

Abstract
Εniminium ions were prepared from the corresponding α,β‐unsaturated carbonyl compounds (enones and enals), and were found to be promoted to their respective triplet states by energy transfer. The photoexcited intermediates underwent intra‐ or intermolecular [2+2] photocycloaddition in good yields (50–78 %) upon irradiation at λ=433 nm or λ=457 nm. Iridium or ruthenium complexes with a sufficiently high triplet energy were identified as efficient catalysts (2.5 mol % catalyst loading) for the reaction. The intermolecular [2+2] photocycloaddition of an eniminium ion derived from a chiral secondary amine proceeded with high enantioselectivity (88 % ee).
Keywords: cycloaddition, enantioselectivity, enones, homogenous catalysis, photochemistry
Whereas the [2+2] photocycloaddition chemistry of α,β‐unsaturated carbonyl compounds (enones and enals) has been extensively explored,1 the related α,β‐unsaturated iminium ions (eniminium ions) have received little attention. Notable studies originate only from the group of Mariano, who investigated the intramolecular [2+2] photocycloaddition of eniminium ions2 in the context of their pioneering work on the photochemistry of iminium ions.3 They found that eniminium ions, upon direct excitation at λ>250 nm, undergo a stereospecific [2+2] photocycloaddition that results from excitation of the respective ππ* transition. Attempts to generate enantioenriched cyclobutanes by using an eniminium ion derived from a chiral secondary amine led to a maximum enantiomeric excess (ee) of 82 % at 40 % conversion.2b
Scheme 1 illustrates the major difference in the photochemical behavior of enones I and eniminium ions II. The lowest‐lying singlet state (S1) of enones is of nπ* character and opens, despite its low absorption coefficient, a convenient entry to populate the reactive triplet state T1 by direct excitation at long wavelengths (λ=300–350 nm) followed by a symmetry‐allowed4 intersystem crossing (ISC). The S1 state of eniminium ions is of ππ* character, and the absorption is shifted hypsochromically relative to the nπ* transition of the enone. As ISC to T1 is notoriously slow4 for the eniminium ion, subsequent reactions occur exclusively from the S1 state. While it has been known for some time5 that E/Z isomerization reactions occur from S1 in photoexcited eniminium ions, it has only recently been disclosed by the Melchiorre group that this state can be quenched with appropriate electron donors to achieve enantioselective alkylation reactions.6
Scheme 1.

Schematic energy diagram for the singlet (S) and triplet (T) states of α,β‐unsaturated carbonyl compounds I and the respective eniminium ions II.
Considering recent interest in the catalysis of photochemical processes by visible‐light‐induced triplet sensitization,7 we have explored the nature of the triplet state T1 of eniminium ions in the present study. We speculated that this state would be accessible by carefully choosing a suitable triplet sensitizer, and we expected it to be an efficient intermediate in [2+2] photocycloaddition reactions. Our preliminary results are described in this Communication.
To investigate our hypothesis, we synthesized 3‐(4‐pentenyl)‐cyclohex‐2‐enone (1),8 which was readily converted into the eniminium salt 2 by treatment with pyrrolidine and azeotropic removal of water9 (Scheme 2). The respective hexafluorophosphate salt precipitated and was recrystallized from ethanol. Enone 1 absorbs at λ=233 nm (ϵ=15 650 m −1 cm−1) and λ=320 nm (ϵ=70 m −1 cm−1). The latter absorption was assigned to the forbidden nπ* transition, and the former absorption to the allowed ππ* transition. The eniminium salt 2 showed an absorption maximum at λ=270 nm (ϵ=21 320 m −1 cm−1). For the reasons mentioned above, the triplet energy of eniminium ion 2 could not be directly measured but its redox potential E1/2 (2 +/2⋅) was determined as −1.39 V vs. SCE.10, 11 As expected from their absorption spectra, neither enone 1 nor the eniminium ion 2 showed any conversion when excited at an irradiation wavelength of λ=420 nm.12 We subsequently attempted to initiate the [2+2] photocycloaddition of eniminium ion 2 by addition of 2.5 mol % of an iridium or ruthenium catalyst (Table 1). The reactions were performed in MeCN solution, and the intermediate iminium ion rac‐3 was hydrolyzed by treatment with aqueous 1 m NaOH solution.
Scheme 2.
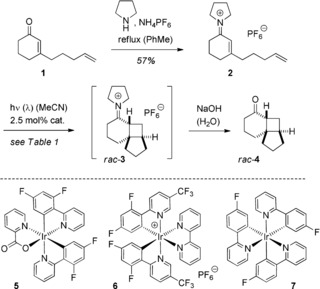
Preparation of eniminium ion 2, its sensitized [2+2] photocycloaddition reaction to products rac‐3 and rac‐4, and structure of iridium complexes 5–7.
Table 1.
[2+2] Photocycloaddition of eniminium salt 2 in the presence of various catalysts (see Scheme 2).
| Entry[a] | λ [a] [nm] | Catalyst | E T [b] [kJ mol−1] | t [a] [h] | Product | Yield[c] [%] |
|---|---|---|---|---|---|---|
| 1 | 420 | 5 | 256 | 20 | rac‐4 | 57 |
| 2 | 420 | 6 | 253 | 20 | rac‐4 | 44 |
| 3 | 420 | 7 | 245 | 20 | –[d] | – |
| 4 | 420 | Ir(ppy)3 | 231 | 20 | –[d] | – |
| 5 | 420 | Ru(bpy)3(PF6)2 | 193 | 20 | –[d] | – |
| 6[e] | 420 | 6 | 253 | 2 | rac‐3 | 75 |
| 7[f] | 433 | 5 | 256 | 2 | rac‐4 | 73 |
| 8[f] | 433 | 6 | 253 | 2 | rac‐4 | 68 |
[a] The reactions were performed at room temperature with a substrate concentration of c=20 mm in MeCN at the indicated wavelength (λ) and for the indicated period of time (t). An 1 m aqueous NaOH solution was used for hydrolysis unless otherwise indicated. [b] Tabulated or measured triplet energies for the respective iridium or ruthenium catalyst (see the main text). [c] Yield of isolated product. [d] No conversion was observed, and the starting material was re‐isolated. [e] No aqueous work‐up was performed. [f] A 3 m aqueous NaOH solution was employed for hydrolysis.
The literature‐reported cyclobutane rac‐4 8b was isolated as a single diastereoisomer from the reactions that were catalyzed by iridium complexes 5 and 6 (entries 1 and 2). The other catalysts, 7, Ir(ppy)3 (ppy=2‐phenylpyridine), and Ru(bpy)3(PF6)2 (bpy=2,2′‐bipyridine), failed to induce the desired transformation (entries 3–5), and the starting material was recovered unchanged. The direct reaction of enone 1 to product rac‐4 was not catalyzed by complex 5 or 6. In combination with the fact that iminium salt rac‐3 could be isolated from the reaction mixture in 75 % yield (entry 6), it is clear that the reaction proceeds via the eniminium ion and not via the enone. Further optimization experiments revealed that ketone rac‐4 was obtained in higher yields upon irradiation at λ=433 nm13 and upon work‐up with 3 m NaOH solution (entries 7 and 8). Under the conditions of entry 7, other eniminium ions 8 reacted equally well, and products rac‐9 were obtained in good yields (Scheme 3).
Scheme 3.

Intramolecular iridium‐catalyzed [2+2] photocycloaddition of eniminium ions 8.
Most of the redox potentials and triplet energies of the photoexcited catalysts shown in Table 1 have been reported.14 The triplet energy of compound 5 was determined from its luminescence emission15 in MeCN solution (see the Supporting Information). We found no correlation between the excited‐state redox potential of the catalysts and their viability in affording the cyclobutane product rac‐4. The strongly reducing iridium complexes 7 and Ir(ppy)3 16 failed to catalyze the [2+2] photocycloaddition while the much weaker reductant 6 [E 1/2(IrIV/IrIII*)14c=−0.55 V] was a very efficient catalyst. When comparing the tabulated triplet energies for the individual complexes (Table 1), there is, however, a very clear correlation. The [2+2] photocycloaddition ceases if the triplet energy ET of the catalyst is below 250 kJ mol−1. Vice versa, this figure provides an estimate for the triplet energy of the elusive T1 state of eniminium ion 2. The observation that enone 1 did not undergo an Ir‐catalyzed [2+2] photocycloaddition (see above) is readily explained by its higher triplet energy compared to that of the eniminium ion.17
To gain more insight into the interaction between the photoexcited catalyst and eniminium salt 2, quenching studies were performed in MeCN solution at a concentration of c=10 μm. Luminescence quenching was observed for catalyst 5 (Figure 1 a), and the Stern–Volmer constant (K SV) extracted from a plot of I 0/I versus the eniminium concentration was 7.49 m −1 (Figure 1 b). The lifetime τ 0 of the excited state of catalyst 5 15 was determined to be 970 ns, and it was shown that a dynamic quenching mechanism applies (see the Supporting Information). The quenching rate constant was calculated from K SV and τ 0 as k q=7.7×106 m −1 s−1. The luminescence intensity of catalyst 6 was similarly quenched by increasing concentrations of eniminium ion 2 (Figure 1 c). The Stern–Volmer constant was calculated to be 7.69 m −1 in the case of catalyst 6. The luminescence of the inactive iridium catalyst Ir(ppy)3 (Table 1, entry 4) was not quenched by addition of eniminium ion 2 (Figure 1 d). These results suggest that the photochemical reaction of eniminium ion 2 is initiated upon energy transfer from compounds 5 and 6 while energy transfer from Ir(ppy)3 is not feasible based on the data from Table 1.
Figure 1.
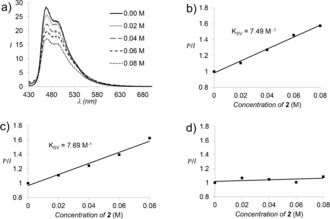
Quenching experiments of photoexcited iridium catalysts 5, 6, and Ir(ppy)3 with eniminium ion 2 in MeCN solution. a) Decrease in the luminescence intensity of compound 5 upon addition of iminium salt 2. Stern–Volmer plots (K SV=Stern–Volmer constant) of the luminescence quenching for b) catalyst 5, c) catalyst 6, and d) Ir(ppy)3.
In a second set of experiments, we studied the intermolecular [2+2] photocycloaddition of eniminium ions derived from cinnamaldehyde (10). Eniminium ion 11 was readily prepared by treatment of aldehyde 10 with trimethylsilyl (TMS) triflate and N‐trimethylsilyl pyrrolidine in diethyl ether (Scheme 4).18 The absorption spectrum of compound 11 shows a maximum at λ=330 nm (ϵ=36 840 m −1 cm−1) but its triplet energy could not be determined. The redox potential E 1/2 (11 +/11 .) was found to be −0.83 V.10 Neither aldehyde 10 nor eniminium ion 11 showed any conversion when their solutions in MeCN were irradiated at λ=457 nm13 in the presence of 2,3‐dimethylbutadiene. To our delight, we found that the desired [2+2] photocycloaddition of eniminium ion 11 could be triggered by the addition of 2.5 mol % of Ru(bpy)3(PF6)2. The reaction was complete after four hours, and product rac‐12 was obtained upon hydrolysis in 69 % as a mixture of two diastereomers. The relative configuration at C3 was different in the two diastereomers, and NOESY studies revealed that the major isomer rac‐12 a places the methyl group in cis orientation relative to the phenyl group. Under the same irradiation conditions, aldehyde 10 underwent hardly any conversion (≤10 %) to product rac‐12.
Scheme 4.

Formation of eniminium ion 11 and its ruthenium‐catalyzed [2+2] photocycloaddition to 2,3‐dimethylbutadiene (d.r.=diastereomeric ratio).
The following observations (see the Supporting Information) provide evidence for the fact that the ruthenium‐catalyzed reaction proceeds via triplet energy transfer and not via electron transfer: a) The ruthenium complex Ru(bpz)3(PF6)2 (bpz=2,2′‐bipyrazine), which is known to be a weak reductant in its excited state [E 1/2(RuIII/RuII*)19=−0.26 V], promoted the [2+2] photocycloaddition as efficiently as Ru(bpy)3(PF6)2. b) Eosin Y (EY), with a triplet energy20 of ET=182 kJ mol−1 and an excited‐state reduction potential of E 1/2(EY.+/EY*)20=−1.11 V, did not catalyze the reaction at λ=512 nm. c) The reaction was catalyzed by typical triplet sensitizers such as benzil and thioxanthone,21 albeit upon irradiation at short wavelengths. d) The regioselectivity of the [2+2] photocycloaddition can only be explained by a triplet pathway that proceeds via a 1,4‐diradical. Addition of an intermediate radical 11. to 2,3‐dimethylbutadiene would lead to the opposite regioisomer.
In a preliminary study of the substrate scope, other olefins were shown to react with eniminium ion 11 (Figure 2). Isoprene gave rac‐13 in a yield that was comparable to the yield recorded for rac‐12. The lower diastereomeric ratio reflects the smaller size of the ethenyl group as compared to the 2‐propenyl group. Likewise, 1,3‐butadiene gave rac‐14 with a low diastereoselectivity at carbon atom C3. Alkynyl‐substituted olefins, such as 2‐methylhex‐1‐en‐3‐yne and (3‐methylbut‐3‐en‐1‐ynyl)trimethylsilane, reacted smoothly to give products rac‐15 and rac‐16.
Figure 2.

[2+2] Photocycloaddition products obtained by the reaction of eniminium ion 11 with different olefins (λ=457 nm, catalyst: 2.5 mol % Ru(bpy)3(PF6)2 in MeCN).
Additionally, we evaluated the potential of the triplet‐sensitized [2+2] photocycloaddition of eniminium ions for enantioselective synthesis.22 To meet this end, the known eniminium ion 17 6 was prepared and subjected to a ruthenium‐catalyzed reaction with 2,3‐dimethylbutadiene (Scheme 5). The reaction was performed at −40 °C with 2.5 mol % of Ru(bpy)3(PF6)2 as the catalyst. Complete conversion was observed after 3.5 h, and product 12 a was obtained essentially as a single diastereoisomer (d.r.=94:6) and with 88 % ee.
Scheme 5.

Enantioselective ruthenium‐catalyzed [2+2] photocycloaddition of eniminium ion 17 (Ar=3,5‐bis(trifluoromethyl)phenyl; TDS=tert‐hexyldimethylsilyl) to cyclobutane 12 a.
In conclusion, we have collected evidence that eniminium ions can be promoted to their triplet states by sensitization with suitable iridium or ruthenium complexes upon irradiation with visible light (λ=433 or 457 nm). This indirect activation mode allows for enantioselective [2+2] photocycloadditions of eniminium ions derived from chiral secondary amines. Transformations of this type could previously be conducted only via the respective singlet intermediate by short‐wavelength irradiation (λ=250–300 nm). More importantly, the hypothesis that the triplet state of an eniminium ion is lower in energy than the triplet state of the respective α,β‐unsaturated carbonyl compound has been substantiated. Under conditions that allowed for sensitized [2+2] photocycloaddition reactions of the eniminium ions 2 and 11, there was no or little conversion of the corresponding carbonyl compounds 1 and 10. Along with thermal iminium ion catalysis23 and photoinduced electron transfer (PET) to iminium ions,6, 24, 25 triplet sensitization seems to offer another promising avenue for the in situ activation of carbonyl groups to explore new reactivity patterns.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by the European Research Council under the European Union's Horizon 2020 research and innovation programme (grant agreement 665951—ELICOS) and the Alexander von Humboldt foundation (postdoctoral fellowship to E.R.) is gratefully acknowledged. We thank M. Grübel for performing the cyclic voltammetry experiments and S. Poplata for his help in the preparation and characterization of products rac‐9.
F. M. Hörmann, T. S. Chung, E. Rodriguez, M. Jakob, T. Bach, Angew. Chem. Int. Ed. 2018, 57, 827.
Contributor Information
M. Sc. Fabian M. Hörmann, http://www.oc1.ch.tum.de/home_en/
Prof. Dr. Thorsten Bach, Email: thorsten.bach@ch.tum.de.
References
- 1.
- 1a. Crimmins M. T., Reinhold T. L., Org. React. 1993, 44, 297–588; [Google Scholar]
- 1b. Margaretha P. in Molecular and Supramolecular Photochemistry, Vol. 12 (Eds.: A. G. Griesbeck, J. Mattay), Dekker, New York, 2005, pp. 211–237; [Google Scholar]
- 1c. Hehn J. P., Müller C., Bach T. in Handbook of Synthetic Photochemistry (Eds.: A. Albini, M. Fagnoni), Wiley-VCH, Weinheim, 2009, pp. 171–215; [Google Scholar]
- 1d. Poplata S., Tröster A., Zou Y. Q., Bach T., Chem. Rev. 2016, 116, 9748–9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Cai X., Chang V., Chen C., Kim H.-J., Mariano P. S., Tetrahedron Lett. 2000, 41, 9445–9449; [Google Scholar]
- 2b. Chen C., Chang V., Cai X., Duesler E., Mariano P. S., J. Am. Chem. Soc. 2001, 123, 6433–6434. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Mariano P. S., Acc. Chem. Res. 1983, 16, 130–137; [Google Scholar]
- 3b. Mariano P. S., Tetrahedron 1983, 39, 3845–3879. [Google Scholar]
- 4.
- 4a. El-Sayed M. A., Acc. Chem. Res. 1968, 1, 8–16; [Google Scholar]
- 4b. Klán P., Wirz J., Photochemistry of Organic Compounds, Wiley, Chichester, 2009, pp. 38–39. [Google Scholar]
- 5.
- 5a. Wald G., Science 1968, 162, 230–239; [DOI] [PubMed] [Google Scholar]
- 5b. Childs R. F., Dickie B. D., Chem. Commun. 1981, 1268–1269; [Google Scholar]
- 5c. Pankratz M., Childs R. F., J. Org. Chem. 1988, 53, 3278–3283; [Google Scholar]
- 5d. Palczewski K., J. Biol. Chem. 2012, 287, 1612–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Silvi M., Verrier C., Rey Y. P., Buzzetti L., Melchiorre P., Nat. Chem. 2017, 9, 868–873. [DOI] [PubMed] [Google Scholar]
- 7.For examples, see:
- 7a. Lu Z., Yoon T. P., Angew. Chem. Int. Ed. 2012, 51, 10329–10332; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10475–10478; [Google Scholar]
- 7b. Zou Y.-Q., Duan S.-W., Meng X.-G., Hu X.-Q., Gao S., Chen J.-R., Xiao W.-J., Tetrahedron 2012, 68, 6914–6919; [Google Scholar]
- 7c. Alonso R., Bach T., Angew. Chem. Int. Ed. 2014, 53, 4368–4371; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4457–4460; [Google Scholar]
- 7d. Liu Q., Zhu F.-P., Jin X.-L., Wang X.-J., Chen H., Wu L.-Z., Chem. Eur. J. 2015, 21, 10326–10329; [DOI] [PubMed] [Google Scholar]
- 7e. Mojr V., Svobodová E., Straková K., Neveselý T., Chudoba J., Dvořáková H., Cibulka R., Chem. Commun. 2015, 51, 12036–12039; [DOI] [PubMed] [Google Scholar]
- 7f. Blum T. R., Miller Z. D., Bates D. M., Guzei I. A., Yoon T. P., Science 2016, 354, 1391–1395; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7g. Tröster A., Alonso R., Bauer A., Bach T., J. Am. Chem. Soc. 2016, 138, 7808–7811; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7h. Zhao J., Brosmer J. L., Tang Q., Yang Z., Houk K. N., Diaconescu P. L., Kwon O., J. Am. Chem. Soc. 2017, 139, 9807–9810; [DOI] [PubMed] [Google Scholar]
- 7i. Edtmüller V., Pöthig A., Bach T., Tetrahedron 2017, 73, 5038–5047; [Google Scholar]
- 7j. Münster N., Parker N., van Dijk L., Paton R. S., Smith M. D., Angew. Chem. Int. Ed. 2017, 56, 9468–9472; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9596–9600; [Google Scholar]
- 7k. Miller Z. D., Lee B. J., Yoon T. P., Angew. Chem. Int. Ed. 2017, 56, 11891–11895; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12053–12057. [Google Scholar]
- 8.
- 8a. de Miguel I., Herrad B., Mann E., Adv. Synth. Catal. 2012, 354, 1731–1736; [Google Scholar]
- 8b. Brenninger C., Pöthig A., Bach T., Angew. Chem. Int. Ed. 2017, 56, 4337–4341; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4401–4405. [Google Scholar]
- 9. Saba S., Vrkic D., Cascella C., DaSilva I., Carta K., Kojtari A., J. Chem. Res. 2008, 301–304. [Google Scholar]
- 10.All redox potentials are reported against a saturated calomel electrode (SCE) to allow for better comparison. Our own measurements were performed against a reference electrode of 0.01 m Ag/AgNO3 in 0.1 m tetrabutylammonium hexafluorophosphate (MeCN, 25 °C). The potential against the SCE was thus obtained by adding 0.30 V to the measured potential; see: Pavlishchuk V. V., Adison A. W., Inorg. Chim. Acta 2000, 298, 97–102. [Google Scholar]
- 11.Potentials originally reported against ferrocenium/ferrocene (Fc+/Fc) were referenced to SCE by addition of 0.42 V to the reported potential; see: Roth H. G., Romero N. A., Nicewicz D. A., Synlett 2016, 27, 714–723. [Google Scholar]
- 12.For the emission spectrum of the light source, see the Supporting Information.
- 13.An LED lamp was used (for the emission spectrum, see the Supporting Information). For the reaction set-up, see:
- 13a. Rackl D., Kais V., Kreitmeier P., Reiser O., Beilstein J. Org. Chem. 2014, 10, 2157–2165; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Lenhart D., Pöthig A., Bach T., Chem. Eur. J. 2016, 22, 6519–6523. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Flamigni L., Barbieri A., Sabatini C., Ventura B., Barigelleti F., Top. Curr. Chem. 2007, 281, 143–203; [Google Scholar]
- 14b. Prier C. K., Rankic D. A., MacMillan D. W. C., Chem. Rev. 2013, 113, 5322–5363; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14c. Singh A., Teegardin K., Kelly M., Prasad K. S., Krishnan S., Weaver J. D., J. Organomet. Chem. 2015, 776, 51–59. [Google Scholar]
- 15.
- 15a. Baranoff E., Curchod B. F. E., Monti F., Steimer F., Accorsi G., Tavernelli I., Rothlisberger U., Scopelliti R., Grätzel M., Nazeeruddin M. K., Inorg. Chem. 2012, 51, 799–811; [DOI] [PubMed] [Google Scholar]
- 15b. Yi S., Kim J.-H., Cho Y.-J., Lee J., Choi T.-S., Cho D. W., Pac C., Han W.-S., Son H.-J., Kang S. O., Inorg. Chem. 2016, 55, 3324–3331. [DOI] [PubMed] [Google Scholar]
- 16.The reported values[14c] for E 1/2(IrIV/IrIII*) against Fc+/Fc are −1.86 V for 7 (−1.44 V vs. SCE)[11] and −1.97 V for Ir(ppy)3 (−1.55 V vs. SCE).[11]
- 17.The triplet energy of the closely related enone 3-methylcyclohex-2-enone was reported as ET=283 kJ mol−1; see: Schuster D. I., Dunn D. A., Heibel G. E., Brown P. B., Rao J. M., Woning J., Bonneau R., J. Am. Chem. Soc. 1991, 113, 6245–6255. [Google Scholar]
- 18. Lakhdar S., Tokuyasu T., Mayr H., Angew. Chem. Int. Ed. 2008, 47, 8723–8726; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 8851–8854. [Google Scholar]
- 19. Haga M., Dodsworth E. S., Eryavec G., Seymour P., Lever A. B. P., Inorg. Chem. 1985, 24, 1901–1906. [Google Scholar]
- 20. Hari D. P., König B., Chem. Commun. 2014, 50, 6688–6699. [DOI] [PubMed] [Google Scholar]
- 21.Triplet sensitization of an iminium ion by xanthone has been suggested to occur; see: Tu C.-L., Mariano P. S., J. Am. Chem. Soc. 1987, 109, 5287–5288. [Google Scholar]
- 22.For a review, see: Brimioulle R., Lenhart D., Maturi M. M., Bach T., Angew. Chem. Int. Ed. 2015, 54, 3872–3890; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3944–3963. [Google Scholar]
- 23.
- 23a. Ahrendt K. A., Borths C. J., MacMillan D. W. C., J. Am. Chem. Soc. 2000, 122, 4243–4244; [Google Scholar]
- 23b. Erkkilä A., Majander I., Pihko P. M., Chem. Rev. 2007, 107, 5416–5470. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Murphy J. J., Bastida D., Paria S., Fagnoni M., Melchiorre P., Nature 2016, 532, 218–222; [DOI] [PubMed] [Google Scholar]
- 24b. Bahamonde A., Murphy J. J., Savarese M., Brémond É., Cavalli A., Melchiorre P., J. Am. Chem. Soc. 2017, 139, 4559–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.For a review, see: Y.-Q. Zou, F. M. Hörmann, T. Bach, Chem. Soc. Rev 2017, DOI: https://doi.org/10.1039/C7CS00509A.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary