

Abstract
Phosphotyrosine (pTyr)-dependent signaling is critical for many cellular processes. It is highly dynamic, as signal output depends not only on phosphorylation and dephosphorylation rates but also on the rates of binding and dissociation of effectors containing phosphotyrosine-dependent binding modules such as Src homology 2 (SH2) and phosphotyrosine-binding (PTB) domains. Previous in vitro studies suggested that binding of SH2 and PTB domains can enhance protein phosphorylation by protecting the sites bound by these domains from phosphatase-mediated dephosphorylation. To test whether this occurs in vivo, we used the binding of growth factor receptor bound 2 (GRB2) to phosphorylated epidermal growth factor receptor (EGFR) as a model system. We analyzed the effects of SH2 domain overexpression on protein tyrosine phosphorylation by quantitative Western and far-Western blotting, mass spectrometry, and computational modeling. We found that SH2 overexpression results in a significant, dose-dependent increase in EGFR tyrosine phosphorylation, particularly of sites corresponding to the binding specificity of the overexpressed SH2 domain. Computational models using experimentally determined EGFR phosphorylation and dephosphorylation rates, and pTyr–EGFR and GRB2 concentrations, recapitulated the experimental findings. Surprisingly, both modeling and biochemical analyses suggested that SH2 domain overexpression does not result in a major decrease in the number of unbound phosphorylated SH2 domain–binding sites. Our results suggest that signaling via SH2 domain binding is buffered over a relatively wide range of effector concentrations and that SH2 domain proteins with overlapping binding specificities are unlikely to compete with one another for phosphosites in vivo.
Keywords: epidermal growth factor receptor (EGFR), growth factor receptor-bound protein 2 (GRB2), phosphorylation, phosphotyrosine, Src homology 2 domain (SH2 domain), crk
Introduction
Src homology 2 (SH2)2 domains are small modular protein domains that bind specifically to tyrosine-phosphorylated sites on proteins (1, 2). In cell signaling, proteins that contain SH2 domains function to “read” post-translational marks that are “written” by activated tyrosine kinases and are “erased” by protein-tyrosine phosphatases (PTPs) (3, 4). Protein complexes mediated by SH2–phosphotyrosine (pTyr) interactions are critical for downstream signaling from a number of important tyrosine-phosphorylated proteins, including activated receptor tyrosine kinases such as the epidermal growth factor receptor (EGFR), and the proteins phosphorylated by receptor tyrosine kinases as well as by non-receptor cytoplasmic tyrosine kinases, such as Abl and Src (5, 6).
In humans, at least 120 unique SH2 domains have been identified in proteins with varying functions, including adaptors, tyrosine kinases and phosphatases, and lipid-modifying enzymes (5, 7). SH2-binding specificity and affinity are determined by the amino acid sequence flanking pTyr in their binding sites. In particular, amino acids at positions −1 through +3 relative to the pTyr have been shown to have the greatest influence. Although the specificity of each SH2 domain is unique, most SH2s bind to one of a few general motifs: +1 D/E, +2 N, +3 P/L/V, and +1 M +3 M (8–10). SH2 domain phosphosite motif specificities and affinities have been elaborated using in vitro methods such as surface plasmon resonance, solution assays, peptide and protein microarrays, and pulldown-based approaches (11–14). In addition to SH2 domains, there are a relatively small number of other pTyr-specific binding modules, which include several phosphotyrosine-binding (PTB) domains (15, 16).
SH2 binding in vivo is highly dynamic (17, 18). Not only do SH2 domains bind to and dissociate from phosphosites rapidly, but phosphosites themselves turn over rapidly, with half-times in the range of seconds; the rate of phosphosite turnover is dependent on both kinase and phosphatase activity (17, 19). Furthermore, multiple SH2 domains are expressed and can compete for binding to phosphosites. Thus, understanding SH2-mediated signal output requires consideration of pTyr flux and local concentrations of SH2-containing proteins, in addition to binding site specificity.
To study the interplay between SH2 domain binding and phosphosite dynamics, we have exploited EGFR, a major docking site for multiple SH2 domain-containing proteins. EGFR kinase activity increases when its ligand, EGF, binds to the extracellular domain of EGFR, inducing structural changes that promote receptor dimerization (20, 21). As a result, cellular levels of EGFR kinase activity can easily be manipulated by varying ligand concentration. Moreover, unlike most tyrosine kinases, EGFR activity does not depend on tyrosine phosphorylation of the so-called activation loop (22, 23). This is important because the effects of SH2 expression on receptor phosphorylation can be assessed independently from phosphorylation-associated receptor activation. Activated dimerized receptors phosphorylate the C-terminal tyrosine residues that serve as binding sites for a set of SH2 and PTB domain-containing proteins, including GRB2, SHCA, PLCγ1, and SHP2 (13, 24, 25). Each SH2 domain is thought to bind preferentially to a specific individual phosphosite or subset of phosphosites based on its individual binding specificity. For example, GRB2 has been shown bind to pYXN motifs (where X can be any amino acid) at EGFR pTyr-1068, pTyr-1086, and pTyr-1114, whereas the SHCA PTB domain has been shown to bind strongly to pTyr-1148, an NPDpY motif (9, 26–28).
Previous studies suggested that SH2 domains could specifically prevent dephosphorylation of their binding partners in vitro (29–33). However, little is known about the impact in living cells, where phosphosite turnover is high and overall occupancy may be low. Here, we use EGFR, as well as constructs containing the GRB2 and CRK (v-Crk avian sarcoma virus CT10 oncogene homolog) SH2 domains, to investigate the interplay between SH2 domain binding and phosphosite dynamics in vivo, focusing specifically on a quantitative analysis of phosphosite protection. By employing biochemical analyses, pTyr-specific mass spectrometry (MS), and computation modeling, we demonstrate that GRB2 can enhance the steady-state tyrosine phosphorylation level of its binding sites in vivo through SH2-dependent protection from PTPs. Our results also suggest that SH2 protection has important implications for our understanding of binding site competition between SH2 domains with similar specificities. Furthermore, SH2-mediated pTyr protection might serve as the basis for a novel method for identifying SH2–pTyr interactions as they occur in vivo.
Results
GRB2 SH2 domain overexpression enhances EGFR phosphorylation
To assess the effect of SH2 protein–phosphosite interaction on tyrosine phosphorylation, we transiently overexpressed full-length wild-type (WT) GRB2 in COS1 cells and monitored cellular tyrosine phosphorylation before and after stimulation with EGF (2.5 ng/ml for 5 min) by anti-pTyr Western blotting (Fig. 1B, 1st to 4th lanes). GRB2 overexpression enhanced the tyrosine phosphorylation of a band corresponding to EGFR (identity based on previous experiments (34) and reactivity with phosphospecific EGFR antibodies, shown below) both before and after EGF treatment. Relative increases in pTyr–EGFR ranged between 1.5- and 4-fold.
Figure 1.

Effect of GRB2 expression in EGF-stimulated COS1 cells. A, diagram of major constructs used for this study: tdEOS-tagged GRB2 SH2, FL WT GRB2, and a chimera of GRB2 SH3 domains and the CRK SH2 domain (GCG). B, GRB2-mediated enhancement of EGFR phosphorylation is SH2-dependent. Representative immunoblot of lysates from COS1 cells transfected with empty vector (ev) or overexpressing GRB2, tdEOS–GRB2 SH2, or GCG constructs before and after stimulation with 2.5 ng/ml EGF. R86K = R86K mutant that cannot bind pTyr sites. Data from three or four biological replicates are shown in bar graph below (error bars, standard error of the mean (S.E.)). Asterisks indicate phosphorylation increases that were statistically significant (paired Student's t test, p < 0.05) when compared with their empty vector control, i.e. EV or EV + EGF. n = 3 for K86R SH2 mutant constructs; n = 4 for other constructs. C, far-Western blotting and immunoblotting of lysates from COS1 cells transfected with GRB2 constructs. In labels on right, “GRB2 FW” and “CRK FW” indicate far-Western blotting with GRB2 and CRK SH2 domains. Anti-tubulin serves as loading control.
GRB2 mediates signaling through a complex series of downstream pathways that could indirectly enhance EGFR phosphorylation, for example by increasing cytoplasmic kinase activity or suppressing phosphatase activity (35–37). To rule out downstream signaling as a driver of GRB2-mediated EGFR phospho-enhancement, we compared protein tyrosine phosphorylation after expression of full-length WT GRB2 and four GRB2-derived constructs as follows: a fluorescently tagged GRB2 SH2 domain (tdEOS–GRB2 SH2), previously shown to be recruited to the plasma membrane of EGF-stimulated cells (34, 38); full-length GRB2 and tdEOS–GRB2 SH2 constructs containing a mutation in the SH2 domain (R86K) previously shown to abrogate phospho-dependent interaction (39, 40); and a chimeric protein in which the SH2 domain of GRB2 is replaced by that from CRK, referred to here as GCG (Fig. 1, A and B, lanes 5–12). GCG was used to ensure any differences relative to GRB2 were due to the SH2 domain alone and not to SH3-mediated effects. Unlike the GRB2 SH2, which binds predominantly to the activated EGFR, the CRK SH2 domain binds predominantly to phosphorylated p130CAS, and only weakly to phosphorylated EGFR (34).
SH2 constructs carrying the R86K mutation failed to increase EGFR phosphorylation, demonstrating that the ability to bind pTyr was required. However, expression of the tdEOS–GRB2 SH2 fusion, which cannot mediate downstream signaling because it lacks the SH3 domains found in full-length GRB2, increased EGFR phosphorylation both before and after EGF stimulation. Compared with the full-length protein, transient expression of this construct resulted in a more pronounced increase in EGFR phosphorylation prior to EGF treatment, but a similar level of phospho-enhancement post-stimulation. By contrast, overexpression of the CRK–GRB2 chimera resulted in a significant increase in phosphorylation of a band corresponding to p130CAS (Fig. S1), with only a minor increase (less than 2-fold) in the phosphorylation of EGFR, consistent with its binding specificity for p130CAS pTyr sites (34, 41–43).
These results showed that overexpression of SH2 domains could specifically increase the tyrosine phosphorylation of their known cellular binding partners. To address more directly the SH2-binding specificity of the phosphosites enhanced by SH2 expression, we performed far-Western blotting on lysates from COS1 cells expressing the various GRB2 constructs and probed with recombinant GRB2 and CRK SH2 domains (Fig. 1C). Expression of constructs containing the GRB2 SH2 domain specifically enhanced binding of the GRB2 SH2 probe, particularly of the band corresponding to EGFR. By contrast, the CRK SH2 probe bound predominantly to the p130CAS band in GCG-expressing cells, although modest increases in binding were also seen for the EGFR band.
GRB2-mediated pTyr–EGFR enhancement depends on GRB2 concentration and EGFR kinase activity
Next, we assessed the dose dependence of the enhancement of EGFR phosphorylation by GRB2 expression. Increasing amounts of cDNA encoding full-length GRB2 were transfected in COS1 cells, and the average GRB2 concentration per cell was then calculated for each dose using a recombinant GST–GRB2 SH2 standard (Fig. 2A and Fig. S2). EGFR phosphorylation increased with GRB2 concentration (Fig. 1, B and C), up to 2–3-fold in both stimulated and serum-starved cells.
Figure 2.

Effect of GRB2 expression and EGF stimulation on GRB2-mediated enhancement. A, representative immunoblot of EGFR tyrosine phosphorylation in cells transfected with empty vector (ev) or increasing amounts of GRB2 construct. GRB2 expression levels were determined using a GRB2 SH2 standard and a GRB2 SH2-specific antibody. Densitometric quantification and fitting of EGFR phosphorylation data from three biological replicates are shown on the right. B, effect of increasing EGF stimulation on enhancement of total EGFR pTyr and EGFR pTyr-1068 (a GRB2 SH2-binding site) in cells overexpressing WT GRB2 or the inactive R86K mutant. Densitometric quantification of the data for total pTyr–EGFR is shown to the right. Error bars represent S.E. for three biological replicates. The increase in phosphorylation associated with GRB2 SH2 expression was significant at all EGF concentrations (paired Student's t test, p < 0.05, *), even though the relative effect size decreases with EGF concentration.
To explore the relationship between phosphosite flux and SH2-mediated phosphosite protection, we modulated EGFR kinase activity in SH2-overexpressing COS1 cells and monitored EGFR phosphorylation. Cells were transfected with tdEOS–GRB2 SH2 or the tdEOS–GRB2 SH2 R86K mutant and then treated with increasing concentrations of EGF (Fig. 2B). Fold enhancement in EGFR tyrosine phosphorylation induced by Grb2 SH2 expression (relative to expression of the mutant SH2) was fairly constant between 0 and 2.5 ng/ml EGF (4–5-fold) but fell off at higher EGF concentrations. The absolute increase in phosphorylation (difference between mutant and wild-type GRB2 pTyr–EGFR signal at a particular EGF concentration) peaked at 25 ng/ml EGF, and dropped off at both higher and lower concentrations. Immunoblots probed with a phosphospecific antibody for EGFR pTyr-1068, an established GRB2-binding site, revealed similar results (Fig. 2B).
Specificity of SH2-dependent phosphotyrosine enhancement
If SH2 domains enhance phosphorylation by protecting their binding sites from dephosphorylation, then protected sites should be enriched for high-affinity sites with canonical binding motifs for the overexpressed SH2 domains (e.g. pYXN motifs for GRB2). To test this, we transfected three constructs (tdEOS–GRB2 SH2, full-length GRB2, and GCG) in COS1 cells and monitored the absolute level of phosphorylation at each EGFR site (with and without treatment with 2.5 ng/ml EGF) using phosphosite-specific antibodies (Fig. 3A). To compare signal levels across multiple antibodies, we created a maximally phosphorylated pTyr–EGFR standard by treating COS1 cells with 200 ng/ml EGF and the phosphatase inhibitor pervanadate for 40 min (Fig. 3A, far right lanes). Using this method, we quantified the percent maximal phosphorylation for seven EGFR phosphosites. Cells expressing each of the three constructs displayed increased EGFR phosphorylation at all of the measured sites to varying degrees. Relative to the overall increase in phosphorylation of EGFR, as measured by anti-pTyr immunoblot, both WT GRB2 and tdEOS–GRB2 SH2 expression resulted in increased phosphorylation of the canonical GRB2-binding site pTyr-1068 (EpYINQ) but had an equal or even greater effect on pTyr-974 (FpYRAL), and it resulted in enhancement of all sites to some extent (Fig. 3A, adjacent bar graphs). As in previous experiments, GCG increased total EGFR phosphorylation by less than 2-fold. In these cells, phosphorylation was shifted in favor of pTyr-992 (EpYLIP), a canonical CRK-binding site, which was increased by ∼5-fold when compared with EGF-treated empty vector controls (Fig. 3A, adjacent bar graphs).
Figure 3.

Site-specific increases in EGFR phosphotyrosine in SH2-overexpressing cells. A, representative anti-pTyr and phosphosite-specific anti-pTyr–EGFR immunoblots from COS1 cells transfected with empty vector (ev), tdEOS–GRB2 SH2, FL GRB2 SH2, or GCG. EGF + pervanadate (200 ng/ml EGF, 100 μm pervanadate, 40 min) was used as a maximally phosphorylated standard and run at 1:10 dilution on the same membranes. Antibodies are indicated to the left; for phosphospecific antibodies, numbers indicate residue number of phosphosite recognized. Percent maximal phosphorylation for each site and total pTyr–EGFR are shown on the panels to right (n = 3 biological replicates, error bars = S.E.). Expression of GRB2 FL and SH2 constructs resulted in statistically significant increases in the total phosphorylation of EGFR as well as the phosphorylation of multiple EGFR phosphotyrosines, including the GRB2-binding site pTyr-1068 (paired Student's t test, p < 0.05, *). An inter-site comparison of fold increase in phosphorylation revealed a statistically significant increase in phosphorylation of pTyr-1068 associated with GRB2 FL expression in non-stimulated cells (ANOVA, Tukey's pairwise comparison, p < 0.05, **). B, relative increase in abundance of EGFR phosphopeptides detected by quantitative mass spectrometry in COS1 lysates from cells expressing indicated SH2 constructs. C, relative increase in abundance of p130CAS phosphopeptides detected by MS as in B. Error bars for B and C represent S.E. for three biological replicates.
Quantitative MS analysis of COS1 cells expressing the same four constructs revealed similar results (Fig. 3, B and C, and Table S1). Overexpression of GRB2 SH2 and full-length GRB2, and to a much lesser extent the GCG construct, resulted in a generalized increase in the relative abundance of the four EGFR phosphosites identified (pTyr-974, -1086, -1148, and -1173) both before and after EGF stimulation (Fig. 3B). Unlike pTyr–EGFR immunoblotting, MS data suggested that tyrosine phosphorylation of EGFR pTyr-974 was only mildly increased by exogenous GRB2 expression. All five of the p130CAS phosphopeptides identified contained the canonical CRK SH2-binding motif, pYDXP. Of these, phosphorylation of four was enhanced by GCG chimera expression (Fig. 3C).
Specificity of GRB2 SH2-mediated phosphosite enhancement is concentration-dependent
Although Western, far-Western, and MS data were broadly consistent with the hypothesis that SH2 domains protect their preferred binding sites from dephosphorylation by phosphatases in vivo, the specificity was rather modest compared with that seen using peptide-based in vitro interaction assays. One reason might be that our experiments were performed using high intracellular concentrations (greater than 4 μm) of SH2-containing constructs to maximize the effect of phosphosite protection. At such high concentrations, SH2 domains are more likely to interact with relatively low affinity sites. To better understand the effect of concentration on specificity in our system, we analyzed lysates of cells expressing a range of GRB2 concentrations by probing with EGFR phosphosite-specific antibodies (Fig. 4A). We found that phosphorylation of the known high-affinity GRB2-binding sites, pTyr-1068 and pTyr-1086, tended to be selectively enhanced at relatively low levels of GRB2 overexpression. By contrast, phosphorylation of sites predicted to bind the GRB2 SH2 with lower affinity, such as pTyr-974, was enhanced only at the highest GRB2 concentrations (Fig. 4B).
Figure 4.
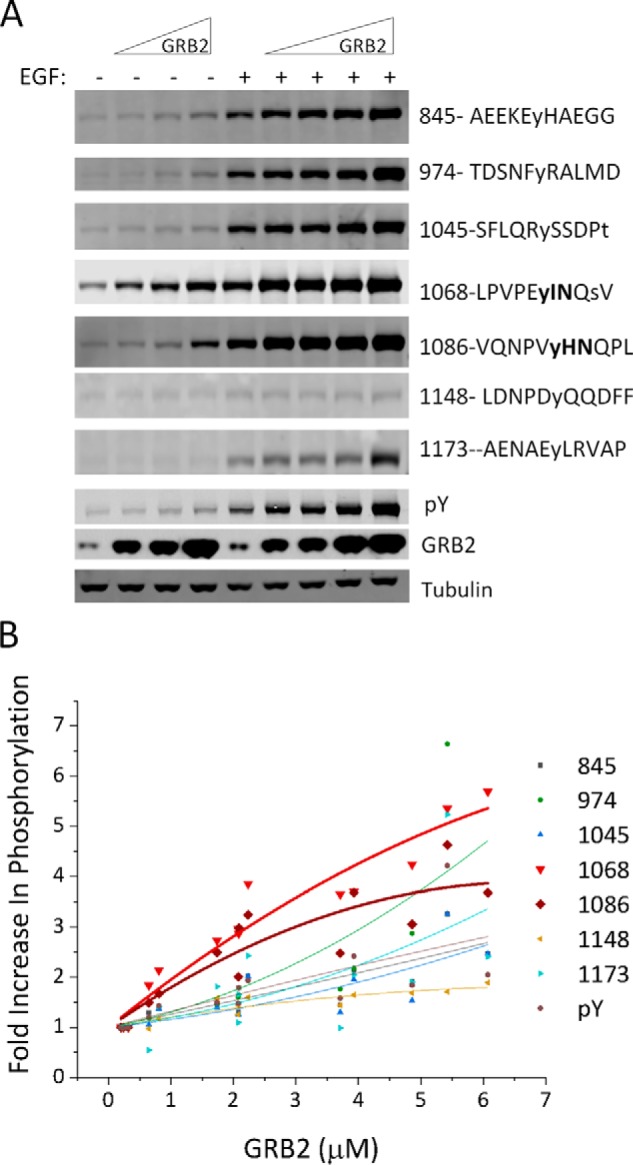
GRB2 specifically enhances its canonical binding motifs in a concentration-dependent manner. A, representative EGFR pTyr-specific immunoblots from COS1 cells expressing an increasing amount of GRB2. For phosphospecific antibodies, residue number and pTyr motifs of phosphosite recognized are indicated on the right. Lysates used were the same as for Fig. 2A, and pTyr, GRB2, and tubulin control blots are duplicated here. B, quantification of EGFR pTyr site-specific phosphorylation following EGF stimulation plotted against overexpressed GRB2 concentration (three biological replicates). Curves represent a polynomial fit of the combined data (R2 values for all curves are >0.84). Data for the two canonical GRB2-binding sites (Tyr-1068 and Tyr-1086) are bolded.
SH2 domain overexpression enhances phosphosites within canonical binding motifs across the phosphoproteome
To delve more deeply into the specificity of phosphosite protection associated with SH2 domain expression, we used our quantitative MS data to examine the sequence specificity of SH2-mediated phosphosite enhancement (34, 44). In total, we identified with high confidence 118 tyrosine-phosphorylated peptides from 79 different proteins (Table 1). The abundance of almost half of these phosphopeptides was significantly increased or decreased in cells overexpressing SH2 domains relative to controls (Fig. 5A). In particular, the phosphosites enhanced by expression of GRB2 or the GRB2 SH2 were enriched for the GRB2 SH2-binding motif (pYXN). When compared with the GRB2 SH2-binding site sequence identified in a recent phosphopeptide array study of 67 SH2 domains performed by Tinti et al. (10), the sites protected by GRB2 are more similar to the Tinti GRB2 SH2 sites than to 88% of the SH2 domains in original study (Fig. 5B and Fig. S3) Likewise, GCG-enhanced sites were enriched for CRK SH2-binding sites (pYXXP). The binding sites enriched by GCG expression matched the CRK1-binding site identified by Tinti et al. (10) more closely than 65% of the SH2 domains assessed by Tinti (Fig. 5C, Fig. S3). This suggests that phosphosite enhancement mediated by the GRB2 and CRK (i.e. GCG) SH2 domains was largely binding site-specific when assessed across the entire tyrosine phosphoproteome.
Figure 5.

Quantitative MS-based analysis of tyrosine phosphoproteome in cells overexpressing SH2-containing proteins. A, bar graph showing the percentage of phosphopeptides in each experimental treatment whose abundance was increased, decreased, or unchanged. B, peptide LOGO of phosphosites whose abundance was enhanced by GRB2 construct expression. Inset shows peptide LOGO for in vitro GRB2-binding phosphopeptide binding data from Tinti et al. (10). Bar graph on the right shows the Euclidean distance between the PWM, derived from data used to create LOGOs. The red lines represent the Euclidean distance between MS-derived GRB2 PWM and the GRB2 PWM from peptide binding data. The blue bars represent comparisons between MS-derived PWM and all other PWMs from Tinti et al. (10). The GRB2–GRB2 similarity is greater than for 88% of all other pairs. C, peptide LOGO of phosphosites whose abundance was enhanced by GCG expression. Inset shows peptide LOGO for CRK-binding phosphopeptides from Tinti et al. (10). Right, bar graph showing the similarity of the MS-derived CRK PWM and all other PWMs from Tinti et al. as in B. The CRK–CRK similarity is greater than for 65% of all other pairs.
Computational model of SH2 phosphosite protection
These results strongly suggested that SH2 domains prevent the dephosphorylation of their binding sites in vivo by shielding those sites from phosphatases. To better understand the behavior of such a system, we generated a quantitative computational model of SH2–pTyr interactions in cells before and after EGF stimulation and compared model predictions with experimental results (Fig. 6A) (45).
Figure 6.

Computational model and parameter determination. A, diagram of the computational model used to quantify the effect of SH2 expression on EGFR phosphorylation. The effect of SH2 domain expression in unstimulated cells utilized the same scheme but used an EGFR kf value obtained in unstimulated cells. B–D, measurement of in vivo phosphorylation and dephosphorylation rates in COS1 cells. B, plot of absolute phosphorylation rate in serum-starved cells treated with pervanadate. C, plot of absolute phosphorylation rate in starved cells treated with pervanadate 5 min after stimulation with 2.5 ng/ml EGF. D, plot of absolute dephosphorylation rate in cells treated with erlotinib 5 min after stimulation with 2.5 ng/ml EGF. Black lines show amount of phosphorylation quantified from experimental data. Red lines show initial rate used to calculate model parameters. pTyr–EGFR concentrations were obtained by comparing signal from anti-pTyr immunoblots of COS1 lysates with a phosphotyrosine standard run on the same blot. E, plot of fitted curves from experimental data for fraction of EGFR pTyr-1068 (blue squares) and total pTyr EGFR (red circles) overlaid on model predictions generated at varying kon values for binding of GRB2 SH2 to EGFR. Plots were created by holding the koff constant at 1 s−1 and varying kon values (black lines).
To generate an accurate and realistic model, we experimentally determined as many parameters as possible in our COS1 cell system (Table 1). For example, we determined the actual steady-state phosphorylation and dephosphorylation rates of EGFR in the absence and presence of EGF. Phosphorylation rates were determined by quantifying anti-pTyr immunoblots of lysates of cells treated with the tyrosine phosphatase inhibitor pervanadate before and after EGF stimulation, whereas dephosphorylation rates were obtained from EGF-stimulated cells treated with the EGFR kinase inhibitor erlotinib. pTyr amounts were quantified by comparison with an absolute standard for pTyr developed in our laboratory (Fig. 6, B–D and Fig. S4) (34, 46). The independently obtained measures of EGFR phosphorylation and dephosphorylation rates in EGF-treated cells were nearly identical, as would be expected of a system at steady state. EGFR phosphorylation was modeled as a first-order substrate-limited reaction, as we were unable to obtain reasonable Vmax values by fitting to the Michaelis-Menten equation. EGFR dephosphorylation was modeled by fitting amounts of phospho-EGFR after erlotinib treatment to the Michaelis-Menten function. The phosphorylation forward rate constant (kf) and dephosphorylation Vmax and Km values were calculated using the measured rates, and the percent pTyr–EGFR values that were obtained in Fig. 3A. Phosphorylation in unstimulated cells was modeled using the same reaction kinetics scheme and the assumption that all EGFR kinases present in the cell had the same basal activity and contributed to substrate domain phosphorylation.
Table 1.
Model parameters
All parameters were defined using experimental data unless otherwise noted below. Cell volumes were approximated from measurements of trypsinized cells. EGF-binding constants were taken from the literature. COS1 cell phosphorylation and dephosphorylation rates were measured using pTyr standard as shown in Fig. 6, B–D. Measurements of in vivo EGFR phosphorylation rates and approximations of EGFR expression in COS1 cells were used to calculate the EGFR kf values. Phosphatase Vmax and Km values were obtained by fitting the quantitative EGFR dephosphorylation data to the Michaelis-Menten function. GRB2 concentrations were calculated via immunoblotting using a purified GRB2 standard run on the same membrane. GRB2 binding constants were determined using data from previously published work and by fitting experimental data to the model (see Fig. 6E).
| Parameter | Description | Values |
|---|---|---|
| Vcyto | Cytoplasmic volume | 2.59 × 103 μm3 |
| Vmem | Approximated membrane volume | 2.05 × 102 μm3 |
| Vextracell | Extracellular volume | 4.0 × 1012 μm3 |
| [EGF] | EGF concentration | 4.0 × 10−5–4 × 10−2 μm |
| kon, EGFa,b | EGF-EGFR binding, forward rate constant | 63.0 μm−1·s−1 |
| koff, EGFa,b | EGF-EGFR binding, reverse rate constant | 0.16 s−1 |
| kf, EGFR–US | Phosphorylation rate constant of unstimulated EGFR | 0.0036 s−1·μm−1 |
| kf, EGF–EGFR | Phosphorylation rate constant of EGF-bound EGFR | 0.54 s−1·μm−1 |
| [EGFR]b | EGFR concentration | 0.81 μm |
| Vmax, PTP | Maximum EGFR dephosphorylation rate in unstimulated cells | 4.6 μm·s−1 |
| Km, PTP | Km of EGFR dephosphorylation | 3.3 μm |
| [GRB2]endo | Endogenous COS1 cell GRB2 concentration | 0.24 μm |
| [GRB2]exo | Total GRB2 construct concentration in transfected cells | 1.7–5.4 μm |
| koff, GRB2–EGFRc | GRB2-EGFR binding, off-rate constant | 1.0 s−1 |
| kon, GRB2–EGFR | GRB2-EGFR binding, on-rate constant | Experimental fit |
| Kd, GRB2–EGFR | GRB2-EGFR dissociation constant | Experimental fit |
EGFR and EGFR substrate concentrations were estimated using published values for EGFR expression in A431 cells and comparing EGFR expression in COS1 and A431 cells by anti-EGFR Western blottings (46). EGF concentrations were those used experimentally. EGF–EGFR binding parameters were previously published and used to define the percent of active EGFR kinases (47–49).
The GRB2 SH2-pTyr–EGFR dissociation rate constant (koff) was obtained from recently published in vivo measurements (34, 38). On-rate (kon) and dissociation constant (Kd) values were then approximated by fitting our experimental GRB2 titration data (see Fig. 2A) for both total pTyr–EGFR and pTyr-1068 to modeling data generated using different kon values (Fig. 6E). The resulting Kd values for pTyr-1068 and pTyr EGFR were approximated at 0.66 and 2.0 μm, near the average of Kd measurements for GRB2 SH2–pTyr interactions generated from solid-phase assays (∼0.2–0.7 μm) and solution methods (2.6 μm) (12, 13, 50, 51). As a compromise, the Kd value of the GRB2–pTyr EGFR interaction was set at 1.0 μm in our models.
Comparison of model predictions and experimental results
We first modeled the effect of GRB2 SH2 overexpression on phosphosite abundance at increasing concentrations of EGF (Fig. 7A, red bars). The size of the effect predicted by the model closely matched experimental results (Fig. 7A, green bars), including the decrease in the relative effect of SH2 domain-mediated protection with increasing EGFR kinase activity.
Figure 7.

Computational modeling recapitulates experimental data. A, comparison of model predictions (red bars) and experimental data (green bars) on the effect of EGF concentration on GRB2 SH2-mediated EGFR phosphosite enhancement relative to GRB2 SH2 R86K mutant-expressing cells (see Fig. 2B). Error bars represent the S.E. of three biological replicates. For modeling data, GRB2 concentration was set at 5.4 μm. There was no statistical difference between the experimental and model data for the effect of EGF on GRB2-mediated pTyr enhancement and between 0 and 2.5 ng/ml EGF (one-way Student's t test, p > 0.05 “NS”). At higher concentrations of EGF (25 and 250 ng/ml), there was a small but statistically significant difference between experimental data and model predictions (one-way Student's t test, p < 0.05, *). B, model predictions of the relationship between SH2-bound phosphosites (pTyr-SH2, brown) and unbound phosphosites (pTyr, blue) in EGF-treated cells at varying SH2 concentrations. C, model predictions showing the effect of an increasing concentration of a GRB2-binding site competitor (SH2competitor) on the amount of pTyr-bound GRB2. Total concentration of GRB2 was held at 0.24 μm. Dark green bars represent pTyr-GRB2 binding in a system with pTyr flux (i.e. rapid phosphorylation and dephosphorylation; Fig. 6A). Light green bars represent pTyr-GRB2 binding data in a system containing a constant 3.6% pTyr EGFR (equal to the amount of pTyr EGFR present in the flux model without any exogenous SH2 present). D, quantification of phosphorylated ERK1 and ERK2 in COS1 cells expressing empty vector or tdEOS–GRB2 SH2 before and after EGF stimulation, using the same lysates as in Fig. 3A. All values are normalized to empty vector-transfected unstimulated cells and total ERK expression. Error bar represents S.E. from three biological replicates. There was no statistically significant difference between the phosphorylation of pERK1/pERK2 in empty vector and GRB2 SH2-expressing cells, before or after EGF stimulation (paired Student's t test, “NS”).
We then modeled the effect of increasing GRB2 concentrations on EGFR phosphorylation in the presence of EGF. Predicted percent maximal phosphorylation values were similar to those found experimentally: ∼5.0% for control and ∼20% for cells overexpressing GRB2 SH2 (compare Figs. 2A and 7B). Somewhat surprisingly, the model predicted that despite the increase in phosphorylation caused by SH2 overexpression, the number of unbound phosphosites (Fig. 7B, blue) was virtually unchanged compared with control. Even at GRB2 concentrations 20 times that of the endogenous protein, resulting in a greater than 4-fold increase in total phosphorylation, the number of unbound sites decreased only by ∼25% (Fig. 7B). This minimal reduction in free phosphosites suggested that protection might minimize the effect of competition between SH2 domains with similar binding specificities.
To test this hypothesis directly, we added a GRB2 competitor SH2 to our model, referred to here as SH2competitor. We set the binding affinity of SH2competitor for phosphorylated EGFR equal to that of the GRB2 SH2, varied its concentration, and calculated the amount of GRB2 bound to EGFR. The model predicted that a 25-fold excess of SH2competitor over endogenous GRB2 would result in only a 22% decrease in the amount of GRB2 bound to EGFR (Fig. 7C, dark bars). We surmised that the surprisingly modest effect of excess competitor must depend on the turnover of phosphosites, which in turn drives protection of SH2-bound sites from dephosphorylation. For comparison, we modeled GRB2–EGFR binding in a system that contained the same initial amount of EGFR pTyr, but without pTyr turnover mediated by kinase and PTP activity (i.e. total phospho-EGFR levels were fixed). In this system, the addition of a similar amount of SH2competitor resulted in a nearly 84% decrease in the amount of GRB2 bound to EGFR (Fig. 7C, light bars).
These results suggested that competing SH2 domains might have little effect on downstream signaling in the cell. To test this, we assessed the phosphorylation of ERK1 and ERK2, well established effectors of EGFR activation (23), in cells overexpressing the GRB2 SH2 domain, which should compete with endogenous GRB2. An ∼30-fold increase in expression of GRB2 SH2 over endogenous GRB2 did not significantly affect the activity of these downstream effectors, with or without EGF treatment (Fig. 7D). These data are consistent with our model predictions, as well as with a recent report showing that overexpression of a GFP-tagged GRB2 construct with an artificially enhanced phosphosite affinity, but not GFP-tagged WT GRB2 SH2, suppressed the growth of EGFR-expressing cell lines in an anchorage-independent growth assay (52).
Discussion
Although the idea that SH2 domains can enhance the phosphorylation of their binding sites by protecting them from dephosphorylation, which we term phosphosite protection, is not a new concept in the field of pTyr signaling, it has not been rigorously investigated. Seminal experiments exploited phosphosite protection to identify specific SH2-binding sites in vitro (26, 28, 30), but its occurrence and significance in living cells are not well understood. Here, we present a focused study of this phenomenon using the EGFR–GRB2 interaction as a model.
We show that GRB2 SH2 overexpression caused dose-dependent enhancement of phosphorylation of its binding sites in EGFR. Replacement of the GRB2 SH2 domain with that of CRK resulted in a shift in protection from EGFR to the focal adhesion protein p130CAS, an established CRK SH2-binding protein (34), suggesting that enhancement depended on SH2 binding specificity and not downstream signaling. We also found that the specificity of phosphosite enhancement depended on SH2 concentration. Expression of GRB2 at concentrations close to the GRB2–EGFR dissociation constant resulted in specific enhancement of canonical GRB2-binding motifs on EGFR; by contrast, very high concentrations of GRB2 (between 4 and 6 μm) enhanced the phosphorylation of both canonical and non-canonical binding sites. This apparent loss of specificity is likely due to increased binding to lower affinity sites at higher SH2 concentrations. Alternatively, SH2 binding might prevent dephosphorylation of unbound phosphorylated sites by sterically blocking phosphatase access to nearby sites. It been suggested that no more than three SH2-containing proteins can bind to EGFR at once (17). In this scenario, as GRB2 SH2 concentration increases and binding nears saturation, phosphorylation of unbound sites increases because they are less accessible to cellular phosphatases even as they remain unbound.
Analysis of the tyrosine phosphoproteome by MS revealed enhancement in the expected canonical SH2-binding motifs in cells overexpressing the GRB2 and CRK SH2 domains (Fig. 5, B and C), consistent with a mechanism dependent on binding-mediated phosphosite protection. Although our experiments mostly identified known interaction partners for the SH2 domains tested, this approach could be useful to identify interaction partners for SH2 domains with more poorly defined specificities, particularly those that are difficult to express and purify for in vitro studies (10, 42, 53). Furthermore, this method would allow for the identification of interactions that occur in vivo, in the cell type of choice, and is significantly simpler than alternative methods utilizing inducible covalent cross-linking or biotinylation, which require mutation and optimization of SH2 domains (54, 55).
To understand better the basis for SH2-mediated phosphosite protection, we created a deterministic ordinary differential equation model using the Virtual Cell reaction modeling software (45). An important feature was the use of experimentally determined steady-state phosphorylation and dephosphorylation rates. These values were determined using a method we recently developed to quantify the absolute amount of pTyr in a sample (46). Although not done in this study, this approach could be combined with percent phosphorylation and receptor concentration data to determine to absolute rates of phosphorylation and dephosphorylation for individual phosphosites.
Model predictions recapitulated experimental data quite accurately, strongly suggesting that phosphosite protection is sufficient to explain increased EGFR phosphorylation upon SH2 domain overexpression in our system. Arguably, a more complex rule-based model incorporating multiple phosphorylated sites might provide more detailed insight into the system (56). However, the simplicity of the model used here makes it more flexible for application to other systems. For example, we and others have previously shown that the SH2 domain of CRK appears to prevent p130CAS tyrosine dephosphorylation (57, 58). This could easily be modeled, using similar methods to quantify protein amounts and phosphorylation/dephosphorylation rates.
One rather counterintuitive prediction from the model is that although phosphosite protection increases steady-state levels of phosphorylation and the amount of SH2–pTyr complex, the amount of free (unbound) phosphosites is virtually unchanged (Fig. 7B). The effect is to reduce competition between SH2 domains with overlapping specificities; in other words, output from one SH2-containing effector can increase or decrease depending on the local concentration of that effector, without greatly affecting output from other SH2-containing effectors that bind to the same sites.
To understand why this is the case, it is helpful to compare the situation where the total number of pTyr-binding sites is fixed versus a more realistic dynamic system in which SH2 binding protects pTyr sites from dephosphorylation. In the former case, a large increase in SH2 domain concentration will cause a modest increase in overall SH2 binding (because the fractional occupancy of phosphosites increases with SH2 concentration), but there is a corresponding decrease in the number of unbound phosphosites. By contrast, in the case of phosphosite protection, a large increase in SH2 concentration causes an increase in overall phosphosite abundance. Therefore, even though the fraction of total phosphosites that are unbound decreases, there is only a minimal decrease in the absolute amount of unbound sites. These unbound sites remain available for binding to competitor domains (see Fig. 7C).
These results imply that SH2 signaling through a specific pTyr motif will be additive, if the following three conditions are met. First, multiple SH2 domains in the system must bind similar pTyr sites. As mentioned above, most SH2-binding motifs fall into several broad classes, suggesting that significant specificity overlap exists (59, 60). Second, phosphatase activity must be high, and as a result, most potential phosphosites must be unphosphorylated at steady state. This premise is consistent with previous studies showing low stoichiometry of tyrosine phosphorylation both in starved and stimulated cells (61) and with the dramatic increase in tyrosine phosphorylation seen after treating cells with the phosphatase inhibitor pervanadate (Fig. 6, B and C). Finally, SH2 domains must have moderate affinities for their pTyr-binding sites (close to their intracellular concentrations), so that binding is not saturated. Kd values for SH2–EGFR interactions measured in vitro generally range between 0.8 and 4 μm, with each phosphosite interacting with multiple SH2 domains (12, 62). A few examples exist of SH2–pTyr interactions with much higher affinities, which may have evolved to drive specific signaling pathways and eliminate significant SH2-dependent signaling cross-talk (63, 64). Recent work that exploited protein structure data to generate SH2 domains with increased affinity suggests there is selective pressure to maintain moderate affinities in most cases (52).
Phosphosite protection may play an important biological role in oncogenic signaling. SH2 proteins that are largely unbound at normal expression levels may behave very differently when highly overexpressed. Viral CRK and its human homolog CRK1 can malignantly transform fibroblasts, despite consisting only of an SH2 and an SH3 domain. Presumably, CRK proteins exploit pTyr flux to create their own binding sites through protection, allowing them to induce excess signaling through pro-growth pathways (30, 65). Likewise, the SH2-containing adaptor GRB7 is often overexpressed in breast cancers overexpressing the EGFR family member HER2. GRB7 overexpression is associated with increased HER2 phosphorylation, which results in activation of pro-oncogenic downstream pathways and promotes cell growth and migration (66, 67). The relative lack of SH2 competition associated with phosphosite protection may also allow cancer cells to maintain homeostatic signaling in the setting of increased expression of oncogenic SH2-containing proteins such as SRC, BCR-ABL, JAK, or STAT.
Finally, it is important to consider phosphosite protection both in experimental design and data interpretation. For example, recently published work has shown that SHCD overexpression results in a significant increase in phosphorylation of EGFR in a PTB-dependent manner. The largest increases occurred at the SHCD PTB-binding site, pTyr-1148, and to a lesser extent at pTyr-1068 and pTyr-1173 (37). These data are highly consistent with phosphosite protection, although this was not considered as a potential mechanism. More generally, fluorescently tagged SH2 domains and SH2 domain-containing proteins are sometimes used as probes to monitor the availability and location of binding sites for their endogenous counterparts in vivo (34, 38). Our current results suggest that SH2-only probes would only marginally affect the binding of endogenous proteins, and therefore they can be used as tracers of SH2–pTyr interactions in vivo without significantly disrupting normal signaling. On the other hand, isolated SH2 domains have also been used as dominant-negative reagents, in principle blocking signaling from phosphosites by preventing the binding of endogenous proteins that normally bind those sites (40, 68–72). Our work shows that SH2-based dominant-negative approaches will not be effective unless expression levels are extremely high, as at more moderate expression levels significant unbound phosphosites remain.
Taken together, the work presented here suggests that phosphosite protection is an inherent characteristic of the pTyr signaling system in vivo. This feature may allow a specific pTyr site to provide both homeostatic signaling and a diverse set of cell type-specific SH2-mediated signaling outputs at the same time. The process is also likely an important feature of signaling through other dynamic post-translational modifications, including serine/threonine phosphorylation and lysine acetylation, both of which can signal through modular reader domains in a similar manner to SH2–pTyr interactions (4). Thus, a complete understanding of signaling from these and other writer–eraser–reader systems requires consideration of the innate ability of reader proteins to participate actively in the signaling process.
Experimental procedures
Plasmids
The tdEOS–GRB2 SH2 construct was cloned as described previously (38). The full-length GRB2 and GCG constructs were cloned into pEBB (40). The GCG construct was creating by replacing the GRB2 SH2 domain (amino acids 58–159) with the chicken CRK SH2 domain (amino acids 1–124).
Antibodies and reagents
Anti-pTyr immunoblot experiments used mouse monoclonal pTyr-100 (Cell Signaling Technology, catalog no. 9411). EGFR was detected using rabbit anti-EGFR (Santa Cruz Biotechnology, Inc., catalog no. sc31157). EGFR phosphosite-specific immunoblots were performed using the following antibodies: pTyr-845 (Santa Cruz Biotechnology, Inc., catalog no. sc-575442); pTyr-974 (Cell Signaling Technology, catalog no. 2641S); pTyr-992 (Cell Signaling Technology, catalog no. 2235P); pTyr-1045 (Cell Signaling Technology, catalog no. 2237P); pTyr-1148 (Cell Signaling Technology, catalog no. 4404S); pTyr-1068 (Cell Signaling Technology, catalog no. 3777P); pTyr-1086 (Cell Signaling Technology, catalog no. 2220S); and pTyr-1173 (Cell Signaling Technology, catalog no. 4407S). pERK1 and pERK2 were detected using rabbit anti-p44/42 pThr-202/pTyr-204 (Cell Signaling Technology, catalog no. 9101S). Full-length GRB2 and GRB2 SH2 were blotted using mouse anti-GRB2 SH2 (R&D Systems, catalog no. 669604). The GCG and full-length GRB2 were detected using a rabbit antiserum raised against GST–GRB2 produced for our laboratory (69). p130 immunodepletion was performed using agarose-conjugated rabbit anti-p130CAS (Santa Cruz Biotechnology, Inc., sc-20029) and blotted using mouse anti-p130CAS (BD Biosciences, catalog 10271). Pervanadate was prepared fresh for each experiment by mixing 1 volume of 100 mm Na3VO4 with 0.32 volume of 30% (w/w) hydrogen peroxide at room temperature for 30–90 min.
Cell culture
COS1 cells, a fibroblast-like African green monkey kidney cell line, were cultured in complete DMEM (10% fetal bovine serum and 1% penicillin/streptomycin). For overnight serum starvation, complete DMEM was aspirated and replaced with starvation DMEM (0% fetal bovine serum and 1% penicillin/streptomycin), and plates were incubated for 6–14 h.
Plasmid expression, Western, and far-Western blotting
The tdEOS–GRB2 SH2, full-length GRB2, GCG, and empty vector constructs were exogenously expressed in COS1 cells using Lipofectamine 2000 (Life Technologies, Inc.). 10 μg of each construct was used for all Western blotting experiments except for the GRB2 dose-dependence experiments. For MS experiments, 30 μg of each construct was used per 10-cm dish. Cells were lysed on ice in Kinase Lysis Buffer (KLB: 150 mm NaCl, 25 mm Tris-HCl, pH 7.4, 5 mm EDTA, 1 mm phenylmethylsulfonyl fluoride (PMSF), 1% Triton X-100, 10% glycerol, 0.1% sodium pyrophosphate, 10 mm β-glycerophosphate, 10 mm NaF, 5 μg/ml aprotinin (Sigma A6279), 50 μm pervanadate), cleared by centrifugation, and processed for Western and far-Western blotting as previously described (34).
GRB2 SH2 quantification
The approximate cellular tdEOS–GRB2 SH2 concentration was determined by comparing tdEOS–GRB2 SH2 expression levels in transfected COS1 cells with a GST–GRB2 SH2 standard of known concentration via immunoblot. Briefly, COS1 cells (30-mm plate) were transfected with 10 μg of tdEOS–GRB2 SH2, incubated for 18 h, lysed in KLB, run by LDS-PAGE along with a serial dilution of the GRB2 standard, transferred to nitrocellulose, and immunoblotted with anti-GRB2 SH2. The average cell volume was calculated from three differential interference contrast images of trypsinized cells.
Determination of site-specific relative EGFR phosphorylation
To determine to relative phosphorylation of each EGFR pTyr site, a maximally phosphorylated pTyr–EGFR control was created by treating COS1 cells with 750 μm pervanadate and 200 ng/ml EGF for 40 min. Lysates from this control were then diluted 1:10, run alongside experimental lysates on LDS-PAGE, transferred to nitrocellulose, Western blotted using the site-specific anti-pTyr–EGFR antibodies, and quantified by using the Odyssey system (LI-COR). The percent of maximum phosphorylation for each site was then determined by comparing its specific Western signal to that of the maximally phosphorylated standard.
EGFR phosphorylation/dephosphorylation rate quantification
To determine the basal EGFR phosphorylation rate, COS1 cells were starved overnight. Media were then replaced with starvation media containing 375 μm pervanadate, and cells were flash-frozen in liquid N2 after rapid media aspiration at the time points shown. To determine the rate of EGF-induced phosphorylation, the cells were treated with 2.5 ng/ml EGF. After 5 min, the EGF media were then replaced with starvation media containing 2.5 ng/ml EGF and 375 μm pervanadate, and the cells were flash-frozen in liquid N2 as above. Cells from each time course were then lysed in KLB and run on LDS-PAGE along with an increasing concentration of the malachite green-quantified pTyr-ABL standard (46), transferred to nitrocellulose, and probed with anti-pTyr-100. The absolute rate of EGFR phosphorylation was then determined using a pTyr-ABL standard curve and determining the initial slope following pervanadate treatment (Fig. 6, B and C). EGFR band was quantified for experimental samples, whereas the entire lane was quantified for pTyr standard.
To determine the rate of dephosphorylation, the cells were treated with 2.5 ng/ml EGF. After 5 min, the EGF media were then replaced with starvation media containing 2.5 ng/ml EGF and 10 μm erlotinib, and the cells were flash-frozen in liquid N2 at the time points shown. The rate of EGFR dephosphorylation was then determined as described above for the phosphorylation rate (Fig. 6D).
Mass spectrometry
Cells were transfected with 30 μg of the constructs listed, stimulated with 2.5 ng/ml EGF for 10 min, and flash-frozen in liquid N2. Cells were lysed with 8 m urea, 1 mm sodium orthovanadate, and protein yield was quantified by BCA assay (Pierce). Samples were reduced with 10 μl of 10 mm DTT in 100 mm ammonium acetate, pH 8.9 (1 h at 56 °C). Samples were alkylated with 75 μl of 55 mm iodoacetamide in 100 mm ammonium acetate, pH 8.9 (1 h at room temperature). 1 ml of 100 mm ammonium acetate and 10 μg of sequencing grade trypsin (Promega, catalog no. V5111) were added, and digestion proceeded for 16 h at room temperature. Samples were acidified with 125 μl of trifluoroacetic acid (TFA) and desalted with C18 spin columns (ProteaBio, catalog no. SP-150). Samples were lyophilized and subsequently labeled with iTRAQ 8plex (AbSciex) per the manufacturer's directions.
Protected consensus sequence determination
The consensus sequence of protected sites was determined from MS data listed in Table 1. The abundance of each phosphosite was compared in cells expressing empty vector or SH2 proteins. Those sites whose abundance was greater than the combined standard deviation of the abundance in each experimental point were determined to be significantly enhanced by SH2 protein expression. Significantly enhanced pTyr sites were then compiled for each experimental treatment, and the abundance of each significantly enhanced peptide was weighted using its fold increase over its empty vector control. This weighted list was then used to create an amino acid sequence LOGO plot using the PhosphositePlus LOGO generator by applying the Frequency Change algorithm and the phospho-Tyr background settings (73, 74). The consensus LOGO for GRB2 was determined using only those pTyr peptides whose abundance was significantly enhanced under all four treatments (i.e. GRB SH2, GRB FL, GRB2 SH2 + EGF, GRB2 FL + EGF). For the CRK consensus sequence LOGO, pTyr-sites enhanced by GCG expression, with and without EGF, were used. Phosphosite LOGOs for the data from Tinti et al. (10) were created using sequences from peptides whose binding signal exceeded the average signal by more than two S.D. (Z score >2). The PhosphoSitePlus LOGO generator was used, and no weighting was applied.
To evaluate the similarity between phosphosites enhanced by SH2 overexpression (“protected sites”) and in vitro SH2-binding sites, the Euclidean distance was calculated between the position weight matrices (PWMs) for the protected sites and the PWMs for the in vitro sites from Tinti et al. (10). Prior to analysis, sequences were trimmed to contain 1 amino acid before and 4 amino acids after the central phosphorylated tyrosine. Any sequences that were shorter were removed from the analysis, and the central phosphorylated tyrosine was excluded. To calculate the PWMs, we modified a function from Wagih et al. (76) to return the log ratio of the result, normalized to the background probabilities (75). For each set of protected sites (Grb2 and GCG), the PWM was compared with those from in vitro phosphopeptide binding, resulting in 67 Euclidean distances for each set of protected sites. Of these 67 distances, the distance between the protected PWM (i.e. Grb2) and the PWM of its in vitro counterpart was compared with all of the remaining distances. For this analysis we used R (77), RStudio (78), and several additional packages (https://CRAN.R-project.org/package=tidyverse; https://CRAN.R-project.org/package=cowplot; https://CRAN.R-project.org/package=matrixcalc (79–81)).3 The script is available on github (https://github.com/lafontaine-uchc/jadwin2017).3
Computational reaction modeling
All reaction modeling was performed using the Virtual Cell version 5.3.17 using the Combined Stiff Solver (IDA/CVCODE). All reaction parameters were approximated as listed in Table 1. Briefly, total cell volume of COS1 cells was calculated from an average diameter of trypsinized cells (∼20 μm). The plasma membrane reaction area was approximated as a hollow sphere 200 nm deep; the nucleus was approximated as an internal sphere with a diameter of 10 μm, and the cytoplasmic volume consisted of the remaining volume. The extracellular volume was set at 4 ml as in our experiments. EGF concentrations were those used in our experiments. EGF–EGFR forward and reverse binding constant values were obtained from Berkers et al. (47) and French et al. (48). EGFR receptor numbers were estimated using previous estimates of EGFR expression in A431 cells and anti-EGFR Western blottings of COS1 and A431 cell lysates (46). The EGFR kf value was determined using the initial phosphorylation rate of EGFR in EGF-stimulated or non-stimulated COS1 cells treated with pervanadate. The PTP Vmax and Km values were obtained by fitting data to the Michaelis-Menten function using OriginPro 2017. The GRB2–EGFR koff value was taken from Oh et al. (38). The GRB2–EGF Kd and kon values were determined by fitting experimental data to modeling data using variable kon values (Fig. 6E). The Virtual Cell BioModel, “spatial protection test- two way competition original” is available from Public BioModels within the Virtual Cell software under the shared username “jjadwin.” The Virtual Cell can be downloaded from http://vcell.org.3 Users have to first download the client software and open the model from within the software.
Statistics
The t test and ANOVA statistics were performed using Mintab 18.1 and Microsoft Excel.
Author contributions
J. A. J. conceived the study, performed all experiments except mass spectrometry, and drafted the manuscript. T. G. C. performed all mass spectrometry analyses. A. T. L. performed statistical analyses related to binding specificity. F. M. W. provided mass spectrometry supervision, conceptual input, and manuscript editing. B. J. M. supervised all except MS experiments and provided conceptual guidance and manuscript editing.
Supplementary Material
Acknowledgments
We thank Ji Yu for critical reading of this manuscript, Kazuya Machida for technical advice with biochemical experiments, Ahmed Elmokadem and Ji Yu for advice on VCell modeling and reaction kinetics, and Shobhit Jain and Gary Bader (University of Toronto, Ontario, Canada) for help and advice on comparing LOGO plots. The Virtual Cell is supported by National Institutes of Health Grant P41 GM103313 from the NIGMS.
This work was supported by National Institutes of Health Grant U01 CA146843 from the NCI. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4 and Table S1.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- SH2
- Src homology 2
- PTB
- phosphotyrosine-binding
- PTP
- protein-tyrosine phosphatase
- EGFR
- epidermal growth factor receptor
- ANOVA
- analysis of variance
- PWM
- position weight matrix
- FL
- full-length.
References
- 1. Jadwin J. A., Ogiue-Ikeda M., and Machida K. (2012) The application of modular protein domains in proteomics. FEBS Lett. 586, 2586–2596 10.1016/j.febslet.2012.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pawson T., Gish G. D., and Nash P. (2001) SH2 domains, interaction modules and cellular wiring. Trends Cell Biol. 11, 504–511 10.1016/S0962-8924(01)02154-7 [DOI] [PubMed] [Google Scholar]
- 3. Lim W. A., and Pawson T. (2010) Phosphotyrosine signaling: evolving a new cellular communication system. Cell 142, 661–667 10.1016/j.cell.2010.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Seet B. T., Dikic I., Zhou M. M., and Pawson T. (2006) Reading protein modifications with interaction domains. Nat. Rev. Mol. Cell Biol. 7, 473–483 10.1038/nrm1960 [DOI] [PubMed] [Google Scholar]
- 5. Pawson T. (2004) Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 116, 191–203 10.1016/S0092-8674(03)01077-8 [DOI] [PubMed] [Google Scholar]
- 6. Wagner M. J., Stacey M. M., Liu B. A., and Pawson T. (2013) Molecular mechanisms of SH2- and PTB-Domain-containing proteins in receptor tyrosine kinase signaling. Cold Spring Harb. Perspect. Biol. 5, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu B. A., Jablonowski K., Raina M., Arcé M., Pawson T., and Nash P. D. (2006) The human and mouse complement of SH2 domain proteins– establishing the boundaries of phosphotyrosine signaling. Mol. Cell 22, 851–868 10.1016/j.molcel.2006.06.001 [DOI] [PubMed] [Google Scholar]
- 8. Huang H., Li L., Wu C., Schibli D., Colwill K., Ma S., Li C., Roy P., Ho K., Songyang Z., Pawson T., Gao Y., and Li S. S. (2008) Defining the specificity space of the human SRC homology 2 domain. Mol. Cell. Proteomics 7, 768–784 10.1074/mcp.M700312-MCP200 [DOI] [PubMed] [Google Scholar]
- 9. Songyang Z., Shoelson S. E., McGlade J., Olivier P., Pawson T., Bustelo X. R., Barbacid M., Sabe H., Hanafusa H., Yi T., Ren R., Baltimore D., Ratnovsky S., Feldman R. A., and Cantley L. C. (1994) Specific motifs recognized by the SH2 domains of Csk, 3BP2, fps/fes, GRB-2, HCP, SHC, Syk, and Vav. Mol. Cell. Biol. 14, 2777–2785 10.1128/MCB.14.4.2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tinti M., Kiemer L., Costa S., Miller M. L., Sacco F., Olsen J. V., Carducci M., Paoluzi S., Langone F., Workman C. T., Blom N., Machida K., Thompson C. M., Schutkowski M., Brunak S., et al. (2013) The human SH2 interaction landscape. Cell Rep. 3, 1293–1305 10.1016/j.celrep.2013.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beebe K. D., Wang P., Arabaci G., and Pei D. (2000) Determination of the binding specificity of the SH2 domains of protein-tyrosine phosphatase SHP-1 through the screening of a combinatorial phosphotyrosyl peptide library. Biochemistry 39, 13251–13260 10.1021/bi0014397 [DOI] [PubMed] [Google Scholar]
- 12. Hause R. J. Jr., Leung K. K., Barkinge J. L., Ciaccio M. F., Chuu C. P., and Jones R. B. (2012) Comprehensive binary interaction mapping of SH2 domains via fluorescence polarization reveals novel functional diversification of ErbB receptors. PLoS ONE 7, e44471 10.1371/journal.pone.0044471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jones R. B., Gordus A., Krall J. A., and MacBeath G. (2006) A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature 439, 168–174 10.1038/nature04177 [DOI] [PubMed] [Google Scholar]
- 14. Schulze W. X., and Mann M. (2004) A novel proteomic screen for peptide-protein interactions. J. Biol. Chem. 279, 10756–10764 10.1074/jbc.M309909200 [DOI] [PubMed] [Google Scholar]
- 15. Kavanaugh W. M., Turck C. W., and Williams L. T. (1995) PTB domain binding to signaling proteins through a sequence motif containing phosphotyrosine. Science 268, 1177–1179 10.1126/science.7539155 [DOI] [PubMed] [Google Scholar]
- 16. Schlessinger J., and Lemmon M. A. (2003) SH2 and PTB domains in tyrosine kinase signaling. Sci. STKE 2003, RE12 [DOI] [PubMed] [Google Scholar]
- 17. Hsieh M. Y., Yang S., Raymond-Stinz M. A., Edwards J. S., and Wilson B. S. (2010) Spatio-temporal modeling of signaling protein recruitment to EGFR. BMC Syst. Biol. 4, 57 10.1186/1752-0509-4-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jones R. B. (2013) Do low-affinity ErbB receptor protein interactions represent the base of a cell signaling iceberg? Expert Rev. Proteomics 10, 115–118 10.1586/epr.12.78 [DOI] [PubMed] [Google Scholar]
- 19. Kleiman L. B., Maiwald T., Conzelmann H., Lauffenburger D. A., and Sorger P. K. (2011) Rapid phospho-turnover by receptor tyrosine kinases impacts downstream signaling and drug binding. Mol. Cell 43, 723–737 10.1016/j.molcel.2011.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lemmon M. A., Schlessinger J., and Ferguson K. M. (2014) The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 6, a020768 10.1101/cshperspect.a020768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kovacs E., Zorn J. A., Huang Y., Barros T., and Kuriyan J. (2015) A structural perspective on the regulation of the epidermal growth factor receptor. Annu. Rev. Biochem. 84, 739–764 10.1146/annurev-biochem-060614-034402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gotoh N., Tojo A., Hino M., Yazaki Y., and Shibuya M. (1992) A highly conserved tyrosine residue at codon 845 within the kinase domain is not required for the transforming activity of human epidermal growth factor receptor. Biochem. Biophys. Res. Commun. 186, 768–774 10.1016/0006-291X(92)90812-Y [DOI] [PubMed] [Google Scholar]
- 23. Zhang X., Gureasko J., Shen K., Cole P. A., and Kuriyan J. (2006) An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149 10.1016/j.cell.2006.05.013 [DOI] [PubMed] [Google Scholar]
- 24. Normanno N., De Luca A., Bianco C., Strizzi L., Mancino M., Maiello M. R., Carotenuto A., De Feo G., Caponigro F., and Salomon D. S. (2006) Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366, 2–16 10.1016/j.gene.2005.10.018 [DOI] [PubMed] [Google Scholar]
- 25. Schulze W. X., Deng L., and Mann M. (2005) Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol. Syst. Biol. 1, 2005.0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Batzer A. G., Blaikie P., Nelson K., Schlessinger J., and Margolis B. (1995) The phosphotyrosine interaction domain of Shc binds an LXNPXY motif on the epidermal growth factor receptor. Mol. Cell. Biol. 15, 4403–4409 10.1128/MCB.15.8.4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Okutani T., Okabayashi Y., Kido Y., Sugimoto Y., Sakaguchi K., Matuoka K., Takenawa T., and Kasuga M. (1994) Grb2/Ash binds directly to tyrosines 1068 and 1086 and indirectly to tyrosine 1148 of activated human epidermal growth factor receptors in intact cells. J. Biol. Chem. 269, 31310–31314 [PubMed] [Google Scholar]
- 28. Ward C. W., Gough K. H., Rashke M., Wan S. S., Tribbick G., and Wang J. (1996) Systematic mapping of potential binding sites for Shc and Grb2 SH2 domains on insulin receptor substrate-1 and the receptors for insulin, epidermal growth factor, platelet-derived growth factor, and fibroblast growth factor. J. Biol. Chem. 271, 5603–5609 10.1074/jbc.271.10.5603 [DOI] [PubMed] [Google Scholar]
- 29. Batzer A. G., Rotin D., Ureña J. M., Skolnik E. Y., and Schlessinger J. (1994) Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol. Cell. Biol. 14, 5192–5201 10.1128/MCB.14.8.5192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Birge R. B., Fajardo J. E., Mayer B. J., and Hanafusa H. (1992) Tyrosine phosphorylated epidermal growth factor receptor and cellular p130 provide high affinity binding substrates to analyze crk-phosphotyrosine-dependent interactions in vitro. J. Biol. Chem. 267, 10588–10595 [PubMed] [Google Scholar]
- 31. Gervais F. G., Chow L. M., Lee J. M., Branton P. E., and Veillette A. (1993) The SH2 domain is required for stable phosphorylation of p56lck at tyrosine 505, the negative regulatory site. Mol. Cell. Biol. 13, 7112–7121 10.1128/MCB.13.11.7112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morris D. L., Cho K. W., Zhou Y., and Rui L. (2009) SH2B1 enhances insulin sensitivity by both stimulating the insulin receptor and inhibiting tyrosine dephosphorylation of insulin receptor substrate proteins. Diabetes 58, 2039–2047 10.2337/db08-1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rotin D., Margolis B., Mohammadi M., Daly R. J., Daum G., Li N., Fischer E. H., Burgess W. H., Ullrich A., and Schlessinger J. (1992) SH2 domains prevent tyrosine dephosphorylation of the EGF receptor: identification of Tyr992 as the high-affinity binding site for SH2 domains of phospholipase Cg. EMBO J. 11, 559–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jadwin J. A., Oh D., Curran T. G., Ogiue-Ikeda M., Jia L., White F. M., Machida K., Yu J., and Mayer B. J. (2016) Time-resolved multimodal analysis of Src homology 2 (SH2) domain binding in signaling by receptor tyrosine kinases. eLife 5, e11835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Biscardi J. S., Maa M. C., Tice D. A., Cox M. E., Leu T. H., and Parsons S. J. (1999) c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J. Biol. Chem. 274, 8335–8343 10.1074/jbc.274.12.8335 [DOI] [PubMed] [Google Scholar]
- 36. Giubellino A., Burke T. R. Jr., and Bottaro D. P. (2008) Grb2 signaling in cell motility and cancer. Expert Opin. Ther. Targets 12, 1021–1033 10.1517/14728222.12.8.1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wills M. K., Lau H. R., and Jones N. (2017) The ShcD phosphotyrosine adaptor subverts canonical EGF receptor trafficking. J. Cell Sci. 130, 2808–2820 10.1242/jcs.198903 [DOI] [PubMed] [Google Scholar]
- 38. Oh D., Ogiue-Ikeda M., Jadwin J. A., Machida K., Mayer B. J., and Yu J. (2012) Fast rebinding increases dwell-time of Src homology 2 (SH2) containing proteins at plasma membrane. Proc. Natl. Acad. Sci. U.S.A. 109, 14024–14029 10.1073/pnas.1203397109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Motto D. G., Ross S. E., Jackman J. K., Sun Q., Olson A. L., Findell P. R., and Koretzky G. A. (1994) In vivo association of Grb2 with pp116, a substrate of the T cell antigen receptor-activated protein tyrosine kinase. J. Biol. Chem. 269, 21608–21613 [PubMed] [Google Scholar]
- 40. Tanaka M., Gupta R., and Mayer B. J. (1995) Differential inhibition of signaling pathways by dominant-negative SH2/SH3 adapter proteins. Mol. Cell. Biol. 15, 6829–6837 10.1128/MCB.15.12.6829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Defilippi P., Di Stefano P., and Cabodi S. (2006) p130Cas: a versatile scaffold in signaling networks. Trends Cell Biol. 16, 257–263 10.1016/j.tcb.2006.03.003 [DOI] [PubMed] [Google Scholar]
- 42. Machida K., Thompson C. M., Dierck K., Jablonowski K., Kärkkäinen S., Liu B., Zhang H., Nash P. D., Newman D. K., Nollau P., Pawson T., Renkema G. H., Saksela K., Schiller M. R., Shin D. G., and Mayer B. J. (2007) High-throughput phosphotyrosine profiling using SH2 domains. Mol. Cell 26, 899–915 10.1016/j.molcel.2007.05.031 [DOI] [PubMed] [Google Scholar]
- 43. Sakai R., Iwamatsu A., Hirano N., Ogawa S., Tanaka T., Mano H., Yazaki Y., and Hirai H. (1994) A novel signaling molecule, p130, forms stable complexes in vivo with v-Crk and v-Src in a tyrosine phosphorylation-dependent manner. EMBO J. 13, 3748–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang Y., Wolf-Yadlin A., Ross P. L., Pappin D. J., Rush J., Lauffenburger D. A., and White F. M. (2005) Time-resolved mass spectrometry of tyrosine phosphorylation sites in the epidermal growth factor receptor signaling network reveals dynamic modules. Mol. Cell. Proteomics 4, 1240–1250 10.1074/mcp.M500089-MCP200 [DOI] [PubMed] [Google Scholar]
- 45. Slepchenko B. M., Schaff J. C., Macara I., and Loew L. M. (2003) Quantitative cell biology with the virtual cell. Trends Cell Biol. 13, 570–576 10.1016/j.tcb.2003.09.002 [DOI] [PubMed] [Google Scholar]
- 46. Thompson C. M., Bloom L. R., Ogiue-Ikeda M., and Machida K. (2015) SH2-PLA: a sensitive in-solution approach for quantification of modular domain binding by proximity ligation and real-time PCR. BMC Biotechnol. 15, 60 10.1186/s12896-015-0169-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Berkers J. A., van Bergen en Henegouwen P. M., and Boonstra J. (1991) Three classes of epidermal growth factor receptors on HeLa cells. J. Biol. Chem. 266, 922–927 [PubMed] [Google Scholar]
- 48. French A. R., Tadaki D. K., Niyogi S. K., and Lauffenburger D. A. (1995) Intracellular trafficking of epidermal growth factor family ligands is directly influenced by the pH sensitivity of the receptor/ligand interaction. J. Biol. Chem. 270, 4334–4340 10.1074/jbc.270.9.4334 [DOI] [PubMed] [Google Scholar]
- 49. Kholodenko B. N. (2006) Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 7, 165–176 10.1038/nrm1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chook Y. M., Gish G. D., Kay C. M., Pai E. F., and Pawson T. (1996) The Grb2-mSos1 complex binds phosphopeptides with higher affinity than Grb2. J. Biol. Chem. 271, 30472–30478 10.1074/jbc.271.48.30472 [DOI] [PubMed] [Google Scholar]
- 51. Lemmon M. A., Ladbury J. E., Mandiyan V., Zhou M., and Schlessinger J. (1994) Independent binding of peptide ligands to the SH2 and SH3 domains of Grb2. J. Biol. Chem. 269, 31653–31658 [PubMed] [Google Scholar]
- 52. Kaneko T., Huang H., Cao X., Li X., Li C., Voss C., Sidhu S. S., and Li S. S. (2012) Superbinder SH2 domains act as antagonists of cell signaling. Sci. Signal. 5, ra68 [DOI] [PubMed] [Google Scholar]
- 53. Jadwin J. A. (2017) in SH2 Domains: Methods and Protocols, (Machida K., and Liu B. A., eds) pp. 477–492, Humana Press Inc., Totowa, NJ [Google Scholar]
- 54. Hino N., Oyama M., Sato A., Mukai T., Iraha F., Hayashi A., Kozuka-Hata H., Yamamoto T., Yokoyama S., and Sakamoto K. (2011) Genetic incorporation of a photo-crosslinkable amino acid reveals novel protein complexes with GRB2 in mammalian cells. J. Mol. Biol. 406, 343–353 10.1016/j.jmb.2010.12.022 [DOI] [PubMed] [Google Scholar]
- 55. Roux K. J., Kim D. I., Raida M., and Burke B. (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 10.1083/jcb.201112098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Creamer M. S., Stites E. C., Aziz M., Cahill J. A., Tan C. W., Berens M. E., Han H., Bussey K. J., Von Hoff D. D., Hlavacek W. S., and Posner R. G. (2012) Specification, annotation, visualization and simulation of a large rule-based model for ERBB receptor signaling. BMC Syst. Biol. 6, 107 10.1186/1752-0509-6-107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Antoku S., and Mayer B. J. (2009) Distinct roles for Crk adaptor isoforms in actin reorganization induced by extracellular signals. J. Cell Sci. 122, 4228–4238 10.1242/jcs.054627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Takino T., Tamura M., Miyamori H., Araki M., Matsumoto K., Sato H., and Yamada K. M. (2003) Tyrosine phosphorylation of the CrkII adaptor protein modulates cell migration. J. Cell Sci. 116, 3145–3155 10.1242/jcs.00632 [DOI] [PubMed] [Google Scholar]
- 59. Liu B. A., Engelmann B. W., and Nash P. D. (2012) The language of SH2 domain interactions defines phosphotyrosine-mediated signal transduction. FEBS Lett. 586, 2597–2605 10.1016/j.febslet.2012.04.054 [DOI] [PubMed] [Google Scholar]
- 60. Liu B. A., Jablonowski K., Shah E. E., Engelmann B. W., Jones R. B., and Nash P. D. (2010) SH2 domains recognize contextual peptide sequence information to determine selectivity. Mol. Cell. Proteomics 9, 2391–2404 10.1074/mcp.M110.001586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sharma K., D'Souza R. C., Tyanova S., Schaab C., Wiśniewski J. R., Cox J., and Mann M. (2014) Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 8, 1583–1594 10.1016/j.celrep.2014.07.036 [DOI] [PubMed] [Google Scholar]
- 62. Kaushansky A., Gordus A., Chang B., Rush J., and MacBeath G. (2008) A quantitative study of the recruitment potential of all intracellular tyrosine residues on EGFR, FGFR1 and IGF1R. Mol. Biosyst. 4, 643–653 10.1039/b801018h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Deindl S., Kadlecek T. A., Cao X., Kuriyan J., and Weiss A. (2009) Stability of an autoinhibitory interface in the structure of the tyrosine kinase ZAP-70 impacts T cell receptor response. Proc. Natl. Acad. Sci. U.S.A. 106, 20699–20704 10.1073/pnas.0911512106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wilson T. J., Garner L. I., Metcalfe C., King E., Margraf S., and Brown M. H. (2014) Fine specificity and molecular competition in SLAM family receptor signalling. PLoS ONE 9, e92184 10.1371/journal.pone.0092184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Birge R. B., Kalodimos C., Inagaki F., and Tanaka S. (2009) Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Commun. Signal. 7, 13 10.1186/1478-811X-7-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bai T., and Luoh S. W. (2008) GRB-7 facilitates HER-2/Neu-mediated signal transduction and tumor formation. Carcinogenesis 29, 473–479 [DOI] [PubMed] [Google Scholar]
- 67. Pradip D., Bouzyk M., Dey N., and Leyland-Jones B. (2013) Dissecting GRB7-mediated signals for proliferation and migration in HER2 overexpressing breast tumor cells: GTP-ase rules. Am. J. Cancer Res. 3, 173–195 [PMC free article] [PubMed] [Google Scholar]
- 68. Carroll D. J., Albay D. T., Terasaki M., Jaffe L. A., and Foltz K. R. (1999) Identification of PLCγ-dependent and -independent events during fertilization of sea urchin eggs. Dev. Biol. 206, 232–247 10.1006/dbio.1998.9145 [DOI] [PubMed] [Google Scholar]
- 69. Gupta R. W., and Mayer B. J. (1998) Dominant negative mutants of the SH2/SH3 adapters Nck and Grb2 inhibit MAP kinase activation and mesoderm-specific gene induction by eFGF in Xenopus. Oncogene 17, 2155–2165 10.1038/sj.onc.1202158 [DOI] [PubMed] [Google Scholar]
- 70. Kontaridis M. I., Swanson K. D., David F. S., Barford D., and Neel B. G. (2006) PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J. Biol. Chem. 281, 6785–6792 10.1074/jbc.M513068200 [DOI] [PubMed] [Google Scholar]
- 71. Northrop J. P., Pustelnik M. J., Lu A. T., and Grove J. R. (1996) Characterization of the roles of SH2 domain-containing proteins in T-lymphocyte activation by using dominant negative SH2 domains. Mol. Cell. Biol. 16, 2255–2263 10.1128/MCB.16.5.2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Qian D., Mollenauer M. N., and Weiss A. (1996) Dominant-negative ζ-associated protein 70 inhibits T cell antigen receptor signaling. J. Exp. Med. 183, 611–620 10.1084/jem.183.2.611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hornbeck P. V., Kornhauser J. M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., and Sullivan M. (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–D270 10.1093/nar/gkr1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Vacic V., Iakoucheva L. M., and Radivojac P. (2006) Two sample logo: a graphical representation of the differences between two sets of sequence alignments. Bioinformatics 22, 1536–1537 10.1093/bioinformatics/btl151 [DOI] [PubMed] [Google Scholar]
- 75. Teyra J., Huang H., Jain S., Guan X., Dong A., Liu Y., Tempel W., Min J., Tong Y., Kim P. M., Bader G. D., and Sidhu S. S. (2017) Comprehensive analysis of the human SH3 domain family reveals a wide variety of non-canonical specificities. Structure 25, 1598–1610 10.1016/j.str.2017.07.017 [DOI] [PubMed] [Google Scholar]
- 76. Wagih O., Reimand J., and Bader G. D. (2015) MIMP: predicting the impact of mutations on kinase-substrate phosphorylation. Nat. Methods 12, 531–533 10.1038/nmeth.3396 [DOI] [PubMed] [Google Scholar]
- 77. RCore Team (2017) R: A Language and Environment for Statistical Computing. Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 78. RStudio Team (2016) RStudio: Integrated Development for R. RStudio, Inc., Boston [Google Scholar]
- 79. Wickham H. (2017) tidyverse: Easily Install and Load 'Tidyverse' Packages, R Package Version 1.1.1 [Google Scholar]
- 80. Novomestky F. (2012) matrixcalc: Collection of Functions for Matrix Calculations, R Package Version 1.0–3 [Google Scholar]
- 81. Wilke C. O. (2017) cowplot: Streamlined Plot Theme and Plot Annotations for ggplot2, R Package Version 0.8.0 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.