

Abstract
A major challenge in photodynamic cancer therapy (PDT) is avoiding PDT-induced hypoxia, which can lead to cancer recurrence and progression through activation of various angiogenic factors and significantly reduce treatment outcomes. Reported here is an acetazolamide (AZ)-conjugated BODIPY photosensitizer (AZ-BPS) designed to mitigate the effects of PDT-based hypoxia by combining the benefits of anti-angiogenesis therapy with PDT. AZ-BPS showed specific affinity to aggressive cancer cells (MDA-MB-231 cells) that overexpress carbonic anhydrase IX (CAIX). It displayed enhanced photocytotoxicity compared to a reference compound, BPS, which is an analogous PDT agent that lacks an acetazolamide unit. AZ-BPS also displayed an enhanced in vivo efficacy in a xenograft mouse tumor regrowth model relative to BPS, an effect attributed to inhibition of tumor angiogenesis by both PDT-induced ROS generation and CAIX knockdown. AZ-BPS was evaluated successfully in clinical samples collected from breast cancer patients. We thus believe that the combined approach described here represents an attractive therapeutic approach to targeting CAIX-overexpressing tumors.
Graphical Abstract
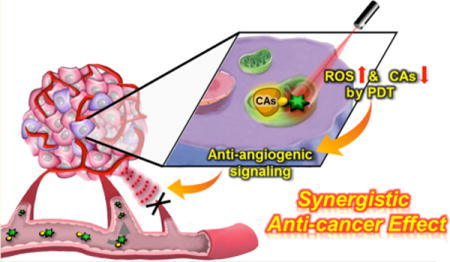
INTRODUCTION
Angiogenesis is a tightly regulated event involving multiple signaling pathways that plays a critical role in a number of physiological processes, including reproduction and wound healing.1,2 Although beneficial for tissue growth and regeneration, aberrant regulation of angiogenesis is a hallmark of many pathological disorders such as cancer, inflammation, and autoimmune disease.3,4 By exploiting angiogenesis mechanisms, malignant cells that might otherwise be starved can enhance their oxygen and nutrients supply. This usurpation of otherwise beneficial mechanisms can exacerbate tumor progression and metastasis. In 1971, Folkman suggested that inhibition of angiogenesis (anti-angiogenesis) would be a promising approach to treating cancer, a seminal idea that inspired intensive scientific activity.5,6 To develop anti-angiogenesis therapy, various inhibitors targeting underlying mechanism of angiogenesis, such as vascular endothelial growth factor (VEGF)7–9 and extracellular matrix (ECM) degradation,10,11 have been developed. This has led to recognized clinical applications.12 Although initial clinical trial results were not encouraging, bevacizumab and pegaptanib have emerged as potent angiogenesis inhibitors approved by the U.S. Food and Drug Administration (FDA).13,14 These agents have validated anti-angiogenesis as a promising approach to anticancer drug development. Nevertheless, improved therapeutic treatments are still needed. Combining angiogenesis-targeting approaches with other modalities could provide this benefit.
Photodynamic therapy (PDT) has emerged as an attractive cancer treatment that is in clinical use.15 In PDT, singlet oxygen (1O2) generated from molecular oxygen by irradiation of photosensitizer (PS) is used to kill cancer cells directly through apoptosis and indirectly by inducing tumor vasculature shutdown and recruitment of immune mediators.16 In principle, PDT offers a number advantages over other more conventional cancer treatment approaches,17 including (i) being minimally invasive compared to other therapies,18 (ii) the use of both a sensitizer and light to improve localization of the therapeutic effect, and (iii) the potential for repeated application without significant side effects.17 Unfortunately, most current PDT agents operate predominantly through an oxygen-dependent type II mechanism, which can lead to severe hypoxia within tumors.19 This, in turn, can limit the therapeutic benefits of photodynamic therapy. PDT-induced hypoxia can arise due to oxygen consumption either directly by the PS or indirectly through vasculature degradation. The resulting hypoxic cells protect themselves from PDT-mediated damage by switching on signaling cascades and releasing pro-angiogenic growth factors, such as vascular endothelial growth factor (VEGF), epidermal growth factor (EGF) and angiopoietin (ANGPT), which trigger angiogenesis and promote tumor regrowth.20 VEGF is a specific regulator of angiogenesis, and its overexpression in a variety of solid tumors is strongly correlated with negative prognoses due to aggressive invasion and metastasis in cancer patients.21–23 The use of PDT in combination with an anti-angiogenesis inhibitor has been suggested as a promising approach to enhancing the benefits of PDT by blocking the counterproductive upregulation of angiogenic factors.12 In fact, using this approach, enhanced PDT effects have been realized.24 Unfortunately, most current angiogenesis inhibitors are antibodies or fusion proteins. Blocking VEGF without resorting to the use of a protein-based inhibitor could overcome this latter limitation. As detailed below, we believe that using an acetazolamide (AZ) moiety to target carbonic anhydrase IX (CAIX) allows the benefits of anti-angiogenic therapy to be combined with those of PDT.
One corollary of PDT-induced hypoxia is that it triggers the hypoxia-inducing factor (HIF)-mediated signaling cascade. This results in upregulation of many regulatory genes that are responsible for tumor progression via the transcriptional activation of angiogenic factors.25,26 Genes encoding for CAIX are among those particularly upregulated by the HIF pathway.27,28 CAIX regulates intra- and extracellular pH and promotes tumor survival and invasion within hypoxic environments. In fact, CAIX is known to be a marker for poor survival and distant metastases in aggressive breast cancer (e.g., MDA-MB-231).29,30 The fact that CAIX is subject to tumor-specific overexpression with highly restricted expression within normal tissues makes it a potentially attractive therapeutic target. Recently, Lou et al. reported that inhibition of CAIX significantly suppresses breast tumor growth and metastasis in vivo.31 An enhanced therapeutic effect was also seen in mouse models after CAIX knockdown and treatment with bevacizumab.32 In addition, Takemoto et al., utilizing a chromophore-assisted light inactivation (CALI) assay, reported that singlet oxygen generation adjacent to target proteins led to efficient inactivation of the proteins in question.33 We thus considered it likely that combining PDT with targeted therapy directed at inhibition of CAIX could give rise to an effective anti-angiogenesis PDT agent. Here, we report preliminary proof-of-principle studies in support of this hypothesis (Figure 1). In vivo, a considerably enhanced PDT effect is seen with an acetazolamide-functionalized boron dipyrromethene (BODI-PY) sensitizer, AZ-BPS, compared to a control system (BPS) lacking the AZ subunit.
Figure 1.
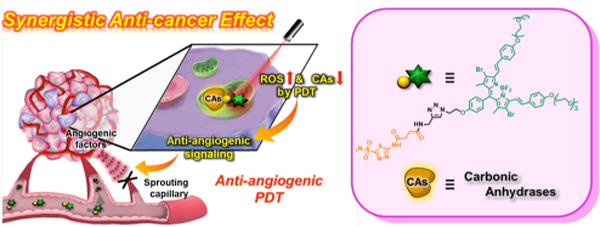
Schematic representation of synergistic anticancer effect by AZ-BPS targeted to CAIX.
RESULTS AND DISCUSSION
Synthesis and Characterization of AZ-BPS and BPS
To create a putative anti-angiogensis PDT agent, an AZ ligand was combined with a BODIPY PS. The AZ moiety was expected to provide CAIX inhibition while promoting efficient tumor targeting of cancer cells overexpressing this key enzyme,34 while the BODIPY subunit was expected to provide for both photodamage and allow for the fluorescence-based tracking of its cellular uptake and distribution.35
The AZ-containing BODIPY PS conjugate (AZ-BPS) of this study and its analogue without AZ (BPS, reference compound) were synthesized as shown in Figure 2. Compound 834 was treated with propagylamine in the presence of 2-(1H-9-azobenzotriazole-1-yl)-1,1,3,3-tetramethylammonium hexafluorophosphate (HATU), diisopropylethylamine (DIPEA), and triethylamine (TEA) in N,N′-dimethylformamide (DMF) to give compound 7 (AZ ligand). Compound 4 was obtained by treating 2,4-dimethylpyrrole with 1036 through a sequence of condensation, oxidation, and boron complexation reactions. Compound 4 was then converted to compound 3 by reaction with sodium azide in DMF. To improve the intersystem crossing rate and hence singlet oxygen generation, compound 3 was brominated (heavy atom effect)35 by adding a mixture of N-bromosuccimide (NBS) in dichloromethane (DCM) to give the dibromo-PS 2. Knoevenagel condensation of 2 with 9 yielded PS 1. AZ-BPS was prepared by reacting PS 1 with the AZ ligand 7 in the presence of CuSO4·5H2O and sodium ascorbate in DMF/methanol (MeOH)/H2O. The analytical data (1H NMR, 13C NMR, and ESI-MS analyses) for AZ-BPS and BPS were fully consistent with the proposed structures (Figures S13–S33, Supporting Information).
Figure 2.
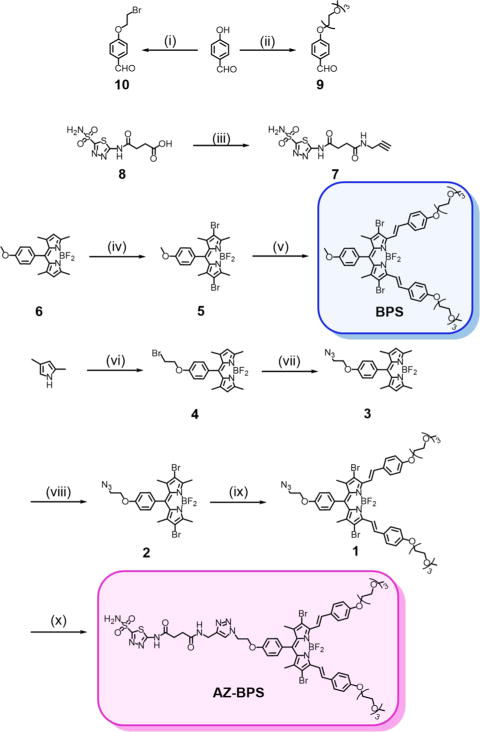
Synthesis of AZ-BPS and BPS. Reagents and conditions: (i) 1,2-dibromoethane, K2CO3, acetonitrile (MeCN), reflux, 6 h, 65%; (ii) CH3(OCH2CH2)3OTs, K2CO3, DMF, reflux, 12 h, 90%; (iii) propargylamine, HATU, TEA, DIPEA, DMF, room temperature (rt), 24 h, 70%; (iv) NBS, DCM, rt, 0.5 h, 95%; (v) 9, Mg(ClO4)2, piperidine, toluene, reflux, 24 h, 31%; (vi) 10, trifluoroacetic acid (TFA), DCM, rt, 12 h; chloranil, BF3·O(C2H5)2, TEA, 1 h, 45%; (vii) NaN3, DMF, 130 °C, 12 h, 91%; (viii) NBS, DCM, rt, 0.5 h, 96%; (ix) 9, Mg(ClO4)2, piperidine, toluene, reflux, 24 h, 44%; (x) 7, CuSO4·5H2O, sodium ascorbate, DMF/MeOH/H2O, 57%.
Spectroscopy
The photophysical properties of AZ-BPS and BPS were analyzed in dimethyl sulfoxide (DMSO). An intense absorption band at 661 nm ascribed to the BODIPY core was seen in the case of both these derivatives (molar extinction coefficients (ε): 5.27 × 104 M−1 cm−1 for AZ-BPS and 4.98 × 104 M−1 cm−1 for BPS) (Figure 3). Upon irradiation at 660 nm, a fluorescence emission feature centered around approximately 690 nm was seen for these two BPSs (λem = 689 vs 688 nm for AZ-BPS and BPS, respectively). These emission maxima lie in the red-to-NIR spectral region and hence are suitable for biological applications. Using Supporting Information eq S1, the fluorescence quantum yields (Φf) of the probes were calculated to be 0.06 for AZ-BPS and 0.05 for BPS. These quantum yields are much lower than those typically seen for BODIPY fluorophores, an effect attributed to the heavy metal effect of bromine atoms, which enhances intersystem crossing.35 The fluorescence spectra of AZ-BPS and BPS showed slight hypsochromic shifts (<10 nm) with increasing solvent polarity (DCM < MeCN < MeOH) (Figure S1, Supporting Information).
Figure 3.
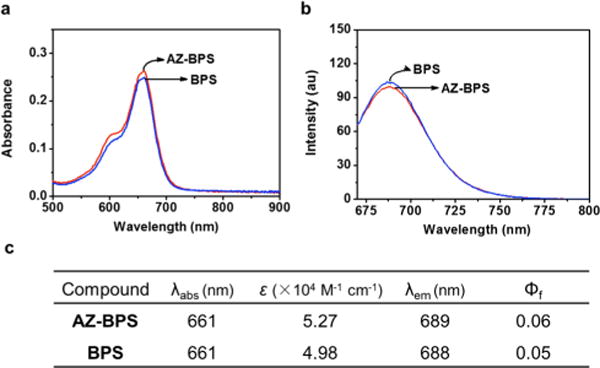
Photophysical features of AZ-BPS and BPS. (a) Absorption and (b) fluorescence spectra of AZ-BPS (red) and BPS (blue) (5 μM for both) in DMSO. (c) Photophysical data for AZ-BPS and BPS. The excitation wavelengths are 660 nm (slit width = 5/5); ε, molar extinction coefficient (×104 M−1 cm−1); λabs, absorption maximum wavelength (nm); λem, emission maximum wavelength (nm); Φf, fluorescence quantum yield.
Singlet Oxygen Generation by AZ-BPS and BPS
The ability of AZ-BPS and BPS to produce singlet oxygen under photosensitizing conditions was studied in DMSO using 1,3-diphenylisobenzofuran (DPBF) as a singlet oxygen trap (Figure 4).37 Irradiation of a solution of AZ-BPS and DPBF with a 660 nm Xe lamp led to dramatic quenching of the DPBF absorption band at 408 nm over a time period of 0–15 min (Figure 4a). Identical behavior was observed in the case of BPS under identical experimental conditions (Figure 4b). Methylene blue (MB) was then used to calculate the singlet oxygen quantum yield (ΦΔ) in accord with an established procedure.38 Using Supporting Information eq S2, ΦΔ values of 0.60 and 0.55 were calculated for AZ-BPS and BPS, respectively. These quantum yields are in line with what is observed for several photosensitizers in clinical use, such as MB (ΦΔ = 0.52 in DMSO, Figure S2, Supporting Information).39 Importantly, both AZ-BPS and BPS were found to work well and to be stable in PBS media (pH 7.4, 10 mM) containing 10% DMSO under conditions of photoirradiation (Figure S3, Supporting Information).
Figure 4.
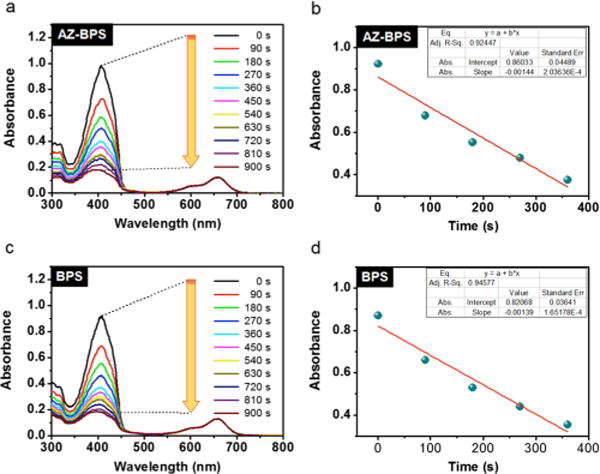
Photosensitized singlet oxygen generation by AZ-BPS and BPS. Time-dependent absorption spectral changes seen for 40 μM DMSO solutions of DPBF and 2 μM of (a) AZ-BPS or (c) BPS upon irradiation at 660 nm (slit width = 15–15, Xe lamp). (b,d) Plots of the change in absorption at 408 nm for the experiments shown in (a,c), respectively.
Isothermal Titration Calorimetric Analyses and Molecular Docking Studies
The binding interactions between AZ-BPS and CAIX were evaluated using isothermal titration calorimetry. These analyses, carried out in a 10% DMSO buffer solution (pH 7.4, 10 mM aqueous PBS), revealed an exothermic reaction characterized by a high dissociation constant, KD = 2.94 ± 0.24 μM (Figure 5b). The corresponding stoichiometry and enthalpy values were found to be 1.27 ± 0.01 and −6.19 ± 0.03 kcal/mol, respectively. On this basis, we conclude that AZ-BPS interacts strongly with CAIX.
Figure 5.
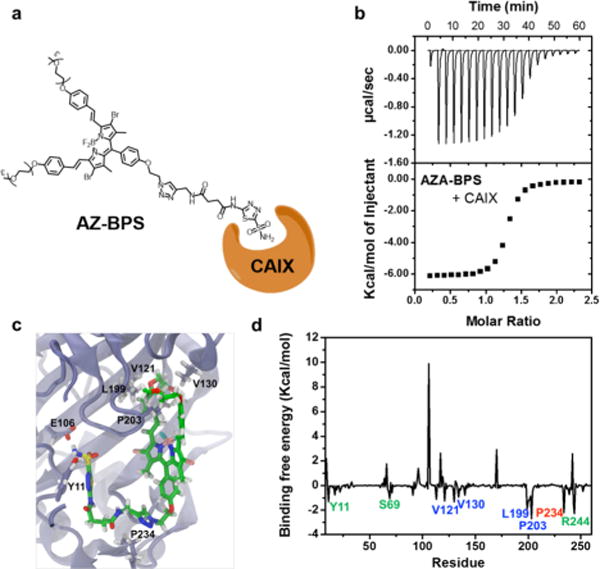
Binding study of AZ-BPS with CAIX. (a) Schematic diagram showing binding of AZ-BPS with CAIX’s active site. (b) Isothermal titration curve for binding interaction of AZ-BPS with CAIX in a 10% DMSO buffer solution (pH 7.4, 10 mM aqueous PBS) (KD = 2.94 ± 0.24 μM; N = 1.27 ± 0.01 sites; ΔH = −6.19 ± 0.03 kcal/mol). (c) Calculated binding site and (d) deconvoluted binding free energy corresponding to the interaction between AZ-BPS and CAIX.
To explore the possible binding modes at the atomic level, docking studies and further hybrid quantum mechanics/molecular mechanics molecular dynamics (QM/MM MD) simulations involving AZ-BPS and CAIX were carried out. Details of the docking calculations and the simulations are provided in the Supporting Information. The binding site and deconvoluted binding free energies between AZ-BPS and each residue of CAIX are shown in Figure 5c,d. The estimated free energy corresponding to the binding between AZ-BPS and CAIX was found to be −35.8 kcal/mol based on the QM/MM MD simulations. Several regions in CAIX, namely, fragments Y11–W17, H68–S69, and S237–R244 (green) and fragments V121–L140, L199–P203 (blue), and G233–P234 (red) were calculated to be involved significantly in the binding interactions. Specifically, the V121–L140 and L199–P203 regions interact with the BPS moiety, the G233–P234 region interacts with the linker part of the overall AZ-BPS construct, whereas the Y11–W17, H68–S69, and S237–R244 regions interact with the AZ moiety.
CAIX-Dependent Specificity of AZ-BPS in Cells
The ability of the AZ ligand to target AZ-BPS to CAIX-positive cells was studied. Prior to the investigation, the endogenous expression of CAIX in various cell lines (BJ, AGS, A549, MDA-MB-231, MCF-7, and HeLa) was confirmed via Western blotting. In agreement with a previous report,40 CAIX expression in MDA-MB-231 cells (p < 0.05) was significantly higher than that with other cell lines, including MCF-7 (cf. Figure 6). Confocal microscopic experiments employing two cell lines, namely, the CAIX/high cancerous MDA-MB-231 and CAIX/low cancerous MCF-7 cell lines. As shown in Figure 7a, the MDA-MB-231 cells showed intense fluorescence intensity for 4 h incubation with AZ-BPS, whereas the MCF-7 cells exhibited little appreciable fluorescence under similar experimental conditions. As expected, a weak fluorescence signal was also observed in normal fibroblast BJ cells (noncancerous cell lines) (Figure S4, Supporting Information). On this basis, we conclude that AZ-BPS is taken up selectively by MDA-MB-231 cells. It was also found that AZ-BPS produced a higher fluorescent signal in the case of MDA-MB-231 spheroids relative to dissociated cells (Figure 7b), a result ascribed to the fact that CAIX expression increases with the degree of hypoxia.32 Note, Figure 7 shows confocal images of both normal 2D- and 3D-cultured systems in an effort to mimic more faithfully the in vivo tumor microenvironment. This desire to examine two kinds of cell forms reflects an appreciation that 2D-cultured cell lines (such as shown in Figure 7a) may not be as good as 3D-culture systems in terms of testing clinically active drugs and potential drug candidates.
Figure 6.
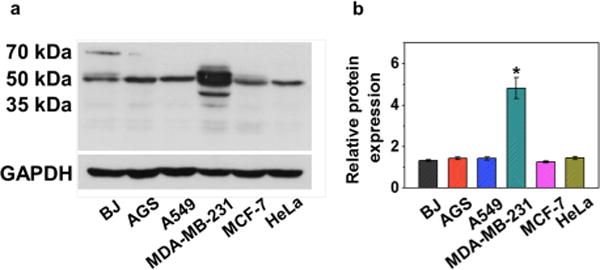
(a) Endogenous expression of CAIX and (b) quantification of endogenous CAIX expression in normal fibroblast (BJ) and various cancer cell lines (AGS, A549, MDA-MB-231, MCF-7, and HeLa).
Figure 7.
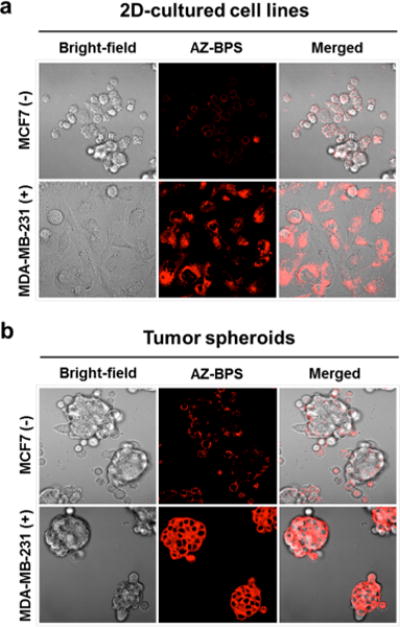
CAIX-dependent specificity of AZ-BPS in adherent cells and tumor spheroids. (a) Confocal microscopic images of MDA-MB-231 cells and MCF-7 cells after incubation with AZ-BPS (5 μM) for 4 h. (b) Confocal microscopic images of tumor spheroids formed from MDA-MB-231 cells and CAIX-negative MCF-7 cells after incubation with AZ-BPS (5 μM) for 4 h. Magnification 400×. To obtain tumor spheroids, 6.0 × 106 cells were seeded in 60 mm Corning ultralow attachment culture dishes (Sigma). AZ-BPS treatment was commenced on the second day following tumor spheroid formation.
To gain insights into the differential cellular uptake of BPS and AZ-BPS by different cell lines under identical experimental condition (Figure S5, Supporting Information), we determined their lipophilicity. This was done by measuring probe partitioning between n-octanol and buffer and calculating the resulting log Poct values, which were found to be 0.76 and 1.38 for AZ-BPS and BPS, respectively. Considering that the cell membranes are composed of lipid bilayers, it has been predicted that more lipophilic sensitizers, such as BPS, should be taken up into cells in preference to more hydrophilic ones, such as AZ-BPS, in the absence of a specific uptake mechanism.41 In fact, a preferential uptake of BPS relative to AZ-BPS was not observed in the case of MDA-MB-231 cells. The higher level of AZ-BPS uptake was confirmed by confocal imaging, while the CAIX expression levels were established by means of Western blots (Figure S6, Supporting Information). On this basis, we conclude that the AZ moiety present in AZ-BPS plays a key role in mediating uptake in the case of the MDA-MB-231 cells.
Photocytotoxicity of BPSs
A successful PDT photosensitizer is one that exhibits efficient off–on cytotoxicity in the absence and presence of photoirradiation. The cytotoxicity of AZ-BPS was thus evaluated under both light and dark conditions. Toward this end, AZ-BPS-treated MDA-MB-231 cells were prestained with propidium iodide (PI) and calcein AM to visualize via fluorescence apoptotic and live cells, respectively. As can be seen from an inspection of Figure 8a, a clear demarcation between regions exists between regions subject or not subject to 660 nm laser irradiation (2.0 W/cm2) for 30 min. The cells kept largely protected from light showed predominantly calcein AM-dependent features, whereas red PI fluorescence signals were seen in the case of photoirradiation. We thus conclude that cells within the irradiated area undergo apoptosis.
Figure 8.

Photocytotoxicity of AZ-BPS in the cells. (a) Confocal microscopic images of MDA-MB-231 cells with AZ-BPS (5 μM) after 660 nm laser irradiation (2.0 W/cm2, 30 min; 3600 J/cm2). (b) Dead cell ratio of MDA-MB-231 and MCF-7 cells (cytotoxicity was measured as a number of PI-stained cells to the total number of cells). (c) Confocal microscopic images of MDA-MB-231 and MCF-7 cells treated with AZ-BPS (5 μM) as a function of 660 nm laser (2.0 W/cm2) irradiation time from 5 to 20 min. Live/dead cells are green/red (calcein AM/PI), respectively. *P < 0.05. Magnification 100×. (d) FACS analysis of MDA-MB-231 cells with AZ-BPS (5 μM) after 660 nm laser irradiation (2.0 W/cm2, 30 min; 3600 J/cm2). Note that AZ-BPS induced significant apoptosis compared to the control and BPS.
Greater cell death is seen in the case of MDA-MB-231 cells than MCF-7 cells upon treatment with AZ-BPS (5 μM) and subjecting to laser irradiation at 660 nm (2.0 W/cm2) for 20 min (ca. 67 vs 27% for these two cell lines, respectively) under similar experimental conditions (Figure 8b). Confocal images corresponding to these experiments were obtained at different laser irradiation times. The MDA-MB-231 cells produced a red fluorescence ascribable to the PI stain that increased with irradiation time over the course of 5–20 min. In contrast, very weak fluorescent signals were observed in the case of the MCF-7 cells (Figure 8c) and also in noncancerous BJ cells (Figure S7, Supporting Information). On this basis, we conclude that the cell killing AZ-BPS-based PDT effect is greater in the case of the MDA-MB-231 cells overexpressing CAIX than in the MCF-7 cells. As noted above, this effect is ascribed to the fact that MDA-MB-231 facilitates the uptake of AZ-BPS, presumably through efficient binding with the AZ ligand.
A comparison of the relative cytotoxicity of AZ-BPS and BPS was also made (Figure S8, Supporting Information). Upon laser irradiation at 660 nm (2.0 W/cm2) for 30 min, with AZ-BPS co-incubated at this higher concentration of 50 μM, a reduction in the viability of the MDA-MB-231 cells of 81% was seen. In contrast, the use of BPS at the same concentration under the same experimental conditions produced only a 27% reduction. No significant cytotoxicity was seen for AZ-BPS in the absence of photoillumination (Figure S8, Supporting Information). As shown in Table S2, the photocytotoxicity index, defined as IC50 (dark)/IC50 (light), against MDA-MB-231 cells was found to be 190 for AZ-BPS and 1.31 for BPS (Table S2, Supporting Information).
Flow cytometry in conjunction with fluorescence-activated cell sorting (FACS) was used to quantify the extent of apoptosis (Figure 8d). Upon laser irradiation of the MDA-MB-231 cells at 660 nm (2.0 W/cm2, 30 min) incubated with AZ-BPS (5 μM), about 92.5% of cell population was seen to be the stages of late apoptosis or necrosis. The corresponding value was only 11.2% in the case of the cells treated with BPS (P < 0.05) (Figure 8d). These results are fully consistent with the conclusion drawn from the confocal microscopy studies shown in Figure 8c, namely, that AZ-BPS is more effective as a PDT sensitizer than BPS, at least in this cell line.
Possible Modes of Anticancer Action
As a first step toward investigating the possible modes of action of AZ-BPS and BPS, fluorescence-based colocalization studies were carried out using different organelle-specific trackers (i.e., Mito-, ER-, and Lyso-Tracker green). As can be seen from an inspection of Figure 9, a fluorescence image of MDA-MB-231 cells co-incubated with AZ-BPS gave rise to a fluorescence profile that overlapped well with that of Mito-Tracker. Although partial overlap with the Lyso- and ER Trackers was also seen, on the basis of these studies, it is concluded that in this CAIX-expressing cell line AZ-BPS localizes predominantly in the mitochondria. This is what would be expected for MDA-MB-231 cells treated with an agent containing an AZ localizing moiety.42 As expected, BPS, lacking the targeting moiety (AZ) present in AZ-BPS, gave rise to a fluorescence response consistent with the agent being distributed over all three organelles (Figure 9). In MCF-7 cells, neither AZ-BPS nor BPS showed profiles consistent with a specific organelle distribution (Figure S9, Supporting Information).
Figure 9.
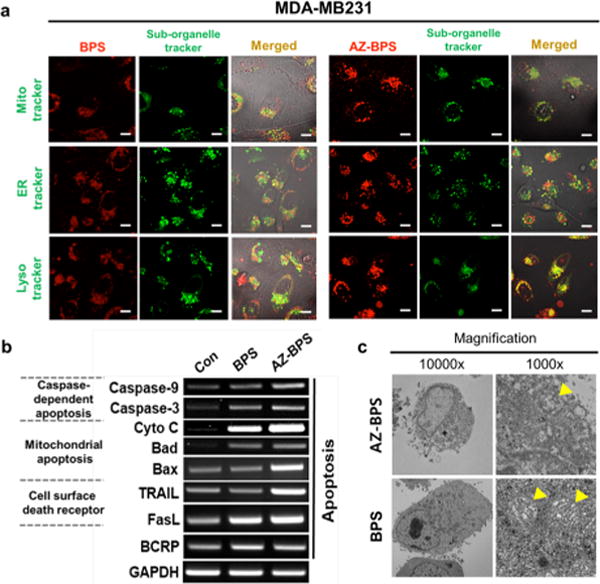
Mechanistic studies of AZ-BPS. (a) Intracellular colocalization of AZ-BPS and BPS: MDA-MB-231 cells were incubated with 5 μM of AZ-BPS and BPS for 4 h. The cells were then washed with PBS, and Mito-, ER-, and Lyso-Trackers (green) were added and the cells incubated for an additional 30 min before the fluorescent images were recorded. Scale bar: 50 μm. (b) Gene expression comparison involving AZ-BPS and BPS. MDA-MB-231 cells were treated 5 μM of AZ-BPS and BPS for 4 h and collected 6 h after being irradiated with 660 nm laser (2.0 W/cm2, 30 min; 3600 J/cm2). Gene expression was determined by RT-PCR. (c) TEM images showing ultrastructure of mitochondria of MDA-MB-231 cells treated separately with either 5 μM AZ-BPS or BPS for 4 h and then subjecting to 660 nm laser irradiation (2.0 W/cm2, 30 min; 3600 J/cm2). The cells for analysis were collected 12 h after photoirradiation. Yellow arrowheads indicate the mitochondria. Magnification 10 000× (right panels) and 1000× (left panels).
Reverse transcription polymerase chain reaction (RT-PCR) and transmission electron microscopy (TEM) studies were then carried out in an effort to gain insights into the mechanisms of PDT-induced cell death. Following NIR photoirradiation of the RT-PCR pattern of NIR-irradiated MDA-MB-231 cells, co-incubated AZ-BPS revealed an increased expression in mitochondria-mediated apoptotic genes (cytochrome c, Bad, and Bax), as well as membrane-located cell death receptors (TRAIL and FasL) (Figure 9b). Expression of these genes leads to mitochondrial dysfunction (including apoptosis) through upregulation of caspase-3 and caspase-9 expression as confirmed by TEM images showing disruption of the normal mitochondrial structure (Figure 9c, yellow arrowheads). While not proof, these findings lead us to suggest that the PDT-induced cell death produced upon photoexcitation of AZ-BPS is mediated in large measure through induced mitochondrial dysfunction.
In Vivo Xenograft Tumor Imaging and Photocyto-toxic Effects of AZ-BPS
In order to examine the in vivo anticancer efficacy of AZ-BPS, MDA-MB-231-inoculated xenograft mice were treated repeatedly with AZ-BPS (administrated nine times through tail vein injections three times a day per week for 3 weeks). In accord with the cancer-localizing design expectations, enhanced in vivo and ex vivo fluorescent signals were observed at the tumor regions relative to other organs (liver, lung, kidney, heart, spleen, and testis; cf. Figure 10a). The mice treated with AZ-BPS and subject to 660 nm photoirradiation (2.0 W/cm2) for 30 min at the tumor regions displayed significant reduction in tumor growth and tumor volumes without affecting the overall body weight compared to control and BPS-treated groups (Figure 10b,c and Figures S10 and S11, Supporting Information).
Figure 10.
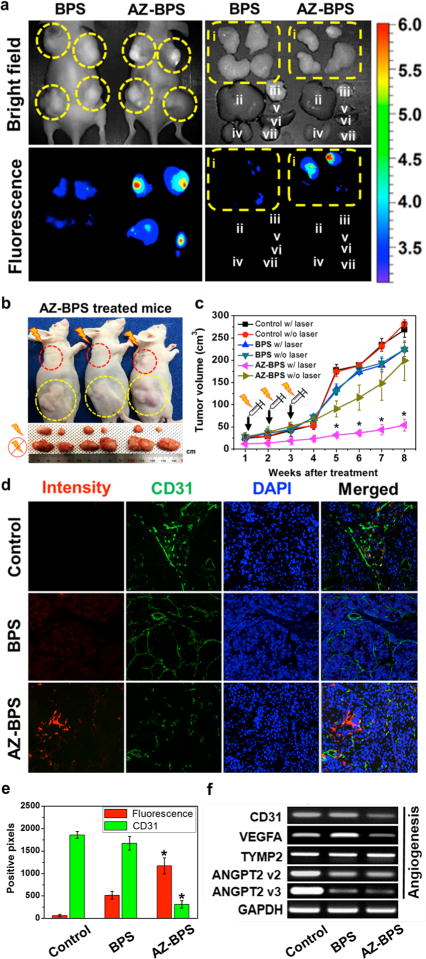
In vivo diagnostic and photocytotoxic effects of BPS. (a) (Left) In vivo images of nude mice 3 h after tail intravenous injection of BPS or AZ-BPS. (Right) Ex vivo images of various organs (i, tumor (yellow circle); ii, liver; iii, lung; iv, kidney; v, heart; vi, spleen; vii, testis) taken from nude mice 6 h after intravenous tail vein injection of BPS or AZ-BPS. (b) (Top) Representative images of nude mice 8 weeks after intravenous tail vein injection of AZ-BPS followed by 660 nm laser irradiation (2.0 W/cm2, 30 min; 3600 J/cm2) of the upper tumor (red circle). The lower tumor (yellow circle) was used as a nonirradiated control. (Bottom) Dissected tumors from each group. (c) Tumor volume of the mice in the BPS or AZ-BPS groups with or without PDT treatment. (d) Fluorescence-based determination of neoangiogenesis in the tumor tissues. Note that significantly decreased CD31-postive pixel area in the tumor tissues of AZ-BPS injected mice with 660 nm laser irradiation (2.0 W/cm2, 30 min; 3600 J/cm2), compared to those of the PBS control and BPS-injected groups. *P < 0.05. Magnification 100×. (e) Bar graph showing extent of neoangiogenesis in the tumor tissues. (f) Expression levels of genes associated with angiogenesis. MDA-MB-231 cells were treated 5 μM of AZ-BPS or BPS for 4 h and collected 6 h after subjecting to 660 nm laser irradiation (2.0 W/cm2, 30 min; 3600 J/cm2). Gene expression was determined by RT-PCR.
The ability of AZ-BPS to inhibit neoangiogenesis was also investigated in vivo. Here, mice bearing MDA-MB-231 xenografts were subject to laser irradiation at 660 nm (2.0 W/cm2) for 30 min after the animals were treated with AZ-BPS or BPS under identical conditions as for the above experiments. The tumors were excised, stained with anti-CD31 antibody (an angiogenesis marker), and subject to cryosection. Tumors analyzed in this way exhibited significantly decreased expression of CD31 as indicated by weak green fluorescence signals compared to control or BPS treatment (Figure 10d,e). These studies thus are consistent with previous studies that led to the suggestion that hypoxia-induced upregulation of CAIX induces a series of proteins associated with neoangiogenesis.26
Angiopoietin-2 (ANGPT2) together with vascular endothelial growth factor A (VEGFA) promotes the initiation of angiogenesis and maturation of new vessels.43 ANGPT2 is known to be expressed by endothelial cells in breast cancer cell lines.44 In the present study, sharply reduced expression levels were seen for both VEGFA and ANGPT2, whereas no change in the expression of thymidine phosphorylase 2 (TYMP2), an enzyme not directly associated with angiogenesis45 (but which has an indirect effect on the angiogenesis by stimulating chemotaxis of endothelial cells in various cancer cell lines), was observed (Figure 10f). On the other hand, increase expression of VEGFA was observed in the BPS-treated groups. This is taken as indirect evidence of resistance under conditions of BODIPY-mediated PDT (Figure 10f). Taken in concert, these results are consistent with the suggestion that AZ-BPS mediates its action in part by enhancing an anti-angiogenesis effect. Such a rationale explains why this agent is more effective than BPS alone; AZ-BPS, but not the control photosensitizer (BPS), can overcome the effects of hypoxia.
Studies of AZ-BPS in Human Breast Cancer Tissue
AZ-BPS was further studied in cryosectioned tissues obtained from breast cancer patients in which CAIX expression is enhanced.46 The results of immunohistochemistry revealed that CAIX-positive cells (green) displayed red fluorescence ascribable to AZ-BPS (Figures 11 and S12, Supporting Information). The Pearson’s coefficient value of yellow pixels (merged green and red) is 0.84, a value we interpret in terms of AZ-BPS localizing via CAIX targeting. In general, CAIX expression is strongly associated with negative prognostic breast cancer and may be correlated to immune-related adverse events in patients with metastasis. It has been known that the 10 year metastasis-free rate is ∼30%, with ∼90% of mortalities (for an estimated overall >10 year survival rate of ∼33%) being due to metastasis.47 Therefore, the strong correlation of AZ-BPS with CAIX leads us to propose that AZ-BPS represents a promising therapeutic candidate for patients with negative prognostic breast cancer.
Figure 11.
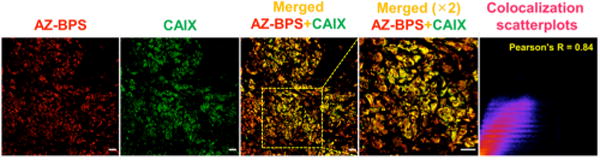
AZ-BPS targeting to CAIX in patient-derived tissue samples. Correlation between CAIX expression as inferred from studies with human anti-CAIX antibody (green) and the fluorescent intensity of AZ-BPS (red) seen in cryosectioned tumor tissues obtained from a human breast cancer patient. A strong degree of colocalization was seen as inferred from the Pearson’s coefficient value (0.84) (yellow color area, randomly selected n = 5 area).
CONCLUSIONS
Among the newly emerging strategies in cancer treatment, the use of angiogenesis inhibitors (anti-angiogenesis) has received considerable attention due to its potential to starve cancer cells with limited nutrients and oxygen supply. To add this putative benefit to those provided by PDT, we have developed the CAIX-targeting photosensitizer AZ-BPS reported here. This system was designed to include two active subunits, namely, (a) an AZ ligand to provide both CAIX inhibition and efficient tumor targeting and (b) a BODIPY moiety to serve as both a singlet oxygen-producing photosensitizer and to allow fluorescence-based tracking of cellular uptake and distribution. In vitro studies revealed that AZ-BPS specifically localizes to the mitochondria of cancer cells that overexpress CAIX. This conjugate also generates singlet oxygen efficiently upon irradiation at 660 nm and induces phototoxicity through mitochondrial dysfunction, as confirmed by TEM images showing disruption of mitochondrial structures. It proved more effective in vitro than the non-targeted BPS reference compound. In vivo studies in xenograft mouse models revealed remarkable tumor suppression. RT-PCR analysis revealed decreased gene expression of various angiogenesis factors, including VEGFA and ANGPT2. AZ-BPS also displayed good CAIX-derived targeting when applied to a clinical sample isolated from a breast cancer patient. The enhanced therapeutic efficacy displayed by AZ-BPS relative to the BPS control was thus attributed to the additive effects of photocytotoxicity and angiogenesis inhibition through CAIX knockdown.
The combination of two modalities, namely, PDT and anti-angiogenesis, provides a benefit that cannot be achieved by either alone. In overcoming the effects of hypoxia, the present approach involving small molecule (AZ) targeting of CAIX and BPS-based photokilling is expected to open up new prospects in the field of PDT while advancing those associated with anti-angiogenesis therapy. It is thus expected to lead to important new developments in the areas of cancer diagnosis and treatment.
Supplementary Material
Acknowledgments
This work was supported by CRI project (No. 2009-0081566, J.S.K.) and the Basic Science Research Programs (2015R1A6A3A04058789, H. S. J, and 2015R1C1A1A02036905, J.H.) from the National Research Foundation of Korea (NRF) funded by the Ministry of Education and by the Ministry of Science, ICT and Future Planning. This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MEST) (2012M3A9B4028636 and 2012M3A9C7050139, J.K.). The work in Austin was supported by the National Institutes of Health (CA68682, J.L.S.) and the Robert A. Welch Foundation.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b02396.
Detailed experimental conditions and methods, synthesis, structural characterization, spectroscopic data, molecular docking studies, and biological experiments (PDF)
ORCID
Hu Shi: 0000-0002-5466-5783
Jonathan L. Sessler: 0000-0002-9576-1325
Jin Yong Lee: 0000-0003-0360-5059
Notes
The authors declare no competing financial interest.
References
- 1.Li J, Zhang YP, Kirsner RS. Microsc Res Tech. 2003;60:107–114. doi: 10.1002/jemt.10249. [DOI] [PubMed] [Google Scholar]
- 2.Lancerotto L, Orgill DP. Adv Wound Care (New Rochelle) 2014;3:626–634. doi: 10.1089/wound.2013.0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carmeliet P, Jain RK. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 4.Folkman J. Nat Rev Drug Discovery. 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 5.Folkman J. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 6.Carmeliet P, Jain RK. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P, Munshi N, Anaissie E, Wilson C, Dhodapkar M, Zeldis J, Barlogie B. N Engl J Med. 1999;341:1565–1571. doi: 10.1056/NEJM199911183412102. [DOI] [PubMed] [Google Scholar]
- 8.O’Donnell A, Padhani A, Hayes C, Kakkar AJ, Leach M, Trigo JM, Scurr M, Raynaud F, Phillips S, Aherne W, Hardcastle A, Workman P, Hannah A, Judson I. Br J Cancer. 2005;93:876–883. doi: 10.1038/sj.bjc.6602797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaheen RM, Davis DW, Liu W, Zebrowski BK, Wilson MR, Bucana CD, McConkey DJ, McMahon G, Ellis LM. Cancer Res. 1999;9:5412–5416. [PubMed] [Google Scholar]
- 10.Ray JM, Stetler-Stevenson WG. Eur Respir J. 1994;7:2062–2072. [PubMed] [Google Scholar]
- 11.Moses MA. Stem Cells. 1997;15:180–189. doi: 10.1002/stem.150180. [DOI] [PubMed] [Google Scholar]
- 12.Rosen L. Oncologist. 2000;5:20–27. doi: 10.1634/theoncologist.5-suppl_1-20. [DOI] [PubMed] [Google Scholar]
- 13.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 14.Gragoudas ES, Adamis AP, Cunningham ET, Jr, Feinsod M, Guyer DR. N Engl J Med. 2004;351:2805–2816. doi: 10.1056/NEJMoa042760. [DOI] [PubMed] [Google Scholar]
- 15.Lucky SS, Soo KC, Zhang Y. Chem Rev. 2015;115:1990–2042. doi: 10.1021/cr5004198. [DOI] [PubMed] [Google Scholar]
- 16.Henderson BW, Dougherty TJ. Photochem Photobiol. 1992;55:145–157. doi: 10.1111/j.1751-1097.1992.tb04222.x. [DOI] [PubMed] [Google Scholar]
- 17.Dolmans DE, Fukumura D, Jain RK. Nat Rev Cancer. 2003;3:380–387. doi: 10.1038/nrc1071. [DOI] [PubMed] [Google Scholar]
- 18.Cheng L, Wang C, Feng LZ, Yang K, Liu Z. Chem Rev. 2014;114:10869–10939. doi: 10.1021/cr400532z. [DOI] [PubMed] [Google Scholar]
- 19.Hung H-I, Klein OJ, Peterson SW, Rokosh SR, Osseiran S, Nowell NH, Evans CL. Sci Rep. 2016;6:33234. doi: 10.1038/srep33234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallagher-Colombo SM, Finlay JC, Busch TM. In: Resistance to Photodynamic Therapy in Cancer. Rapozzi V, Jori G, editors. Springer; Switzerland: 2015. [Google Scholar]
- 21.Kaya M, Wada T, Akatsuka T, Kawaguchi S, Nagoya S, Shindoh M, Higashino F, Mezawa F, Okada F, Ishii S. Clin Cancer Res. 2000;6:572–577. [PubMed] [Google Scholar]
- 22.Ishigami SI, Arii S, Furutani M, Niwano M, Harada T, Mizumoto M, Mori A, Onodera H, Imamura M. Br J Cancer. 1998;78:1379–1384. doi: 10.1038/bjc.1998.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inoue K, Ozeki Y, Suganuma T, Sugiura Y, Tanaka S. Cancer. 1997;79:206–213. doi: 10.1002/(sici)1097-0142(19970115)79:2<206::aid-cncr2>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 24.Olivo M, Bhuvaneswari R, Lucky SS, Dendukuri N, Soo-Ping Thong P. Pharmaceuticals. 2010;3:1507–1529. doi: 10.3390/ph3051507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrario A, von Tiehl KF, Rucker N, Schwarz MA, Gill PS, Gomer CJ. Cancer Res. 2000;60:4066–4069. [PubMed] [Google Scholar]
- 26.Giatromanolaki A, Koukourakis MI, Sivridis E, Pastorek J, Wykoff CC, Gatter KC, Harris AL. Cancer Res. 2001;61:7992–7998. [PubMed] [Google Scholar]
- 27.Kaluz S, Kaluzova M, Liao SY, Lerman M, Stanbridge EJ. Biochim Biophys Acta, Rev Cancer. 2009;1795:162–172. doi: 10.1016/j.bbcan.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDonald PC, Winum JY, Supuran CT, Dedhar S. Oncotarget. 2010;1:84–97. doi: 10.18632/oncotarget.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lock FE, McDonald PC, Lou Y, Serrano I, Chafe SC, Ostlund C, Aparicio S, Winum J-Y, Supuran CT, Dedhar S. Oncogene. 2013;32:5210–5219. doi: 10.1038/onc.2012.550. [DOI] [PubMed] [Google Scholar]
- 30.Supuran CT. Nat Rev Drug Discovery. 2008;7:168–181. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- 31.Lou Y, McDonald PC, Oloumi A, Chia S, Ostlund C, Ahmadi A, Kyle A, Auf dem Keller U, Leung S, Huntsman D, Clarke B, Sutherland BW, Waterhouse D, Bally M, Roskelley C, Overall CM, Minchinton A, Pacchiano F, Carta F, Scozzafava A, Touisni N, Winum J-Y, Supuran CT, Dedhar S. Cancer Res. 2011;71:3364–3376. doi: 10.1158/0008-5472.CAN-10-4261. [DOI] [PubMed] [Google Scholar]
- 32.McIntyre A, Patiar S, Wigfield S, Li J-l, Ledaki I, Turley H, Leek R, Snell C, Gatter K, Sly WS, Vaughan-Jones RD, Swietach P, Harris AL. Clin Cancer Res. 2012;18:3100–3111. doi: 10.1158/1078-0432.CCR-11-1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takemoto K, Matsuda T, McDougall M, Klaubert DH, Hasegawa A, Los GV, Wood KV, Miyawaki A, Nagai T. ACS Chem Biol. 2011;6:401–406. doi: 10.1021/cb100431e. [DOI] [PubMed] [Google Scholar]
- 34.Cecchi A, Hulikova A, Pastorek J, Pastoreková S, Scozzafava A, Winum J-Y, Montero J-L, Supuran CT. J Med Chem. 2005;48:4834–4841. doi: 10.1021/jm0501073. [DOI] [PubMed] [Google Scholar]
- 35.Kamkaew A, Lim SH, Lee HB, Kiew LV, Chung LY, Burgess K. Chem Soc Rev. 2013;42:77–88. doi: 10.1039/c2cs35216h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sundriyal S, Viswanad B, Ramarao P, Chakraborti AK, Bharatam PV. Bioorg Med Chem Lett. 2008;18:4959–4962. doi: 10.1016/j.bmcl.2008.08.028. [DOI] [PubMed] [Google Scholar]
- 37.Morone M, Beverina L, Abbotto A, Silvestri F, Collini E, Ferrante C, Bozio R, Pagani GA. Org Lett. 2006;8:2719–2722. doi: 10.1021/ol060742i. [DOI] [PubMed] [Google Scholar]
- 38.Adarsh N, Avirah RR, Ramaiah D. Org Lett. 2010;12:5720–5723. doi: 10.1021/ol102562k. [DOI] [PubMed] [Google Scholar]
- 39.Nowak-Sliwinska P, Karocki A, Elas M, Pawlak A, Stochel G, Urbanska K. Biochem Biophys Res Commun. 2006;349:549–555. doi: 10.1016/j.bbrc.2006.08.060. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Wang H, Oosterwijk E, Tu C, Shiverick KT, Silverman DN, Frost SC. Cancer Invest. 2009;27:613–623. doi: 10.1080/07357900802653464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peng Q, Warloe T, Berg K, Moan J, Kongshaug M, Giercksky KE, Nesland JM. Cancer. 1997;79:2282–2308. doi: 10.1002/(sici)1097-0142(19970615)79:12<2282::aid-cncr2>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 42.Nishimori I, Vullo D, Innocenti A, Scozzafava A, Mastrolorenzo A, Supuran CT. J Med Chem. 2005;48:7860–7866. doi: 10.1021/jm050483n. [DOI] [PubMed] [Google Scholar]
- 43.Huang H, Bhat A, Woodnutt G, Lappe R. Nat Rev Cancer. 2010;10:575–585. doi: 10.1038/nrc2894. [DOI] [PubMed] [Google Scholar]
- 44.Harrell JC, Pfefferle AD, Zalles N, Prat A, Fan C, Khramtsov A, Olopade OI, Troester MA, Dudley AC, Perou CM. Clin Exp Metastasis. 2014;31:33–45. doi: 10.1007/s10585-013-9607-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miyadera K, Sumizawa T, Haraguchi M, Yoshida H, Konstanty W, Yamada Y, Akiyama S, Yamada Y. Cancer Res. 1995;55:1687–1690. [PubMed] [Google Scholar]
- 46.Tafreshi NK, Bui MM, Bishop K, Lloyd MC, Enkemann SA, Lopez AS, Abrahams D, Carter BW, Vagner J, Grobmyer SR, Gillies RJ, Morse DL. Clin Cancer Res. 2012;18:207–219. doi: 10.1158/1078-0432.CCR-11-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rahman M, Mohammed S. Oncol Lett. 2015;10:1233–1239. doi: 10.3892/ol.2015.3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.