

Abstract
In eukaryotes, DNA replication initiates from multiple origins of replication for timely genome duplication. These sites are selected by origin licensing, during which the core enzyme of the eukaryotic DNA replicative helicase, the Mcm2-7 (minichromosome maintenance) complex, is loaded at each origin. This origin licensing requires loading two Mcm2-7 helicases around origin DNA in a head-to-head orientation. Current models suggest that the origin–recognition complex (ORC) and cell-division cycle 6 (Cdc6) proteins recognize and encircle origin DNA and assemble an Mcm2-7 double-hexamer around adjacent double-stranded DNA. To test this model and assess the location of Mcm2-7 initial loading, we placed DNA–protein roadblocks at defined positions adjacent to the essential ORC-binding site within Saccharomyces cerevisiae origin DNA. Roadblocks were made either by covalent cross-linking of the HpaII methyltransferase to DNA or through binding of a transcription activator-like effector (TALE) protein. Contrary to the sites of Mcm2-7 recruitment being precisely defined, only single roadblocks that inhibited ORC–DNA binding showed helicase loading defects. We observed inhibition of helicase loading without inhibition of ORC–DNA binding only when roadblocks were placed on both sides of the origin to restrict sliding of a helicase-loading intermediate. Consistent with a sliding helicase-loading intermediate, when either one of the flanking roadblocks was eliminated, the remaining roadblock had no effect on helicase loading. Interestingly, either origin-flanking nucleosomes or roadblocks resulted in helicase loading being dependent on an additional origin sequence known to be a weaker ORC–DNA-binding site. Together, our findings support a model in which sliding helicase-loading intermediates increase the flexibility of the DNA sequence requirements for origin licensing.
Keywords: ATPases associated with diverse cellular activities (AAA), cell cycle, DNA helicase, DNA replication, DNA–protein interaction, nucleosome, Saccharomyces cerevisiae, cdt1, helicase loading, origin of replication
Introduction
In eukaryotic organisms, DNA replication initiates from multiple origins of replication to ensure the genome replicates in a timely manner. These sites are selected by origin licensing, during which the core enzyme of the eukaryotic DNA helicase, the Mcm2-7 complex, is loaded at each origin of replication. Three proteins are required for Mcm2-7 loading: the origin recognition complex (ORC),6 Cdc6, and Cdt1 (1). Two Mcm2-7 complexes are loaded in a head-to-head conformation at each origin (2, 3). Although inactive, the loaded helicases are the first marker of bidirectional initiation as they are poised to leave the origin in opposite directions upon their activation in S phase (4).
In Saccharomyces cerevisiae cells, origins of replication are defined by a combination of specific sequences and local chromatin. All S. cerevisiae origins include an essential 11-bp ARS consensus sequence (ACS) (5). In addition to the ACS, S. cerevisiae origins also require AT-rich B elements (B1, B2, and sometimes B3) that further facilitate origin function in vivo (6–8). Mutation of any individual B element reduces origin function, but when all B elements are mutated simultaneously, origin function is lost (7). Along with the ACS, the B1 element forms a bipartite ORC-binding site (9, 10), which is reflected in B1-overlapping sequences being conserved at ORC-binding sites (11, 12). B3 is only present at a subset of origins and is a binding site for the nucleosome-positioning protein Abf1 (13). The B2 element, although lacking sequence conservation, has been identified at all origins examined in detail (7, 8, 14–16). Despite differences in sequence and length, identified B2 elements can be swapped between origins and their function preserved (6, 17). Although originally hypothesized to function as a DNA-unwinding element (18), helical stability does not correlate with B2 element function when mutations are introduced into ARS1 (19). Instead, B2 has been proposed to be either a second ORC-binding site (19, 20) or an Mcm2-7-binding site (21, 22).
To facilitate replication protein binding, origin sequences reside within nucleosome-free regions (NFRs) of ∼125 bp, around which are regularly-spaced nucleosomes (11, 23). This positioning depends on ORC and, when a B3 element is present, Abf1 (11, 22). The nucleosomes adjacent to the origin are dynamic and enriched for H2A.z (24), and their relative location impacts helicase loading and activation (22, 25, 26).
The location of initial Mcm2-7 loading remains unclear. The B2 element has been proposed to be a site of initial Mcm2-7 recruitment because disruption of this element in vivo specifically inhibits Mcm2-7 loading but not ORC binding to ARS1 (21, 22). Despite these observations, binding of Mcm2-7 to B2 has not been reported. Structural studies have identified helicase-loading intermediates with one ORC adjacent to either a single or double Mcm2-7 hexamer (27–29). Importantly, dsDNA passes through the ring-shaped ORC–Cdc6 and Mcm2-7 complexes in these structures (27–29). Together with single-molecule studies of helicase loading (30), these structures have led to a model in which one ORC sequentially loads two Mcm2-7 complexes on the adjacent DNA. An alternative model suggests two ORC proteins bound in opposite orientation sequentially load two Mcm2-7 complexes that subsequently come together to form a double hexamer (20). This model is supported by evidence that sequences with only one potential ORC-binding site are poorly functional in vitro and inactive in vivo, and an MCM3 mutant that interferes with the Mcm–ORC interaction is defective for both the first and second Mcm2-7 loading events (20, 31).
To gain additional insights into the mechanism and location of Mcm2-7 loading, we assessed the impact of site-specific protein–DNA adducts along the origin DNA on helicase loading. We found that only adducts that interfered with ORC–DNA binding prevented Mcm2-7 loading. These findings suggest that ORC occupies DNA that is subsequently encircled by the first loaded Mcm2-7. In addition, there must be significant flexibility for the loading site of the second Mcm2-7. Creating origin DNA templates with roadblocks on either side of the origin showed a strikingly different pattern. Addition of a second roadblock predicted to limit sliding of helicase-loading intermediates identified several protein–DNA adducts near the B2 element that inhibited Mcm2-7 loading without impacting ORC–DNA binding or initial Mcm2-7 recruitment. Interestingly, although B2 mutations had no effect on helicase loading for origins with one or no roadblock, origin-flanking roadblocks made this element essential. Our findings reveal that one or more helicase-loading intermediate is capable of sliding on DNA in a manner that expands the sequences that can act as an origin.
Results
Site-specific protein–DNA adducts disrupt helicase loading
Current models based on structural evidence suggest ORC/Cdc6 and Mcm2-7/Cdt1 encircle ∼25 and ∼30 bp of DNA, respectively. However, the structures available do not reveal the sequence of the associated DNA within either the ORC/Cdc6 or Mcm2-7 ring (29). Although current biochemical evidence indicates that ORC/Cdc6 binds the ACS (32, 33), where Mcm2-7/Cdt1 first interacts with ORC/Cdc6 within the asymmetric origin sequence is unknown.
To investigate the location of initial Mcm2-7/Cdt1 binding within the origin sequence, we examined the effects of protein roadblocks using an in vitro helicase-loading assay (Fig. 1A). In this assay, DNA templates composed primarily of the ARS1 origin and flanking yeast DNA (Fig. S1) are attached to magnetic beads and incubated with purified helicase-loading proteins (ORC, Cdc6, and Mcm2-7/Cdt1). After loading, templates were washed, and the remaining DNA-associated proteins were analyzed by SDS-PAGE. Roadblocks were formed by covalent cross-linking between the DNA and the HpaII DNA methyltransferase (M.HpaII). Substitution of a 5-fluoro-2′-deoxycytidine (5FdC) at the second position of the M.HpaII-binding motif (CCGG) covalently cross-linked M.HpaII to the corresponding location within the origin DNA (34). M.HpaII–DNA adducts are known to act as barriers to Mcm2-7 passage even when present on single-stranded DNA (35–37). Comparison of the structure of M. HhaI, a smaller but related methyltransferase bound to DNA, illustrates why DNA-bound M.HpaII cannot be accommodated in the Mcm2-7 central channel (Fig. 1B). Thus, we hypothesized that placing an M.HpaII roadblock at a site that would normally be encircled by Mcm2-7 during loading will interfere with Mcm2-7 recruitment.
Figure 1.

M.HpaII modification at a subset of locations within ARS1 affects Mcm2-7 helicase loading. A, rationale for using M.HpaII to inhibit helicase loading. Current models suggest ORC/Cdc6 binds to ∼25 bp of DNA, with the addition of each Mcm2-7 hexamer occupying an additional 30 bp of DNA. Binding of M.HpaII to a location in the origin DNA sequence where Mcm2-7 interacts should prevent stable helicase loading. B, structures of the Mcm2-7 ring (PDB code 3JA8) and a smaller but related S-adenosylmethionine-dependent methyltransferase (HhaI methyltransferase, PDB code 2Z6A) illustrate that the M.HpaII methyltransferase adduct is not expected to be accommodated within the Mcm2-7 central channel once the Mcm2-7 ring has closed around dsDNA. C, diagram showing the relative locations of the M.HpaII consensus sequence in the 12 different templates tested. Sites of M.HpaII cross-linking are shown as red “stop” signs (each template included only one modification). Changes at the origin sequence level are shown in Fig. S2A. Important origin sequences are indicated. All templates tested contained a TALE protein (black) bound to its consensus motif at the non-bead-attached end of the template to prevent helicases from sliding off the DNA. D, different M.HpaII-containing templates were assessed for their ability to load salt-stable Mcm2-7 complexes. Templates were either treated with M.HpaII (+) or left untreated (−). M.HpaII binding to 5FdC-modified DNA is quantitative (Fig. S2B). The location of the 5FdC-modified M.HpaII recognition motif is indicated above each pair of lanes. To eliminate any differences in DNA bead preparation or from introduction of the M.HpaII consensus sequence, relative changes between identical DNA templates either treated or untreated with M.HpaII were compared and quantified in E. E, comparative levels of Mcm2-7 loading across origins containing a 5FdC-modified M.HpaII recognition motif at the indicated location. Amount of helicase loading is reported as a fraction of loading when treated with M.HpaII relative to the level of loading on the same template without M.HpaII treatment. Error bars are the standard deviation from the mean calculated from three independent experiments.
We cross-linked M.HpaII to 12 different locations spanning both sides of the ORC-binding site by substituting the 5FdC-modified M.HpaII consensus sequence for a 4-bp sequence in the origin (Fig. 1C and Fig. S2A). We indicate the location of these sites relative to the conserved ACS at ARS1. In all our experiments, the +1 position is the 1st bp of the T-rich strand of the ACS. Positive numbers indicate adduct locations in the B element region (referred to as downstream of the ACS), whereas negative numbers indicate adducts on the other side of the ACS (referred to as upstream of the ACS).
Based on structural studies, we expected the first Mcm2-7 hexamer to interact with origin DNA ∼30 bp on one side of the ORC-binding site. Thus, we placed roadblocks over >30-bp regions on either side of the ORC-binding site (Fig. 1C and Fig. S2A) (28). We avoided modifying key residues important for origin function to ensure helicase loading would still occur in the absence of M.HpaII (7, 38). Consistent with this goal, all M.HpaII recognition sequence-containing templates loaded Mcm2-7 in the absence of M.HpaII cross-linking (Fig. 1D). M.HpaII cross-linking to DNA was highly efficient (Fig. S2B), but addition of M.HpaII to DNA without a C(5FdC)GG motif had no effect on helicase loading (Fig. 1D, compare lanes 1 and 2). Mutations in the ACS and B2 sequence elements (A-B2-) eliminated helicase loading, confirming that our assays were fully dependent on a functional origin sequence and that helicase loading was occurring at the origin DNA (Fig. 1D, compare lanes 1 and 13).
Because loaded Mcm2-7 can slide on DNA (2, 3), we sought to prevent loaded helicases from sliding off the end of the DNA templates. To this end, we included the binding site for an engineered sequence-specific transcription activator-like effector (TALE) protein at the free end of the template (Fig. 1C) (2, 3). TALE protein binding depended on the presence of the TALE sequence motif and remained bound through the high-salt washes used to detect stable helicase loading (Fig. S3, A and C) (39, 40). Importantly, addition of TALE to these templates inhibits sliding of loaded Mcm2-7 complexes off the DNA to a similar extent as a permanent M.HpaII cross-link (Fig. S3B).
Helicase loading was inhibited by M.HpaII adducts placed between −15 and +38 bp (Fig. 1, D and E). The strongest M.HpaII–adduct inhibition was observed for the +14, +23, and +31 templates (Fig. 1D, lanes 3–6 and 27–28 and quantified in E), which are located in and around the ACS and B1 element of ARS1 (Fig. 1C and Fig. S2A). Approximately 2-fold defects were observed for the +38, −5, and −10 protein adducts (Fig. 1D, lanes 7–8, 23–26 and quantified in E) and a weak but reproducible defect was observed when the M.HpaII adduct was placed at −15 (Fig. 1D, lanes 21 and 22 and quantified in E). Protein adducts located outside of the −15- and +38-bp region showed no defects. Doubling the size of the M.HpaII protein by fusion to the maltose-binding protein did not change the range of adducts that inhibited helicase loading (Fig. S4), arguing against the hypothesis that ORC/Cdc6 or Mcm2-7 can accommodate unmodified M.HpaII within their central channels.
Inhibitory M.HpaII adducts prevent ORC–DNA binding
We investigated which step during origin licensing was inhibited by the different sites of M.HpaII modification. To this end, we replaced ATP with ATPγS in the helicase-loading assays, which arrests helicase loading after the initial association of all of the helicase-loading proteins and the first Mcm2-7 complex (Fig. 2) (28, 40). This intermediate is referred to as the OCCM complex due to the presence of one copy of ORC, Cdc6, Cdt1, and Mcm2-7. Unlike loaded Mcm2-7 that can slide off the end of the template, binding of the TALE protein at the end of the template did not increase Mcm2-7 retention in ATPγS and thus was not included in these assays (Fig. S5). As with helicase loading, OCCM formation was dependent on a functional origin (Fig. 2B, compare lanes 1 and 3 and quantified in C). A similar subset of M.HpaII adducts affected OCCM formation. Strong defects in OCCM formation were observed when M.HpaII was placed at the +14, +23, +31, and +38 locations (Fig. 2B, lanes 17–24 and quantified in C). Adducts at the −5 and −10 positions caused a 2-fold or less defect in OCCM formation (Fig. 2B, lanes 13–16 and quantified in C) and the remaining adducts exhibited no defect.
Figure 2.

M.HpaII adducts at the −5 through +38 locations affect both ORC and Mcm2-7 association in ATPγS. A, diagram showing the relative locations of the M.HpaII consensus sequence in the 12 different templates tested for OCCM formation. Locations are the same as those shown in Fig. 1. Because TALE binding did not aid OCCM formation (Fig. S5), it was not included. B, effect of M.HpaII-cross-linked templates on OCCM complex formation in ATPγS. Templates were either treated with M.HpaII (+) or left untreated (−) as indicated. The location of the 5FdC-modified M.HpaII recognition sequence is noted above each pair of lanes. The associated proteins were detected by fluorescent protein staining. The Mcm2-7/Orc1 and Orc2 bands are marked. C, quantitation of different sites of M.HpaII cross-linking on Mcm2-7 and Orc2 origin DNA association in the presence of ATPγS. In each case, the relative amount of Mcm2-7 and Orc2 association are reported as the fraction of DNA association for M.HpaII-treated templates (+) relative to untreated templates (−). Error bars are the standard deviation from the mean calculated for three independent experiments.
The defects in OCCM formation reflected a corresponding defect in ORC–DNA binding. At all locations displaying a helicase-recruitment defect (−5, +14, +23, +31, and +38), a decrease in ORC–DNA association was also observed (Fig. 2B, lanes 15–24, compare Orc1 or Orc2 bands, and quantified in C). This observation suggested loss of ORC binding was the primary reason for the defects in helicase loading observed in Fig. 1. To test this hypothesis, we measured ORC binding to M.HpaII-modified templates using an electrophoretic mobility shift assay (EMSA) (9). Radiolabeled ARS1 DNA probes (∼260 bp) containing the same set of modified M.HpaII consensus-motif insertions were incubated with the indicated amounts of ORC in the presence or absence of M.HpaII cross-linking (Fig. 3). The EMSA experiments identified ORC–DNA binding defects in the same subset of M.HpaII adducts as the ATPγS and ATP helicase-loading experiments (Figs. 1 and 2), indicating that the major impact of the single M.HpaII–DNA adducts is the inhibition of ORC binding to the origin.
Figure 3.

M.HpaII modification at the −5 through +38 locations inhibits ORC DNA binding. A–G, electrophoretic mobility shift assays were used to compare ORC DNA binding to ARS1 DNA templates with or without M.HpaII cross-linking at the indicated positions. Control experiments with wild-type (WT) unmodified ARS1 DNA or a mutant ARS1 DNA lacking ORC-binding sites (A-B2-) are shown in A. For the remaining panels, the site of the 5FdC-modified M.HpaII-binding site and templates that have been cross-linked to M.HpaII are indicated. Note that binding to M.HpaII-modified DNA results in a supershifted complex relative to ORC binding to the same DNA without M.HpaII cross-linking (see C and E). Low levels of shifted DNA in the M.HpaII-cross-linked samples at high levels of ORC in D and F (marked with an asterisk) are due to ORC binding to a small amount of unmodified DNA present in these DNA preparations (note the lack of a supershift relative to the unmodified DNA samples).
Upstream roadblocks cause additional M.HpaII–DNA adducts to inhibit helicase loading
The results of our initial M.HpaII cross-linking studies are inconsistent with a model for helicase loading in which ORC remains bound to the ACS as it sequentially recruits two Mcm2-7 complexes to encircle the adjacent ∼60 bp of DNA. If this model was correct, regardless of which side of ORC the Mcm2-7 proteins bind, several of the tested M.HpaII adducts should inhibit Mcm2-7 loading without affecting ORC DNA binding. Instead, we identified an ∼50-bp region (+38 to −10) where M.HpaII–DNA adducts inhibit ORC–DNA binding and helicase loading (due to the ORC–DNA-binding defects). Although recent studies suggested a second ORC bound downstream of the first loads the second Mcm2-7, this model cannot explain why only single roadblocks that interfere with ORC binding prevent helicase loading (20). These studies found that an M.HpaII–DNA adduct between the two sites of ORC binding (as would be the case for +47, +58, +67, and +79 M.HpaII adducts) strongly inhibited helicase loading. Because we did not observe inhibition by these adducts, we hypothesized that at least one Mcm2-7-containing intermediate complex formed during helicase loading can slide along dsDNA after initial sequence-specific ORC–DNA binding. Such a mobile intermediate could slide upstream of the ACS and away from the M.HpaII adduct to avoid interference with Mcm2-7 recruitment and loading (Fig. 4A). If this is the case, placement of a roadblock at a position predicted to prevent this sliding would change the set of M.HpaII adducts that inhibit helicase loading.
Figure 4.

Placing roadblocks on both sides of the ACS causes synthetic defects in helicase loading. A, diagram showing the logic behind the creation of double-roadblock DNA templates. In the presence of a single roadblock, we propose a helicase intermediate can slide away to accommodate the loading of a second Mcm2-7. In contrast, the presence of two roadblocks should prevent this movement, allowing the identification of modifications that affect helicase only when a second roadblock is present and sliding is restricted. B, diagram showing the location of TALE binding and M.HpaII cross-linking used in C. Each ARS1–DNA template includes one TALE-binding sequence (black object) at one of three locations upstream of the ACS and one site of M.HpaII cross-linking (red stop signs) at one of five locations downstream of the ACS. C, ARS1 DNA with a downstream M.HpaII cross-linked DNA and with or without an upstream roadblock were tested. The indicated modified ARS1–DNA templates were assayed for the formation of salt-stable Mcm2-7 association. All templates were treated first with M.HpaII and then either treated (+) or not treated (−) with TALE protein as indicated above each lane. The location of the 5FdC-modified M.HpaII recognition motif (M.HpaII-seq) and the location of the TALE-binding sequence (Tal-seq) are noted. D, quantification of the levels of Mcm2-7 loading with and without TALE binding. For each of the tested sites of M.HpaII cross-linking, we report the fraction of Mcm2-7 loading with and without TALE protein binding. Error bars are the standard deviation from the mean calculated for three independent experiments.
To block sliding of helicase-loading intermediates, we added a TALE-binding sequence (used at the free end of our DNA templates in previous assays) at positions −25, −35, or −45 for several of the M.HpaII-modified templates (Fig. 4A and Figs. S6 and S7). Having two orthogonal protein–DNA adducts allowed us to test whether having a TALE blockage upstream of the ACS altered the impact of an M.HpaII adduct placed downstream of the ACS. Importantly, all the TALE-binding sites and the M.HpaII–DNA adducts tested in combination were located outside of DNA regions identified as important for ORC binding and OCCM formation with individual blocks and did not inhibit helicase loading individually (Figs. 1–3 and 4C and Fig. S8). In contrast, combining templates with M.HpaII cross-linked at +47, +58, or +67 with TALE binding to any of the three locations tested (−25, −35, or −45) inhibited helicase loading 5–10-fold (Fig. 4D). These defects were not due to inhibition of initial ORC–DNA binding or Mcm2-7/Cdt1 association, because OCCM formation was unaffected (Fig. S8). Increasing the spacing between the two roadblocks attenuated the helicase-loading defects. Moving the M.HpaII adduct to the +79 location or the end of the DNA template (∼+280) showed no inhibition of helicase loading with the TALE protein bound at any of the three locations (Fig. 4C, lanes 7 and 8, 14–18, and 25–30). Similarly, the M.HpaII–DNA adduct at −25 shows a stronger effect relative to the two more distal sites of TALE binding.
B2 element of ARS1 is required in the presence of origin-flanking roadblocks
Although a minimum distance between the roadblocks might be required for helicase loading, examination of the distances between various pairs of roadblocks showed that this was not the sole determinant for successful loading in our assay. The −25 TALE/+79 M.HpaII, −35 TALE/+67 M.HpaII, and −45 TALE/+58 M.HpaII templates each have the two roadblocks spaced ∼100 bp apart, yet only one of these templates (−25/+79) successfully loaded Mcm2-7. Interestingly, of these three templates, only the −25/+79 combination included the entire B2 element of ARS1 between the two adducts.
To examine the role of the B2 element in our assays, we first tested whether B2 contributed to helicase loading on DNA templates without M.HpaII adducts (Fig. 5, A and B, and Fig. S9). Although mutation of the ACS (A-) showed a severe defect in Mcm2-7 loading (Fig. 5A, compare lanes 1 and 3 and quantified in B), mutation of the B2 element (B2-) showed only an ∼10% decrease in helicase loading (Fig. 5A, compare lanes 1 and 4 and quantified in B). Despite the fact that the B2 element can act as an ORC-binding site under certain conditions (19, 33, 41), simultaneous mutation of the ACS and B2 elements did not significantly change helicase loading compared with mutation of the ACS alone (Fig. 5A, compare lanes 2 and 3 and quantified in B). Thus, the B2 element did not significantly contribute to Mcm2-7 loading on unmodified templates.
Figure 5.

ARS1-flanking roadblocks lead to enhanced B2 requirement for Mcm2-7 loading. A, Mcm2-7 loading following a high-salt wash using the wild-type and mutant ARS1 DNA templates. Comparison of the sequences between different mutant origin templates are shown in Fig. S9. B, quantification of the relative amount of helicase loading for mutant ARS1 DNA templates. The level of Mcm2-7 loading for wild-type ARS1 DNA was set to 1, and the relative ratio of Mcm2-7 loading for each mutant DNA is reported. Error bars are the standard deviation from the mean calculated for three independent experiments. C, impact of B2 mutation on Mcm2-7 loading onto ARS1 DNA with or without ARS1-flanking DNA roadblocks. ARS1 DNA templates with or without a mutation in B2 were cross-linked to M.HpaII either at +79 or the DNA end. These templates were tested for salt-stable Mcm2-7 loading with or without TALE binding to one of three sites as indicated. The location of the 5FdC-modified M.HpaII recognition motif (M.HpaII-seq) and the location of the TALE-binding sequence (Tal-seq) are noted above each pair of lanes. D, quantitation of relative Mcm2-7 loading with or without ARS1-flanking DNA roadblocks. The ratio of Mcm2-7 loading with and without a second (TALE) roadblock is reported for ARS1–DNA templates with M.HpaII cross-linked at position +79 with or without a B2 mutation. Error bars are the standard deviation from the mean calculated for three independent experiments.
We next asked whether B2 was important when the origin was flanked by protein roadblocks. We mutated the B2 element in the presence of the +79 M.HpaII modification and assessed whether TALE binding at −25, −35, or −45 influenced helicase loading (Fig. 5C). We observed no defects in Mcm2-7 loading when the B2 element and both roadblocks were present. In contrast, mutation of the B2 element strongly inhibited helicase loading in the −25/+79 and −35/+79 double-blocked templates (Fig. 5C, lanes 1–8). The −45/+79 template showed an ∼50% reduction in stable helicase loading in the presence of both blockages (Fig. 5C, lanes 9–12), suggesting that increasing the available DNA reduces the need for B2.
The B2 dependence of helicase loading requires roadblocks on both sides of the origin. In the absence of TALE protein, the M.HpaII-bound +79 template showed strong helicase loading with or without B2 (Fig. 5C, lanes 1 and 3, for example). In contrast, TALE addition to the same templates resulted in helicase loading that was dependent on B2 (Fig. 5C, compare lanes 2 and 4, for example). These B2-dependent defects also required placement of the two adducts close to the origin. A TALE-bound template where the M.HpaII was cross-linked at the end of the template (∼280 bp from the origin sequence) showed no B2-dependent defect (Fig. 5C, lanes 13–16, and Fig. S10). Initial association of ORC and Mcm2-7 in ATPγS was unaffected by the mutation of the B2 element even with both roadblocks (Fig. S11), indicating the B2-dependent defect we observed occurs downstream of initial Mcm2-7 recruitment.
Nucleosomal DNA templates require the B2 element for helicase loading
Although the artificial roadblocks used above are not present in vivo, origins of replication are consistently found within NFRs flanked by positioned nucleosomes (11, 23). We hypothesized that restricting DNA access with nucleosomes would also reveal an important role for the B2 element. To test this hypothesis, we assembled chromatinized templates containing wild-type and mutant ARS1 DNAs (Fig. 6A and Fig. S12A). Incorporation of DNA into nucleosomes unmasked a strong B2 element dependence for helicase loading similar to that observed with our artificial roadblocks and in vivo. Although naked DNA templates did not show B2-dependent loading, nucleosomal DNA templates showed strong defects when B2 was mutated (Fig. 6B). In contrast, the levels of OCCM formation on templates with or without nucleosomes were largely unchanged, similar to what was observed with artificial protein roadblocks, (Fig. S12B). This observation further emphasizes that the B2 element is not required for OCCM formation.
Figure 6.

B2 element is important when nucleosomes flank ARS1 DNA. A, reaction scheme for helicase loading assay performed in B. ORC-bound ARS1–DNA templates were either assembled into nucleosomes or not. These templates were tested for salt-stable helicase loading. DNA-associated proteins were detected by immunoblot. B, ARS1–DNA-associated Mcm2-7 and H2B amounts were detected by immunoblot. WT and B2- mutant ARS1 DNA are compared. C, comparison of origin-proximal nucleosome positioning for in vitro assembled ARS1 WT (red) and ARS1 B2- (blue) nucleosomal DNA templates. Origin-proximal nucleosome positioning was determined by high-throughput MNase-Seq of pUC19 ARS1-WT or pUC19 ARS1-B2- plasmids assembled into nucleosomes. Peaks represent the location of the midpoint of the protected nucleosomal DNA.
Nucleosome encroachment has been implicated previously to impact helicase loading (22, 25, 26). Importantly, the NFR region around the ACS was unchanged between the WT and B2 templates, indicating that changes in nucleosome positioning were not the cause of the helicase-loading defects observed (Fig. 6C). Together, our findings show that the importance of the B2 element is dependent on restricting access to origin-flanking DNA.
Discussion
Our use of protein–DNA adducts to explore the role of different regions of origin DNA during helicase loading reveals that the sites of initial Mcm2-7 loading are either also involved in ORC DNA binding (first Mcm2-7) or can occur at many locations (second Mcm2-7). Our findings also reveal that at least one intermediate in helicase loading is able to slide on DNA. This dynamic feature of helicase loading allows exploration of additional DNA sequences to facilitate the loading process. When this sliding is restricted, however, we observe a strong requirement for the B2 element, consistent with models suggesting two ORC-binding sites facilitate helicase loading (20). The positioned nucleosomes that tightly flank S. cerevisiae origins (11, 23) would significantly limit the sequences that could function during helicase loading; however, such a mobile intermediate in higher organisms could explain the lack of well-defined sites of replication initiation.
The same roadblocks inhibit ORC–DNA binding and OCCM formation
Our finding that ORC binding is inhibited by roadblocks outside of the ACS and B1 sequences (−5 and +38, Fig. 3) is consistent with previous studies suggesting that ORC binds a more extensive region of DNA. ORC and ORC–Cdc6 footprinting and ORC–DNA cross-linking experiments both found that ORC protects or cross-links to DNA outside of the ACS and B1 regions (10, 32, 33, 41). The region identified is significantly larger than the ∼25 bp of DNA encircled by ORC–Cdc6 in the OCCM structure and DNA-binding models based on ORC crystal structures (29, 42, 43). The differences between the structural and biochemical data are likely due to both the elimination of parts of ORC in structural studies and changes in ORC–DNA binding during helicase loading. For example, most of Orc6 and regions of Orc1, Orc2, and Orc3 were eliminated or were not detected in structural studies of ORC (29, 42, 43). Consistent with Orc6 extending the region of DNA bound by ORC, Orc6 is related to the DNA-binding protein TFIIB (44), and elimination of Orc6 allows two ORC molecules to bind DNA closer to one another on a single DNA than the complete ORC1–6 complex (19).
Our findings strongly suggest that ORC initially occupies DNA that is encircled by the first recruited Mcm2-7. Structural studies indicate that ORC and the first Mcm2-7 encircle adjacent DNA (28, 29). Despite this, we did not identify any modified DNA that inhibited OCCM formation (which includes the first Mcm2-7) without impacting ORC–DNA binding. Instead, we observed a one-to-one correspondence between mutants that inhibited these events. Unlike double-hexamer formation, addition of a second roadblock did not expand the set of roadblocks that inhibited OCCM formation. Because the same set of DNA–protein adducts interferes with ORC binding and OCCM formation, we propose that recruitment of the first Mcm2-7 displaces ORC bound to origin DNA outside of the ACS (i.e. ∼ +15 to +45, Fig. 7). This hypothesis is supported further by the similar size of DNA encircled by the OCCM (50–55 bp) and the region of DNA that when cross-linked to M.HpaII inhibited ORC DNA binding and OCCM formation (at least 48 bp, between −10 and +38, Fig. 3). This overlap has the advantage that initial ORC–DNA binding would require enough space for subsequent recruitment of the first Mcm2-7.
Figure 7.
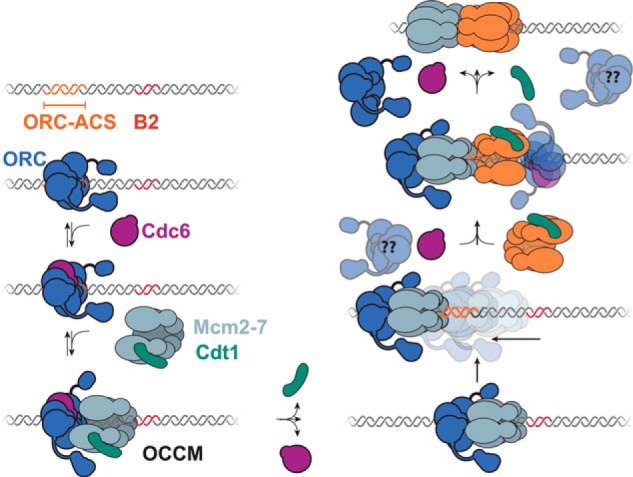
Proposed model for Mcm2-7 helicase loading. ORC binds to the ACS, recruiting Cdc6 and Mcm2-7/Cdt1 to form the OCCM. Cdc6 and Cdt1 are sequentially released from the OCCM (30). Prior to the loading of the second hexamer, sliding of a helicase-loading intermediate away from the B2 element occurs (illustrated as an ORC-Mcm2-7 complex). This allows space for the loading of the second Mcm2-7 hexamer, possibly involving a second ORC molecule interacting in the B2 sequence element. After recruitment of the second Mcm2-7–Cdt1 complex, release of Cdc6 followed by ORC and Cdt1 occurs (30, 46).
Although we do not know how ORC's interaction with the DNA changes as the first Mcm2-7 is recruited, one likely alteration would involve Orc6 releasing any DNA interactions. Biochemical studies suggest that Orc6 interacts with Cdt1 in the context of the OCCM (29, 45). Because Cdt1 and the primary Orc6 interaction with the rest of ORC are both located outside of the ORC/Cdc6 and Mcm2-7 ring in the OCCM structure, it is unlikely that Orc6 would be able to maintain DNA contacts in this intermediate. Thus, Orc6 is unlikely to have DNA access in the OCCM. In addition to Orc6, ORC–DNA cross-linking studies also implicate Orc2 and Orc4 as being proximal to the DNA between beyond the B1 element (41). Thus, these subunits could also change their interaction with DNA upon Mcm2-7 recruitment.
Sliding intermediate during helicase loading
The lack of individual protein–DNA adducts that interfered with Mcm2-7 recruitment without preventing ORC–DNA binding indicates that helicase loading does not involve tightly defined sites of Mcm2-7 recruitment. If both Mcm2-7 hexamers were sequentially recruited while remaining associated with ORC bound at the ACS, then protein–DNA adducts extending over ∼85 bp (Fig. 1A) would interfere with helicase loading. Instead, only adducts between −10 and +38 that interfered with ORC binding showed helicase-loading defects. One possibility that we considered to explain these findings was DNA looping between ORC/Cdc6 and Mcm2-7-bound DNA. Structural studies of the OCCM argue against a model in which looping occurs between ORC and the first recruited Mcm2-7 (28, 29). Although it is possible that the second Mcm2-7 could be recruited through a looping mechanism, such a model does not explain why M.HpaII–DNA adducts and TALE protein binding that have no effect on their own would eliminate helicase loading in combination.
Instead, our data support sliding of a helicase-loading intermediate mediating the ability of helicase loading to accommodate protein–DNA adducts that do not prevent initial ORC binding. Sliding of an intermediate containing the first Mcm2-7 to a position upstream of the site of initial OCCM formation (away from the B elements) would allow the loading process to avoid M.HpaII–DNA adducts located at downstream positions that would otherwise prevent loading of a second Mcm2-7 adjacent to a static first hexamer (Fig. 7). Consistent with this idea, upstream roadblocks (DNA-bound TALE protein) that would prevent sliding of intermediates away from M.HpaII–DNA adducts expand the number of deleterious downstream roadblocks (e.g. +47 and +58). Importantly, like similarly positioned M.HpaII–DNA adducts (Fig. 1), the upstream TALE roadblocks have no effect on their own (Fig. 4). Consistent with restriction of a sliding intermediate, the TALE and M.HpaII roadblocks only inhibit helicase loading when they closely flank core origin sequences. Placing both roadblocks either on the same side of the origin (e.g. TALE at +280 (end) and M.HpaII at +79, Fig. 1) or flanking roadblocks placed far apart (e.g. M.HpaII at +280 (end) and TALE at −35, Fig. 4) has no impact on helicase loading.
Our studies do not identify the sliding intermediate but do restrict the possibilities. Consistent with sliding occurring after recruitment of the first Mcm2-7 (Fig. 7), addition of a second roadblock does not affect OCCM formation (Fig. S11). In addition, we found that the OCCM was not able to slide on the DNA (Fig. S5), indicating that the sliding intermediate is formed after this step. Given that Mcm2-7 double hexamers are known to slide on the DNA, it is possible that the intermediate is the first Mcm2-7 on its own. It has recently been proposed that the two Mcm2-7 complexes are loaded separately from one another and move toward one another in an ATP-dependent manner to form a double hexamer (Fig. S13A) (20). Although possible, the predicted direction of ATP-dependent movement of an Mcm2-7 complex (with the N-terminal domain leading, (4)) would move the first Mcm2-7 toward the B element roadblocks further restricting loading. Instead, the loading intermediate would need to move away from the M.HpaII adducts and the B elements in our experiments to create space for a second Mcm2-7 loading (Fig. S13B). Because this direction of movement is the opposite predicted for active Mcm2-7 movement (4), we suggest that the movement is passive sliding rather than directional. It is also possible that one or more helicase-loading proteins are associated with the first Mcm2-7 during sliding. Because Cdc6 and Cdt1 are rapidly released from the OCCM but ORC is retained until the end of helicase loading (30, 46), the most likely such intermediate would contain ORC and Mcm2-7 (Fig. 7). Although the OCCM does not slide, release of Cdc6 or Cdt1 and/or a change in ORC DNA associations upon recruitment of the first Mcm2-7 could allow this intermediate to slide (for example due to changes in the ORC DNA association described above).
Flanking roadblocks restrict the sequence requirements for helicase loading
Restricting access to DNA adjacent to the origin also changed the sequence requirements for helicase loading. When flanking DNA is made inaccessible either by roadblocks or nucleosomes in vitro, we observed a strong requirement for the B2 sequence (Figs. 5 and 6). In contrast, no B2 requirement was observed with single roadblocks or in the absence of roadblocks. A simple explanation for these observations is that when helicase loading intermediates are allowed to slide, other possible B2 elements can be explored. It is interesting to note that ORC cannot simultaneously bind the ACS and B2 at the ARS1 origin tested here (33), suggesting this origin would require sliding of the first intermediate for any ORC binding to B2. That such sequences could be identified in the <1-kb template used on our assays is supported by the weak sequence requirements for B2 (16, 19). This hypothesis is consistent with studies of artificial origins with a single B2-like sequence that found a strong B2 dependence (20).
How is B2 functioning in these assays? The most likely explanation is that it acts as a second ORC-binding site involved in recruiting the second Mcm2-7. We saw no defect in initial OCCM formation in ATPγS when the B2 element was mutated, indicating a role downstream of this intermediate. Early studies found that B2 acts as a second inverted ORC-binding site (19, 33). Strong support for such a role comes from studies showing that a second inverted ORC-binding site is required for helicase loading and origin function (20). These authors proposed a model in which ORC complexes bound at the ACS and B2 use a similar mechanism to load helicase in opposite directions that subsequently come together to form a double hexamer. These findings contrast with single-molecule studies that found one ORC is sufficient for helicase loading and that loading of the first and second Mcm2-7 complexes is coupled rather than independent (30, 46). What can explain these differences? Several trivial explanations can be eliminated. The presence of unlabeled ORC molecules was eliminated in the original single-molecule studies (30). Similarly, a short-lived second ORC DNA-binding event that was not detected is unlikely as the Cdc6 that is associated with the second loading event is long-lived and easily detected.
A more likely explanation of the differences between the bulk and single-molecule experiments is that the low concentration of ORC needed to observe individual ORC–DNA-binding events in single-molecule experiments reduces the importance of a second ORC-binding event. One likely possibility is that recruitment of the second Mcm2-7 involves interactions with the first Mcm2-7 as well as binding to ORC and Cdc6. In this regard, it is noteworthy that endogenous origins typically contain one strong, essential ORC-binding site and one or more weaker B2-like ORC-binding sites. This distinction is consistent with the second loading event involving the weaker ORC-binding site requiring additional interactions. The different kinetics of the first and second loading events are also consistent with this model (30). Such a model would suggest that an intermediate with the first Mcm2-7 would need to move to allow the second Mcm2-7-loading event to be successful unless the B2 element was precisely positioned relative to the ACS. Such a requirement could explain why the presence of an M.HpaII adduct between two ORC-binding sites would inhibit helicase loading (20). Finally, an alternative possibility is that the same ORC interacts with both the ACS and the B2 element. The identification of in vitro conditions that reveal B2 function will provide a powerful tool to investigate the role of B2 during helicase loading.
Experimental procedures
Yeast strains
All yeast strains were derived from W303 background and were constructed and manipulated by standard genetic techniques; detailed genotypes are listed in Table S1.
Purification of M.HpaII
M.HpaII and MBP-M.HpaII were purified similarly to Ref. 36. Briefly, the M.HpaII-expression plasmid (pM15MH or pM15MH-MBP) was transformed into BL21-DE3 Escherichia coli (New England Biolabs, Ipswich, MA). After 1 h of growth in Super Optimal broth with catabolite repression (SOC), the transformation cells were transferred to 100 ml of LB containing 100 μg/ml ampicillin (LB-AMP) and grown overnight. The resulting cells were transferred to 1 liter of LB-AMP to an A600 nm of 0.7–0.8. Expression of M.HpaII was induced by the addition of 0.5 mm IPTG for 2.5 h at 37 °C. Following induction, cells were harvested and resuspended in 25 ml of M.HpaII Loading Buffer (20 mm Tris-Cl, pH 8.5, 500 mm KCl, 10% glycerol, 10 mm imidazole) with cOmpleteTM protease inhibitor mixture (Roche Applied Science, Basel, Switzerland), snap-frozen in liquid nitrogen, and stored at −80 °C. Cells were thawed at room temperature by rotating for 20 min, and the total volume was raised to 50 ml with M.HpaII Loading Buffer containing cOmplete protease inhibitors (Roche Applied Science). Cells lysis was initiated by adding lysozyme to 100 μg/ml followed by incubation at 4 °C for 30 min while rotating. The lysozyme-treated cells were sonicated (Branson 250 with microtip; amplitude 35% for 2 min with 5 s on and 5 s off duty cycle), and the resultant extract was centrifuged at 125,000 × g for 35 min. The resulting supernatant was incubated with 2 ml of Ni-NTA resin (HisPur, Thermo Fisher Scientific) equilibrated with M.HpaII Loading Buffer at 4 °C and rotating for 2 h. The collected resin was washed with 12 column volumes of M.HpaII Wash Buffer (20 mm Tris-Cl, pH 8.5, 500 mm KCl, 10% glycerol, 30 mm imidazole) and eluted with M.HpaII Elution Buffer (20 mm Tris-Cl, pH 8.5, 100 mm KCl, 10% glycerol, 1 mm dithiothreitol (DTT)) with 500 mm imidazole. Fractions containing eluted protein were pooled and concentrated to a final volume of 1 ml using a centrifugal concentrator (Vivaspin 6, 10,000 MWCO, VivaProducts), and the resulting sample was separated on a Superdex 75 size-exclusion column equilibrated with M.HpaII Elution Buffer (without imidazole). Fractions containing M.HpaII or M.HpaII-MBP were concentrated and exchanged into M.HpaII Elution Buffer containing 30% glycerol using the same centrifugal concentrator.
Purification of helicase-loading proteins
ORC and Mcm2-7/Cdt1 were purified from yeast strains (ySK100 and yST144, respectively; see Table S1) that simultaneously overexpress all subunits of the indicated complexes as described previously (47). Cdc6 was expressed in bacteria and purified as described previously (47).
Purification of TALE protein
A plasmid expressing TALE (pSNAP-TALE) was transformed into Rosetta II cells (Novagen, Madison, WI) and grown in 1 ml of SOC for 1 h. These cells were transferred into 50 ml of LB containing 35 μg/ml chloramphenicol and 100 μg/ml ampicillin (LB-CAM-AMP) and grown overnight at 37 °C. The resulting cells were transferred to 1 liter of LB-CAM-AMP and grown at 37 °C to A600 nm of ∼0.6 followed by addition of 0.5 mm IPTG and further growth for 3 h at 37 °C. Cells were harvested and resuspended in 25 ml of TALE Lysis Buffer (50 mm Tris-HCl, pH 8.0, 500 mm NaCl, 10 mm imidazole, 10% glycerol, 0.5 mm TCEP) with cOmplete protease inhibitors (Roche Applied Science), snap-frozen in liquid nitrogen, and stored at −80 °C. Cells were thawed at room temperature by rotating for 20 min, and the total volume was raised to 50 ml with TALE Lysis Buffer. Cells were lysed, and the extract was clarified as described for the M.HpaII purification. The resulting extract was added to 1-ml packed volume of Ni-NTA resin (Thermo Fisher Scientific) equilibrated with TALE Lysis Buffer and incubated while rotating for 1 h at 4 °C. The resin was collected, washed with TALE Lysis Buffer, and eluted with TALE Elution Buffer (50 mm Tris-HCl, pH 7.5, 500 mm NaCl, 10% glycerol, 0.5 mm TCEP) with 250 mm imidazole. Fractions containing TALE protein were pooled and spin concentrated to 1 ml (Vivaspin 6, 10,000 MWCO), and the resulting sample was fractionated on a Superdex 200 size-exclusion column equilibrated with TALE Elution Buffer (lacking imidazole). TALE-containing fractions were pooled, spin-concentrated, and diluted 1:1 with TALE Storage Buffer (80% glycerol, 6 mm NaCl, 50 mm Tris-HCl, pH 7.5, 0.5 mm TCEP) and stored at −80 °C.
Construction of M.HpaII-modified DNA
DNA templates containing 5FdC-modified M.HpaII-binding sites were constructed by linking “short” and “long” PCR products that overlapped at the site of modification (Fig. S14). To generate the short PCR product, oligonucleotides (oligos, Integrated DNA Technologies) were designed that included the 4-bp M.HpaII consensus motif (CCGG) at specific locations across the ARS1 origin sequence. Each of these oligos was used to perform PCR with a second oligo either containing the TALE-binding sequence (SNAP-tal end) or without a TALE sequence (Nco Pri1) that annealed to an adjacent region of pUC19 ARS1. A list of oligos and plasmids used in this study is provided in Tables S2 and S3. A second set of oligos that were complementary to the first set of M.HpaII consensus motif-containing oligos were synthesized but included 5FdC in the M.HpaII consensus motif (Bio-synthesis, Lewisville, TX). These oligos were used to generate the long PCR product using the same pUC19 ARS1 template and a second biotinylated oligo (biopuc19 seqA, Integrated DNA Technologies). The complementarity of the two M.HpaII consensus motif-containing oligos ensures that the two PCR products have a 30-bp region of identity within the region of ARS1 coinciding with the location of the M.HpaII motif substitution mutant. The resulting PCR products were purified using a PCR clean-up column (TaKaRa, Clontech).
The ends of the corresponding short and long PCR products were resected by treatment with 1.5 units of T4 DNA polymerase (New England Biolabs) in T4 DNA ligase buffer (New England Biolabs) in the absence of dNTPs at 20 °C for 30 min (long PCR fragment) or 20 min (short PCR fragment). These reactions were stopped by addition of dCTP (1 mm). The resulting 5′-overhangs were phosphorylated by addition of 10 units of T4 polynucleotide kinase (New England Biolabs) at 37 °C for 15 min. The complementary 3′-ends of the resected long and short PCR products were annealed to one another by mixing in a 1:5 molar ratio, followed by a 10-min incubation at 80 °C and slow cooling to 25 °C. The annealed templates were then incubated with 0.3 units/pmol T4 DNA polymerase and 20 units/pmol T4 DNA ligase (New England Biolabs) in buffer containing 100 μm dNTPs and 1 mm ATP for 2 h at 16 °C. Production of full-length (∼1 kb, Fig. S1) ARS1 DNA was assayed by agarose gel. For some templates, the T4 DNA polymerase resection, annealing, and fill-in steps were repeated. In all cases, the final DNA preparation was >90% full-length template. The resulting 5FdC-containing templates were coupled to streptavidin-coated magnetic beads overnight (Dynabead M280, Invitrogen), and the amount of bead-coupled DNA was assayed by EcoRI digestion of a fraction of the beads (2–5 μl) followed by quantification of the released DNA on an agarose gel.
M.HpaII coupling to 5FdC-modified origin DNA
Magnetic beads coupled to 5FdC-modified origin-containing DNA (0.75 pmol) were incubated in M.HpaII Binding Buffer (50 mm Tris-HCl (pH 7.5), 0.5 mm β-mercaptoethanol, 10 mm EDTA) with or without a 10-fold molar excess of M.HpaII or MBP-M.HpaII for 1.5 h at 37 °C in the presence of 80 μm S-adenosylmethionine (New England Biolabs). Excess M.HpaII was removed by three successive washes with H buffer (50 mm HEPES-KOH (pH 7.6), 5 mm MgOAc, 1 mm ZnOAc, 1 mm DTT, 10% glycerol, 0.1 mm EDTA, 0.1 mm EGTA, 0.02% Nonidet P-40) with 300 mm potassium glutamate (H/300).
TALE binding to origin DNA
Magnetic beads coupled to 5FdC-modified origin-containing DNA (0.75 pmol) were incubated with a 10-fold molar excess of purified TALE protein at 25 °C for 15 min. Excess protein was removed by two successive washes with H/300 buffer.
Helicase-loading assays
Helicase-loading assays were performed as described in Ref. 47 with the following modifications. Magnetic beads coupled to 0.75 pmol of origin-containing DNA (either untreated or pre-treated with M.HpaII and/or TALE protein, as indicated) were incubated with 0.17 pmol of ORC, 3 pmol of Cdc6, and 3 pmol of Mcm2-7/Cdt1 protein in Reaction Buffer (25 mm HEPES-KOH, pH 7.6, 12 mm MgOAc, 50 μm ZnOAc, 350 mm potassium glutamate (KGlut), 3 mm ATP, 4 mg/ml BSA) for 30 min at 25 °C in a thermo-mixer (Eppendorf, Westbury, NY) at 1200 rpm. Following incubation, the beads were washed once with H/300, once with H buffer containing 500 mm NaCl (H/500), and a final time with H/300. DNA-associated proteins were released from the beads by treatment with 1 unit of DNase (Worthington) in DNase Buffer (H buffer with 5 mm CaCl2, 5 mm MgOAc, 50 mm KCl) at 25 °C and 1200 rpm for 10 min. After capture of the magnetic beads, the resulting supernatant was boiled in SDS-PAGE sample buffer for 5 min and separated on an 8% SDS-polyacrylamide gel. Proteins were stained with fluorescent protein stain (Krypton, Thermo Fisher Scientific) according to the manufacturer's instructions.
Helicase association in ATPγS
Association assays were performed identically to helicase-loading assays with following changes: 1 pmol of ORC was used, 5 mm ATPγS was used instead of ATP, and three successive washes with H/300 were performed following the 30-min incubation with the helicase-loading proteins.
Insertion of TAL sequence at a location near the ACS
An oligonucleotide containing the TAL-binding sequence (5′-TGAACTTGACTATATCTTATA-3′) along with an AatII restriction site was inserted into the pUC19 ARS1 plasmid by site-directed mutagenesis (Quikchange Lightning, Agilent). Plasmids containing TALE-sequence insertions were then used as the template for PCR to create origin-containing DNA templates with 5FdC-modified M.HpaII-consensus-site insertions as described above.
Radiolabeled DNA templates
A plasmid containing two NcoI cleavage sites at −136 and +117 flanking the ARS1 origin (pUC19 ARS1 NcoI) was created using site-directed mutagenesis. The resulting plasmid was used as a template to create origin-containing DNA templates with 5FdC-modified M.HpaII consensus-site insertions as described above except the biotinylated oligo was replaced with an oligo of the same sequence lacking the biotin modification (pUC19 seqA). Approximately 2 μg of full-length 5FdC-containing template was digested with NcoI and purified by phenol/chloroform extraction and ethanol precipitation. The resulting DNA was resuspended in 10 μl of TE and radiolabeled by filling in the DNA ends using T4 DNA polymerase (New England Biolabs) and 0.1 mm dGTP, dTTP, and dATP with 4 μCi of [α-32P]dCTP. The resulting radiolabeled DNA was gel-purified on a 5% TBE polyacrylamide gel.
ORC electrophoretic mobility shift assays (EMSA)
Radiolabeled DNA probes (2000 cpm) were coupled to M.HpaII as described above. ORC and the DNA probe were incubated for 10 min at 25 °C followed by 10 min at 4 °C in G buffer (25 mm Tris-Cl, pH 7.8, 5 mm MgCl2, 0.8 mm ATP, 2 mg/ml BSA, 5 mm DTT, 5% glycerol) with 100 mm KCl and 2 μg of poly(dG/dC) competitor DNA (Sigma). The ORC–DNA samples were separated and analyzed as described previously (9) except that 80 μg/μl BSA was included in the polyacrylamide gel and running buffer.
M.HpaII and TALE-bound DNA templates
PCR was carried out using a plasmid containing the TALE-binding sequence (Tal-seq) at either the −25, −35, or −45 locations using a biotinylated oligo (Biopuc19A) and an oligo containing a 5FdC-modified M.HpaII methylation sequence (as described above) to create a large PCR fragment containing a TALE-binding sequence. The resulting DNA was purified using a PCR clean-up column (TaKaRa Clontech) and coupled to streptavidin-coated magnetic beads (Dynabeads M280, Invitrogen). Templates were first incubated with M.HpaII and then with TALE protein as described above.
Nucleosomal DNA templates and helicase-loading assays
Bead-associated nucleosomal DNA templates were assembled using ISW1a, Nap1, and purified yeast histone octamers as described previously (25) with the following modifications: a DNA/histone octamer ratio of 1:2.5 was used during the assembly reaction, and the assembly reactions were performed in the presence of 6 nm purified ORC. Naked templates were incubated with 6 nm ORC in buffers lacking ISW1a, hNap1, and histone octamers. After assembly, both naked DNA and nucleosomal DNA-bound magnetic beads were washed three times with 120 μl of A, 0.3 KGlut (25 mm HEPES-KOH, pH 7.6, 0.5 mm EGTA, 0.1 mm EDTA, 5 mm MgCl2, 10% glycerol, 0.02% Nonidet P-40, 0.1 mg/ml BSA, and 0.3 m KGlut) to remove excess nucleosome-assembly factors and ORC. Helicase-loading assays using bead-bound nucleosomal DNA templates bound to ORC were initiated by addition of 9 nm Cdc6 and 18 nm Mcm2-7/Cdt1 in a 20-μl reaction containing 60 fmol of bead-coupled 3.8-kb ARS1 DNA (with or without nucleosomes) in chromatin-loading buffer (25 mm HEPES-KOH, pH 7.6, 12.5 mm magnesium acetate, 300 mm KGlut, 20 μm creatine phosphate, 0.02% Nonidet P-40, 10% glycerol, 3 mm ATP, 1 mm DTT, and 2 μg creatine kinase). Reactions were briefly vortexed and then incubated at 25 °C at 1250 rpm for 30 min in a Thermomixer. Beads were then washed as described for the helicase-loading assay. DNA-bound proteins were eluted from the beads by incubation at 95 °C for 20 min after addition of 15 μl of 2× SDS-PAGE sample buffer (120 mm Tris, pH 6.8, 4% SDS, 20% glycerol, 5% β-mercaptoethanol). Eluted proteins were separated by SDS-PAGE and analyzed by immunoblotting.
Nucleosome mapping
Nucleosomes were assembled onto 1 pmol of pUC19 ARS1 plasmid DNA in the presence of 2 pmol of ISW1a, 80 pmol of Nap1, 2.5 pmol of ORC, and 20 pmol of histone octamers as described above. Free histones and assembly proteins were removed by gel filtration using a 5-ml Sephacryl S-500 column equilibrated in H buffer with 100 mm KGlut. ORC-bound chromatinized plasmids were then subjected to complete MNase digestion, and the resulting DNA was purified by phenol/chloroform extraction followed by ethanol precipitation. The purified DNA was separated on a 1.5% agarose gel electrophoresis and stained with ethidium bromide. DNA fragments in the size range that includes mononucleosomes (140–160 bp) were extracted from the gel and purified using spin columns (Freeze and Squeeze DNA Gel Extraction Spin Columns, Bio-Rad). DNA samples were end-repaired and adaptor-ligated. Libraries were quantified using the Fragment Analyzer (Advanced Analytical) and qPCR before being loaded for paired-end sequencing using an Illumina HiSeq 2000 (MIT BioMicro Center). Paired-end sequencing reads were aligned to the plasmid sequence, and nucleosome density was determined as described previously (Eaton et al. (11).
Author contributions
S. P. B. and M. D. W. conceived and coordinated the study. M. D. W. designed, performed, and analyzed the experiments shown in Figs. 1–5. M. D. W., S. K., and I. F. A. designed, performed, and analyzed the experiments shown in Fig. 6. Y. Z. engineered the TALE protein used in experiments shown in Figs. 1 and 3–5. M. D. W. and S. P. B. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Jeff Gelles for the TALE protein expression construct. The M.HpaII expression construct was a generous gift from New England Biolabs. We thank Johannes Walter and Julien Duxin for help with the M.HpaII expression protocol and David MacAlpine and Matthew Eaton for assistance with the analysis of nucleosome-positioning data. We thank the members of the Bell Laboratory for helpful discussions. We thank David Phizicky and Bob Sauer for comments on the manuscript. We thank the Genomics and Biopolymers cores of the Koch Institute Swanson Biotechnology Center for technical support.
This work was supported in part by the Koch Institute Support Grant P30-CA14051 from the NCI, National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S14, Tables S1–S3, and supporting references 1–8.
- ORC
- origin recognition complex
- ACS
- ARS consensus sequence
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- PDB
- Protein Data Bank
- NFR
- nucleosome-free region
- TALE
- transcription activator-like effector
- TCEP
- tris(2-carboxyethyl)phosphine
- 5FdC
- 5-fluoro-2′deoxycytidine
- KGlut
- potassium glutamate
- oligo
- oligonucleotide
- AMP
- ampicillin
- CAM
- chloramphenicol
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- Ni-NTA
- nickel-nitrilotriacetic acid
- MNase
- micrococcal nuclease.
References
- 1. Bell S. P., and Labib K. (2016) Chromosome duplication in Saccharomyces cerevisiae. Genetics 203, 1027–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Evrin C., Clarke P., Zech J., Lurz R., Sun J., Uhle S., Li H., Stillman B., and Speck C. (2009) A double-hexameric MCM2–7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl. Acad. Sci. U.S.A. 106, 20240–20245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Remus D., Beuron F., Tolun G., Griffith J. D., Morris E. P., and Diffley J. F. X. (2009) Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell 139, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Georgescu R., Yuan Z., Bai L., de Luna Almeida Santos R., Sun J., Zhang D., Yurieva O., Li H., and O'Donnell M. E. (2017) Structure of eukaryotic CMG helicase at a replication fork and implications to replisome architecture and origin initiation. Proc. Natl. Acad. Sci. U.S.A. 114, E697–E706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Broach J. R., Li Y. Y., Feldman J., Jayaram M., Abraham J., Nasmyth K. A., and Hicks J. B. (1983) Localization and sequence analysis of yeast origins of DNA replication. Cold Spring Harb. Symp. Quant. Biol. 47, Pt: 2, 1165–1173 [DOI] [PubMed] [Google Scholar]
- 6. Rao H., Marahrens Y., and Stillman B. (1994) Functional conservation of multiple elements in yeast chromosomal replicators. Mol. Cell. Biol. 14, 7643–7651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marahrens Y., and Stillman B. (1992) A yeast chromosomal origin of DNA replication defined by multiple functional elements. Science 255, 817–823 [DOI] [PubMed] [Google Scholar]
- 8. Theis J. F., and Newlon C. S. (1994) Domain B of ARS307 contains two functional elements and contributes to chromosomal replication origin function. Mol. Cell. Biol. 14, 7652–7659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rao H., and Stillman B. (1995) The origin recognition complex interacts with a bipartite DNA binding site within yeast replicators. Proc. Natl. Acad. Sci. U.S.A. 92, 2224–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rowley A., Cocker J. H., Harwood J., and Diffley J. F. (1995) Initiation complex assembly at budding yeast replication origins begins with the recognition of a bipartite sequence by limiting amounts of the initiator, ORC. EMBO J. 14, 2631–2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eaton M. L., Galani K., Kang S., Bell S. P., and MacAlpine D. M. (2010) Conserved nucleosome positioning defines replication origins. Genes Dev. 24, 748–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu W., Aparicio J. G., Aparicio O. M., and Tavaré S. (2006) Genome-wide mapping of ORC and Mcm2p binding sites on tiling arrays and identification of essential ARS consensus sequences in S. cerevisiae. BMC Genomics 7, 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Diffley J. F., and Stillman B. (1988) Purification of a yeast protein that binds to origins of DNA replication and a transcriptional silencer. Proc. Natl. Acad. Sci. U.S.A. 85, 2120–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chang F., Theis J. F., Miller J., Nieduszynski C. A., Newlon C. S., and Weinreich M. (2008) Analysis of chromosome III replicators reveals an unusual structure for the ARS318 silencer origin and a conserved WTW sequence within the origin recognition complex binding site. Mol. Cell. Biol. 28, 5071–5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crampton A., Chang F., Pappas D. L. Jr., Frisch R. L., and Weinreich M. (2008) An ARS element inhibits DNA replication through a SIR2-dependent mechanism. Mol. Cell 30, 156–166 [DOI] [PubMed] [Google Scholar]
- 16. Chang F., May C. D., Hoggard T., Miller J., Fox C. A., and Weinreich M. (2011) High-resolution analysis of four efficient yeast replication origins reveals new insights into the ORC and putative MCM binding elements. Nucleic Acids Res. 39, 6523–6535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin S., and Kowalski D. (1997) Functional equivalency and diversity of cis-acting elements among yeast replication origins. Mol. Cell. Biol. 17, 5473–5484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang R. Y., and Kowalski D. (1993) A DNA unwinding element and an ARS consensus comprise a replication origin within a yeast chromosome. EMBO J. 12, 4521–4531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wilmes G. M., and Bell S. P. (2002) The B2 element of the Saccharomyces cerevisiae ARS1 origin of replication requires specific sequences to facilitate pre-RC formation. Proc. Natl. Acad. Sci. U.S.A. 99, 101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Coster G., and Diffley J. F. X. (2017) Bidirectional eukaryotic DNA replication is established by quasi-symmetrical helicase loading. Science 357, 314–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zou L., and Stillman B. (2000) Assembly of a complex containing Cdc45p, replication protein A, and Mcm2p at replication origins controlled by S-Phase cyclin-dependent kinases and Cdc7p-Dbf4p kinase. Mol. Cell Biol. 20, 3086–3096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lipford J. R., and Bell S. P. (2001) Nucleosomes positioned by ORC facilitate the initiation of DNA replication. Mol. Cell 7, 21–30 [DOI] [PubMed] [Google Scholar]
- 23. Berbenetz N. M., Nislow C., and Brown G. W. (2010) Diversity of eukaryotic DNA replication origins revealed by genome-wide analysis of chromatin structure. PLoS Genet. 6, e1001092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dion M. F., Kaplan T., Kim M., Buratowski S., Friedman N., and Rando O. J. (2007) Dynamics of replication-independent histone turnover in budding yeast. Science 315, 1405–1408 [DOI] [PubMed] [Google Scholar]
- 25. Azmi I. F., Watanabe S., Maloney M. F., Kang S., Belsky J. A., MacAlpine D. M., Peterson C. L., and Bell S. P. (2017) Nucleosomes influence multiple steps during replication initiation. Elife 6, e22512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Simpson R. T. (1990) Nucleosome positioning can affect the function of a cis-acting DNA element in vivo. Nature 343, 387–389 [DOI] [PubMed] [Google Scholar]
- 27. Sun J., Fernandez-Cid A., Riera A., Tognetti S., Yuan Z., Stillman B., Speck C., and Li H. (2014) Structural and mechanistic insights into Mcm2-7 double-hexamer assembly and function. Genes Dev. 28, 2291–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sun J., Evrin C., Samel S. A., Fernández-Cid A., Riera A., Kawakami H., Stillman B., Speck C., and Li H. (2013) Cryo-EM structure of a helicase loading intermediate containing ORC–Cdc6-Cdt1-MCM2–7 bound to DNA. Nat. Struct. Mol. Biol. 20, 944–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yuan Z., Riera A., Bai L., Sun J., Nandi S., Spanos C., Chen Z. A., Barbon M., Rappsilber J., Stillman B., Speck C., and Li H. (2017) Structural basis of Mcm2-7 replicative helicase loading by ORC–Cdc6 and Cdt1. Nat. Struct. Mol. Biol. 24, 316–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ticau S., Friedman L. J., Ivica N. A., Gelles J., and Bell S. P. (2015) Single-molecule studies of origin licensing reveal mechanisms ensuring bidirectional helicase loading. Cell 161, 513–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frigola J., Remus D., Mehanna A., and Diffley J. F. (2013) ATPase-dependent quality control of DNA replication origin licensing. Nature 495, 339–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Speck C., Chen Z., Li H., and Stillman B. (2005) ATPase-dependent cooperative binding of ORC and Cdc6 to origin DNA. Nat. Struct. Mol. Biol. 12, 965–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bell S. P., and Stillman B. (1992) ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature 357, 128–134 [DOI] [PubMed] [Google Scholar]
- 34. Chen L., MacMillan A. M., Chang W., Ezaz-Nikpay K., Lane W. S., and Verdine G. L. (1991) Direct identification of the active-site nucleophile in a DNA (cytosine-5)-methyltransferase. Biochemistry 30, 11018–11025 [DOI] [PubMed] [Google Scholar]
- 35. Fu Y. V., Yardimci H., Long D. T., Ho T. V., Guainazzi A., Bermudez V. P., Hurwitz J., van Oijen A., Schärer O. D., and Walter J. C. (2011) Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell 146, 931–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Duxin J. P., Dewar J. M., Yardimci H., and Walter J. C. (2014) Repair of a DNA-protein cross-link by replication-coupled proteolysis. Cell 159, 346–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Long D. T., Joukov V., Budzowska M., and Walter J. C. (2014) BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol. Cell 56, 174–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liachko I., Youngblood R. A., Keich U., and Dunham M. J. (2013) High-resolution mapping, characterization, and optimization of autonomously replicating sequences in yeast. Genome Res. 23, 698–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Donovan S., Harwood J., Drury L. S., and Diffley J. F. (1997) Cdc6p-dependent loading of Mcm proteins onto pre-replicative chromatin in budding yeast. Proc. Natl. Acad. Sci. U.S.A. 94, 5611–5616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Randell J. C., Bowers J. L., Rodríguez H. K., and Bell S. P. (2006) Sequential ATP hydrolysis by Cdc6 and ORC directs loading of the Mcm2-7 helicase. Mol. Cell 21, 29–39 [DOI] [PubMed] [Google Scholar]
- 41. Lee D. G., and Bell S. P. (1997) Architecture of the yeast origin recognition complex bound to origins of DNA replication. Mol. Cell. Biol. 17, 7159–7168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tocilj A., On K. F., Yuan Z., Sun J., Elkayam E., Li H., Stillman B., and Joshua-Tor L. (2017) Structure of the active form of human origin recognition complex and its ATPase motor module. Elife 6, e20818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bleichert F., Botchan M. R., and Berger J. M. (2015) Crystal structure of the eukaryotic origin recognition complex. Nature 519, 321–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bleichert F., Balasov M., Chesnokov I., Nogales E., Botchan M. R., and Berger J. M. (2013) A Meier-Gorlin syndrome mutation in a conserved C-terminal helix of Orc6 impedes origin recognition complex formation. Elife. 2, e00882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen S., and Bell S. P. (2011) CDK prevents Mcm2-7 helicase loading by inhibiting Cdt1 interaction with Orc6. Genes Dev. 25, 363–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ticau S., Friedman L. J., Champasa K., Corrêa I. R. Jr., Gelles J., and Bell S. P. (2017) Mechanism and timing of Mcm2-7 ring closure during DNA replication origin licensing. Nat. Struct. Mol. Biol. 24, 309–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kang S., Warner M. D., and Bell S. P. (2014) Multiple functions for Mcm2-7 ATPase motifs during replication initiation. Mol. Cell 55, 655–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.