542 10.1038/nrc1649 [DOI] [PubMed] [Google Scholar]
- 11. Wadström T., and Ljungh A. (1999) Glycosaminoglycan-binding microbial proteins in tissue adhesion and invasion: key events in microbial pathogenicity. J. Med. Microbiol. 48, 223–233 10.1099/00222615-48-3-223 [DOI] [PubMed] [Google Scholar]
- 12. Bartlett A. H., and Park P. W. (2010) Proteoglycans in host-pathogen interactions: molecular mechanisms and therapeutic implications. Expert Rev. Mol. Med. 12, e5 10.1017/S1462399409001367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chung C. S., Hsiao J. C., Chang Y. S., and Chang W. (1998) A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J. Virol. 72, 1577–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hsiao J. C., Chung C. S., and Chang W. (1998) Cell surface proteoglycans are necessary for A27L protein-mediated cell fusion: identification of the N-terminal region of A27L protein as the glycosaminoglycan-binding domain. J. Virol. 72, 8374–8379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin C. L., Chung C. S., Heine H. G., and Chang W. (2000) Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection in vitro and in vivo. J. Virol. 74, 3353–3365 10.1128/JVI.74.7.3353-3365.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hsiao J. C., Chung C. S., and Chang W. (1999) Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol. 73, 8750–8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mikami T., and Kitagawa H. (2013) Biosynthesis and function of chondroitin sulfate. Biochim. Biophys. Acta 1830, 4719–4733 10.1016/j.bbagen.2013.06.006 [DOI] [PubMed] [Google Scholar]
- 18. Matho M. H., de Val N., Miller G. M., Brown J., Schlossman A., Meng X., Crotty S., Peters B., Xiang Y., Hsieh-Wilson L. C., Ward A. B., and Zajonc D. M. (2014) Murine anti-vaccinia virus D8 antibodies target different epitopes and differ in their ability to block D8 binding to CS-E. PLoS Pathog. 10, e1004495 10.1371/journal.ppat.1004495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Matho M. H., Maybeno M., Benhnia M. R., Becker D., Meng X., Xiang Y., Crotty S., Peters B., and Zajonc D. M. (2012) Structural and biochemical characterization of the vaccinia virus envelope protein D8 and its recognition by the antibody LA5. J. Virol. 86, 8050–8058 10.1128/JVI.00836-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meng X., Zhong Y., Embry A., Yan B., Lu S., Zhong G., and Xiang Y. (2011) Generation and characterization of a large panel of murine monoclonal antibodies against vaccinia virus. Virology 409, 271–279 10.1016/j.virol.2010.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sela-Culang I., Benhnia M. R., Matho M. H., Kaever T., Maybeno M., Schlossman A., Nimrod G., Li S., Xiang Y., Zajonc D., Crotty S., Ofran Y., and Peters B. (2014) Using a combined computational-experimental approach to predict antibody-specific B cell epitopes. Structure 22, 646–657 10.1016/j.str.2014.02.003 [DOI] [PubMed] [Google Scholar]
- 22. Kaever T., Matho M. H., Meng X., Crickard L., Schlossman A., Xiang Y., Crotty S., Peters B., and Zajonc D. M. (2016) Linear epitopes in vaccinia virus A27 are targets of protective antibodies induced by vaccination against smallpox. J. Virol. 90, 4334–4345 10.1128/JVI.02878-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaever T., Meng X., Matho M. H., Schlossman A., Li S., Sela-Culang I., Ofran Y., Buller M., Crump R. W., Parker S., Frazier A., Crotty S., Zajonc D. M., Peters B., and Xiang Y. (2014) Potent neutralization of vaccinia virus by divergent murine antibodies targeting a common site of vulnerability in L1 protein. J. Virol. 88, 11339–11355 10.1128/JVI.01491-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matho M. H., Schlossman A., Meng X., Benhnia M. R., Kaever T., Buller M., Doronin K., Parker S., Peters B., Crotty S., Xiang Y., and Zajonc D. M. (2015) Structural and functional characterization of anti-A33 antibodies reveal a potent cross-species orthopoxviruses neutralizer. PLoS Pathog. 11, e1005148 10.1371/journal.ppat.1005148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Su H. P., Golden J. W., Gittis A. G., Hooper J. W., and Garboczi D. N. (2007) Structural basis for the binding of the neutralizing antibody, 7D11, to the poxvirus L1 protein. Virology 368, 331–341 10.1016/j.virol.2007.06.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gilchuk I., Gilchuk P., Sapparapu G., Lampley R., Singh V., Kose N., Blum D. L., Hughes L. J., Satheshkumar P. S., Townsend M. B., Kondas A. V., Reed Z., Weiner Z., Olson V. A., Hammarlund E., et al. (2016) Cross-Neutralizing and Protective Human Antibody Specificities to Poxvirus Infections. Cell 167, 684–694.e9 10.1016/j.cell.2016.09.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. ten Dam G. B., van de Westerlo E. M., Purushothaman A., Stan R. V., Bulten J., Sweep F. C., Massuger L. F., Sugahara K., and van Kuppevelt T. H. (2007) Antibody GD3G7 selected against embryonic glycosaminoglycans defines chondroitin sulfate-E domains highly up-regulated in ovarian cancer and involved in vascular endothelial growth factor binding. Am. J. Pathol. 171, 1324–1333 10.2353/ajpath.2007.070111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Esko J. D., and Sharon N. (2009) Microbial Lectins: Hemagglutinins, Adhesins, and Toxins. in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., and Etzler M. E., eds) 2nd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 29. Esko J. D., and Linhardt R. J. (2009) Proteins that Bind Sulfated Glycosaminoglycans. in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., and Etzler M. E., eds) 2nd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 30. Benhnia M. R., McCausland M. M., Su H. P., Singh K., Hoffmann J., Davies D. H., Felgner P. L., Head S., Sette A., Garboczi D. N., and Crotty S. (2008) Redundancy and plasticity of neutralizing antibody responses are cornerstone attributes of the human immune response to the smallpox vaccine. J. Virol. 82, 3751–3768 10.1128/JVI.02244-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., and Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Uyama T., Kitagawa H., and Sugahara K. (2007) Biosynthesis of glycosaminoglycans and proteoglycans. in Comprehensive Glycoscience (Kamerling J. P., ed) pp. 79–104, Elsevier, New York [Google Scholar]
- 33. Vallen M. J., van Tilborg A. A., Tesselaar M. H., ten Dam G. B., Bulten J., van Kuppevelt T. H., and Massuger L. F. (2014) Novel single-chain antibody GD3A10 defines a chondroitin sulfate biomarker for ovarian cancer. Biomarkers Med. 8, 699–711 10.2217/bmm.14.6 [DOI] [PubMed] [Google Scholar]
- 34. Joo E. J., Weyers A., Li G., Gasimli L., Li L., Choi W. J., Lee K. B., and Linhardt R. J. (2014) Carbohydrate-containing molecules as potential biomarkers in colon cancer. Omics 18, 231–241 10.1089/omi.2013.0128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Salanti A., Clausen T. M., Agerbaek M. Ø., Al Nakouzi N., Dählback M., Oo H. Z., Lee S., Gustavsson T., Rich J. R., Hedberg B. J., Mao Y., Barington L., Pereira M. A., LoBello J., Endo M., et al. (2015) Targeting human cancer by a glycosaminoglycan binding malaria protein. Cancer Cell 28, 500–514 10.1016/j.ccell.2015.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tully S. E., Rawat M., and Hsieh-Wilson L. C. (2006) Discovery of a TNF-alpha antagonist using chondroitin sulfate microarrays. J. Am. Chem. Soc. 128, 7740–7741 10.1021/ja061906t [DOI] [PubMed] [Google Scholar]
- 37. Deepa S. S., Yamada S., Fukui S., and Sugahara K. (2007) Structural determination of novel sulfated octasaccharides isolated from chondroitin sulfate of shark cartilage and their application for characterizing monoclonal antibody epitopes. Glycobiology 17, 631–645 10.1093/glycob/cwm021 [DOI] [PubMed] [Google Scholar]
- 38. Brown J. M., Xia J., Zhuang B., Cho K. S., Rogers C. J., Gama C. I., Rawat M., Tully S. E., Uetani N., Mason D. E., Tremblay M. L., Peters E. C., Habuchi O., Chen D. F., and Hsieh-Wilson L. C. (2012) A sulfated carbohydrate epitope inhibits axon regeneration after injury. Proc. Natl. Acad. Sci. U.S.A. 109, 4768–4773 10.1073/pnas.1121318109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Watanabe I., Hikita T., Mizuno H., Sekita R., Minami A., Ishii A., Minamisawa Y., Suzuki K., Maeda H., Hidari K. I., and Suzuki T. (2015) Isolation and characterization of monoclonal antibodies specific for chondroitin sulfate E. Glycobiology 25, 953–962 10.1093/glycob/cwv039 [DOI] [PubMed] [Google Scholar]
- 40. Purushothaman A., Fukuda J., Mizumoto S., ten Dam G. B., van Kuppevelt T. H., Kitagawa H., Mikami T., and Sugahara K. (2007) Functions of chondroitin sulfate/dermatan sulfate chains in brain development: critical roles of E and iE disaccharide units recognized by a single chain antibody GD3G7. J. Biol. Chem. 282, 19442–19452 10.1074/jbc.M700630200 [DOI] [PubMed] [Google Scholar]
- 41. Weitkamp J. H., Kallewaard N., Kusuhara K., Feigelstock D., Feng N., Greenberg H. B., and Crowe J. E. Jr. (2003) Generation of recombinant human monoclonal antibodies to rotavirus from single antigen-specific B cells selected with fluorescent virus-like particles. J. Immunol. Methods 275, 223–237 10.1016/S0022-1759(03)00013-9 [DOI] [PubMed] [Google Scholar]
- 42. Benhnia M. R., McCausland M. M., Laudenslager J., Granger S. W., Rickert S., Koriazova L., Tahara T., Kubo R. T., Kato S., and Crotty S. (2009) Heavily isotype-dependent protective activities of human antibodies against vaccinia virus extracellular virion antigen B5. J. Virol. 83, 12355–12367 10.1128/JVI.01593-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rogers C. J., Clark P. M., Tully S. E., Abrol R., Garcia K. C., Goddard W. A. 3rd, and Hsieh-Wilson L. C. (2011) Elucidating glycosaminoglycan-protein-protein interactions using carbohydrate microarray and computational approaches. Proc. Natl. Acad. Sci. U.S.A. 108, 9747–9752 10.1073/pnas.1102962108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shipp E. L., and Hsieh-Wilson L. C. (2007) Profiling the sulfation specificities of glycosaminoglycan interactions with growth factors and chemotactic proteins using microarrays. Chem. Biol. 14, 195–208 10.1016/j.chembiol.2006.12.009 [DOI] [PubMed] [Google Scholar]
- 45. Kabsch W. (2010) Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McCoy A. J., Grosse-Kunstleve R. W., Storoni L. C., and Read R. J. (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr. D Biol. Crystallogr. 61, 458–464 10.1107/S0907444905001617 [DOI] [PubMed] [Google Scholar]
- 47. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 49. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Winn M. D., Isupov M. N., and Murshudov G. N. (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57, 122–133 10.1107/S0907444900014736 [DOI] [PubMed] [Google Scholar]
- 51. Lovell S. C., Davis I. W., Arendall W. B. 3rd, de Bakker P. I., Word J. M., Prisant M. G., Richardson J. S., and Richardson D. C. (2003) Structure validation by Cα geometry: φ,ψ and Cβ deviation. Proteins 50, 437–450 10.1002/prot.10286 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.