

Abstract
A short history of Campbell's primordial soup: In this essay we try to disclose some of the historical connections between the studies that have contributed to our current understanding of the emergence of catalytic RNA molecules and their components from an inanimate matter.

Keywords: nucleosides, nucleotides, oligonucleotides, prebiotic chemistry, RNA
The development of some disciplines follows a straight path that is based on a shared body of progressing knowledge. Well‐established branches of science follow this trend. Other disciplines enjoy frequent rediscoveries. Possibly due to its interdisciplinary character, prebiotic chemistry is one of these fields.
Interdisciplinarity is, in principle, an advantage. Scholars of genetics are well acquainted with the phenomenon dubbed “the strength of the hybrid”: mixing distant characteristics often results in increased fitness. Prebiotic chemistry encompasses the inorganic or organic chemistry in a natural environment before the advent of life on Earth. Its main drive is to understand how the simple molecules present on the early Earth might have given rise to the complex systems and processes of contemporary biology. This quest profits from the ingenuity of scientists with backgrounds as different as astrochemistry, organic and quantum chemistry, geology, paleobiology, molecular genetics, theoretical physics, and cosmology.
One drawback of this state of things is that sometimes principles, facts, and reactions are revisited or rediscovered, the difference between the two terms being essentially that revisitation entails appropriate credit to the original discoverers and the development of their findings within the frame of prebiotic chemistry aims and purports, whereas rediscoveries encompass a loss of time (sometimes decades) and candid repetitions. With this in mind, a glimpse of a few central aspects of prebiotic chemistry could be of some interest. Here, we briefly travel over recent progress in the abiotic generation of key components of potentially biogenic compounds, such as nucleobases, nucleosides, nucleotides and oligonucleotides. However, we only focus on studies that are directly related to an RNA‐based origin concept. Thus, although they are equally important, breakthrough studies related to alternative genetic systems1 and the design of self‐replicating systems2 are not addressed in this work.
1. Nucleobases
Famously, the first abiotic synthesis of a nucleobase was reported by Oró from 1960 to 1962,3 describing the reaction of a concentrated solution of ammonia and HCN to form adenine. The formation of guanine in a comparable chemical system was subsequently reported.4 These pioneering discoveries were followed 12 years later by a report of the synthesis of purine from formamide.5 Simply heating formamide at 160 °C yields low amounts of purine; adenine can be synthesized in trace amounts when the reaction is performed in the presence of added HCN.
The first documented rediscovery in the prebiotic chemistry literature was related to the synthesis of pyrimidine bases, such as cytosine, and, curiously, involved two of the greatest minds working in the field, Stanley Miller and Leslie Orgel. Cytosine was synthesized for the first time in Orgel's laboratory in the late 1960s from the reaction of cyanoacetylene with urea.6 Almost 30 years later, Robertson and Miller reported essentially the same chemistry with the difference that cyanoacetaldehyde was used instead of cyanoacetylene in the synthesis.7 This report was followed by a short correction that gave credit to the first discovery and pointed out that the earlier reported cyanoacetylene‐based synthesis most likely proceeded through cyanoacetaldehyde.8
The field experienced little progress for decades. Scepticism about the worth of this bottom‐up approach starting from one‐carbon precursors prevailed to the point that the very possibility of abiotically obtaining a pyrimidine as important as cytosine became a subject of debate.9
The field was revisited in from 200110 (a gap of 23 years) in a paper that focused on the chemistry of formamide, which rapidly provided evidence for the nonfastidious synthesis of all the biotically relevant nucleobases11 (reviewed in ref. 12).
2. Nucleosides
Understanding the circumstances enabling the prebiotic synthesis of nucleosides is notoriously difficult. Their formation might, in principle, be reconstructed through different approaches: 1) synthesizing the sugar starting at the appropriate position of the base; 2) synthesizing the nucleobase by stepwise assembly of the heterocycle at the appropriate position of the sugar; or 3) forming the connecting β‐glycosidic bond between preformed sugar and nucleobase moieties. The difficulty of linking nucleobases and carbohydrates via an N‐glycosidic bond has recently been reviewed13 and is a major problem in understanding the prebiotic origin of nucleosides.
For de novo synthetic approaches, pioneering studies suggesting the potential of oxazolines for nucleoside synthesis were reported in the late 1950s14 (reviewed in ref. 15) and were applied in a prebiotic context in 1970.16 In this last work, it was shown that pyrimidine ribonucleosides can be obtained from oxazoline chemistry. This chemistry was revisited 40 years later.17 In these systems, formamide acts as solvent for the thiolysis of anhydronucleoside intermediates and for their phosphorylation.17c In another recent work, purine ribonucleosides were synthesized through formamido pyrimidine (FPy) chemistry,18 which required formamide for the N‐formylation of amino pyrimidine intermediates for purine ring closure. This reaction is the reverse of the previously reported formation of FPy by treating purine nucleosides with formamide.21 Remarkably, formamide plays a role in both pathways.
3. Nucleotides
Notwithstanding the many successes in other areas, the possibility of prebiotic phosphorylation was a focus of scepticism. The central role of phosphates in biology (discussed by Westheimer in ref. 23) seemed at odds with the difficult identification of the prebiotic source of phosphates for prebiological chemistry24 (critically reviewed in ref. 25). This feeling prevailed in spite of the pioneering description of urea–inorganic phosphate mixtures as prebiotic phosphorylating agents.26 This discovery was followed by resolutory observations made by Schoffstall between 1976 and 1988,19, 20, 27 who showed that phosphorylation could occur through formamide chemistry, starting from free phosphates and/or a phosphate mineral as common as hydroxyapatite. His approach was revisited 20 years later22, 28 in papers that detailed the heat dependence of the phosphorylation reaction and showed that organic phosphorylation of nucleosides in formamide may occur at every possible position of the sugar moiety and can lead to cyclic nucleotides. Furthermore, they illustrated that numerous phosphate‐containing minerals are plausible sources of phosphate for the generation of nucleotides.
The field has recently been revisited,29 and the range of prebiotically plausible reaction media that could drive the organic phosphorylation of nucleosides has been extended to urea/ammonium formate/water (UAFW) eutectic solutions. Upon heating, the UAFW mixtures are partially converted to formamide.29 For a summary of the studies overviewed in this and the preceding paragraphs, see Figure 1.
Figure 1.
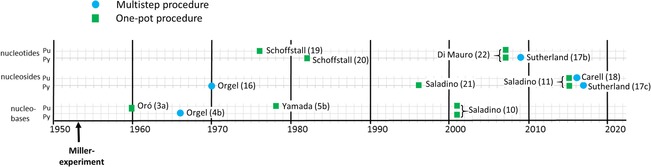
Chronology of selected historically innovative one‐pot and multistep syntheses of nucleic acid building blocks. Oró (1960):3a ammonium hydroxide, HCN, H2O at 90 °C. Orgel (1966):4 step 1: formamidine, NH2CH(CN)2, EtOH at reflux; step 2: imidazole intermediate, formamidine, methyl cellosolve at reflux. Orgel (1970):16 step 1: sugar monophosphate, NCNH2, H2O; step 2: oxazoline intermediate, N≡CC≡CH; step 3: photochemistry. Schoffstall (1976):19 purine nucleoside, KH2PO4, formamide at 70 °C. Yamada (1978):5b formamide, HCN, sealed vial at 160 °C. Schoffstall (1982):20 pyrimidine nucleoside, KH2PO4, formamide at 125 °C. Saladino (1996):21 formamide at 160 °C. Saladino (2001):10 formamide, minerals at 160 °C. Di Mauro (2007):22 nucleoside, formamide, KH2PO4 or mineral phosphates at 90 °C; Sutherland (2009):17b step 1: glycolaldehyde, NCNH2, H2O; step 2: 2‐aminooxazole intermediate, glyceraldehyde, phosphate buffer; step 3: pentose aminooxazoline intermediate, N≡CC≡CH, phosphate buffer; step 4: anhydroarabinonucleoside intermediate, phosphate buffer. Saladino (2015):11 formamide, minerals, proton beam at 25 °C; Carell (2016):18 step 1: guanidine and aminocyanoacetamide; step 2: aminopyrimidinone intermediate, formic acid or formamide at reflux; step 3: formamide pyrimidinone intermediate, ribose, dry state followed by treatment with borax at 100 °C or NH3, or other conditions. Sutherland (2017):17c step 1: 2‐aminooxazole intermediate, and glyceraldehyde; step 2: pentose aminooxazoline intermediate, hydrosulfide hydrate, sodium hydrosulfide hydrate in formamide at 50 °C for 7 h; step 3: 2‐thioribocytidine intermediate, hydrosulfide hydrate, degassed water, irradiation at 254 nm for 2.5 days; step 4: NH4H2PO4, urea in formamide at 100 °C for 6 days. Pu: purines, Py: pyrimidines.
4. Abiotic RNA Polymerizations
The precursors for abiotic RNA polymerization in prebiotic environments could hardly have been based on highly activated compounds. The logic underpinning this consideration derives from thermodynamic and kinetic considerations: the higher the chemical potential of a compound, the lower its expected half‐life. On the other hand, when starting from much too stable monomers, an activation step must be incorporated in the pathway leading to oligonucleotides, thus decreasing the chance that such chemistry would have taken place in a prebiotic context. Circumventing this difficulty, Orgel analyzed the possibility that cyclic nucleotides could be appropriate precursors for nonenzymatic RNA polymerization. The results, reported in 1973,30 showed that limited oligomerization could be achieved under alkaline conditions in dry state in the presence of diamines or amino alcohols serving as activators. The RNA oligomerization reaction starting from 2′,3′‐cyclic nucleotides was tested and resulted in short oligomers, the monomers of which were connected by a mixture of 3′,5′‐ and 2′,5′‐phosphodiester bonds. With the notable exception of two well‐known subsequent studies by Usher and McHale,31 which showed the possible prevalence of 3′,5′‐phosphodiester bonds, the analysis of cyclic nucleotide‐based polymerization was then abandoned, and abiotic RNA syntheses starting from highly preactivated phosphoimidazolides were developed. The use of these compounds, introduced as early as 1968,32 became the preferred methodology and stimulated the development of powerful nonenzymatic polymerization analyses.33 The worth of cyclic nucleotides as precursors for prebiotic RNA synthesis pioneered by Orgel was only revisited again from 2009, 36 years after it was first proposed, and is now an actively developing field of study.34 A recent investigation demonstrated that 3′,5′‐cGMPs have the intrinsic propensity of selectively forming 3′,5′‐linked oligonucleotides.34d Thus, the emergence of the 3′,5′‐linkage selectivity of modern RNAs is not necessarily the consequence of sophisticated repair mechanisms, such as those suggested in ref. 35.
Shortly after the discovery of the nonenzymatic oligomerization of 2′,3′‐cyclic nucleotides, in 1975, conditions leading to the formation of aminoacyl nucleotides and oligopeptides were described.36 The chemistry was elaborated in 2015 when a reaction network utilizing carbodiimide activation and leading to peptidyl‐RNA chains was reported.37 A completely different, RNA‐catalyzed aminoacylation involving a phenylalanyl‐adenosine monophosphate substrate was described by the Yarus group.38 Ten years later, essentially the same chemistry was used to demonstrate the transfer of an aminoacyl group from the phosphate group of a 3′‐aminoacylated nucleoside 3′‐phosphate to the 2′‐hydroxy group of the same nucleotide.39
5. From RNA Catalysis to the Ribosome
The largely accepted RNA‐world40 scenario relies on the (self‐)catalytic properties of RNA polymers. These were discovered by Cech and collaborators in 1981,41 who described the in vitro splicing of the ribosomal RNA precursors of Tetrahymena, based on the involvement of a guanosine nucleotide in the excision of the intervening sequence. The reaction was modelized,42 which showed that it could be performed by simpler model molecules. Recently, a similar reaction was observed for abiotically synthesized oligonucleotides afforded by cyclic ribonucleotide precursors.43
The recognition that the central catalytic core of the ribosome is a ribozyme44 is considered to be the proof of principle that today's protein world had to be preceded by an RNA world,40 in which RNA served as a carrier of genetic information and as a catalyst. It has been suggested that the modern ribosome evolved from an ancient protoribosome that catalyzed noncoded peptide bond formation.45 This simple molecular machine could consist of two symmetry‐related oligonucleotides consisting of 60–90 nucleotides. It has been suggested that pocket‐like entities formed in this way are able to accommodate amino acids bound to short oligonucleotides.45, 46 In this context, genetic coding (i.e., amino acid selectivity) is the consequence of the suitability of certain amino acid sequences for catalyzing some essentially important reactions or for increasing the stability of the molecular machinery. (For a chronological overview of selected studies related to the emergence of catalytically active oligonucleotides, see Figure 2.)
Figure 2.
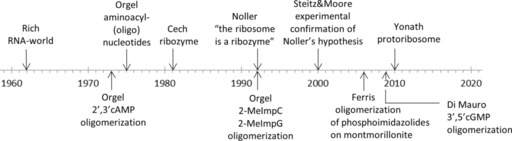
Chronological order of selected studies related to the emergence of catalytically active oligonucleotides from simple nucleotide precursors.
6. Concluding Remarks
Whereas the synthesis of building blocks usually proceeds on timescales of a couple of hours or days, self‐assembling—the prerequisite of every oligomerization—apparently requires more time. Thus, when progressing towards higher molecular complexities, time and statistics become increasingly more important experimental factors. This makes prebiotic studies related to the emergence of catalytic activity or oligomerization technically more difficult and scarce compared to those aimed at reconstructing the synthesis of building blocks. The incomparably higher amount of information available on the basic chemistry leading to life's building blocks might be one of the reasons why reinventions and revisitations are more common in this topic. Nevertheless, time and statistics should not be put aside even in this case: experts working on the synthesis of building blocks must also acknowledge that laboratory reconstructions of life's origin inherently lack a very important parameter: the time that enabled nature to make a sufficiently large number of attempts to reach the goal. For this reason, every piece of the picture, regardless of whether it is a high‐yield or low‐yield process, is potentially an important contribution because it represents a snapshot of the whole pool of the numerous possibilities from which life evolved.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
Financial support from the project STARS in the CAOS (Simulation Tools for Astrochemical Reactivity and Spectroscopy in the Cyberinfrastructure for Astrochemical Organic Species) and from the project GAČR 17–05076S is gratefully acknowledged.
R. Saladino, J. E. Šponer, J. Šponer, E. Di Mauro, ChemBioChem 2018, 19, 22.
Contributor Information
Prof. Raffaele Saladino, Email: saladino@unitus.it.
Dr. Judit E. Šponer, Email: judit@ncbr.muni.cz.
References
- 1.
- 1a. Pitsch S., Krishnamurthy R., Bolli M., Wendeborn S., Holzner A., Minton M., Lesueur C., Schlönvogt I., Jaun B., Eschenmoser A., Helv. Chim. Acta 1995, 78, 1621–1635; [Google Scholar]
- 1b. Schöning K.-U., Scholz P., Guntha S., Wu X., Krishnamurthy R., Eschenmoser A., Science 2000, 290, 1347–1351; [DOI] [PubMed] [Google Scholar]
- 1c. Bolli M., Micura R., Eschenmoser A., Chem. Biol. 1997, 4, 309–320; [DOI] [PubMed] [Google Scholar]
- 1d. Hud N. V., Cafferty B. J., Krishnamurthy R., Williams L. D., Chem. Biol. 2013, 20, 466–474. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Sievers D., von Kiedrowski G., Nature 1994, 369, 221–224; [DOI] [PubMed] [Google Scholar]
- 2b. Tjivikua T., Ballester P., Rebek J., J. Am. Chem. Soc. 1990, 112, 1249–1250; [Google Scholar]
- 2c. Bissette A. J., Fletcher S. P., Angew. Chem. Int. Ed. 2013, 52, 12800–12826; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 13034–13061. [Google Scholar]
- 3.
- 3a. Oró J., Biochem. Biophys. Res. Commun. 1960, 2, 407–412; [Google Scholar]
- 3b. Oró J., Nature 1961, 191, 1193–1194; [DOI] [PubMed] [Google Scholar]
- 3c. Oró J., Kimball A. P., Arch. Biochem. Biophys. 1961, 94, 217–227; [DOI] [PubMed] [Google Scholar]
- 3d. Oró J., Kimball A. P., Arch. Biochem. Biophys. 1962, 96, 293–313. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Sanchez R., Ferris J. P., Orgel L. E., Science 1966, 153, 72–73; [DOI] [PubMed] [Google Scholar]
- 4b. Ferris J. P., Orgel L. E., J. Am. Chem. Soc. 1966, 88, 1074–1074. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Yamada H., Hirobe M., Higashiyama K., Takahashi H., Suzuki K. T., Tetrahedron Lett. 1978, 19, 4039–4042; [Google Scholar]
- 5b. Yamada H., Hirobe M., Higashiyama K., Takahashi H., Suzuki K. T., J. Am. Chem. Soc. 1978, 100, 4617–4618. [Google Scholar]
- 6. Ferris J. P., Sanchez R. A., Orgel L. E., J. Mol. Biol. 1968, 33, 693–704. [DOI] [PubMed] [Google Scholar]
- 7. Robertson M. P., Miller S. L., Nature 1995, 375, 772–774. [DOI] [PubMed] [Google Scholar]
- 8. Robertson M. P., Miller S. L., Nature 1995, 377, 257–257. [Google Scholar]
- 9. Shapiro R., Proc. Natl. Acad. Sci. USA 1999, 96, 4396–4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saladino R., Crestini C., Costanzo G., Negri R., Di Mauro E., Bioorg. Med. Chem. 2001, 9, 1249–1253. [DOI] [PubMed] [Google Scholar]
- 11. Saladino R., Carota E., Botta G., Kapralov M., Timoshenko G. N., Rozanov A. Y., Krasavin E., Di Mauro E., Proc. Natl. Acad. Sci. USA 2015, 112, E2746–E2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Saladino R., Crestini C., Pino S., Costanzo G., Di Mauro E., Phys. Life Rev. 2012, 9, 84–104; [DOI] [PubMed] [Google Scholar]
- 12b. Saladino R., Botta G., Pino S., Costanzo G., Di Mauro E., Chem. Soc. Rev. 2012, 41, 5526–5565; [DOI] [PubMed] [Google Scholar]
- 12c. Carota E., Botta G., Rotelli L., Di Mauro E., Saladino R., Curr. Org. Chem. 2015, 19, 1963–1979. [Google Scholar]
- 13. Sutherland J. D., Angew. Chem. Int. Ed. 2016, 55, 104–121; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 108–126. [Google Scholar]
- 14.
- 14a. Micheel F., Kochling H., Chem. Ber. 1957, 90, 1597–1598; [Google Scholar]
- 14b. Konstas S., Photaki I., Zervas L., Chem. Ber. 1959, 92, 1288–1293. [Google Scholar]
- 15. Wolfrom M. L., Winkley M. W., McWain P. in Synthetic Procedures in Nucleic Acid Chemistry (Eds.: W. W. Zorbach, R. S. Tipson), Wiley, New York, 1968, pp. 239–241. [Google Scholar]
- 16. Sanchez R. A., Orgel L. E., J. Mol. Biol. 1970, 47, 531–543. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Powner M. W., Sutherland J. D., Angew. Chem. Int. Ed. 2010, 49, 4641–4643; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 4745–4747; [Google Scholar]
- 17b. Powner M. W., Gerland B., Sutherland J. D., Nature 2009, 459, 239–242; [DOI] [PubMed] [Google Scholar]
- 17c. Xu J. F., Tsanakopoulou M., Magnani C. J., Szabla R., Šponer J. E., Šponer J., Góra R. W., Sutherland J. D., Nat. Chem. 2017, 9, 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Becker S., Thoma I., Deutsch A., Gehrke T., Mayer P., Zipse H., Carell T., Science 2016, 352, 833–836. [DOI] [PubMed] [Google Scholar]
- 19. Schoffstall A. M., Orig. Life 1976, 7, 399–412. [DOI] [PubMed] [Google Scholar]
- 20. Schoffstall A. M., Barto R. J., Ramos D. L., Orig. Life Evol. Biosph. 1982, 12, 143–151. [DOI] [PubMed] [Google Scholar]
- 21. Saladino R., Mincione E., Crestini C., Negri R., Di Mauro E., Costanzo G., J. Am. Chem. Soc. 1996, 118, 5615–5619. [Google Scholar]
- 22. Costanzo G., Saladino R., Crestini C., Ciciriello F., Di Mauro E., J. Biol. Chem. 2007, 282, 16729–16735. [DOI] [PubMed] [Google Scholar]
- 23. Westheimer F. H., Science 1987, 235, 1173–1178. [DOI] [PubMed] [Google Scholar]
- 24. Yamagata Y., Watanabe H., Saitoh M., Namba T., Nature 1991, 352, 516–519. [DOI] [PubMed] [Google Scholar]
- 25. Orgel L. E., Crit. Rev. Biochem. Mol. Biol. 2004, 39, 99–123. [DOI] [PubMed] [Google Scholar]
- 26. Lohrmann R., Orgel L. E., Science 1971, 171, 490–494. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Schoffstall A. M., Laing E. M., Orig. Life Evol. Biosph. 1985, 15, 141–150; [Google Scholar]
- 27b. Schoffstall A. M., Mahone S. M., Orig. Life Evol. Biosph. 1988, 18, 389–396. [DOI] [PubMed] [Google Scholar]
- 28. Saladino R., Crestini C., Ciciriello F., Pino S., Costanzo G., Di Mauro E., Res. Microbiol. 2009, 160, 441–448. [DOI] [PubMed] [Google Scholar]
- 29. Burcar B., Pasek M., Gull M., Cafferty B. J., Velasco F., Hud N. V., Menor-Salvan C., Angew. Chem. Int. Ed. 2016, 55, 13249–13253; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13443–13447. [Google Scholar]
- 30. Verlander M. S., Lohrmann R., Orgel L. E., J. Mol. Evol. 1973, 2, 303–316. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Usher D. A., McHale A. H., Science 1976, 192, 53–54; [DOI] [PubMed] [Google Scholar]
- 31b. Usher D. A., McHale A. H., Proc. Natl. Acad. Sci. USA 1976, 73, 1149–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weimann B. J., Lohrmann R., Orgel L. E., Schneide H., Sulston J. E., Science 1968, 161, 387–387. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a. Joyce G. F., Orgel L. E., J. Mol. Biol. 1986, 188, 433–441; [DOI] [PubMed] [Google Scholar]
- 33b. Wu T., Orgel L. E., J. Am. Chem. Soc. 1992, 114, 7963–7969; [DOI] [PubMed] [Google Scholar]
- 33c. Ferris J. P., Orig. Life Evol. Biosph. 2002, 32, 311–332; [DOI] [PubMed] [Google Scholar]
- 33d. Huang W. H., Ferris J. P., J. Am. Chem. Soc. 2006, 128, 8914–8919; [DOI] [PubMed] [Google Scholar]
- 33e. Mansy S. S., Schrum J. P., Krishnamurthy M., Tobe S., Treco D. A., Szostak J. W., Nature 2008, 454, 122–125; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33f. Schrum J. P., Ricardo A., Krishnamurthy M., Blain J. C., Szostak J. W., J. Am. Chem. Soc. 2009, 131, 14560–14570; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33g. Taran O., Thoennessen O., Achilles K., von Kiedrowski G., J. Syst. Chem. 2010, 1, 9; [Google Scholar]
- 33h. Deck C., Jauker M., Richert C., Nat. Chem. 2011, 3, 603–608; [DOI] [PubMed] [Google Scholar]
- 33i. Egetenmeyer S., Richert C., Chem. Eur. J. 2011, 17, 11813–11827; [DOI] [PubMed] [Google Scholar]
- 33j. Kaiser A., Spies S., Lommel T., Richert C., Angew. Chem. Int. Ed. 2012, 51, 8299–8303; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8424–8428; [Google Scholar]
- 33k. Coari K. M., Martin R. C., Jain K., McGown L. B., Orig. Life Evol. Biosph. 2017, 47, 305–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.
- 34a. Costanzo G., Pino S., Ciciriello F., Di Mauro E., J. Biol. Chem. 2009, 284, 33206–33216; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34b. Costanzo G., Saladino R., Botta G., Giorgi A., Scipioni A., Pino S., Di Mauro E., ChemBioChem 2012, 13, 999–1008; [DOI] [PubMed] [Google Scholar]
- 34c. Morasch M., Mast C. B., Langer J. K., Schilcher P., Braun D., ChemBioChem 2014, 15, 879–883; [DOI] [PubMed] [Google Scholar]
- 34d. Šponer J. E., Šponer J., Giorgi A., Di Mauro E., Pino S., Costanzo G., J. Phys. Chem. B 2015, 119, 2979–2989; [DOI] [PubMed] [Google Scholar]
- 34e. Costanzo G., Pino S., Timperio A. M., Šponer J. E., Šponer J., Nováková O., Šedo O., Zdráhal Z., Di Mauro E., PLoS One 2016, 11, e0165723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mariani A., Sutherland J. D., Angew. Chem. Int. Ed. 2017, 56, 6563–6566; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 6663–6666. [Google Scholar]
- 36.
- 36a. Lohrmann R., Ranganathan R., Sawai H., Orgel L. E., J. Mol. Evol. 1975, 5, 57–73; [DOI] [PubMed] [Google Scholar]
- 36b. Sawai H., Lohrmann R., Orgel L. E., J. Mol. Evol. 1975, 6, 165–184; [DOI] [PubMed] [Google Scholar]
- 36c. Sawai H., Orgel L. E., J. Mol. Evol. 1975, 6, 185–197. [DOI] [PubMed] [Google Scholar]
- 37. Jauker M., Griesser H., Richert C., Angew. Chem. Int. Ed. 2015, 54, 14564–14569; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14772–14777. [Google Scholar]
- 38. Illangasekare M., Sanchez G., Nickles T., Yarus M., Science 1995, 267, 643–647. [DOI] [PubMed] [Google Scholar]
- 39. Biron J.-P., Parkes A. L., Pascal R., Sutherland J. D., Angew. Chem. Int. Ed. 2005, 44, 6731–6734; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 6889–6892. [Google Scholar]
- 40.
- 40a. Rich A. in Horizons in Biochemistry (Eds.: M. Kasha, B. Pullman), Academic Press, New York, 1962, pp. 103–126; [Google Scholar]
- 40b. Gilbert W., Nature 1986, 319, 618–618. [Google Scholar]
- 41. Cech T. R., Zaug A. J., Grabowski P. J., Cell 1981, 27, 487–496. [DOI] [PubMed] [Google Scholar]
- 42. Zaug A. J., Cech T. R., Science 1986, 231, 470–475. [DOI] [PubMed] [Google Scholar]
- 43.
- 43a. Pino S., Costanzo G., Giorgi A., Šponer J., Šponer J. E., Di Mauro E., Entropy 2013, 15, 5362–5383; [Google Scholar]
- 43b. Stadlbauer P., Šponer J., Costanzo G., Di Mauro E., Pino S., Šponer J. E., Chem. Eur. J. 2015, 21, 3596–3604. [DOI] [PubMed] [Google Scholar]
- 44.
- 44a. Noller H. F., Hoffarth V., Zimniak L., Science 1992, 256, 1416–1419; [DOI] [PubMed] [Google Scholar]
- 44b. Nissen P., Hansen J., Ban N., Moore P. B., Steitz T. A., Science 2000, 289, 920–930. [DOI] [PubMed] [Google Scholar]
- 45. Davidovich C., Belousoff M., Wekselman I., Shapira T., Krupkin M., Zimmerman E., Bashan A., Yonath A., Isr. J. Chem. 2010, 50, 29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Davidovich C., Belousoff M., Bashan A., Yonath A., Res. Microbiol. 2009, 160, 487–492. [DOI] [PubMed] [Google Scholar]