

Abstract
Biofilm formation by pathogenic bacteria is a hallmark of chronic infections. In many cases, lectins play key roles in establishing biofilms. The pathogen Pseudomonas aeruginosa often exhibiting various drug resistances employs its lectins LecA and LecB as virulence factors and biofilm building blocks. Therefore, inhibition of the function of these proteins is thought to have potential in developing “pathoblockers” preventing biofilm formation and virulence. A covalent lectin inhibitor specific to a carbohydrate binding site is described for the first time. Its application in the LecA‐specific in vitro imaging of biofilms formed by P. aeruginosa is also reported.
Keywords: biofilms, carbohydrates, glycomimetics, lectins, Pseudomonas aeruginosa
Lectins are carbohydrate‐binding proteins with very diverse functions that are found in all domains of life.1 These proteins play crucial roles in numerous processes such as cell–cell recognition, infection processes, and immune defense. They are generally characterized by an intermediate to low affinity towards their carbohydrate ligands that is often overcome by Nature through multivalency of both the lectin receptors and their carbohydrate ligands resulting in avidity with an increase in apparent affinity.
Because these carbohydrate‐binding proteins play essential roles in a number of pathological processes, they have become attractive targets for therapy. However, the fact that lectins display moderate affinities to their ligands renders this class of proteins as difficult targets for drugs.2 Despite this drawback, a number of recent success stories impressively demonstrated their potential for therapy: the selectin antagonist GMI‐1070 is currently in phase III clinical trials, and various FimH inhibitors are in the late preclinical stage.2, 3
Lectins are involved in infections with the Gram‐negative bacterium P. aeruginosa, one important member of the often highly drug‐resistant ESKAPE pathogens, which are Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumanii, P. aeruginosa, and Enterobacter species, and currently cause most of the severe hospital infections in western countries.4 The two bacterial lectins,5 LecA and LecB, are virulence factors and important for bacterial adhesion and biofilm formation.6 The latter is especially problematic as resistance against antibiotics inside a biofilm is increased by a factor of 10–1000.7 Thus, the inhibition of these lectins provides a promising way to dismantle the bacterium from the protective biofilm environment and restore immune defense and activity of antibiotics.8 Current approaches to inhibit both lectins range from small molecules to multivalent structures and are summarized in recent reviews.9 We focus on the development of small molecules and recently published various potent glycomimetic inhibitors for the high affinity lectin LecB as inhibitors of P. aeruginosa adhesion.10 In contrast, LecA only has an intermediate affinity for its monovalent d‐galactose‐derived ligands in the 50–100 μm range.5, 11 Phenyl β‐d‐galactosides and derivatives showed an increased affinity of approximately 10 μm, for example compound 1 (Figure 1), but despite a high number of derivatives analyzed, no further significant increase in potency could be achieved.12
Figure 1.
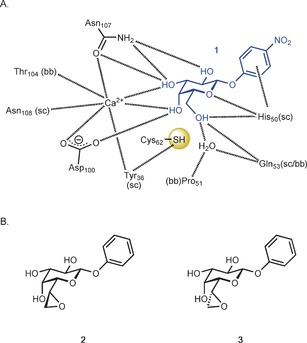
A) Galactoside recognition by the bacterial lectin LecA (pdb: 3ZYF[15]); B) Electrophilic epoxide derivatives 2 and 3 for the targeting of Cys62. The distances from Cys62‐S to C6 and O6 of 1 are between 4.1 and 4.3 Å. (sc=side chain, bb=backbone).
Covalent inhibition is one strategy to avoid dissociation of the inhibitor from the target and thus to persistently inactivate proteins. To date, a specific covalent inhibition of the carbohydrate binding site in a lectin has not been achieved despite attempts using squaric acid to target FimH13 or photoactivatable substituents for the targeting of galectins.14 The latter probes covalently bind to unspecific residues of the protein in proximity (3 Å) to the photoactivated center. The crystal structure of LecA11a, 15 reveals the presence of one cysteine residue (Cys62) in the carbohydrate binding domain (Figure 1). The specific targeting of cysteine residues with electrophilic warheads is a general strategy in the search for cysteine protease inhibitors,16 but has never been addressed in carbohydrate recognition domains. To target Cys62, we designed the two diastereoisomeric galactose‐derived epoxides 2 and 3 (Figure 1) as potential covalent active site inhibitors of LecA.
The 8‐step synthesis of epoxides 2 and 3 started from d‐galactose (4), with epoxidation as the last step (Scheme 1). Galactose 4 was protected as diacetonide (5) and the free primary hydroxy group was oxidized under Swern conditions (6) in good yields. Then, heptose 7 was established in a Wittig reaction followed by a change in protecting groups from acetonides to acetates (8). Lewis acid catalyzed glycosylation of phenol with 8 gave phenyl β‐glycoside 9 in good yield. Glycoside 9 was deprotected in a Zemplén transesterification reaction to give olefin 10 in 97 % yield. Late‐stage oxidation with mCPBA yielded the two diasteroisomeric epoxides 2 and 3 in 9 % and 19 %, respectively. The stereochemistry of 3 was established by X‐ray crystallography in complex with LecA (see Figure 3).
Scheme 1.
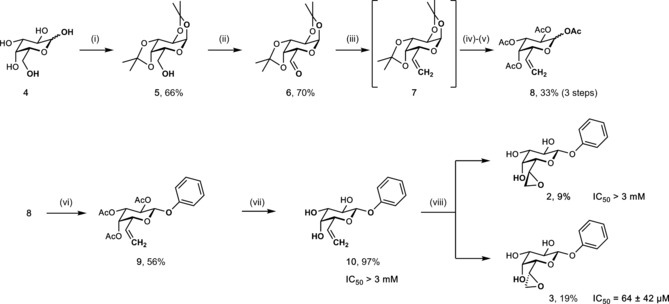
Synthesis of LecA‐directed epoxides and competitive binding to LecA. i) acetone, ZnCl2, H2SO4; ii) (COCl)2, DMSO, NEt3, CH2Cl2, −78 °C to 0 °C; iii) PPh3*MeI, NaH, DMSO; iv) 70 % HOAc aq.; v) Ac2O, pyridine; vi) PhOH, BF3*Et2O, CH2Cl2, −20 °C–r.t.; vii) NaOMe, MeOH, r.t.; viii) mCPBA, NaHCO3, CH2Cl2/MeOH;.
Figure 3.
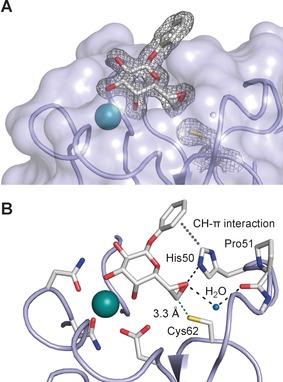
Crystal structure of epoxide 3 in complex with LecA at 1.80 Å resolution in the non‐covalent binding mode obtained at pH 4.6 (pdb code 5MIH). A) Electron density displayed at 1σ for ligand and Cys62 side chain. B) Interaction of the ligand with LecA: the epoxy oxygen atom accepts hydrogen bonds from His50 and one protein‐bound water molecule. Furthermore, His50 established a CH–π interaction with the phenyl agylycon. In the crystal structure, the sulfur atom of Cys62 is 3.3 Å away from C7 of ligand 3.
Both epoxides 2 and 3 and olefin 10 were then tested in a recently developed competitive binding assay12d for inhibition of LecA. No inhibition was observed for olefin 10 up to 3 mm. In contrast, the epoxides showed a strong diastereoselectivity for inhibition of LecA: 3 was a good inhibitor with an IC50=64 μm, whereas its diastereomer 2 was not recognized (IC50 >3 mm), indicating a specific binding of epoxide 3 to LecA. Covalent inhibitors usually show a time dependent reduction of IC50s due to the accumulation at the protein. We have therefore studied the time dependency of the binding of 2, 3, and phenyl β‐d‐galactoside to LecA (Supporting Information, Figure S21). The IC50 of the latter non‐covalent inhibitor stayed constant over time, inactive diastereomer 2 remained inactive, whereas the binding epoxide diastereomer 3 showed a time dependent decrease of the IC50 values, indicative for covalent binding.
To assess the binding mode of 3 with LecA, we analyzed LecA in presence and absence of 3 by mass spectrometry (Figure 2). LC‐MS measurements on intact protein level showed a mass shift of 268.1 Da when incubated with 3 and thus prove a covalent binding of the epoxide to LecA (Figure 2 A,B). Attempts to enzymatically or chemically digest the LecA complex failed due to the extraordinary stability of LecA. To localize the binding site of the epoxide, an MS‐based sequencing using MALDI in source decay (ISD) was therefore performed for both samples (Figure 2 C). MALDI‐ISD experiments with c‐ion series annotated in a range from 5000 to 8000 m/z were instrumental to identify the peptide sequence ranging from Arg 48 to Thr 74 and Asn 71, respectively. For the LecA sample co‐incubated with 3, a mass increase of 268 Da to the c62 ion indicated the binding of compound 3 to Cys62 of LecA, with some unspecific binding to Cys57 also detected. Based on the mass increase of 268 Da to the c62 ion, 3 showed covalent binding to LecA and the nucleophile for the epoxide ring‐opening was the sulfhydryl group of Cys62 in the carbohydrate recognition domain.
Figure 2.
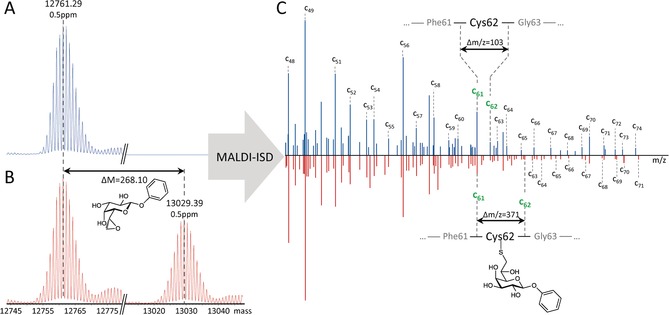
Covalent binding mode of 3 with LecA established by mass spectrometry. Deconvoluted intact protein MS spectra of LecA: A) without inhibitor, and B) with inhibitor 3; C) MALDI‐ISD experiments with c‐ion series annotated in a range from 5000 to 8000 m/z.
We then crystallized LecA in complex with epoxide 3 and solved the structure by X‐ray crystallography (Figure 3). In this complex, 3 adopts a coordination to the calcium ion bound to LecA as it had been reported for other galactosides before.11a, 15 Surprisingly, despite its orientation towards Cys62, the epoxide moiety in 3 is still intact and the covalent adduct could not be observed in this structure. These differences to the covalent adduct detected by mass spectrometry are likely a result of the different pH values of the buffers employed: lectin binding assays and mass spectrometry were performed at a physiologically buffered pH (7.4), whereas LecA was crystallized at pH 4.6. Numerous attempts to obtain LecA crystals with 3 as a covalent adduct by cocrystallization or soaking at neutral pH have been unsuccessful to date. All data collection of protein crystals incubated with diastereomeric epoxide 2 led to empty binding sites confirming the low affinity of 2 for LecA.
To exploit this unique covalent lectin ligand for biological applications such as lectin specific staining, we synthesized alkyne‐bearing derivatives that were then coupled to a fluorescent azide in a Huisgen dipolar cycloaddition (Scheme 2). Glycosyl donor 8 was reacted under Lewis acid catalysis with the acceptor monopropargyl hydroquinone to give the glycoside 11. Here, we first oxidized the peracetylated olefin 11 using mCPBA and the two diasteromeric epoxides 12 and 13 were obtained after chromatographic separation in 21 % and 46 % yield, respectively. Subsequently, the acetates were removed in a Zemplén type reaction to individually give 14 or 15. Both were then tested for inhibition of LecA showing a comparable diastereoselectivity as observed for the unsubstituted phenyl derivatives 2 and 3 before: 6d epoxide 15 inhibited LecA with an IC50 of 109 μm, whereas the diastereomeric 6l epoxide 14 was inactive (IC50>3 mm). The stereochemistry of 14 and 15 was unambiguously assigned by combining the activity data and NMR chemical shift and coupling constant analysis and comparison with analogues 2 and 3. The active diasteromer 15 was then coupled in a copper(II)‐catalyzed click reaction to the azide 16 17 to give fluorescent probe 17 in good yields.
Scheme 2.
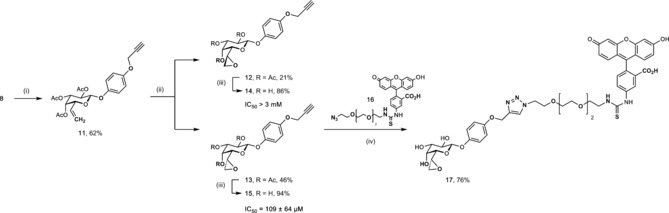
Synthesis of LecA‐directed propargylated epoxides with LecA inhibition data and synthesis of fluorescent derivative 17. i) hydoquinone monopropargyl ether, BF3*Et2O, CH2Cl2, −20 °C–r.t.; ii) mCPBA, NaHCO3, CH2Cl2, 0 °C–r.t.; iii) NaOMe, MeOH, 0 °C; iv) CuSO4, sodium ascorbate, H2O, DMF, r.t.
The covalent nature of the binding of fluoresceine‐derivative 17 was then further studied by incubation with LecA. Complexes of LecA with 17 or with a non‐covalent analogue 18 were performed as observed by high fluorescence polarization. In contrast to the covalent complex of 17 with LecA, 18 could be completely displaced from LecA with the competitive inhibitor methyl galactoside (Supporting Information, Figure S22). Furthermore, the complex of LecA with 17 was analyzed by polyacrylamide gel electrophoresis under denaturing conditions (Figure 4). LecA incubated with 17 gave a single fluorescent band that was also stained by Coomassie corresponding to the molecular weight of the denatured monomer of LecA and thus yields further evidence for covalent binding.
Figure 4.
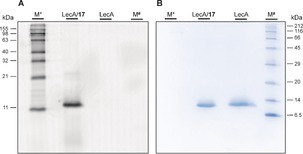
Covalent binding of 17 to LecA established by SDS‐PAGE. A) Fluorescence imaging; B) Coomassie staining; M* BenchMark fluorescent protein standard (Thermo), M# protein marker III (Applichem).
Tools to visualize the presence of carbapenem‐resistant bacterial pathogens in a test tube by specific activity‐based probes have recently been reported.18 The visualization of bacterial biofilm structures is of outstanding current interest, both, in vitro and in vivo.19 LecA is involved in the biofilm formation of P. aeruginosa and LecA‐deficient strains were shown to have thinner biofilms with a reduced biomass.6b, 20 Because the expression of LecA is upregulated in biofilms and it is located extracellularly,6b, 21 this protein is a promising target for the imaging of biofilms. We therefore explored whether the LecA‐directed epoxides reported here can specifically stain biofilms of P. aeruginosa. Bacterial biofilms were grown using mCherry‐expressing PAO1 wildtype bacteria and the corresponding LecA‐deficient strain PAO1 ΔlecA and then analyzed by confocal fluorescence microscopy (Figures 5, 6). Under shaking growth conditions, bacterial aggregates22 of the biofilm were observed in the PAO1 wildtype strain, whereas the ΔlecA strain generally showed a heavily reduced number of aggregates with smaller sizes and therefore also a higher number of planktonic bacteria since bacterial growth is comparable (Figures 5, 6; Supporting Information, Figure S23). After addition of the LecA‐directed dye 17 to the bacterial cultures, a specific staining of the wildtype biofilm aggregates was detected and no staining was visible in case of the aggregates formed by the ΔlecA strain. The green fluorescence originating from 17 was observed on the entire structure of the wildtype bacterial aggregates, whereas no or only a very faint color on the surface of the ΔlecA strain aggregates was observed without any detectable fluorophore inside these aggregates. The largest aggregate found for the ΔlecA strain was also analyzed (Supporting Information, Figures S24–S26) and staining was LecA‐specific and independent of aggregate size. Thus, the bacterial lectin LecA can be exploited as a target to visualize biofilms of P. aeruginosa using conjugates of LecA‐ligands, such as the galactose‐derived epoxide 17.
Figure 5.
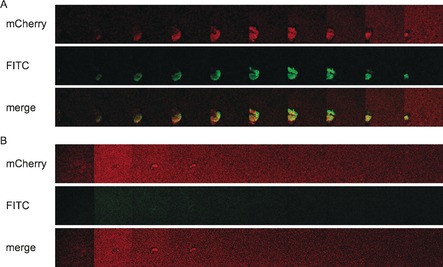
Galleries of LecA‐dependent staining of P. aeruginosa biofilms with 17. A) P. aeruginosa PAO1 wt or B) the lecA knockout (ΔlecA) mutant, both expressing mCherry from pMP7605, were incubated at 37 °C for 24 h with agitation (180 rpm). Biofilms were stained with the covalent LecA ligand fused to fluorescein (17) for 10–30 min. Z‐stacks (232×232 μm) were recorded every 2 μm at 561 nm for mCherry (red, A and B, upper panels) and 488 nm for fluorescein (green, A and B middle panels). The galleries show every 4th z‐stack recorded. Lower panels show merged images of both channels (488 nm and 561 nm).
Figure 6.

Three‐dimensional imaging of LecA‐dependent staining of P. aeruginosa PAO1 biofilms with 17. A) P. aeruginosa PAO1 wt or B) the lecA knockout (ΔlecA) mutant expressing mCherry from pMP7605 were incubated at 37 °C for 24 h with agitation (180 rpm). Biofilms were stained with 17 for 10–30 min. Z‐stacks (232×232 μm) were recorded every 2 μm at 561 nm for mCherry (red) and 488 nm for fluorescein (green). The 3D images show merged images of both channels (488 nm and 561 nm) from top and side views.
In summary, we have developed the first covalent inhibitor of carbohydrate binding sites by rational structure‐based design. Both diastereomers of the epoxygalactoheptoside 2 and 3 were synthesized and biologically evaluated. LecA displayed a strong diastereoselectivity for the 6d epimer 3 over its 6l isomer 2. The binding site and its covalent nature at physiological pH was established using mass spectrometry‐based sequencing and the non‐covalent crystal structure of 3 in complex with LecA was solved at pH 4.6. Finally, we used the fluoresceine‐derivative 17 for the LecA‐specific staining of P. aeruginosa biofilms. Such conjugates may lead to the development of pathogen‐specific imaging agents to localize bacterial biofilm‐associated infections inside an infected host enabling pathogen‐ and tissue‐directed therapy.23
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors acknowledge the continuous support of Prof. Rolf W. Hartmann (HIPS). We are further grateful to Prof. Bodo Philipp (University of Münster) and Prof. Arthur F. J. Ram (Leiden University) for providing bacterial strains and plasmids used in this study, to Sarah Henrikus (Saarland University) for the synthesis of 16 and to Laura Becker (University of Konstanz) for initial synthetic experiments towards the galactose‐derived epoxides and Dr. Josef Zapp (Saarland University) for acquisition of NMR spectra. Emilie Gillon prepared LecA for crystallization and performed some crystallization trials. A.I. and A.V. acknowledge support from the ANR projects Glyco@Alps (ANR‐15‐IDEX‐02) and Labex ARCANE (ANR‐11‐LABX‐003). A.V. acknowledges financial support from the European Community's Seventh Framework Programme (FP7/2007–2013) under BioStruct‐X (grant agreement No. 283570). The authors thank the EMBL‐Grenoble staff for assistance in using the High Throughput Crystallization (HTX) facility and the European Synchrotron Radiation Facility, Grenoble, France for the access and technical support to beamline BM30A‐FIP. A.T. acknowledges the following funding agencies for generous funding: the Helmholtz‐Association (grant no. VH‐NG‐934), the Deutsche Forschungsgemeinschaft (grant no. Ti 756/2‐1), and the European Research Council for an ERC Starting Grant (SWEETBULLETS).
S. Wagner, D. Hauck, M. Hoffmann, R. Sommer, I. Joachim, R. Müller, A. Imberty, A. Varrot, A. Titz, Angew. Chem. Int. Ed. 2017, 56, 16559.
References
- 1.
- 1a. Lis H., Sharon N., Chem. Rev. 1998, 98, 637–674; [DOI] [PubMed] [Google Scholar]
- 1b. Varki A. Essentials of Glycobiology , 2nd ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N. Y., 2009. [PubMed] [Google Scholar]
- 2. Ernst B., Magnani J. L., Nat. Rev. Drug Discovery 2009, 8, 661–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Chang J., Patton J. T., Sarkar A., Ernst B., Magnani J. L., Frenette P. S., Blood 2010, 116, 1779–1786; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Klein T., Abgottspon D., Wittwer M., Rabbani S., Herold J., Jiang X., Kleeb S., Lüthi C., Scharenberg M., Bezençon J., Gubler E., Pang L., Smiesko M., Cutting B., Schwardt O., Ernst B., J. Med. Chem. 2010, 53, 8627–8641; [DOI] [PubMed] [Google Scholar]
- 3c. Cusumano C. K., Pinkner J. S., Han Z., Greene S. E., Ford B. A., Crowley J. R., Henderson J. P., Janetka J. W., Hultgren S. J., Sci. Transl. Med. 2011, 3, 109ra115; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Telen M. J., Wun T., McCavit T. L., De Castro L. M., Krishnamurti L., Lanzkron S., Hsu L. L., Smith W. R., Rhee S., Magnani J. L., Thackray H., Blood 2015, 125, 2656–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Boucher H. W., Talbot G. H., Bradley J. S., Edwards J. E., Gilbert D., Rice L. B., Scheld M., Spellberg B., Bartlett J., Clin. Infect. Dis. 2009, 48, 1–12; [DOI] [PubMed] [Google Scholar]
- 4b. Rice L. B., J. Infect. Dis. 2008, 197, 1079–1081. [DOI] [PubMed] [Google Scholar]
- 5. Gilboa-Garber N., Methods Enzymol. 1982, 83, 378–385. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Tielker D., Hacker S., Loris R., Strathmann M., Wingender J., Wilhelm S., Rosenau F., Jaeger K.-E., Microbiology 2005, 151, 1313–1323; [DOI] [PubMed] [Google Scholar]
- 6b. Diggle S. P., Stacey R. E., Dodd C., Cámara M., Williams P., Winzer K., Environ. Microbiol. 2006, 8, 1095–1104. [DOI] [PubMed] [Google Scholar]
- 7. Davies D., Nat. Rev. Drug Discovery 2003, 2, 114–122. [DOI] [PubMed] [Google Scholar]
- 8. Michaud G., Visini R., Bergmann M., Salerno G., Bosco R., Gillon E., Richichi B., Nativi C., Imberty A., Stocker A., Darbre T., Reymond J.-L., Chem. Sci. 2016, 7, 166–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Reymond J.-L., Bergmann M., Darbre T., Chem. Soc. Rev. 2013, 42, 4814–4822; [DOI] [PubMed] [Google Scholar]
- 9b. Bernardi A., Jiménez-Barbero J., Casnati A., De Castro C., Darbre T., Fieschi F., Finne J., Funken H., Jaeger K.-E., Lahmann M., Lindhorst T. K., Marradi M., Messner P., Molinaro A., Murphy P. V., Nativi C., Oscarson S., Penadés S., Peri F., Pieters R. J., Renaudet O., Reymond J.-L., Richichi B., Rojo J., Sansone F., Schäffer C., Turnbull W. B., Velasco-Torrijos T., Vidal S., Vincent S., Wennekes T., Zuilhof H., Imberty A., Chem. Soc. Rev. 2013, 42, 4709–4727; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9c. Titz A., in Topics in Medicinal Chemistry, Vol. 12 (Carbohydrates as Drugs) (Eds.: P. H. Seeberger, C. Rademacher), Springer, Berlin, Heidelberg, 2014, p. 169–186; [Google Scholar]
- 9d. Sommer R., Joachim I., Wagner S., Titz A., CHIMIA 2013, 67, 286–290; [DOI] [PubMed] [Google Scholar]
- 9e. Wagner S., Sommer R., Hinsberger S., Lu C., Hartmann R. W., Empting M., Titz A., J. Med. Chem. 2016, 59, 5929–5969. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Hauck D., Joachim I., Frommeyer B., Varrot A., Philipp B., Möller H. M., Imberty A., Exner T. E., Titz A., ACS Chem. Biol. 2013, 8, 1775–1784; [DOI] [PubMed] [Google Scholar]
- 10b. Sommer R., Exner T. E., Titz A., PLoS One 2014, 9, e112822; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c. Sommer R., Hauck D., Varrot A., Wagner S., Audfray A., Prestel A., Möller H. M., Imberty A., Titz A., ChemistryOpen 2015, 4, 756–767; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10d. Hofmann A., Sommer R., Hauck D., Stifel J., Göttker-Schnetmann I., Titz A., Carbohydr. Res. 2015, 412, 34–42; [DOI] [PubMed] [Google Scholar]
- 10e. Beshr G., Sommer R., Hauck D., Siebert D. C. B., Hofmann A., Imberty A., Titz A., Med. Chem. Commun. 2016, 7, 519–530; [Google Scholar]
- 10f. Sommer R., Wagner S., Varrot A., Nycholat C. M., Khaledi A., Häussler S., Paulson J. C., Imberty A., Titz A., Chem. Sci. 2016, 7, 4990–5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Cioci G., Mitchell E. P., Gautier C., Wimmerová M., Sudakevitz D., Pérez S., Gilboa-Garber N., Imberty A., FEBS Lett. 2003, 555, 297–301; [DOI] [PubMed] [Google Scholar]
- 11b. Blanchard B., Nurisso A., Hollville E., Tétaud C., Wiels J., Pokorná M., Wimmerová M., Varrot A., Imberty A., J. Mol. Biol. 2008, 383, 837–853. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Rodrigue J., Ganne G., Blanchard B., Saucier C., Giguère D., Shiao T. C., Varrot A., Imberty A., Roy R., Org. Biomol. Chem. 2013, 11, 6906–6918; [DOI] [PubMed] [Google Scholar]
- 12b. Cecioni S., Praly J.-P., Matthews S. E., Wimmerová M., Imberty A., Vidal S., Chem. Eur. J. 2012, 18, 6250–6263; [DOI] [PubMed] [Google Scholar]
- 12c. Kadam R. U., Garg D., Schwartz J., Visini R., Sattler M., Stocker A., Darbre T., Reymond J.-L., ACS Chem. Biol. 2013, 8, 1925–1930; [DOI] [PubMed] [Google Scholar]
- 12d. Joachim I., Rikker S., Hauck D., Ponader D., Boden S., Sommer R., Hartmann L., Titz A., Org. Biomol. Chem. 2016, 14, 7933–7948. [DOI] [PubMed] [Google Scholar]
- 13. Grabosch C., Hartmann M., Schmidt-Lassen J., Lindhorst T. K., ChemBioChem 2011, 12, 1066–1074. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. van Scherpenzeel M., Moret E. E., Ballell L., Liskamp R. M. J., Nilsson U. J., Leffler H., Pieters R. J., ChemBioChem 2009, 10, 1724–1733; [DOI] [PubMed] [Google Scholar]
- 14b. Ballell L., van Scherpenzeel M., Buchalova K., Liskamp R. M. J., Pieters R. J., Org. Biomol. Chem. 2006, 4, 4387–4394; [DOI] [PubMed] [Google Scholar]
- 14c. Ballell L., Alink K. J., Slijper M., Versluis C., Liskamp R. M. J., Pieters R. J., ChemBioChem 2005, 6, 291–295. [DOI] [PubMed] [Google Scholar]
- 15. Kadam R. U., Bergmann M., Hurley M., Garg D., Cacciarini M., Swiderska M. A., Nativi C., Sattler M., Smyth A. R., Williams P., Cámara M., Stocker A., Darbre T., Reymond J.-L., Angew. Chem. Int. Ed. 2011, 50, 10631–10635; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10819–10823. [Google Scholar]
- 16. Otto H., Schirmeister T., Chem. Rev. 1997, 97, 133–171. [DOI] [PubMed] [Google Scholar]
- 17. Loison S., Cottet M., Orcel H., Adihou H., Rahmeh R., Lamarque L., Trinquet E., Kellenberger E., Hibert M., Durroux T., Mouillac B., Bonnet D., J. Med. Chem. 2012, 55, 8588–8602. [DOI] [PubMed] [Google Scholar]
- 18. Mao W., Xia L., Xie H., Angew. Chem. Int. Ed. 2017, 56, 4468–4472; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4539–4543. [Google Scholar]
- 19.
- 19a. Kim J.-Y., Sahu S., Yau Y.-H., Wang X., Shochat S. G., Nielsen P. H., Dueholm M. S., Otzen D. E., Lee J., Delos Santos M. M. S., Yam J. K. H., Kang N.-Y., Park S.-J., Kwon H., Seviour T., Yang L., Givskov M., Chang Y.-T., J. Am. Chem. Soc. 2016, 138, 402–407; [DOI] [PubMed] [Google Scholar]
- 19b. Garrido V., Collantes M., Barberán M., Peñuelas I., Arbizu J., Amorena B., Grilló M.-J., Antimicrob. Agents Chemother. 2014, 58, 6660–6667; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19c. Mills B., Bradley M., Dhaliwal K., Clin. Transl. Imaging 2016, 4, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ligeour C., Vidal O., Dupin L., Casoni F., Gillon E., Meyer A., Vidal S., Vergoten G., Lacroix J.-M., Souteyrand E., Imberty A., Vasseur J.-J., Chevolot Y., Morvan F., Org. Biomol. Chem. 2015, 13, 8433–8444. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Dötsch A., Eckweiler D., Schniederjans M., Zimmermann A., Jensen V., Scharfe M., Geffers R., Häussler S., PLoS One 2012, 7, e31092; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21b. Hentzer M., Eberl L., Givskov M., Biofilms 2005, 2, 37–61. [Google Scholar]
- 22. Kragh K. N., Hutchison J. B., Melaugh G., Rodesney C., Roberts A. E. L., Irie Y., Jensen P. Ø., Diggle S. P., Allen R. J., Gordon V., Bjarnsholt T., MBio 2016, 7, e00237-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The Supporting Information contains experimental details, 1H and 13C NMR spectra, and LC-MS traces of 16 and 17 Crystallographic data of the complex of LecA with 3 are given in Table S1; binding kinetics experiments and inhibitor displacement experiments, biofilm fluorescence microscopy data prior to staining, stained unprocessed raw data, and processed data of the largest biofilm aggregate of the ΔlecA strain are also given.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary