

Abstract
CTLA-4 is a cell surface receptor on T cells that functions as an immune checkpoint molecule to enforce tolerance to cognate antigens. Anti-CTLA-4 immunotherapy is highly effective at reactivating T cell responses against melanoma, which is postulated to be due to targeting CTLA-4 on T cells. Here we report that CTLA-4 is also highly expressed by most human melanoma cell lines, as well as in normal human melanocytes. Interferon-gamma (IFNG) signaling activated the expression of the human CTLA-4 gene in a melanocyte and melanoma cell-specific manner. Mechanistically, IFNG activated CTLA-4 expression through JAK1/2-dependent phosphorylation of STAT1, which bound a specific gamma-activated sequence (GAS) site on the CTLA-4 promoter, thereby licensing CBP/p300-mediated histone acetylation and local chromatin opening. In melanoma cell lines, elevated baseline expression relied upon constitutive activation of the MAPK pathway. Notably, RNA-seq analyses of melanoma specimens obtained from patients who had received anti-CTLA-4 immunotherapy (ipilimumab) showed upregulation of an IFNG-response gene expression signature, including CTLA-4 itself, which correlated significantly with durable response. Taken together, our results raise the possibility that CTLA-4 targeting on melanoma cells may contribute to the clinical immunobiology of anti-CTLA-4 responses.
Keywords: Melanoma, Melanocytes, Interferon-γ, CTLA-4, Signaling, Immunotherapy, Ipilimumab, Vemurafenib, Ruxolitinib, BRAF, NRAS
INTRODUCTION
T cells require two simultaneous signals from antigen presenting cells (APC) for optimal activation; T cell receptor (TCR) interaction with a peptide antigen presented by the major histocompatibility complex (MHC) class I molecules, and the engagement of T cell costimulatory molecules, e.g. CD28, with those of APCs, e.g. B7-1 and B7-2 (CD80 and CD86, respectively) (1). However, a number of coinhibitory molecules function to limit T cell activity in order to prevent tissue damage due to immune over-activation (1). Cytotoxic T Lymphocyte Antigen 4 (CTLA4, CD152) is a coinhibitory molecule that was originally identified as a homodimeric glycoprotein of the Ig family expressed on the surface of activated T cells (2). CTLA4 is the most potent inhibitor of the cytotoxic T lymphocyte (CTL) activation and thus a key negative regulator of anti-tumor activity by contributing to T cell tolerance and anergy, leading to tumor immunoevasion (2). Anti-CTLA4 monoclonal antibody (Ipilimumab) therapy functions by restoring the activity of the tumor antigen-specific CTLs (1). The best characterized application of anti-CTLA4 immunotherapy has been in the therapeutic management of melanoma, albeit with limited response rates (3). CTLA4 is conventionally thought to be expressed predominantly by the T cell lineage, and the therapeutic action of Ipilimumab is thought to be antagonistic to a mechanism whereby either CTLs themselves or the immunosuppressive regulatory T cell subtype (Treg) limit the activity of T cells (4). Notably, however, CTLA4 expression has also been reported in some non-lymphoid cell lineages such as tumor cells, which highlights the potential of CTLA4 as a therapeutic target beyond the T cell compartment (5,6). There is evidence suggesting that CTLA4 is expressed on melanoma cells and is involved in tumor immune escape (2,7–9).
In order to uncover the molecular mechanisms underlying ultraviolet radiation (UVR)-induced melanomagenesis, we previously investigated the genomic response of melanocytes to UVR in vivo (10). We showed that in addition to damaging DNA, UVR alters gene expression in exposed melanocytes that drives their interactions with elements of the microenvironment to remodel damaged skin and escape destruction. These results implicated a UVR-induced pro-tumorigenic inflammatory cascade, whereby UVB directly upregulated melanocytic expression of ligands to the chemokine receptor CCR2, which recruited macrophages into the neonatal skin microenvironment. A subset of these macrophages produced interferon-gamma (IFNG), which elicited a positive feedback type activation of expression in stimulated melanocytes of a putative “survival signature”, consisting of genes involved in immunoevasive mechanisms (10). Intriguingly, CTLA4 was the highest upregulated gene prominently clustered among this IFNG-induced gene expression signature (10), prompting us to hypothesize that CTLA4 is a novel direct downstream target gene regulated by the IFNG-induced signaling pathway in melanocytes.
Here we show that CTLA4 is expressed in human primary melanocytes and is highly overexpressed in melanoma cell lines, but not in non-melanoma tumor cell lines. Concordantly, we have found that the promoter region of CTLA4 exhibits open chromatin configuration in melanocytes and melanoma cells, akin to T cells, but not in other cell types. Most interestingly, we have identified CTLA4 as a novel downstream target gene of IFNG signaling via activation of STAT1-mediated signaling, which recruits CBP and POLII to the CTLA4 promoter and modulates histone acetylation. We have also shown evidence that overexpression of CTLA4 in human melanoma cell lines is driven by constitutive activation of the MAPK pathway, which is independent of the IFNG pathway activation. An analysis of previously published RNA-seq datasets of melanoma patients treated with ipilimumab showed that patients that exhibited an IFNG-responsive gene expression signature, including overexpression of CTLA4, demonstrated better clinical response than those that did not express this signature.
MATERIALS AND METHODS
Cell culture
The human primary neonatal foreskin melanocytes: HEMn-LP (from lightly pigmented donor), HEMn-MP (from moderately pigmented donor) and HEMn-DP (from darkly pigmented donor) were cultured at 37°C in medium 254 supplemented with HMGS-2 (PMA-free) and Gentamycin (50ug/ml) with 5% CO2. The human epidermal neonatal keratinocytes (HEKn) were cultured at 37°C in EpiLife medium supplemented with HKGS and Gentamycin (50ug/ml) with 5% CO2. All cells, media and supplements listed above were purchase from Life Technology.
The melanoma cell line Hs 936.T was purchased from ATCC; the melanoma cell lines A2058 and COLO679 were obtained from Dr. Glenn Merlino (NCI); the melanoma cell lines WM983(B), 451 Lu, WM3918 and WM3912 were obtained from Dr. Meenhard Herlyn (Wistar Inst); the melanoma cell line UACC1273 was obtained from Dr. Ashani Weeraratna (Wistar Inst); the human colon carcinoma cell lines RKO and HCT116 were obtained from Dr. Jean-Pierre Issa (Temple Univ); the human embryonic kidney cell line HEK293 and the human osteosarcoma cell line U-2 OS were obtained from Dr. Richard Pomerantz (Temple); The human fibroblast cell line FS2, the human hepatocellular carcinoma cell line FOCUS, the human ovarian adenocarcinoma cell lines SK-OV-3 and OVCAR429, the human ovarian teratocarcinoma cell line PA-1, the human prostate carcinoma cell lines HTB-81 and PC-3, the human osteosarcoma cell line MG63, the human breast adenocarcinoma cell lines MCF7 and MDA-MB-231, the human acute lymphoblastic leukemia cell line CEM, the human Burkitt’s Lymphoma Daudi, and the human lung carcinoma cell line A549 were obtained from Dr. Raghbir Athwal (Temple). All tumor cell lines were cultured at 37°C in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), L-alanyl-L-Glutamine (2mM) and Gentamycin (50ug/ml) at 5% CO2. DMEM, FBS and L-alanyl-L-Glutamine were purchased from Corning, Cellgro.
IFNG Treatment
Human recombinant IFNG (with carrier) was purchased from Cell Signaling Technology (catalog #8901). The concentration used was 10ng/ml except Fig. 2B, where the concentrations are indicated within the figure. According to MSDS provided by the manufacturer, the bioactivity of h-IFNG was determined in a virus protection assay. The ED50 of each lot is between 0.3–1.2ng/ml. The conversion of 10ng/ml to biological activity is 8.33U/ml-33.33U/ml.
Quantitative RT-PCR
qRT-PCR analysis was performed to quantitatively measure the mRNA abundance. Total cellular RNA was extracted using the Trizol method (Life Technologies) and RNeasy Mini Kit (QIAGEN). RNA concentration was determined by Nanodrop. 500ng-5μg total RNA was used to generate cDNA by GoTaq 2-step RT system (QIAGEN). qRT-PCR analysis of human CTLA4, IRF1, GAPDH, NR4A3, STAT1, PSMB9 and TAP1 were performed with the Power SYBR-Green PCR Master Mix (Fisher Scientific) and the ABI StepOnePlus PCR system. 18s rRNA was used as the reference gene. The ΔΔCT method was used to calculate relative expression levels. The sequence of primers for amplification of different genes were: Human CTLA4 (Forward 5′-AGCCAGGTGACTGAAGTCTG-3′, Reverse 5′-CATAAATCTGGGTTCCGTTG-3′); Human IRF1 (Forward 5′-AGTGATCTGTACAACTTCCAGG-3′, Reverse 5′-CCTTCCTCATCCTCATCTGTTG-3′); Human GAPDH (Forward 5′-CTTTGTCAAGCTCATTTCCTGG -3′, Reverse 5′-TCTTCCTCTTGTGCTCTTGC -3′); Human NR4R3 (Forward 5′-AGTGTCTCAGTGTTGGAATGG-3′, Reverse: 5′-AGGAGAAGGTGGAGAGGG-3′); Human STAT1 (Forward 5′-TGAACTTACCCAGAATGCCC -3′, Reverse 5′-CAGACTCTCCGCAACTATAGTG -3′); Human PSMB9 (Forward 5′-GAGAGGACTTGTCTGCACATC-3′, Reverse 5′- GCATCCACATAACCATAGATAAAGG-3′); Human TAP1 (Forward 5′- AGAAGGTGGGAAAATGGTACC-3′, Reverse: 5′-GTTGGCAAAGCTTCGAACTG-3′); 18s rRNA (Forward 5′-CTTAGAGGGACAAGTGGCG-3′, Reverse 5′-ACGCTGAGCCAGTCAGTGTA-3′).
Immunofluorescence staining
Human melanocytes and melanoma cells were fixed with 4% PFA for 10 min and permeabilized with 0.25% Triton X-100 in PBS, then incubated with a blocking buffer (1% BSA, 22.52 mg/ml glycine in PBST) for 30 min at room temperature, which was followed by overnight incubation with mouse anti-human-CTLA4 (1:100, BNI3, BD Biosciences) antibody at 4°C. The control groups were only incubated with antibody dilution buffer (1% BSA in PBST). After washing, the fixed cells on cover slips were incubated with Alexa Fluor 488–conjugated goat anti-mouse secondary antibody (1:400, Life Technology) for 1h at room temperature. The cover slips were mounted with VECTASHIELD mounting medium with DAPI (Vector Laboratories Inc) overnight and imaged with Leica TCS SP8 Confocal microscope at the specified magnification.
Flow cytometry
For intracellular staining, human melanocytes and melanoma cells (1×106) were fixed and permeabilized by BD Fixation/Permeabilization Solution Kit, followed by manufacture’s protocol. Cells were then stained with a mouse anti-human CTLA4-PE (BD Biosciences, cat#555853) or isotype control anti-mouse IgG-PE (BD Biosciences, cat#554648) for 30 min at 4°C. Cells were then washed and analyzed on a BD FACSCalibur. The data were analyzed using FlowJo software.
Ruxolitinib treatment
HEMn-MP cells were seeded into 60mm dishes at 5×105 per dish. After one day, these cells were pretreated with Ruxolitinib (5μM, Selleckchem) for 4h, then cultured in the presence or absence of rIFNG for indicated time periods. The cells were harvested to assess CTLA4 and IRF1 mRNA expression by qRT-PCR. Hs 936.T, A2058, and WM983(B) cells were seeded into 60mm dishes at 5×105 per dish. After one day, these cells were treated with Ruxolitinib (5μM) for 1 day and harvested to assess CTLA4, IRF1, STAT1, TAP1 and PSMB9 expression by qRT-PCR.
Western blotting
Human melanocytes and melanoma cells were lysed in Pierce RIPA buffer (Thermo Scientific) containing 1× Halt protease inhibitor cocktail (100×, Thermo Scientific) and 1× Halt phosphatase inhibitor cocktail (100×, Thermo Scientific) and the protein concentration was measured with the Bio-Rad Protein Assay following manufacturer’s protocol. The same amounts of protein extracts were subjected to polyacrylamide gel electrophoresis using the 4%–20% Mini-Protean TGX gel system (Bio-Rad), transferred to PVDF (0.45 um pore size, Millipore) membranes, and immunoblotted using antibodies that specifically recognize STAT1 (1:1000, Cell Signaling Technology), pSTAT1 (Y701, 58D6, 1:1000, Cell Signaling Technology), pSTAT1 (Y727, D3B7, 1:2000, Cell Signaling Technology), STAT3 (124H6, 1:1000, Cell Signaling Technology), pSTAT3 (Y705, D3A7, 1:2000, Cell Signaling Technology), GAPDH-HRP (D16H11, 1:1000, Cell Signaling Technology), IRF1 (D5E4, 1:1000, Cell Signaling Technology), Histone 3 (#ab1791, 1:3000, Abcam), Histone 4 (#39269, 1:1000, Active Motif), pan-acetyl-Histone 3 (#06-599, 1:10,000, Millipore), pan-acetyl-Histone 4 (#06-866, 1:5000, Millipore). The secondary antibodies used for detection were HRP-conjugated goat anti-mouse and goat anti-rabbit IgG (1:5000, Thermo Scientific). The blots were incubated with Luminata Western HRP substrate (Millipore) for 5 min. Band intensities of Tiff images were quantified by using Image J software.
siRNA-mediated knockdown
UACC1273 melanoma cells were transfected with indicated siRNAs (10pM) or scramble (Scr) siRNA (10pM) with Lipofectamine RNAiMAX Reagent (Invitrogen) according to the manufacturer’s protocol. Transfection efficiency was assessed by measuring the amounts of the proteins of interest. The siRNAs are listed below: Scramble siRNA (Scr, Silencer® Select Negative Control No. 1 siRNA, Catalog number: 4390843, Ambion); STAT1 siRNA S1-1, sense, 5′-CGGUUGAACCCUACACGAATT-3′, ID:s278, Ambion; STAT1 siRNA S1-2, sense, 5′-CCUACGAACAUGACCCUAUTT -3′, ID:s277, Ambion; STAT3 siRNA S3-1, sense, 5′- GCACCUUCCUGCUAAGAUUtt-3′, ID:s745, Ambion; STAT3 siRNA S3-2, sense, 5′-GCCUCAAGAUUGACCUAGATT -3′, ID:s743, Ambion; IRF1 siRNA IRF1-1, sense, 5′-GCAGAUUAAUUCCAACCAATT -3′, ID:s7502, Ambion; IRF1 siRNA IRF1-2, sense, 5′- CCUCUGAAGCUACAACAGATT-3′, ID:s7501, Ambion. All siRNAs were purchased from Thermo Scientific.
Plasmid construction
The 1021-base pair (bp) segment of CTLA4 promoter luciferase construct containing four putative GAS sites was synthesized and cloned into a firefly luciferase vector (pGL4.20 [luc2/Puro], Promega) by Genescript gene synthesis and cloning service. Site-directed mutagenesis was performed by QuikChange II site-directed Mutagenesis Kit (Agilent Technologies) and generated mutants with deletion of each putative GAS sites by manufacture’s protocol. Human CTLA4 ORF cDNA vector was purchased from Origene.
Luciferase reporter assays
HEMn-MP cells (1×106) were transiently transfected with different CTLA4 promoter firefly luciferase reporter constructs with Nucleofector Kits for Human Melanocytes (NHEM-neo) following manufacturer’s protocol. The cells were also co-transfected with a Renilla luciferase vector (pGL4.74 [hRluc/TK], Promega) using Nucleofector I device. Two days after transfection, the cells were treated with IFNG, or mock-treated, for 7 days, then were harvested and analyzed. The luciferase activity of each samples was measured with the Dual-Luciferase Reporter Assay System (Promega) with a Promega Glomax detection system according to the manufacturer’s protocol.
Chromatin Immunoprecipitation (ChIP) assays
Chromatin immunoprecipitation (ChIP) was performed with the modified protocol provided by Upstate Biotechnology with minor modifications (11). Briefly, HEMn-MP cells were cultured in the presence or absence of 10 ng/ml rIFNG for 7 days, then fixed in 1% formaldehyde for 15 min, and neutralized by addition of 0.125M glycine (pH 7.0) for 5 min. Cells were lysed in 100 μL SDS lysis buffer containing proteinase and phosphatase inhibitors per 1×106 cells and then sonicated using a sonic dismembrator (Fisher Scientific) to shear chromatin into 200–300bp DNA fragments. The chromatin fragments were subjected to immunoprecipitation with the following antibodies: Rabbit IgG (5ug, Millipore), STAT1 (1:50, Cell Signaling Technology), pSTAT1 (Y701, 58D6, 1:100, Cell Signaling Technology), Histone 3 (#ab1791, 5ug, Abcam), Histone 4 (62-141-13, 5ug, Millipore), pan-acetylated-Histone 3 (5ug, Millipore), pan-acetylated-Histone 4 (5ug, Millipore), CBP (D6C5, 1:25, Cell Signaling Technology), and RNA polymerase II (8WG16, 5ug, Millipore). The coimmunoprecipitated DNA was purified with GeneJET PCR Purification Kit (Thermo Scientific). Occupancy of the promoter and other regulatory regions was measured by qRT-PCR with the following primer sets by using 1/100 of the ChIP DNA. The base pair location is in reference to transcription start site (TSS) of CTLA4: R1 (−885bp to −736bp) Forward 5′-ATTCAATCCTAAGTGCACAGAATTC-3′, Reverse 5′-TGTAGACAGGACCAATGATCTAAC-3′; R2 (−576bp to −480bp) Forward 5′-TTGTCTCTGTTGAGTTAAGGC-3′, Reverse 5′- CACAAGAAATAAACTGAAAATAGGCG-3′; R3 (−319bp to −240bp) Forward 5′-GCTCAGAAAGTTAGCAGCCTAGTAG-3′, Reverse 5′-CAATCTTCTGGGCATCCTTAACC-3′; R4 (+530bp to +621bp) Forward 5′-CAGGCAATTTCAGACCCTTCTATG-3′, Reverse 5′-CCTGAAACCCAGCTCAAATG-3′.
CBP inhibitor treatment
HEMn-DP and UACC1273 were seeded into 60mm dishes at 5×105 cells per dish. After one day, these cells were pretreated with SGC-CBP30 (5μM or 10μM, Selleckchem) or PF-CBP1 (10μM or 20μM, Selleckchem) for 4h, then cultured in the presence or absence of rIFNG for indicated time periods. The cells were harvested to assess CTLA4 and IRF1 mRNA expression by qRT-PCR analysis, and p-STAT1 (Y701), STAT1, IRF1 and GAPDH protein expression by western blot.
MAPK pathway inhibitor treatment
A2058, WM983(B), UACC1273, Hs 936.T, SK-MEL-2, WM3912 and WM3918 cells were seeded into 60mm dishes at 5×105 cells per dish. After one day, these cells were treated with either BRAFV600E inhibitor Vemurafenib (10 μM, Selleckchem) or MEK inhibitor PD0325901 (5 μM, Selleckchem) for 1d and then harvested to assess CTLA4 mRNA expression by qRT-PCR analysis.
CCLE Analysis
The Cancer Cell Line Encyclopedia (CCLE) mRNA data were downloaded from the CCLE website (http://www.broadinstitute.org/ccle) from the file CTLA4_file6805.gct. The figure was generated by GraphPad Prism. RMA gene expression values of cell lines were used to generate the figure.
RNA-seq data of ipilimumab-treated melanoma patients
Ipilimumab-treated melanoma patients were described previously (12,13). RNA-seq data on tumors collected pre- and post-treatment were downloaded from dbGaP (phs001038). Cluster analysis is performed on anti-CTLA4 treated melanoma patients using Heat Mapper. Average linkage clustering method and Euclidean distance measurement method are applied in the hierarchical clustering (14).
Statistical analysis
All experiments were performed in triplicate and data are presented as mean±SEM, and graphs were prepared with GraphPad Prism. To analyze statistical difference between two groups, two-tailed unpaired Student’s t-test was used. Comparisons involving multiple groups were assessed by one-way ANOVA with post-hoc Tukey analysis. For survival analysis, Kaplan-Meier plots and log-rank tests were performed to determine the significance of differences in cumulative survival. For gene expression correlation analyses, the Pearson correlation coefficient was used. P value <0.05 was considered as statistically significant.
RESULTS
CTLA4 is overexpressed in human melanoma cells
We had previously found CTLA4 to be highly expressed in mouse melanoma allograft tumors with distinct expression in melanoma cells (10). We therefore analyzed the Broad-Novartis Cancer Cell Line Encyclopedia (CCLE) database for CTLA4 expression in its large panel of approximately 1000 cell lines of a variety of human cancer types, including 61 melanoma cell lines (15,16). Human melanoma cell lines exhibited the greatest mean CTLA4 expression among all the cancer cell types (Fig. 1A). In fact, this mean expression was even greater than that of 180 of the hematologic and lymphoid cancer cell lines, with the difference being statistically significant (Fig. 1A). This prompted us to validate the mRNA expression of CTLA4 in human primary melanocytes and melanoma cells. Three different primary human epidermal melanocyte (HEMn) cell populations, but not human epidermal keratinocytes (HEKn), exhibited low but readily detectable expression of CTLA4 tested by quantitative RT-PCR (qRT-PCR) (Fig. 1B). On the other hand, all of the 13 human melanoma cell lines expressed CTLA4 at ~30-10,000-fold greater levels than HEMn cells (Fig. 1B). We also tested 15 human non-melanoma cancer cell lines by qRT-PCR. Only U2OS and MG63 osteosarcoma cell lines, and MDA-MB-231 breast adenocarcinoma showed low but detectable levels of CTLA4 expression that were at par with the levels seen in primary melanocytes (Supplementary Fig. S1A,B).
Figure 1.
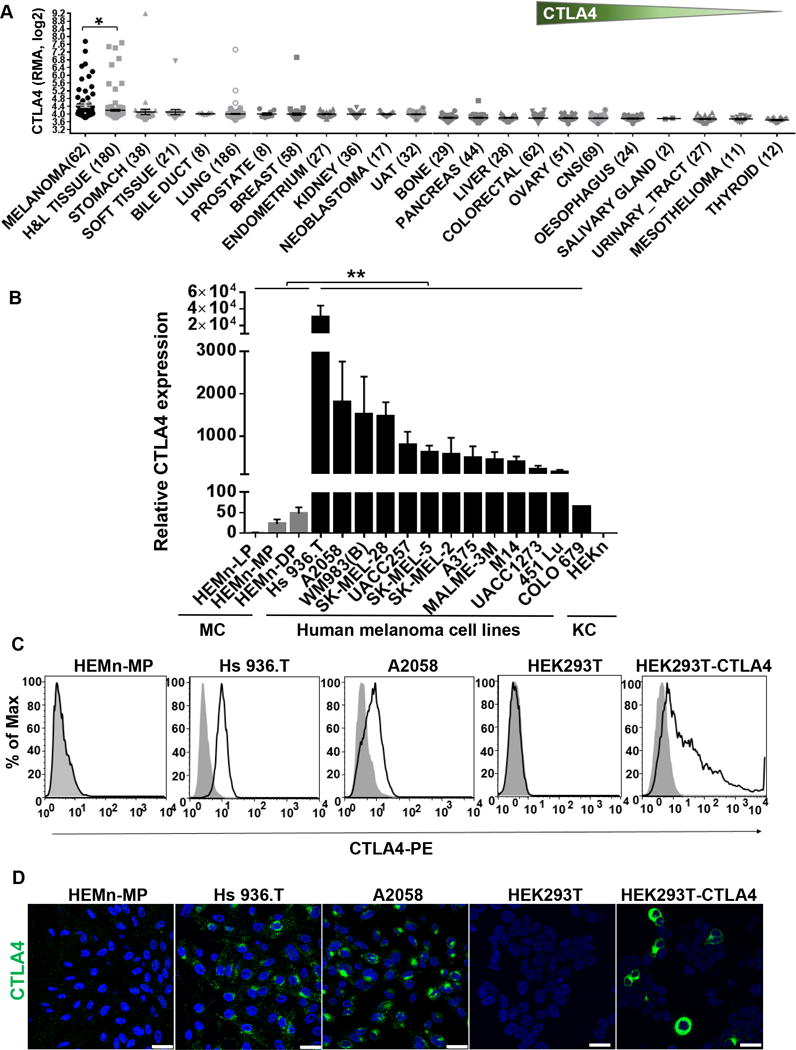
CTLA4 is highly overexpressed in human melanoma cells. A, CTLA4 mRNA expression pattern across different types of cancer cell lines in the Cancer Cell Line Encyclopedia (CCLE) database. Sample numbers (n) are indicated in parentheses. H&L, hematopoietic and lymphoid tumor cell lines; UAT, upper aerodigestive tract tumor cell lines; CNS, central nervous system tumor cell lines. Each dot represents one tumor cell line. *p<0.05. Unpaired t-test with Welch’s correction. Y axis, RMA (Robust Multi-Array Average) was used to normalize the expression data and then converted to log2. B, CTLA4 mRNA expression in human primary melanocytes (MC), human melanoma cells, and human primary keratinocyte (KC) was determined by qRT-PCR, and plotted relative to HEMn-LP melanocytes. Data presented as mean±SEM of three to six independent experiments. C, Total CTLA4 protein expression in fixed and permeabilized human melanocytes, melanoma cell lines, HEK293T and CTLA4-overexpressing HEK293T cells stained with either mouse anti-human CTLA4-PE (BNI3) or isotype-PE control antibody, were analyzed by flow cytometry. D, Confocal photomicrographs of CTLA4 immunostaining (green) in melanocytes, melanoma cell lines, HEK293T and CTLA4-overexpressed HEK293T cells. Blue, DAPI. Images are representative of 3–5 independent experiments. Scale bar=25μm.
Flow cytometric analysis failed to detect cell surface expression of CTLA4 in HEMn and melanoma cell lines. This is in accord with previously published expression pattern of CTLA4 seen in T cells, where it is also rarely expressed on the membrane and gets rapidly internalized into cytoplasm via clathrin-mediated endocytosis (17). Intracellular immunostaining readily detected CTLA4 protein expression in melanoma cells, but the expression in HEMn cells was barely detectable by flow cytometry (Fig. 1C). On the other hand, immunofluorescence staining of HEMn cells showed very low levels of diffuse punctate localization throughout cytoplasm, but not on the cell surface, which is consistent with endosomal/lysosomal vesicular localization within cytoplasm (Fig. 1D). Human melanoma cell lines showed much greater cytoplasmic punctate localization, but a large majority of the overexpressed CTLA4 protein was observed to be localized in perinuclear areas, presumably in the trans-Golgi network (Fig. 1D), as previously reported in T cells (18–20). As a positive control for the specificity of anti-CTLA4 antibody, we ectopically expressed human CTLA4 in HEK293T cells, which do not express endogenous CTLA4. The mouse anti-human-CTLA4 antibody (clone BNI3) readily and specifically detected CTLA4 in the ectopic CTLA4-expressing cells by both flow analysis of intracellular immunostaining and immunofluorescence, but not in the untransfected control HEK293T cells (Fig. 1C,D).
IFNG activates expression of CTLA4 in melanocytes and melanoma cells
Since we had identified CTLA4 to be the highest upregulated gene in the context of UVB-mediated IFNG-induced gene expression signature in melanocytes (10), we postulated that CTLA4 may be regulated by the IFNG signaling pathway in melanocytes and melanoma cells. Indeed, we found that treatment of HEMn cells with relatively low concentrations of recombinant IFNG (1–10 ng/ml; 1.3–13.3 U/ml) robustly activated the expression of CTLA4, with a plateau of response reached between 10 ng/ml and 100 ng/ml concentrations (Fig. 2A,B). This IFNG-mediated induction of expression was statistically significant within 30 min, and continually increased in extent to approximately 200-fold by 3 wk timepoint (Fig. 2A). IFNG treatment also increased CTLA4 in human melanoma cells at both mRNA and protein levels, albeit to a lesser degree (<4-fold change) than in HEMn, presumably due to the already relatively high baseline expression in these cells (Fig. 2C). Flow cytometric analysis also showed robust and statistically significant induction of intracellular CTLA4 protein expression by IFNG in melanocytes and melanoma cells (Fig. 2D,E). However, the IFNG-mediated induction of CTLA4 expression was restricted to the melanocytes and melanoma cells, as neither the other primary or normal cells, i.e. keratinocytes, human embryonic kidney cells, and fibroblasts, nor several non-melanoma solid tumor cell lines expressed CTLA4 before or after IFNG treatment (Fig. 2F). Immunofluorescence analysis readily showed upregulation of CTLA4 in the HEMn-MP melanocytes and UACC1273 melanoma cells (Fig. 2G). Once again, the CTLA4 expression was absent from the cell membrane, but was diffusely spread throughout cytoplasm in the distinct punctate pattern, with dominant trans-Golgi network localization (Fig. 2G).
Figure 2.
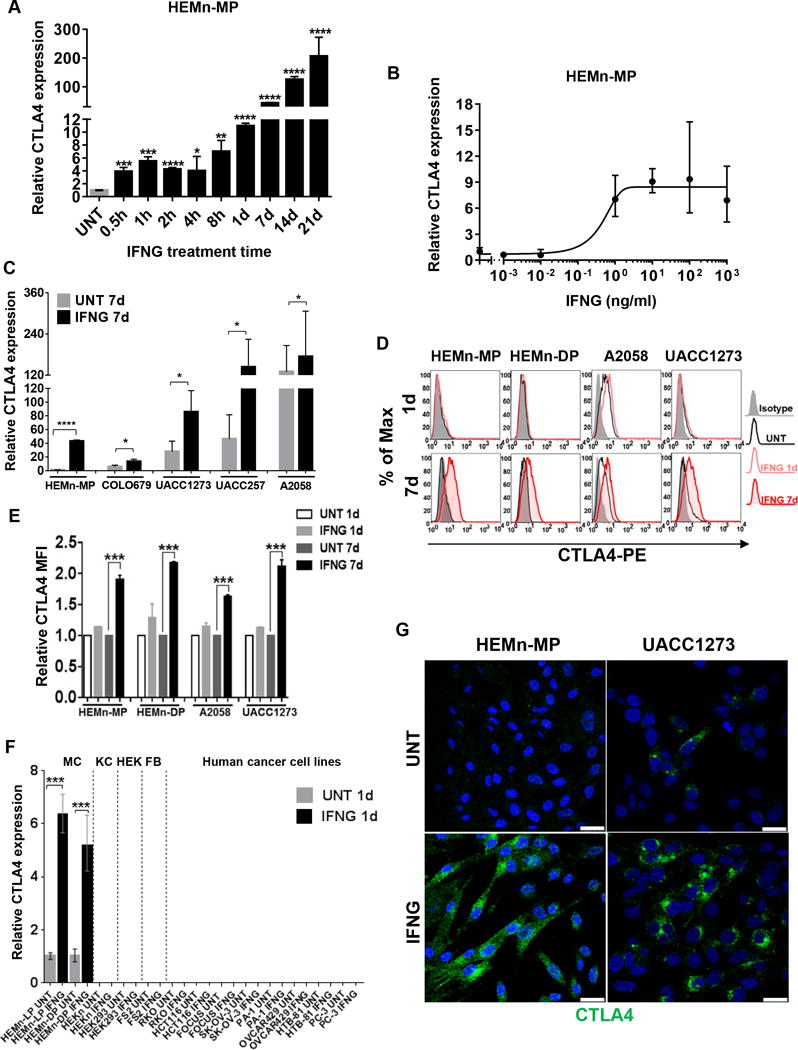
IFNG induces CTLA4 expression in human primary melanocytes and melanoma cell lines. A, qRT-PCR analysis of CTLA4 mRNA expression in HEMn-MP cells cultured in the presence or absence of 10 ng/ml IFNG is shown at indicated time points. B, qRT-PCR analysis of CTLA4 mRNA expression in HEMn-MP cells cultured in the presence or absence of indicated concentrations of IFNG for 1d. C, qRT-PCR analysis of CTLA4 mRNA expression in cells that were cultured for 7 days in the presence or absence of 10 ng/ml IFNG. D, Total CTLA4 protein expression in fixed and permeabilized human melanocytes and melanoma cell lines cultured in the presence or absence of 10 ng/ml IFNG for either 1d or 7d, immunostained with either mouse anti-human CTLA4-PE (BNI3) or isotype-PE control antibody, and analyzed by flow cytometry. Data are representative of at least three independent experiments. E, Histogram representing the average MFI±SEM of three independent experiments. Y axis, fold-change of CTLA4 MFI is compared to the untreated (UNT) group. F, qRT-PCR analysis of CTLA4 expression in human primary neonatal melanocytes (MC); human epidermal neonatal keratinocytes (KC); human embryonic kidney cells (HEK); human fibroblast cell line (FB); and the indicated human solid cancer cell lines. RKO/HCT116, human colon carcinoma cell lines; FOCUS, human hepatocellular carcinoma; SK-OV-3/OVCAR429, human ovarian adenocarcinoma cell lines; PA-1, human ovarian teratocarcinoma cell line; HTB-81/PC-3, human prostate carcinoma cell lines. G, Confocal photomicrographs of CTLA4 immunostaining (green) in HEMn-MP and UACC1273 cells that were cultured in presence or absence of 10 ng/ml recombinant IFNG for 7d. Blue, DAPI. Images are representative of three independent experiments. Scale bar=25μm. All graphed data are presented as mean±SEM of three biological replicates. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
IFNG induces CTLA4 expression via STAT1-mediated canonical signaling
The canonical IFNG cytokine signaling downstream of the IFNG receptors is mediated by activation of Janus Kinases 1 and 2 (JAK1/2), which phosphorylate the Signal Transducer and Activator of Transcription 1 (STAT1) transcription factor prompting its homodimerization (21). The phosphorylated STAT1 (pSTAT1) homodimers then shuttle to the nucleus and activate transcription of genes whose promoters harbor the IFN-gamma-activated sequence (GAS) DNA-binding motif(s) (21). One of the classical primary response genes activated by IFNG signaling is the one that encodes the transcription factor Interferon Regulatory Factor 1 (IRF1), which subsequently transcribes numerous secondary IFNG-response genes (22). Additionally, IFNG signaling also results in phosphorylation of transcription factor STAT3 in a cell type-specific and context-dependent manner (23). We sought to delineate the role of the canonical IFNG signaling and the specific downstream transcription factor(s) responsible for the activation of CTLA4 expression in melanocytes and melanoma cells.
We started by testing the ability of JAK1/2 inhibitor drug ruxolitinib (24) to block IFNG-induced CTLA4 expression in melanocytes. Indeed, ruxolitinib (5uM) completely inhibited activation of CTLA4 in response to IFNG treatment of HEMn-MP melanocytes (Fig. 3A). The induction of IRF1 expression by IFNG treatment was also completely abolished, indicating blockade of the IFNG signaling pathway (Fig. 3B).
Figure 3.
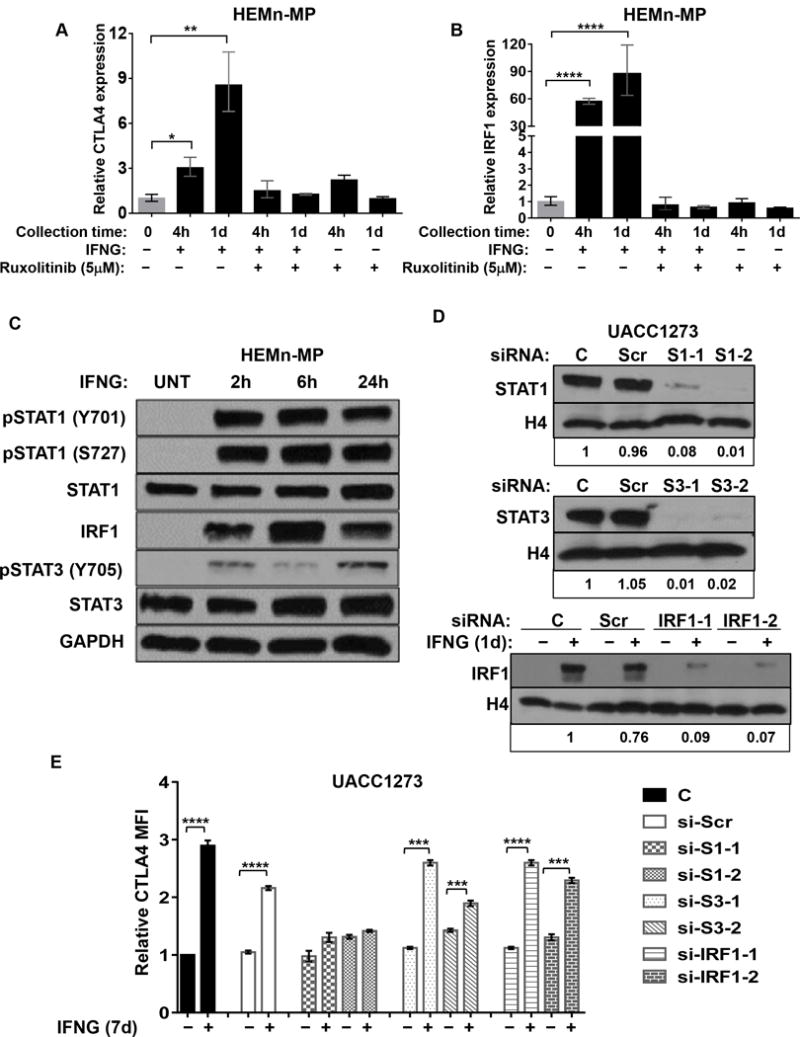
IFNG-induced CTLA4 expression is mediated by JAK-STAT1 pathway. A and B, Ruxolitinib treatment blocks IFNG-induced CTLA4 and IRF1 expression. HEMn-MP cells were pretreated with Ruxolitinib (5uM) for 4h before IFNG treatment. qRT-PCR analysis of CTLA4 (A) and IRF1 (B) expression in HEMn-MP cells after indicated treatments. Data are presented as mean±SEM of three biological replicates. *P<0.05; **P<0.01 compared to both IFNG and Ruxolitinib-untreated groups. C, Western blot analysis of activation of pSTAT1, pSTAT3, and IRF1 by IFNG treatment in human melanocyte. HEMn-MP cells were cultured in the presence or absence of 10 ng/ml IFNG for indicated time intervals, then pSTAT1 (Y701), pSTAT1 (Y727), total STAT1, IRF1, pSTAT3 (Y705), total STAT3 and GAPDH expression by western blot. D, siRNA-mediated knockdown of STAT1, STAT3, and IRF1 in UACC1273 melanoma cell line with either scrambled siRNA (Scr), si-STAT1 (S1-1, S1-2, two different siRNA-mediated KD), si-STAT3 (S3-1, S3-2, two different siRNAs), or si-IRF1 (IRF1-1 and IRF1-2, two different siRNAs) for 2d, then assessed for STAT1, STAT3, or IRF1 protein levels by western blotting. Histone 4 (H4) protein was used as loading control. Densitometry-based calculations of fold changes, normalized to control group, are shown at the bottom of the blot images. Band intensities of Tiff images were quantified by Image J. Immunoblotting images are representative of 2 independent experiments. E, UACC1273 cells were transfected with indicated siRNA for 2d, then cultured in the presence or absence of 10 ng/ml IFNG treated for 7d before measuring CTLA4 protein expression by flow cytometry. Y axis, fold change of CTLA4 mean MFI compared to no-siRNA control (C) cells without IFNG treatment. ***P<0.001; ****P<0.0001. The histogram represents MFI±SEM of three biological replicates.
IFNG treatment of HEMn cells robustly increased phosphorylation of STAT1 at both tyrosine 701 (pSTAT1-Y701) and serine 727 (pSTAT1-S727), both of which are important for pSTAT1-mediated transcriptional activation. These phosphorylation events were detectable immediately and sustained for as long as the cells were kept exposed to IFNG treatment (Fig. 3C). Since S727 phosphorylation requires nuclear translocation and chromatin-binding of pSTAT1-Y701 (25), these results indicate robust IFNG-induced STAT1 activation response in melanocytes. Interestingly, STAT3 also exhibited sustained phosphorylation (pSTAT3-Y705) in response to IFNG in HEMn-MP cells (Fig. 3C). Strong activation of IRF1 expression was also confirmed (Fig. 3C).
In order to determine which of the transcription factors (STAT1, STAT3, or IRF1) is primarily responsible for IFNG-induced CTLA4 transactivation, we generated siRNA-mediated knockdowns (KD) of STAT1, STAT3, and IRF1 in the UACC1273 melanoma cell line. Two siRNAs were used for each of the three knockdowns, with >90% reduction in expression, along with parental cells (C) and scrambled (Scr) siRNA-transfected cells as controls (Fig. 3D). All of the KD cells were treated with IFNG for 7 days, and then analyzed for intracellular CTLA4 expression by flow cytometry. While the UACC1273 cells with STAT3-KD and IRF1-KD exhibited significant increase in CTLA4 protein expression in response to IFNG treatment, it was completely inhibited in the STAT1-KD cells (Fig. 3E). These results clearly indicate that STAT1 is the principal transcription factor responsible for mediating the IFNG-induced CTLA4 expression in melanocytes and melanoma cells, and that CTLA4 is one of the primary response genes downstream of IFNG signaling pathway.
CTLA4 promoter is transcriptionally active in melanocytes and melanoma cells
To determine the chromatin characteristics of the CTLA4 gene promoter, we inspected the cell type-specific DNAse I hypersensitivity of this DNA region through the ENCODE Project (National Human Genome Research Institute, National Institutes of Health) (26). As expected, T cells, CD4+ helper T cells, and CD8+ αβ T cells showed DNAse I hypersensitivity peaks in the CTLA4 promoter region (Fig. 4A, solid box). Interestingly, human foreskin epidermal melanocytes and melanoma cell lines (SK-MEL-5, MEL-2183, and COLO829) exhibited DNAse I hypersensitivity peaks of similar intensity. In contrast, RPMI8226 myeloma cell line, human foreskin keratinocytes and fibroblasts, mammary epithelial cells, PC-3 prostate cancer cell line, MG63 osteosarcoma cell line, and RKO colon carcinoma cell line did not show DNAse I hypersensitivity in this region (Fig. 4A, dotted box). These results are consistent with a transcriptionally active open chromatin (euchromatin) configuration in the CTLA4-expressing T cells and melanocyte/melanoma cells, but a heterochromatic (repressive) chromatin in the CTLA4 promoter regions of CTLA4-non-expresser cell types. Concordantly, ENCODE RNA-seq analysis of human foreskin melanocytes showed a good correlation of CTLA4 transcriptional profile with that of the DNAse I hypersensitivity in the CTLA4 promoter region (Fig. 4B, solid box).
Figure 4.
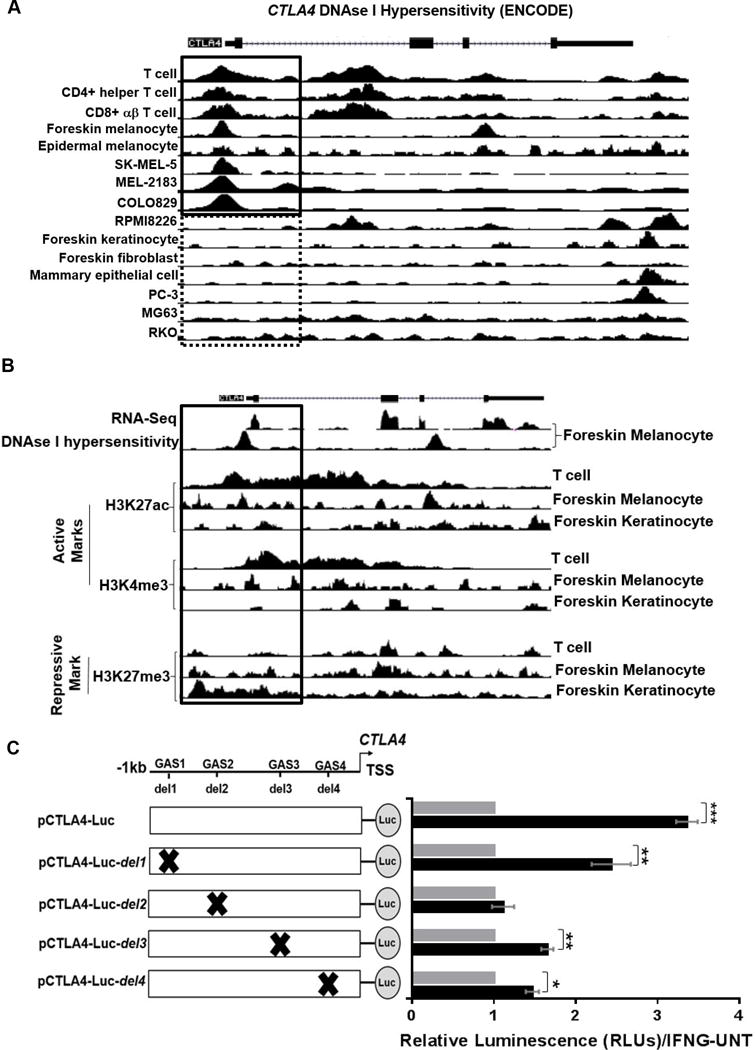
Promoter analysis of human CTLA4. A, UCSC genome browser view shows the human CTLA4 locus (GRCh37/hg19). DNase I hypersensitivity by DNase-seq analysis for human T cells, CD4+ helper T cell, CD8+ αβ T cells, foreskin melanocytes, epidermal melanocytes, SK-MEL-5, MEL-2183, COLO829, RPMI8226, foreskin keratinocytes, foreskin fibroblasts, mammary epithelial cells, PC-3, MG63 and RKO were generated by ENCODE Project. Boxes indicate the CTLA4 promoter area. B, UCSC genome browser view shows RNA-seq of foreskin melanocytes plus strand signal, DNase-seq signal of foreskin melanocytes and the ChIP-seq fold change signals over control for active promoter marks (H3K27ac, H3K4me3) and repressive mark H3K27me3 in the human T cells, foreskin melanocyte and foreskin keratinocyte and the promoter region around CTLA4 (GRCh37/hg19). C, Left: diagrams of firefly luciferase constructs containing putative GAS sites in −1021 bp upstream of TSS. pCTLA4-Luc contains all four putative GAS sites (−1021bp-TSS), whereas pCTLA4-Luc-del constructs contain single GAS deletions (e.g. del1 for GAS1 deletion) by site-directed mutagenesis. Right: dual luciferase assays were performed in HEMn-MP cells co-transfected with indicated firefly luciferase and Renilla luciferase constructs for 2d, then cultured in the presence or absence of IFNG for 7d. Renilla luciferase activity was used as transfection control. Data are shown as the fold-change of luciferase activity in cells transfected with indicated constructs and treated with IFNG for 7d to that of untreated transfected cells. Data represent mean±SEM of three biological replicates. *P < 0.05; **P< 0.01; ***P< 0.001.
Histone modifications play a dominant role in the regulation of chromatin architecture, with specific modifications closely associated with transcriptionally active or inactive chromatin. In this context, two of the most common histone modifications linked with active chromatin within 1 kilobases (−1 kb) of the promoter transcription start sites (TSS) are histone 3 lysine 4 trimethylation (H3K4me3) and histone 3 lysine 27 acetylation (H3K27ac) (27). The ENCODE ChIP-seq data (28) showed high levels of H3K4me3 and H3K27Ac enrichment in the CTLA4 promoter region in both human T cells and foreskin melanocytes, but not in foreskin keratinocytes (Fig. 4B). In contrast, the most common repressive histone mark associated with inactive chromatin, H3K27me3 (27), showed no or low enrichment in the CTLA4 promoter region in T cells and melanocytes, but high enrichment in keratinocytes (Fig. 4B). Altogether, the results obtained from the ENCODE analysis of chromatin state at the CTLA4 promoter locus in different cell types are in congruence with their respective CTLA4 expression patterns, and clearly indicate that CTLA4 promoter harbors a transcriptionally active chromatin configuration in melanocytes and melanoma cells.
We next inspected the human CTLA4 gene sequence upstream and downstream the TSS and found four putative GAS motifs (Supplementary Fig. S2A–C) (29). To determine the validity of these putative GAS sites and their contribution to IFNG-induced activation of CTLA4 expression, we generated reporter constructs with CTLA4 promoter (−1kb to TSS) driving firefly luciferase (pCTLA4-Luc), with four different variants that individually harbored deletions of the four putative GAS motifs (pCTLA4-Luc-del1-4) (Fig. 4C). These five constructs were transiently transfected in HEMn-MP cells, along with the control Renilla luciferase construct, and their luciferase activities were tested following 7d of IFNG treatment. The construct with wildtype CTLA4 promoter (pCTLA4-Luc) showed robust and significant luciferase activity in response to IFNG treatment (Fig. 4C). The constructs with the deletion of the most distal putative GAS site (pCTLA4-Luc-del1), and two proximal GAS deletions (pCTLA4-Luc-del3 and -del4) activated intermediate but significant levels of luciferase activity. In contrast, deletion of the second putative GAS site (GAS2 at approximately −850 bp from TSS) (pCTLA4-Luc-del2) completely abolished induction of luciferase in response to IFNG treatment, clearly identifying this particular site as the principal IFNG-responsive GAS motif in the CTLA4 promoter region (Fig. 4C).
IFNG signaling recruits the transcriptional machinery to the CTLA4 promoter
We next sought to characterize the recruitment of the IFNG-induced transcriptional machinery onto the CTLA4 promoter region in melanocytes. Guided by the results of the DNAse I hypersensitivity analysis and the promoter analysis above, we designed primer sets to test, by ChIP-qPCR, recruitment of pSTAT1 to three different regions of the CTLA4 promoter and a region downstream of the TSS (Regions R1-4) (Fig. 5A and Supplementary Fig. S2B,C). These regions were strategically selected in regard to the localization of DNAse I hypersensitivity and modified histone markers, expected fragmentation size of sonicated DNA, and optimum detection of pSTAT1 recruitment to the CTLA4 TSS (Supplementary Fig. S3). ChIP-qPCR assays with pSTAT1- and total STAT1-specific antibodies confirmed robust recruitment of pSTAT1 to CTLA4 promoter in an IFNG-dependent manner (Fig. 5B and Supplementary Fig. S4A).
Figure 5.
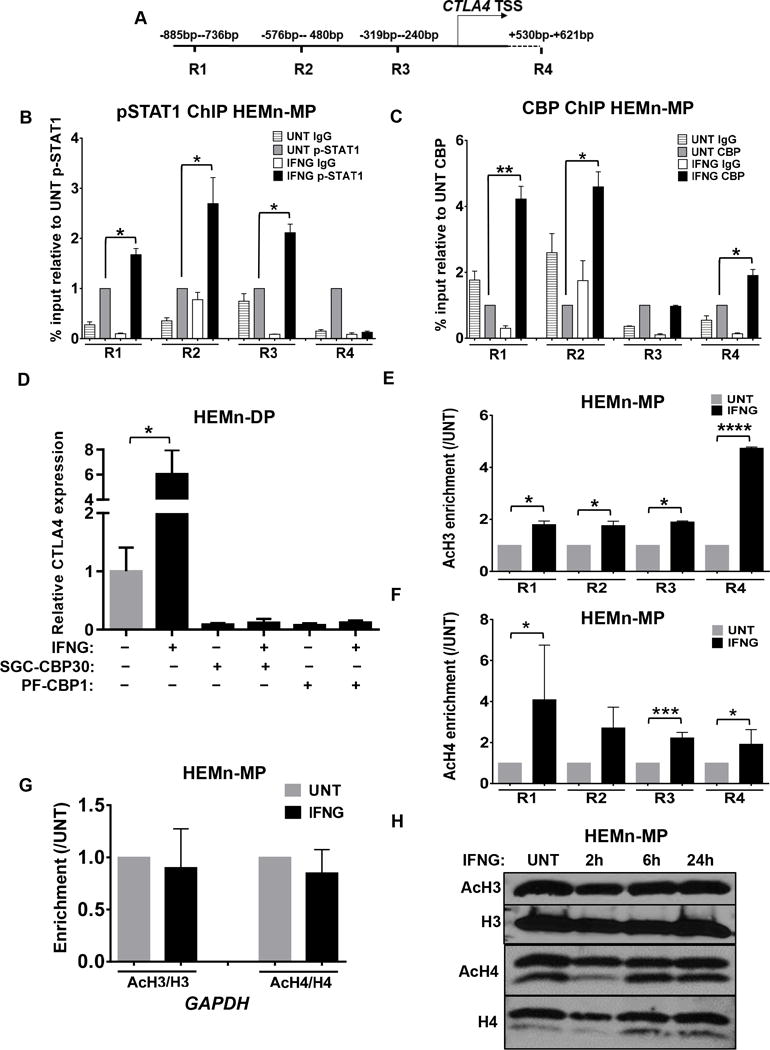
IFNG/pSTAT1-mediated recruitment of CBP/p300 to the CTLA4 promoter and accompanied acetylation of histone 3 (H3) and histone 4 (H4). A, Schematic representation of primer sets used to detect the CTLA4 promoter regions (R1–R4). HEMn-MP cells were cultured in the presence or absence of IFNG for 7d before being subjected to chromatin immunoprecipitation (ChIP) and qRT-PCR assay with B, anti-pSTAT1 (Y705), and C, anti-CBP antibodies to measure recruitment to the CTLA4 promoter. Sonicated nuclear extracts before treatment with antibody were used as input. Relative abundance was calculated as % input and compared to the IFNG-untreated (UNT) cells. D, HEMn-DP cells were pretreated with CBP inhibitors SGC-CBP30 (10μM) and PF-CBP1 (20μM) for 4h before IFNG treatment for 1d. qRT-PCR analysis of CTLA4 mRNA expression in HEMn-DP cells after indicated treatments is shown with the data presented as mean±SEM of three biological replicates. E and F, Enrichment of AcH3 and AcH4 around CTLA4 promoter. G, Enrichments of AcH3 and AcH4 at the promoter of GAPDH. The enrichment of acetylated histones was normalized to total histone and compared to the IFNG-UNT cells. H, The effect of IFNG on global levels of AcH3 and AcH4. HEMn-MP cells were cultured in the presence or absence of IFNG for indicated time, then the global AcH3 and AcH4 levels were detected by western blotting. Immunoblotting images are representative of 3 independent experiments. *P < 0.05; **P < 0.01; ***P<0.001.
Histone acetylation mediated by histone acetyltransferase CREB-binding protein (CBP)/p300 has been shown to be necessary for transcriptional activation by pSTAT1 (30). Therefore, we assessed the recruitment of CBP/p300 to CTLA4 promoter by ChIP-qPCR analysis, and found that indeed CBP/p300 was recruited there in an IFNG-dependent fashion (Fig. 5C). This was accompanied by IFNG-dependent recruitment of RNA polymerase II (POL II) to the promoter, as shown by ChIP-qPCR assay with anti-POL II antibody (Supplementary Fig. S4B).
Pretreatment of melanocytes with two different CBP/p300 inhibitors (SGC-CBP30 and PF-CBP1) completely abolished IFNG-induced CTLA4 expression in HEMn-DP cells (Fig. 5D). Both inhibitor drugs also blocked expression of another CBP/p300-dependent gene, NR4A3, in melanocytes (Supplementary Fig. S5A). However, they did not affect expression of the housekeeping gene GAPDH, which is not dependent on CBP/p300, and IFNG-induced STAT1 phosphorylation or IRF1 expression, indicating lack of cellular toxicity (Supplementary Fig. S5B,C).
IFNG signaling induces histone acetylation at CTLA4 promoter
IFNG-mediated recruitment of pSTAT1/CBP complex to the CTLA4 promoter and consequent transcriptional activation in melanocytes was accompanied by enrichment of acetylation of both histones 3 and 4 (AcH3 and AcH4), as shown by ChIP-qPCR analysis with AcH3- and AcH4-specific antibodies (Fig. 5E,F). These results raised the question whether the IFNG-induced histone acetylation was regional or global. Histone acetylation at the GAPDH locus was found to be unaffected by treatment of melanocytes with IFNG, indicating that the effect is selective, regional, and perhaps promoter-specific (Fig. 5G). Indeed, global H3 and H4 acetylation were also unaffected by IFNG treatment of melanocytes, as determined by western blot analysis (Fig. 5H and Supplementary Fig. S6A,B).
Overexpression of CTLA4 in melanoma cell lines is regulated by MAPK pathway
While human melanoma cell lines overexpressed CTLA4, it was unclear whether this overexpression was a result of aberrant activation of the IFNG pathway. Inhibition of the IFNG pathway by ruxolitinib treatment did not affect CTLA4 expression in human melanoma cell lines with high baseline CTLA4 expression (Fig. 6A). The baseline expression of other known IFNG target genes also remained unaffected by ruxolitinib, indicating that these melanoma cell lines do not have an intrinsic upregulation of the IFNG pathway (Supplementary Fig. S7A–D).
Figure 6.
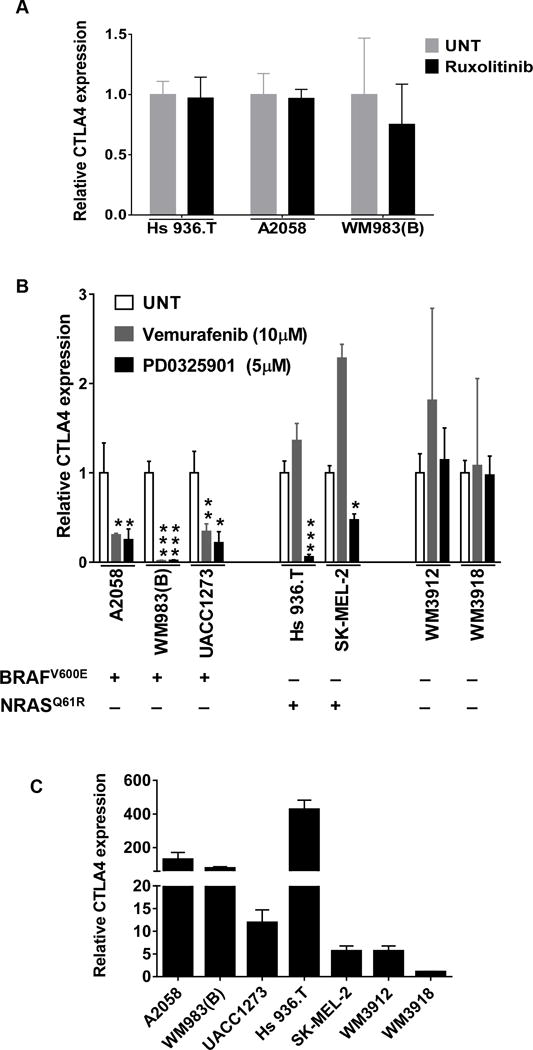
MAPK pathway regulates basal CTLA4 expression in human melanoma cells. A, Ruxolitinib treatment does not affect CTLA4 expression in human melanoma cell lines. The indicated cell lines were treated with Ruxolitinib (5uM) for 1d and qRT-PCR analysis of CTLA4 expression was performed. UNT, untreated. B, The indicated cells were treated with either BRAFV600E inhibitor vemurafenib or MEK inhibitor PD0325901 at indicated concentration for 1d and CTLA4 expression was measured by qRT-PCR. Data presented as mean±SEM of three biological replicates. *P<0.05; **P<0.01; ***P<0.001 over untreated group. The mutational status of each cell line for BRAFV600E and NRASQ61R is given below; +, mutation present; -, mutation absent. C, The basal expression of CTLA4 in human melanoma cell lines. Data presented as mean±SEM of three biological replicates for each.
We next asked whether the constitutive activation of the MAPK pathway, driven by oncogenic mutations of BRAF or NRAS, was responsible for the baseline overexpression of CTLA4 in human melanoma cell lines. We utilized the BRAFV600E-specific inhibitor drug vemurafenib (BRAFi) and the MEK inhibitor (MEKi) drug PD0325901 (31). While both vemurafenib and PD0325901 inhibited CTLA4 expression in the BRAFV600E mutant cell lines (but WT for NRAS), only PD0325901 inhibited CTLA4 expression in cell lines carrying mutant NRASQ61K (but either BRAFWT or BRAFN581K) (Fig. 6B). On the other hand, both drugs did not affect the low baseline CTLA4 expression levels of cell lines that did not harbor either BRAFV600E or NRAS mutations (Fig. 6B). Interestingly, the BRAFV600E or NRAS-mutant cell lines expressed CTLA4 at a much greater (40-fold on average) levels than the cell lines wildtype for BRAFV600 and NRAS (Fig. 6C). These results clearly indicate that constitutive upregulation of the MAPK pathway in melanoma cells enhances CTLA4 expression independent of the IFNG pathway.
IFNG-induced gene signature in melanoma correlates with response to ipilimumab
Having determined that CTLA4 is a bona-fide primary IFNG-responsive gene in melanocytes and melanoma cells, we asked the question whether its expression correlated with that of other known IFNG target genes in melanoma. We analyzed RNA-seq transcriptome data of melanoma tissues obtained from 20 patients, previously reported by Snyder et al. and Chiappinelli et al. (12,13). This cohort of 20 patients was part of a larger cohort that had received ipilimumab treatment, and showed either long-term benefit (8 patients with stable disease or better for >6 mo), or no/minimal benefit (12 patients with stable disease for <6 mo or disease progression) (12). Analysis of the transcriptome of these whole melanoma tissues showed that indeed CTLA4 expression significantly correlated with known classical IFNG target genes, such as STAT1, IRF1, TAP2, GBP2, and HLA-DRB5 (Fig. 7A). Interestingly, CTLA4 expression was also significantly associated with expression of melanoma-associated antigen 11 (MAGEA11), and other immune checkpoints PD-L1, TIM-3 and LAG-3 (Fig. 7A). Intriguingly, clustering of gene expression data of these patients with respect to IFNG responsive gene expression signature (32) showed that high expression of the IFNG response signature, including CTLA4, in whole tumor tissue was associated with long-term benefit from ipilimumab treatment (Fig. 7B). In fact, the patients that showed long-term survival benefit from ipilimumab treatment expressed CTLA4 at significantly higher levels than the non-responders (Fig. 7C,D).
Figure 7.

Relationship between CTLA4 expression in melanoma cells and response to anti-CTLA4 immunotherapy. A, Correlation of CTLA4 expression with IFNG-response genes STAT1, IRF1, TAP2, GBP2, and HLA-DRB5, a melanoma-specific antigen MAGEA11 and other immune checkpoints TIM-3, LAG3 and PDL1 in human melanoma tumors. X, Y axis, FPKM values of indicated genes. Spearman correlation coefficients (R2), Pearson correlation R, and P values are listed. *P < 0.05; **P < 0.01; ****P < 0.0001. B, Clustering analysis was performed on IFNG-regulated genes and other immune inhibitory receptors TIM-3, LAG3 and PDL1 from anti-CTLA4-treated metastatic melanoma patients with either long-term benefit (blue; n=8) or no/minimal benefit (orange; n=12). C, CTLA4 expression in tumor tissues of patients with long-term benefit (blue; n=8) or no/minimal benefit (orange; n=12). D, Kaplan-Meier overall survival analysis between metastatic melanoma patients with long-term benefit from anti-CTLA4 immunotherapy treatment and no/minimal benefit patients. P is via log-rank test.
DISCUSSION
The high expression of CTLA4 in human melanoma cells and its regulation by the IFNG/JAK/STAT1-mediated signaling pathway may have important functional consequences in the clinical response to anti-CTLA4 immunotherapy of melanoma. These findings have potential implications for the conventional and prototypical roles of the IFNG signaling pathway and CTLA4 in tumor immunosurveillance and tumor immunoevasion. Interferons are cytokines best known for their regulation of immune responses involved in host defense against viral and bacterial infections, and have long been associated with cytostatic/cytotoxic and antitumor immune surveillance (33). IFNG has been postulated to be intimately involved in the elimination stage of the immunoediting paradigm (34,35). However, we have previously suggested that it may also be important at the equilibrium and/or evasion stages, in potential roles that are pro-melanomagenic (10,21). If so, what are the molecular mechanisms for these counter-dogmatic roles? One clue may be in its homeostatic function. While IFNG activates an inflammatory cascade, it also plays a crucial role in limiting the destruction of tissues in the aftermath of inflammation. IFNG signaling can plausibly act to protect normal cells from the collateral damage associated with inflammation-associated tissue remodeling. Concomitantly, these same mechanisms may allow cells harboring oncogenic mutations to evade destruction and survive in a state of equilibrium until they become transformed. In effect, IFNG-mediated inflammation may lead to an immunosuppressive and tolerogenic tissue or tumor microenvironment, which may be mechanistically achieved by IFNG-induced enhancement of immune checkpoints, mediated by molecules like CTLA4 and Programmed Death 1 and its ligand (PD-1 and PD-L1) in the tissue/tumor microenvironment. Indeed, PD-L1 is a known downstream target gene of IFNG in many tumor types, including melanoma, and contributes to T cell inhibition by interacting with B7-1 (CD80) (36–38). Here we have reported significant correlation of CTLA4 expression with those of the immune checkpoints PD-L1, TIM-3, and LAG-3. It is plausible that IFNG-induced CTLA4 expression in melanocytes and melanoma cells contributes to microenvironmental immunosuppression by direct melanocyte/melanoma cell-mediated inhibition of T cell cytotoxicity, which would aid melanomagenesis. If found to be true, it would challenge the current paradigm of the route of action of anti-CTLA4 immunotherapy, which is thought to go through inhibition of CTLA4 expressed by the T cells. It is possible that the mechanism of action of anti-CTLA4 immunotherapy at least partially depends upon inhibition of CTLA4 expressed by the melanoma cells, which would interfere with a melanoma cell-mediated T cell deactivation. Further mechanistic studies will be required to delineate such a melanoma cell-T cell inhibitory crosstalk.
We have shown evidence that the overexpression of CTLA4 in human melanoma cell lines is driven by the constitutively activated MAPK/ERK pathway, as targeted inhibitors of BRAFV600E and MEK were able to abolish CTLA4 overexpression, specifically in the cell lines harboring activating BRAF or NRAS mutations. These intriguing results add to the complexity of the crosstalk between the mutational landscape of melanoma cells and the immune profile of the tumor microenvironment. There is currently considerable interest in potential therapeutic strategies combining the molecularly targeted inhibitors of MAPK pathway and immunotherapies. Several ongoing clinical trials are exploring the efficacy of concomitant treatment of advanced melanoma with BRAF and MEK inhibitors, anti-CTLA4 and/or anti-PD-1 antibodies (e.g. NCT02224781, NCT01940809). It has been suggested that BRAF/MEK inhibition can enhance T cell infiltration and immune recognition of melanoma cells by increasing the expression levels of melanoma-specific antigens, which would in turn enhance efficacy of anti-melanoma immunotherapy (39–41). Activated MAPK-driven CTLA4 expression within melanoma cells may play a substantial role in determining the responses to such combination therapeutic regimens. These potential molecular circuits warrant further investigation.
We have also identified an IFNG-response gene expression signature in human melanoma tissues, including CTLA4, as a potential biomarker for response to anti-CTLA4 immunotherapy. There are two important caveats with these conclusions. First, these results are based on a very small cohort of 20 patients, and warrant further larger-scale studies to verify clinical applicability. Secondly, the gene expression data were derived from whole tumor tissues, which leaves open the possibility that the source of the differential expression of IFNG signature and CTLA4 is the difference in tumor-associated immune cells, e.g. tumor infiltrating lymphocytes (TILs) and macrophages. Snyder et al. had reported that in this cohort of patient tissues, there was no difference between the responders and non-responders with respect to tumor-associated CD45+ leukocytes, CD8+ T cells, and FOXP3+ Treg cells, which seemingly rules out a quantitative difference in immune cell infiltrate as an explanation for this finding (12). However, qualitative differences in the immune infiltrate cannot be ruled out. For example, we have previously reported that 70% of human melanomas harbor macrophages that secrete IFNG (10), which would explain the different levels of IFNG in the tumor microenvironment and the consequent IFNG-responsive signature in the tumor tissues. The functional status of TILs in melanoma tissues remains poorly characterized and their relative contribution to therapeutic response is poorly understood (42). Therefore, whether it is the CTLA4 expression in melanoma cells or tumor-associated immune cells that is the defining factor in patient response to ipilimumab remains an open question. The latter seems to be the most likely explanation, especially in light of our results that show concordance of CTLA4 expression with other immune checkpoints. However, overexpression of CTLA4 in melanoma cell lines suggests that it may be a plausible alternative or complementary target of ipilimumab. Further studies will be required to assess the effects of melanocytic expression of immune checkpoints on the TIL function in the tumor microenvironment and response to immune checkpoint inhibitors.
We had discovered CTLA4 overexpression in melanocytes in the context of erythemal inflammatory response in UVB-irradiated neonatal mouse skin (10). What could be the possible function of CTLA4 expression in melanocytes? Melanocytes are built for enhanced survival, to withstand both UVR exposure ensuring the continued synthesis of melanin, and the chemical stresses associated with the presence of melanin itself. This is accomplished in part through BCL2, an anti-apoptosis protein whose expression is regulated by the melanocyte lineage survival factor MITF (43). Expression of immunoevasive molecules like CTLA4 and PD-L1 could be a complementary survival mechanism in the aftermath of UVR assault. These immunoevasive elements may play an integral role in further protecting melanocytes from eradication by the UVB-induced inflammatory response, which is otherwise designed to remodel all damaged portions of the skin. Melanoma could then be branded as an opportunistic cancer taking advantage of this built-in circuitry to develop into one of the most immunoevasive cancers. The fact that this circuitry converges on IFNG epitomizes the importance of this signaling pathway to melanocytic survival mechanisms, and provides a novel perspective regarding its dual role in melanomagenesis, and perhaps in tumorigenesis in general.
Supplementary Material
Acknowledgments
We thank Drs. Glenn Merlino, Richard Pomerantz, Raghbir Athwal, Meenhard Herlyn, Ashani Weeraratna, and Jean-Pierre Issa for providing cell lines. Thanks to Anna-Mariya Kukuyan and Dr. Joseph Testa for providing access to instrumentation. Thanks to Drs. Merlino, Issa, and Chi-Ping Day for critical reading of the manuscript.
Financial Support: This work was supported by National Cancer Institute, National Institutes of Health grant number R01CA193711 to M. Raza Zaidi.
Footnotes
Conflicts of Interest: The authors declare no potential conflicts of interest.
References
- 1.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nature reviews Immunology. 2013;13:227–42. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weber JS. Tumor evasion may occur via expression of regulatory molecules: a case for CTLA-4 in melanoma. The Journal of investigative dermatology. 2008;128:2750–2. doi: 10.1038/jid.2008.341. [DOI] [PubMed] [Google Scholar]
- 3.Callahan MK, Postow MA, Wolchok JD. CTLA-4 and PD-1 Pathway Blockade: Combinations in the Clinic. Frontiers in oncology. 2014;4:385. doi: 10.3389/fonc.2014.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolchok JD, Saenger Y. The mechanism of anti-CTLA-4 activity and the negative regulation of T-cell activation. The oncologist. 2008;13(Suppl 4):2–9. doi: 10.1634/theoncologist.13-S4-2. [DOI] [PubMed] [Google Scholar]
- 5.Contardi E, Palmisano GL, Tazzari PL, Martelli AM, Fala F, Fabbi M, et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. International journal of cancer. 2005;117:538–50. doi: 10.1002/ijc.21155. [DOI] [PubMed] [Google Scholar]
- 6.Pistillo MP, Tazzari PL, Palmisano GL, Pierri I, Bolognesi A, Ferlito F, et al. CTLA-4 is not restricted to the lymphoid cell lineage and can function as a target molecule for apoptosis induction of leukemic cells. Blood. 2003;101:202–9. doi: 10.1182/blood-2002-06-1668. [DOI] [PubMed] [Google Scholar]
- 7.Egen JG, Kuhns MS, Allison JP. CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nature immunology. 2002;3:611–8. doi: 10.1038/ni0702-611. [DOI] [PubMed] [Google Scholar]
- 8.Attia P, Phan GQ, Maker AV, Robinson MR, Quezado MM, Yang JC, et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J Clin Oncol. 2005;23:6043–53. doi: 10.1200/JCO.2005.06.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shah KV, Chien AJ, Yee C, Moon RT. CTLA-4 is a direct target of Wnt/beta-catenin signaling and is expressed in human melanoma tumors. The Journal of investigative dermatology. 2008;128:2870–9. doi: 10.1038/jid.2008.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaidi MR, Davis S, Noonan FP, Graff-Cherry C, Hawley TS, Walker RL, et al. Interferon-gamma links ultraviolet radiation to melanomagenesis in mice. Nature. 2011;469:548–53. doi: 10.1038/nature09666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin KA, Lupey LN, Tempera I. Epstein-Barr Virus Oncoprotein LMP1 Mediates Epigenetic Changes in Host Gene Expression through PARP1. Journal of virology. 2016;90:8520–30. doi: 10.1128/JVI.01180-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. The New England journal of medicine. 2014;371:2189–99. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell. 2015;162:974–86. doi: 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Babicki S, Arndt D, Marcu A, Liang Y, Grant JR, Maciejewski A, et al. Heatmapper: web-enabled heat mapping for all. Nucleic acids research. 2016;44:W147–53. doi: 10.1093/nar/gkw419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cancer Cell Line Encyclopedia C, Genomics of Drug Sensitivity in Cancer C. Pharmacogenomic agreement between two cancer cell line data sets. Nature. 2015;528:84–7. doi: 10.1038/nature15736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qureshi OS, Kaur S, Hou TZ, Jeffery LE, Poulter NS, Briggs Z, et al. Constitutive clathrin-mediated endocytosis of CTLA-4 persists during T cell activation. The Journal of biological chemistry. 2012;287:9429–40. doi: 10.1074/jbc.M111.304329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leung HT, Bradshaw J, Cleaveland JS, Linsley PS. Cytotoxic T lymphocyte-associated molecule-4, a high-avidity receptor for CD80 and CD86, contains an intracellular localization motif in its cytoplasmic tail. The Journal of biological chemistry. 1995;270:25107–14. doi: 10.1074/jbc.270.42.25107. [DOI] [PubMed] [Google Scholar]
- 19.Mead KI, Zheng Y, Manzotti CN, Perry LC, Liu MK, Burke F, et al. Exocytosis of CTLA-4 is dependent on phospholipase D and ADP ribosylation factor-1 and stimulated during activation of regulatory T cells. Journal of immunology. 2005;174:4803–11. doi: 10.4049/jimmunol.174.8.4803. [DOI] [PubMed] [Google Scholar]
- 20.Valk E, Leung R, Kang H, Kaneko K, Rudd CE, Schneider H. T cell receptor-interacting molecule acts as a chaperone to modulate surface expression of the CTLA-4 coreceptor. Immunity. 2006;25:807–21. doi: 10.1016/j.immuni.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 21.Zaidi MR, Merlino G. The two faces of interferon-gamma in cancer. Clin Cancer Res. 2011;17:6118–24. doi: 10.1158/1078-0432.CCR-11-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–95. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- 23.Caldenhoven E, Buitenhuis M, van Dijk TB, Raaijmakers JA, Lammers JW, Koenderman L, et al. Lineage-specific activation of STAT3 by interferon-gamma in human neutrophils. Journal of leukocyte biology. 1999;65:391–6. doi: 10.1002/jlb.65.3.391. [DOI] [PubMed] [Google Scholar]
- 24.Mesa RA, Yasothan U, Kirkpatrick P. Ruxolitinib. Nature reviews Drug discovery. 2012;11:103–4. doi: 10.1038/nrd3652. [DOI] [PubMed] [Google Scholar]
- 25.Sadzak I, Schiff M, Gattermeier I, Glinitzer R, Sauer I, Saalmuller A, et al. Recruitment of Stat1 to chromatin is required for interferon-induced serine phosphorylation of Stat1 transactivation domain. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:8944–9. doi: 10.1073/pnas.0801794105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nature reviews Genetics. 2014;15:272–86. doi: 10.1038/nrg3682. [DOI] [PubMed] [Google Scholar]
- 28.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Begitt A, Droescher M, Meyer T, Schmid CD, Baker M, Antunes F, et al. STAT1-cooperative DNA binding distinguishes type 1 from type 2 interferon signaling. Nature immunology. 2014;15:168–76. doi: 10.1038/ni.2794. [DOI] [PubMed] [Google Scholar]
- 30.Zhang JJ, Vinkemeier U, Gu W, Chakravarti D, Horvath CM, Darnell JE., Jr Two contact regions between Stat1 and CBP/p300 in interferon gamma signaling. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:15092–6. doi: 10.1073/pnas.93.26.15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boussemart L, Malka-Mahieu H, Girault I, Allard D, Hemmingsson O, Tomasic G, et al. eIF4F is a nexus of resistance to anti-BRAF and anti-MEK cancer therapies. Nature. 2014;513:105–9. doi: 10.1038/nature13572. [DOI] [PubMed] [Google Scholar]
- 32.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. Journal of leukocyte biology. 2004;75:163–89. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 33.Brown TJ, Lioubin MN, Marquardt H. Purification and characterization of cytostatic lymphokines produced by activated human T lymphocytes. Synergistic antiproliferative activity of transforming growth factor beta 1, interferon-gamma, and oncostatin M for human melanoma cells. Journal of immunology. 1987;139:2977–83. [PubMed] [Google Scholar]
- 34.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nature reviews Immunology. 2006;6:836–48. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 35.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 36.Ritprajak P, Azuma M. Intrinsic and extrinsic control of expression of the immunoregulatory molecule PD-L1 in epithelial cells and squamous cell carcinoma. Oral oncology. 2015;51:221–8. doi: 10.1016/j.oraloncology.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 37.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–22. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell reports. 2017;19:1189–201. doi: 10.1016/j.celrep.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19:1225–31. doi: 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci Transl Med. 2015;7:279ra41. doi: 10.1126/scitranslmed.aaa4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18:1386–94. doi: 10.1158/1078-0432.CCR-11-2479. [DOI] [PubMed] [Google Scholar]
- 42.Luke JJ, Flaherty KT, Ribas A, Long GV. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nature reviews Clinical oncology. 2017;14:463–82. doi: 10.1038/nrclinonc.2017.43. [DOI] [PubMed] [Google Scholar]
- 43.McGill GG, Horstmann M, Widlund HR, Du J, Motyckova G, Nishimura EK, et al. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell. 2002;109:707–18. doi: 10.1016/s0092-8674(02)00762-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.