

Abstract
Current disjunct patterns can result from long‐distance dispersal or postglacial contraction. We herein investigate the evolutionary history of Triplostegia to elucidate the disjunction between the Himalaya–Hengduan Mountain region (HHM) and Taiwan (TW). Genetic structure of Triplostegia was investigated for 48 populations using sequences from five chloroplast loci and the ribosomal nuclear internal transcribed spacer. Divergence time estimation, ancestral area reconstruction, and species distribution modeling (SDM) were employed to examine the biogeographic history of Triplostegia. Substantial genetic differentiation among populations from southwestern China (SW), Central China (CC), and TW was detected. Triplostegia was inferred to have originated in SW, and diversification began during the late Miocene; CC was colonized in the mid‐Pliocene, and TW was finally colonized in the early Pleistocene. SDM suggested an expansion of climatically suitable areas during the Last Glacial Maximum and range contraction during the Last interglacial in Triplostegia. Disjunction between HHM and TW in Triplostegia is most likely the consequence of topographic isolation and postglacial contraction. The potential climatic suitability areas for Triplostegia by 2070s (2061–2080) are predicted to slightly shrink and move northward. With continued global warming and human‐induced deforestation, extinction risk may increase for the cold‐adapted species, and appropriate strategies should be employed for ecosystem conservation.
Keywords: conservation, Himalaya–Hengduan Mountain region, phylogeography, species distribution modeling, Taiwan, Triplostegia
1. INTRODUCTION
Organismal genetic structure and distributions have been greatly impacted by climatic oscillations, especially the Quaternary glacial/interglacial cycles (Comes & Kadereit, 1998; Hewitt, 2000; Lomolino, Riddle, Whittaker, & Brown, 2010). Compared with North America and northern Europe, East Asia was inferred to be less affected by glaciation except in the higher mountains (Pinot et al., 1999; Shi & Yao, 2002). Numerous species show disjunct distributions in the high mountains of East Asia (Li et al., 2016; Liu et al., 2014; Meng et al., 2017; Wang, 1989; Wang et al., 2013). In particular, disjunctions between the Himalaya–Hengduan Mountain region (HHM) and Taiwan (TW) have received extensive attention because of the geographically distant separation of the two regions by the Taiwan Strait and a terrestrial distance of over 2,000 km (Chen, Ying, & Lu, 2012; Wu & Wang, 2001; Ye, Chen, Liu, Qin, & Yang, 2012).
Himalaya–Hengduan Mountain region and TW are two of the most important biodiversity hotspots worldwide, with high species richness and endemism (Lu, Peng, Cheng, Hong, & Chiang, 2001; Myers, Mittermeier, Mittermeier, Da Fonseca, & Kent, 2000; Tang, Wang, Zheng, & Fang, 2006; Wei, Yang, Li, & Wang, 2010). HHM is located in southwestern China and includes the eastern Qinghai–Tibetan Plateau (QTP), Hengduan Mountains (Mts.), and adjacent areas with an average elevation of more than 4,000 m (Peng, Wan, & Luo, 2000; Zhang, Li, & Zheng, 2002). Dramatic climatic change events have occurred in HHM with the accelerated uplift of the QTP during the Cenozoic (Favre et al., 2015; Wen, Zhang, Nie, Zhong, & Sun, 2014). Taiwan is the largest subtropical mountainous island situated at the Tropic of Cancer in the monsoonal western Pacific region, which began to emerge during the late Miocene (c. 9–5 million years ago, Ma; Sibuet & Hsu, 1997, 2004). There are over 100 species pairs that show disjunct distributions between HHM and TW (Ye et al., 2012). Based on previous studies (Lu et al., 2001; Wang et al., 2013; Ye, Zhu, Chen, Zhang, & Bu, 2014), two hypotheses have been proposed to elucidate the formation of the HHM‐TW disjunction: long‐distance dispersal and postglacial contraction.
Long‐distance dispersal in plants usually involves rare events driven by dispersal vectors such as animals, currents, winds, or water (Nathan, 2006). Phylogeographic study of Cunninghamia revealed that populations in Taiwan were derived from continental Asia via long‐distance seed dispersal (Lu et al., 2001). The postglacial contraction hypothesis states that many species had a continuous distribution from Mainland China to TW during the Quaternary glacial period, and disjunction between HHM and TW occurred when taxa migrated to higher altitudes as the Earth became warmer (Chen et al., 2012; Wang, 1989; Wang et al., 2013; Ye et al., 2014). Most disjunct genera and species between HHM and TW are herbaceous (Chen, Ying, & Lu, 2012; Ye et al., 2012). However, few studies on the HHM‐TW disjunction focused on herbaceous plants, so it remains unclear whether similar mechanisms and timing shaped the disjunct patterns of woody and herbaceous plants between HHM and TW.
Recently, species distribution modeling (SDM) has been used to infer range shifts of plants and animals in response to the Quaternary climate oscillations (Jakob, Ihlow, & Blattner, 2007; Luo et al., 2016; Peterson, Ammann, & Diniz‐Filho, 2013). In general, most temperate species retracted into smaller refugial areas during glacial periods and would experience range expansion during interglacial periods (Bueno et al., 2016; Hewitt, 1996, 1999, 2000). However, several recent studies suggested a different biogeographic history for cold‐adapted coniferous species that inhabit high mountains, with range expansion during glacial periods and contraction during interglacial periods (Liu et al., 2013; Shao & Xiang, 2015; Tian, López‐Pujol, Wang, Ge, & Zhang, 2010; Wang et al., 2017). Compared with lowland species, mountainous species might be more likely to survive during climatic oscillations, because short‐distance vertical migrations allow them to track their optimal temperature niche (Hoorn, Mosbrugger, Mulch, & Antonelli, 2013; Sandel et al., 2011). Nevertheless, continued global warming in the near future may be a major threat to cold‐adapted species, especially those that are already confined to mountaintops or islands, because migration to higher elevations may be not possible or range shifts are not fast enough to track suitable climate (Wiens, 2016). How organisms responded to the Quaternary climatic oscillations and predictions of response to future climate can likely be inferred with short‐lived herbaceous plants, which have far more life cycles within a given time period and thus may respond more quickly to rapidly changing environments (Comes & Kadereit, 1998; Faye et al., 2016; Zanne et al., 2014).
As an herbaceous genus of Caprifoliaceae s.l. (The Angiosperm Phylogeny Group, 2016), Triplostegia Wall. ex DC. has a disjunct distribution between HHM and TW (see Figure S1), with only a few sporadic records in Indonesia (Hong, Ma, & Barrie, 2011; Zhang, Chen, Li, Chen, & Tang, 2003). Triplostegia is sensitive to habitat and climatic changes and requires a specific forest habitat that has fertile soil and an altitude ranging from 1,500 to 4,000 m; these features make this genus an ideal model for studying formation of HHM‐TW disjunctions and how herbs respond to Quaternary climate oscillations and future global warming. By sequencing five chloroplast DNA (cpDNA) regions and the ribosomal nuclear internal transcribed spacer (ITS) for 397 individuals from 48 populations, estimating divergence time, reconstructing ancestral areas, and conducting SDM, we (1) investigated the genetic structure and identified possible refugia for Triplostegia; (2) reconstructed the biogeographic history of Triplostegia and explored how the HHM‐TW disjunction formed; and (3) predicted the future climatically suitable areas of the cold‐adapted Triplostegia under a global warming scenario.
2. MATERIALS AND METHODS
2.1. Population sampling
Triplostegia includes two species: T. glandulifera Wall. ex DC. and T. grandiflora Gagnep. (Peng, Tobe, & Takahashi, 1995; Pyck & Smets, 2004; Zhang, Chen, Li, Chen, & Tang, 2003). Triplostegia grandiflora is endemic to China (confined to Yunnan and Sichuan Provinces), and its geographical range overlaps with that of T. glandulifera (Hong, Ma, & Barrie, 2011). Both species are perennial herbs only with minor differences in flower size. We herein treated Triplostegia as a monotypic genus based on the evidence from pollen morphology and phylogenomic analysis of nine complete chloroplast genomes (Niu et al., unpublished).
From September 2011 to August 2014, we collected 397 individuals from 48 natural populations, which covered the entire geographical range of Triplostegia in China. Generally, 10–12 individuals were arbitrarily selected from a population, whereas three to six individuals were collected from populations with few individuals (Table S1). For each individual, leaves were collected and dried in silica gel for DNA extraction. Voucher specimens were deposited at the Herbarium of Institute of Botany, Chinese Academy of Sciences (PE).
2.2. DNA extraction, amplification, and sequencing
Total genomic DNA was extracted from silica gel‐dried leaves using a plant total genomic DNA kit following the manufacturer's protocol (Biomed, Beijing, China). Five cpDNA regions (psbK–psbI, rpl20–rps12, trnH–psbA, trnL–F, and trnS–trnG) and ITS were amplified with primers described in Baldwin (1993), Hamilton (1999), Lahaye, Savolainen, Duthoit, Maurin, and Van der Bank (2008), Sang, Crawford, and Stuessy (1997), Soejima and Wen (2006), Taberlet, Gielly, Pautou, and Bouvet (1991), and White, Bruns, Lee, and Taylor (1990). PCR primers and amplification profiles for each DNA region are shown in Table S2. The PCR products were purified and sequenced by Beijing Biomed Gene Technique Company (http://www.biomed168.com/goods). The forward and reverse sequences were assembled in GENEIOUS 9.1.4 (http://www.geneious.com; Kearse et al., 2012). Sequences were initially aligned using MAFFT 1.3.5 (Katoh, Misawa, Kuma, & Miyata, 2002) and then manually adjusted in GENEIOUS.
2.3. Genetic structure analyses
The number of haplotypes, haplotype diversity (H d), and nucleotide diversity (π) were calculated in DNASP 5.0.0 (Librado & Rozas, 2009). All haplotype sequences identified in this study were deposited in GenBank (https://www.ncbi.nlm.nih.gov/; see Table S3 for accession numbers). Genealogical relationships among haplotypes were investigated using the median‐joining network method as implemented in NETWORK 5.0.0 (Bandelt, Forster, & Röhl, 1999).
The average gene diversity within populations (H S), total gene diversity (H T), and the coefficients of differentiation (G ST, a population differentiation estimate based solely on haplotype frequencies, and N ST, a parameter that considers both haplotype frequencies and their genetic divergence) were estimated using PERMUT 1.0.0 (Pons & Petit, 1996) with 1,000 random permutations. When N ST is larger than G ST, closely related haplotypes occur more often in the same area than less closely related haplotypes, which indicates strong phylogeographic structure.
Spatial genetic structure of cpDNA and ITS haplotypes was analyzed by SAMOVA 1.0 (Dupanloup, Schneider, & Excoffier, 2002). This program uses a simulated annealing approach to identify groups of populations (K) that are geographically homogeneous and maximally differentiated from each other. To maximize the proportion of total genetic variance observed between groups (Φ CT), 1,000 iterations were run for K = (2, …, 8) from each of 100 random initial conditions. To further quantify genetic differentiation among populations and groups, the hierarchical analysis of molecular variance (AMOVA) was estimated in ARLEQUIN 3.5.1 (Excoffier & Lischer, 2010) with significance tests based on 1,000 permutations.
2.4. Demographic reconstruction
Mismatch distribution analysis with a spatial expansion model (Rogers & Harpending, 1992) was conducted in ARLEQUIN to infer the historical demography of Triplostegia based on both cpDNA and ITS datasets. The goodness of fit based on the sum of squared deviation (SSD) and Harpending's raggedness index (H Rag) was tested using 1,000 parametric bootstrap replicates. If the expansion model was not rejected, the formula t = τ/2u was used to estimate the expansion time (t), where u = μkg [μ, substitution rate, substitutions per site per year, subs/site/year; k, average sequence length of cpDNA; g, generation time in years (i.e., age of first reproduction; approximated as 1 year, J. F. Ye, personal observation)]. In addition, statistical significance of population expansion was further examined with two neutrality tests, Tajima's D (Tajima, 1989) and Fu's F S (Fu, 1997), as implemented in DNASP, in which significantly negative values indicate population expansion.
2.5. Phylogenetic relationships among haplotypes
Phylogenetic analyses for cpDNA and ITS haplotypes were performed using the maximum likelihood (ML), Bayesian inference (BI), and neighbor‐joining (NJ) methods, with gaps treated as missing data. The ML analysis was conducted using RAxML 8.2.8 (Stamatakis, 2006, 2014) using a GTR + G substitution model with 1,000 bootstrap (BS) replicates (Stamatakis, Hoover, & Rougemont, 2008) on the CIPRES Science Gateway Portal (https://www.phylo.org/portal2/; Miller, Pfeiffer, & Schwartz, 2010). The best‐fitting model of nucleotide substitutions was selected in JMODELTEST 2.1.4 (Darriba, Taboada, Doallo, & Posada, 2012) under the Akaike information criterion (AIC). BI analyses were conducted in MRBAYES 3.2 (Ronquist & Huelsenbeck, 2003; Ronquist et al., 2012) with the best‐fit model determined above. Two Markov chain Monte Carlo (MCMC) chains were run for two million generations with one tree sampled every 1,000 generations. The average standard deviation of the split frequencies approached 0.01, which indicated that two runs converged to a stationary distribution. After discarding the first 25% trees as burn‐in, a 50% majority‐rule consensus tree was constructed from the remaining trees to estimate posterior probabilities (PP). NJ analysis for ITS was carried out in MEGA 6.0.0 (Tamura, Stecher, Peterson, Filipski, & Kumar, 2013) with 1,000 BS replicates.
2.6. Divergence time estimation
To estimate the crown age of Triplostegia, we downloaded sequences of three cpDNA regions (trnH–psbA, trnL–F, and trnS–trnG) for 41 Dipsacales species (including five Adoxaceae species as out‐groups) from GenBank (Table S4) that represent all major lineages of Dipsacales (Bell & Donoghue, 2005; Carlson, Linder, & Donoghue, 2012; Wang, Landrein et al., 2015). Sequences were assembled, with 16 Triplostegia haplotypes identified from the same three cpDNA regions, to represent all 20 cpDNA haplotypes identified in the phylogeographic analyses. Bayesian searches for tree topologies and node ages of this cpDNA dataset were conducted in BEAST 1.7.3 (Drummond & Rambaut, 2007), which was run on the CIPRES Science Gateway Portal. An uncorrelated lognormal relaxed clock (Drummond, Nicholls, Rodrigo, & Solomon, 2002) was applied with the GTR + G substitution model and a Yule process tree prior. A secondary calibration point (52.8–89.0 Ma; Magallón, Gómez‐Acevedo, Sánchez‐Reyes, & Hernández‐Hernández, 2015) was used to constrain the crown group of Dipsacales that assumed a normal distribution with a mean (± SD) of 70.94 ± 11 Ma (node 1 in Figure S2).
Three fossil dates were used to assign minimum age constraints on three internal nodes of Dipsacales with a lognormal distribution (nodes 2–4 in Figure S2). The earliest fossil record of Viburnum from the late Paleocene to early Eocene (c. 56 Ma; Baskin et al., 2006; Wing et al., 1995) was used to constrain the crown group of Adoxaceae (node 2 in Figure S2; mean = 0, SD = 1.0, offset = 56 Ma) following Moore and Donoghue (2007). Fossil seeds of Weigela have been reported from the Miocene and Pliocene in Poland (Łańcucka‐Środoniowa, 1967), the Miocene in Mammoth Mts., Eastern Russia, the Oligocene and Miocene of Western Siberia (Dorofeev, 1963), and the Miocene in Denmark (Friis, 1985). We thus constrained divergence between Weigela and its sister Diervilla to 23 Ma (node 3 in Figure S2; mean = 0, SD = 1.0, offset = 23 Ma) following Wang, Landrein et al. (2015). Finally, we used the fossil fruits of Diplodipelta from the late Eocene Florissant flora of Colorado (36–35 Ma; Manchester & Donoghue, 1995) to constrain the stem age of Dipelta (node 4 in Figure S2; mean = 0, SD = 1.0, offset = 36 Ma) following Bell and Donoghue (2005) and Wang, Landrein et al. (2015). We assessed estimate robustness by sequentially removing each calibration while retaining the other three calibrations (Wahlberg, Wheat, & Peña, 2013).
To determine the intraspecific node ages within Triplostegia, we applied a similar BEAST analysis to a dataset that included all 20 haplotypes of five cpDNA regions, with Dipsacus asper as the out‐group. We used the median stem age and crown age of Triplostegia obtained from the Dipsacales chronogram to constrain the root and crown age of Triplostegia, respectively (nodes A and B in Figure S2). The normal distribution and a constant‐size coalescent tree prior were applied. MCMC runs were performed, each of 108 generations, with sampling every 104 generations, following a burn‐in of the initial 10% of cycles. We used TRACER 1.5.1 (Raymond & Rousset, 2009) to examine the stationarity of runs. Trees were then compiled into a maximum clade credibility tree using TREEANNOTATOR 1.7.5 (Drummond, Suchard, Xie, & Rambaut, 2012). FIGTREE 1.4.0 (Rambaut, 2009) was used to display mean ages and highest posterior density intervals at 95% for each node.
2.7. Ancestral area reconstruction
We used the Bayesian Binary MCMC (BBM) method implemented in RASP 3.0.0 to estimate the possible ancestral areas of Triplostegia with the trees obtained from the intraspecific BEAST analysis (Yu, Harris, Blair, & He, 2015). Three geographical regions that represent the current distribution of Triplostegia were defined: southwestern China (SW; East Himalayas and Hengduan Mts.); Central China (CC; surrounding areas of the Sichuan Basin); and Taiwan (TW; Taiwan mountain ranges). SW and CC were defined as two separate regions based on their unique haplotypes and altitudinal gradients, despite their geographical proximity. Triplostegia distribution was inferred based on the specimen information from the Chinese Virtual Herbarium (CVH, http://www.cvh.org.cn/), Global Biodiversity Information Facility (GBIF, http://www.gbif.org/), and Specimens Database of Native Plants in Taiwan (SDNPT, http://www.hast.biodiv.tw/Announce/newsC.aspx). The out‐group (Dipsacus asper) is found in both SW and CC, so we set its distribution as SW/CC. The number of maximum areas at each node was set to three. To account for phylogenetic uncertainty, 1,000 trees from BEAST were randomly chosen for BBM analysis. We set the root distribution to null, applied 10 MCMC chains with the JC + G model, and sampled the posterior distribution every 100 generations for 106 generations.
2.8. Species distribution modeling
We used MaxEnt species distribution modeling (henceafter MaxEnt) to predict past, current, and future climatically suitable areas for Triplostegia (Phillips, Anderson, Dudík, Schapire, & Blair, 2017). MaxEnt is one of the most popular tools to model species distributions based on occurrence records and relevant climatic variables, and it was conceptualized in Merow, Smith, and Silander (2013) and Phillips, Anderson, and Schapire (2006) for details.
We first downloaded 19 bioclimatic variables with the 2.5 arc‐minute spatial resolution available from database of WorldClim 1.4 (http://www.worldclim.org; Hijmans, Cameron, Parra, Jones, & Jarvis, 2005). To eliminate multicollinearity effects in the parameter estimates of SDM, we excluded variables with |Pearson's R| ≥ .85, and selected nine variables as climatic predictors to model the past, current, and future climatically suitable areas of Triplostegia (Camargo, Werneck, Morando, Sites, & Avila, 2013). The resultant climatic variables included annual mean temperature (Bio1; °C), mean diurnal range (Bio2; °C), isothermality (Bio3; dimensionless), temperature seasonality (Bio4; Standard Deviation*100), max temperature of warmest month (Bio5; °C), mean temperature of coldest month (Bio6; °C), mean temperature of driest quarter (Bio9; °C), annual precipitation (Bio12; mm), and precipitation seasonality (Bio15; Coefficient of Variation). These nine climatic variables (i.e., the maximum, minimum, mean and standard deviation of temperature and precipitation) can influence the distribution and physiological performance of plant species. Altitude is an important factor to predict suitable habitats for Triplostegia, but we did not take it into consideration due to its close relationship with temperature and precipitation.
Current distribution of Triplostegia was developed using the same nine bioclimatic variables for current climate (1960–1990). To model the paleoclimatic distribution of Triplostegia, we used three global climate models (GCMs: CCSM4, MIROC‐ESM, and MPI‐ESM‐P) for the Last Glacial Maximum (LGM; 0.021 Ma), but only considered NCAR‐CCSM for the Last interglacial (LIG; 0.12–0.14 Ma) because it is the sole GCM available in the database of WorldClim. To predict the future climatically suitable areas of Triplostegia in 2050s (average for 2041–2060) and 2070s (average for 2061–2080), we used three GCMs (CCSM4, MIROC‐ESM and MPI‐ESM‐LR) and two greenhouse gas concentration scenarios as representative concentration pathways (RCPs) of 4.5 (mean, 780 ppm; range, 595–1,005 by 2100) and 8.5 (mean, 1,685 ppm; range, 1,415–1,910 by 2100), representing the low and high gas concentration scenarios, respectively (http://www.ipcc.ch/; http://www.ccafs-climate.org/). RCP 8.5 may have larger cumulative concentrations of carbon dioxide than RCP 4.5, resulting in a different pattern of future climate change in response to various anthropogenic concentrations of greenhouse gases and other pollutants (http://www.ipcc.ch/report/ar5/).
We corrected all the occurrence data to reduce the negative effect of sampling bias on the performance of the SDMs. For records from GBIF, CVH, and SDNPT, we excluded the duplicated entries within a 2.5 arc‐minute pixel (4.3 km at the equator) and omitted entries with implausible geographical coordinates using ArcGIS 10.2 (ESRI Inc., Redlands, CA, USA; http://www.esri.com/software/arcgis/arcgis-for-desktop) and Google Earth (http://earth.google.com/; Gueta & Carmel, 2016; Maldonado et al., 2015). We also checked voucher photos for records from CVH and SDNPT to make sure that the species are correctly identified. Finally, a total of 250 cleaned occurrence records were obtained from GBIF, CVH, SDNPT, and our own collections (Table S1 and Figure S3), which well covered the distribution range of Triplostegia (Hong, Ma, & Barrie, 2011).
As suggested by previous studies (Halvorsen et al., 2016; Martinez‐Freiria, Velo‐Antón, & Brito, 2015; Merow, Smith, & Silander, 2013; Phillips et al., 2017; Radosavljevic, Anderson, & Araújo, 2014), our modeling was set as follows: (1) the regularization multiplier level was at two to produce smooth and general response curves that represent a biologically realistic model; (2) cloglog output format was used to evaluate distribution probability of Triplostegia; (3) ten independent replicates of cross‐validation procedures for model testing; (4) the maximum number of background points was set to 104; and (5) jackknife analyses of the regularized gain using training data were conducted to elucidate the importance of each predictor (Fawcett, 2006). The other settings followed the suggestions of Merow, Smith, and Silander (2013). No obvious differences were detected among the predictions based on the three GCMs (Figure S4). To decrease the prediction uncertainty, mean consensus for the pixels of distribution probabilities modeled by three GCMs was calculated to assess the distributions of Triplostegia during LGM and the future (RCPs 4.5 and 8.5 for both 2050s and 2070s).
We evaluated the model performance by calculating the area under the receiver operating characteristic (ROC) curve (AUC). AUC was used to assess the accuracy of predictions, and models with AUC values larger than 0.7 were considered useful for our study (Phillips & Dudík, 2008). However, using the traditional AUC may be not sufficient to assess the performance of SDMs. We herein used a binomial test (based on the training omission rate) for model validation based on the occurrence data only. The training omission rate is the proportion of training occurrence records among pixels of predicted absences (Phillips & Dudík, 2008; Phillips, Anderson, & Schapire, 2006). One‐sided test was used for the null hypothesis, and the test points are predicted no better than those by a random prediction (Phillips & Dudík, 2008; Phillips, Anderson, & Schapire, 2006). We used the 11 common thresholds defaulted by MaxEnt to define binomial probabilities (Phillips, Anderson, & Schapire, 2006). The models with <17% training omission rate were useful for our study (Phillips, Anderson, & Schapire, 2006).
3. RESULTS
3.1. Genetic diversity and population differentiation
We obtained 2,812 bp of cpDNA sequences with 430 bp of psbK–psbI, 718 bp of rpl20–rps12, 201 bp of trnH–psbA, 788 bp of trnL–F, and 675 bp of trnS–trnG. There were 20 haplotypes defined by 33 polymorphic sites (Table S1) and three haplotype clusters that corresponded to three geographical regions: SW, CC, and TW (Figure 1a). H d and π for the cpDNA dataset were estimated to be 0.87 and 0.24 × 10−2, respectively (Table S1). H T and H S were 0.88 and 0.18, respectively. The coefficients of differentiation measured over the 48 populations were G ST = 0.80 and N ST = 0.96. Permutation test revealed that N ST was significantly higher than G ST (p < .05), which indicates strong phylogeographic structure for the Triplostegia cpDNA haplotypes. The highest levels of cpDNA sequence diversity (π) were recorded within the SW group and the lowest levels in the CC group (Table S1). Differences in cpDNA sequences were highly significant among the three geographical regions (F CT = 0.85, p < .001, AMOVA; Table 1), populations within regions (F SC = 0.77, p < .001, AMOVA; Table 1), and among all populations (F ST = 0.97, p < .001, AMOVA; Table 1). Differences in cpDNA sequences were significant between every two of the three geographical regions (Table S5). The highest differentiation index was found between SW and TW (F CT = 0.88***), and the lowest between CC and TW (F CT = 0.68***).
Figure 1.
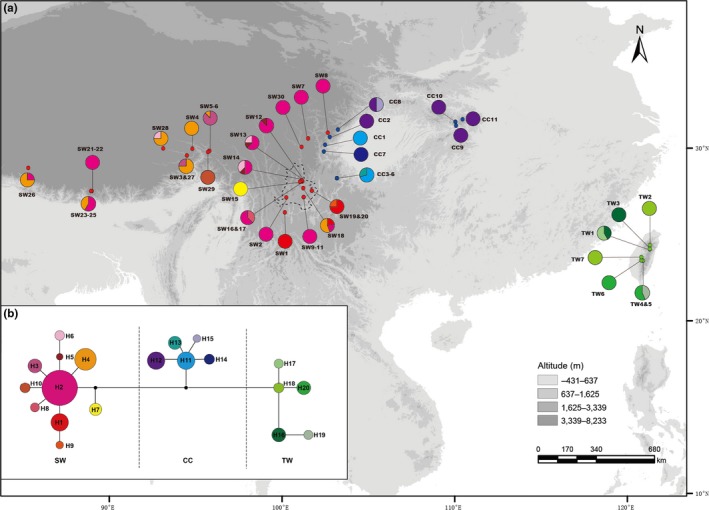
(a) Geographical distribution of the cpDNA haplotypes detected in Triplostegia (see Table S1 for population codes) and (b) network of genealogical relationships among 20 haplotypes. Each circle represents a single haplotype with sizes in proportion to frequency. Black dots in network represent missing haplotypes. Potential interglacial refugia recognized by this study are encircled by dashed lines
Table 1.
Molecular variances of cpDNA and ITS for 48 Triplostegia populations
| Gene type | Source of variation | df | SS | VC | PV | Fixation index |
|---|---|---|---|---|---|---|
| cpDNA | Among regions | 2 | 1,167.64 | 5.46 | 85.21 | F CT = 0.85*** |
| Among populations | 45 | 280.45 | 0.73 | 11.38 | F SC = 0.77*** | |
| Within populations | 349 | 76.29 | 0.22 | 3.14 | F ST = 0.97*** | |
| Total | 396 | 1,524.38 | 6.40 | |||
| ITS | Among regions | 2 | 1,070.74 | 5.01 | 88.48 | F CT = 0.89*** |
| Among populations | 45 | 237.82 | 0.64 | 11.29 | F SC = 0.98*** | |
| Within populations | 349 | 4.64 | 0.013 | 0.23 | F ST = 0.99*** | |
| Total | 396 | 1,313.20 | 5.66 |
df, degree of freedom; SS, sum of squares; VC, variance components; PV, percentage of variation; ***, p < .001, 1,000 permutations.
The matrix for aligned ITS had 573 bp with 36 polymorphic sites and 14 ITS haplotypes (see Table S3 for accession numbers). H d and π for ITS were estimated to be 0.67 and 0.011, respectively (Table S1). Network and geographical distribution analysis based on ITS also recognized three groups in SW, CC, and TW (Figure 2). The highest level of ITS π was recorded within the SW group and the lowest in the CC group (Table S1). Differences in ITS sequences were highly significant among the three geographical regions (F CT = 0.89, p < .001, AMOVA; Table 1), between populations within regions (F SC = 0.98, p < .001), and among all populations (F ST = 0.99, p < .001). Differences in ITS sequences were significant between each pair of geographical regions (Table S6). The highest differentiation index was observed between SW and TW (F CT = 0.91***) and the lowest between CC and TW (F CT = 0.84***). Analysis of the ITS variation across all populations revealed that the total gene diversity (H T = 0.67) was much higher than the within‐population diversity (H S = 0.025). The coefficients of differentiation measured over the 48 populations were G ST = 0.96 and N ST = 0.99. Permutation test revealed that N ST was significantly higher than G ST (p < .05), which indicated a strong phylogeographic structure for ITS haplotypes. SAMOVA indicated that values of Φ CT reached a plateau at 0.88 for three groups (K = 3; Figure 2c), which corresponded to the three clades in SW, CC, and TW.
Figure 2.
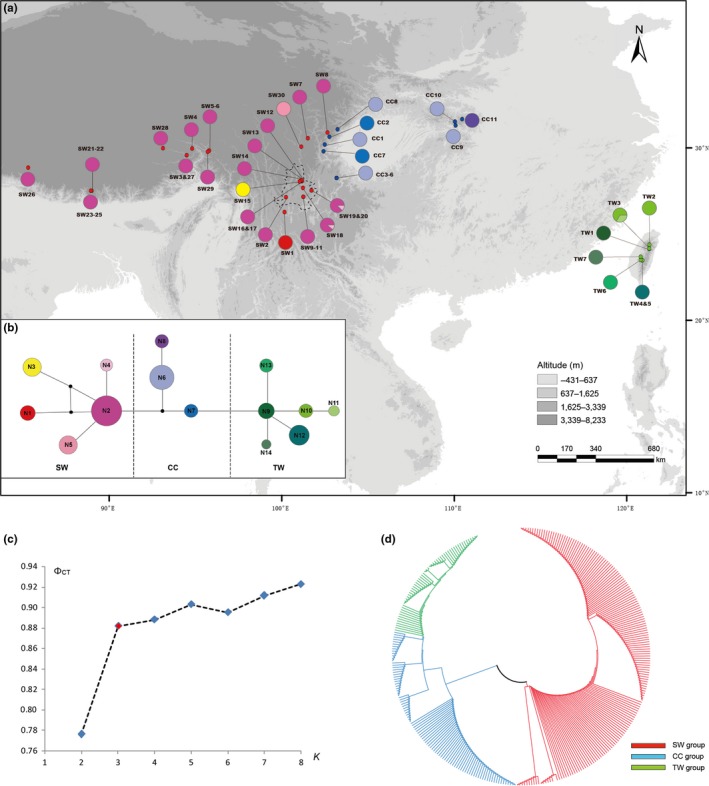
Analysis based on ITS dataset. (a) Geographical distribution of haplotypes in Triplostegia (see Table S1 for population codes); (b) network of genealogical relationships between 14 haplotypes. Each circle represents a single haplotype sized in proportion to its frequency. Black dots in the network represent missing haplotypes; (c) results of SAMOVA (the red point indicated the optimized number of groups K); and (d) neighbor‐joining tree without out‐group. Potential interglacial refugia recognized by this study are encircled by dashed lines
3.2. Phylogenetic relationships and divergence times
Both ML and BI analyses based on the 20 cpDNA haplotypes supported three lineages within Triplostegia that corresponded to their distributions in SW, CC, and TW (Figure S5). However, incongruence existed regarding the relationships among three lineages between the BI (SW(CC, TW)) and ML (TW(SW,CC)) analyses, although the support values were not high. The ML, BI, and NJ analyses based on ITS retrieved the same topology (SW(CC, TW)) as the BI tree based on cpDNA (Figure S5).
The chronogram for Dipsacales derived from cpDNA supported the monophyly of the 16 Triplostegia haplotypes (PP = 1.00; Figure S2). The stem age of Triplostegia was estimated to be c. 48.29 Ma (95% highest posterior density, HPD: 31.65–66.45 Ma; node A; Table S7) and crown age of Triplostegia as c. 10.92 Ma (95% HPD: 4.28–23.76 Ma; node B; Table S7). The divergence time within Triplostegia showed that the coalescent time of all 20 haplotypes was during the late Miocene (6.79 Ma, 95% HPD: 2.50–13.6 Ma; node I in Figure S5). Within the CC–TW lineage, CC and TW group split c. 4.03 Ma (95% HPD: 1.31–8.62 Ma; node II in Figure S5). Split within the three groups occurred during the early Pleistocene: SW (c. 1.98 Ma, 95% HPD: 0.62–4.83 Ma; node III in Figure S5), CC (c. 1.41 Ma, 95% HPD: 0.26–3.85 Ma; node IV in Figure S5), and TW (c. 2.15 Ma, 95% HPD: 0.48–5.32 Ma; node V in Figure S5). For this chronogram, BEAST provided an average substitution rate of 0.49 × 10−9 subs/site/year, which is similar to that of Tetrastigma hemsleyanum derived from three cpDNA markers (petL–psbE, trnK–matK, and rbcL; 0.50 × 10−9 subs/site/year; Wang, Jiang et al., 2015).
3.3. Demographical analyses
The overall observed mismatch distribution of pairwise nucleotide differences was not unimodal and rejected the hypothesis that there was recent demographic population growth in Triplostegia (Figures 3a and S6). However, for each subregion (SW, CC, and TW), populations had a unimodal shape in the mismatch distribution. This supported the hypothesis that there had been recent demographic population expansion in the genus (Figures 3b–d and S6b–d). However, based on the SSD and H Rag statistics of cpDNA sequences (Table 2), only SW had a good statistical support for the spatial expansion model (p > .05). In addition, Tajima's D and Fu's F S values of cpDNA and ITS sequences were all negative in SW, which further indicated that populations in SW experienced sudden expansion (Table 2 & Figure S6). Based on the value of τ and assuming a substitution rate of 0.49 × 10−9 subs/site/year for cpDNA, the demographic expansion in SW populations was inferred to have occurred c. 0.33 Ma (Table 2).
Figure 3.
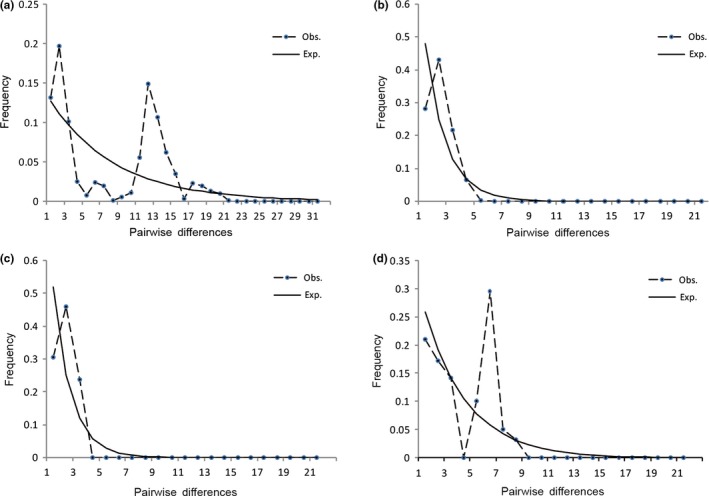
Number of pairwise nucleotide differences in Triplostegia based on chloroplast DNA sequences in all areas (a), southwestern China (b), Central China (c), and Taiwan (d). Black dots and dashed line show observed values (Obs.); solid lines indicate expected values (Exp.) under a model of sudden (stepwise) population expansion
Table 2.
Results of mismatch distribution analysis and neutrality tests for pooled populations of Triplostegia lineages
| Region | τ | θ0 | θ1 | SSD (p value) | H Rag (p value) | D | F S | t |
|---|---|---|---|---|---|---|---|---|
| Southwestern China | 0.94 | 0.11 | 30,000.62 | .16 | .30 | −0.79 | −2.38 | 0.33 |
| Central China | 0.63 | 0.08 | 0.60 | .013* | .027* | 0.38 | 0.22 | — |
| Taiwan | 1.53 | 0.00 | 14,285.81 | .031* | .16 | 2.26* | 3.93 | — |
τ = 2ut, where t is the expansion time and u is the mutation rate per generation; θ0, pre‐expansion population size; θ1, postexpansion population size; SSD, sum of squared deviations; H Rag, Harpending's raggedness index; t, time since expansion (Ma); *, p < .05.
3.4. Ancestral area reconstruction
Based on the topology of the intraspecific chronogram (Figure S5), BBM analysis supported an ancestral distribution in SW (node I in Figure 4). BBM analysis inferred that Triplostegia colonized CC in the mid‐Pliocene (c. 4.03 Ma; 95% HPD: 1.30–8.60 Ma; node II in Figures 4 and S5) and arrived TW in the early Pleistocene (c. 1.98 Ma, 95% HPD: 0.62–4.83 Ma; node III in Figures 4 and S5). The three clades, which corresponded to their distributions in SW, CC, and TW, each had their own most recent common ancestors (nodes III, IV, and V in Figure 4), which indicated possible in situ diversification in the three regions.
Figure 4.
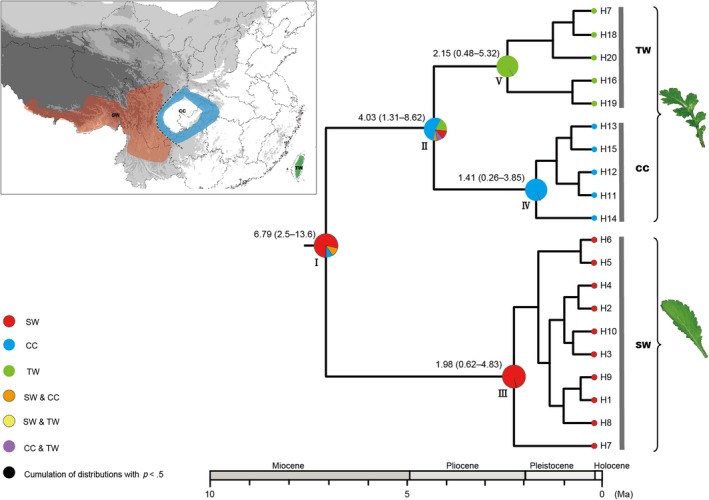
Ancestral area reconstructions based on the Bayesian binary Markov chain Monte Carlo (BBM) method implemented in RASP using the BEAST‐derived chronogram of Triplostegia. The upper left map shows major distribution divisions of Triplostegia in China: southwestern (SW), Central (CC), and Taiwan (TW). Pie chart at each node illustrates the marginal probabilities for each alternative ancestral area derived from BBM with the maximum area number set to three. The legend denotes possible ancestral ranges at different nodes. Ages of key nodes are labeled above branches. The out‐group (Dipsacus asper, see Figure S5) was not shown here
3.5. Species distribution modeling
The training and test AUC values of the models were larger than 0.9, and all the training omission rates were <17%, indicating a good performance of the predictive model. The jackknife results showed that temperature was the most influential factor.
During the LIG, high climatic suitability areas for Triplostegia were more restricted than its current range in southwestern China (e.g., the eastern Himalayas, Hengduan mountain regions; Figure 5a). The LGM modeling predicted a more widespread of high climatic suitability areas than current in East China, extending southward into Taiwan, plus a higher suitability of HHM regions (Figure 5b). The predicted suitable habitats of Triplostegia under current conditions were generally similar to the actual species distribution (Figure 5c). High climatic suitability areas for Triplostegia by 2050s (Figure S7a,b) and 2070s (Figures 5d and S7c) may experience northward range shift in Mainland China, and the suitability slightly decline in southern Hengduan Mountain regions and Taiwan.
Figure 5.
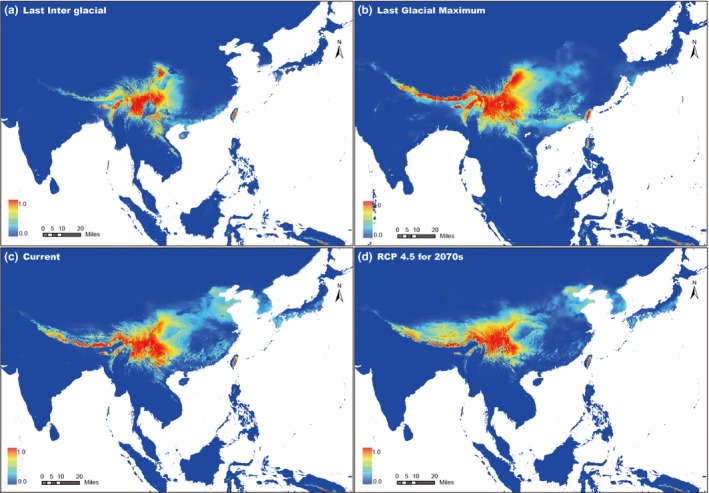
Species distribution models showing climatic suitability for Triplostegia in East Asia: (a) the Last interglacial (LIG; 0.12–0.14 Ma); (b) the Last Glacial Maximum (LGM; 0.021 Ma); (c) current conditions (current, 1960–1990); and (d) 2070s (2061–2080) with the representative concentration pathways (RCP) of 4.5. Climatic suitability increases with color from blue to red. Resolution for the potential distribution map is 2.5 arc‐minutes
4. DISCUSSION
4.1. Genetic diversity and potential refugia
Results from PERMUT (N ST > G ST, p < .05), AMOVA (Table 1), and SAMOVA (Figure 2c) revealed strong genetic structure for the Triplostegia populations from SW, CC, and TW (Figures 1 and 2). The three distinct haplotype groups could be distinguished by a strong underlying phylogenetic hierarchy that supports three major clades in Triplostegia that correspond to their distributions in SW, CC, and TW (Figures 2d and S5). The strong genetic structure among Triplostegia populations is consistent with the three great topographic steps in China. SW populations are all from the first‐step terrain in China with an average elevation above 3,144 m, and have 10 cpDNA and five ITS haplotypes (Table S1). CC populations are distributed on the second‐step terrain around Sichuan Basin (average elevation: 2,238 m), and share five cpDNA and three ITS haplotypes. TW populations are all from the central mountain range of Taiwan (average elevation: 2,321 m) and possess five cpDNA and six ITS haplotypes. Strong geography‐haplotype‐correlated genetic structures were also detected in birds (Qu et al., 2015; Wang et al., 2013), insects (Ye et al., 2014), and plants (Gao et al., 2007; Meng et al., 2017), which highlight the importance of topographic complexity in promoting species differentiation both by increasing habitat diversity and limiting gene flow between elevation‐restricted populations (Hoorn, Mosbrugger, Mulch, & Antonelli, 2013; Verboom, Bergh, Haiden, Hoffmann, & Britton, 2015).
Potential refugia are generally predicted to have a widespread ancestral haplotype, and other mutationally derived and unique haplotypes (Crandall & Templeton, 1993). According to coalescent theory, most ancient haplotypes should be located at the interior nodes of a haplotype network, whereas the most recent haplotypes should be at the tips (Posada & Crandall, 2001). In the cpDNA network of Triplostegia, haplotype H2 from SW is at the central position with high frequency (Figure 1). The ancestral haplotype occurs widely across SW, which indicates that the species had persisted over a longer period of time in SW during the glacial epochs. Three populations (SW13, SW14, and SW18) from Muli County, Sichuan Province, have higher haplotype and nucleotide diversity (SW13: H d = 0.61, π = 0.34 × 10−3; SW14: H d = 0.68, π = 0.40 × 10−3; SW18: H d = 0.67, π = 0.34 × 10−3) and contain additional derived and unique haplotypes (Figure 1). Muli County is located in the central Hengduan Mts., with numerous north‐south trending valleys and ridges extending from 1,000 m to over 6,000 m in elevation (Peng, Wan, & Luo, 2000), which might have provided suitable habitats for Triplostegia during the Pleistocene climate oscillations. Multiple refugia have been inferred in the Hengduan Mts. for many animals and plants during the Pleistocene glacial periods (Gao et al., 2007; Sun et al., 2014; Wen et al., 2014; Ye et al., 2014; Zhang et al., 2015).
4.2. Biogeographic history
Our analyses revealed that coalescence of all Triplostegia cpDNA haplotypes is most likely to have occurred in SW, and diversification initiated during the late Miocene (c. 6.79 Ma; 95% HPD: 2.50–13.6 Ma; node I in Figures 4 and S5). The late Miocene has been recognized as an important period of diversification for both woody and herbaceous plants (e.g., Heterobalanus, Meng et al., 2017; Cercidiphyllum, Qi et al., 2012; Tetracentron, Sun et al., 2014; Tetrastigma, Wang, Jiang et al., 2015). Differentiation within Triplostegia might have been initiated and driven by both uplift of the Hengduan Mts. and global cooling after the late Miocene (Nagalingum et al., 2011; Zachos, Dickens, & Zeebe, 2008). The Hengduan Mts., at the southeastern margin of the QTP, are proposed to have experienced rapid uplift during the late Miocene (Clark et al., 2005; Kirby et al., 2002). By investigating the evolutionary histories of multiple plant groups, Xing and Ree (2017) detected an increase in the rate of in situ diversification in the Hengduan Mts. during the late Miocene (c. 8.00 Ma). The SW origin of Triplostegia is further supported by our phylogenetic reconstruction based on nine complete chloroplast genomes (Niu et al., unpublished) that represented populations from SW, CC, and TW, which strongly supported (PP = 1.00; BS = 100%) initial divergence of the SW clade, and CC and TW populations grouped together (Figure S8). In addition, previous phylogenetic and phylogeographic studies generally inferred that extant populations in TW are descendants from Mainland China (e.g., Taiwania, Chou, Thomas, Ge, LePage, & Wang, 2011; Cunninghamia konishii, Lu et al., 2001; Sassafras, Nie, Wen, & Sun, 2007; Dichocarpum arisanense, Xiang et al., 2017), with no reported cases that supported a reverse dispersal direction.
Ancestral area reconstruction revealed that Triplostegia might have colonized CC during the Pliocene (c. 4.03 Ma; 95% HPD: 1.30–8.60 Ma; node II in Figure 4) as the global temperature continued to decrease. Concurrently, accelerated uplift of the eastern QTP during the late Miocene and Pliocene induced dramatic geomorphological changes in HHM (Favre et al., 2015; Ge et al., 2013; Mulch & Chamberlain, 2006). The absence of shared haplotypes between SW and CC indicates a lack of gene flow between the two regions that was probably due to habitat isolation caused by elevation gradients (average elevation for populations in SW is c. 900 m higher than those from CC; Table S1). Morphologically, CC populations also differed from SW populations based on the presence of a serrate leaf blade (vs. pinnatifid leaf blade in SW; Figure 4). Divergence between Mainland China and Taiwan populations in Triplostegia (c. 2.15 Ma, 95% HPD: 0.50–5.30 Ma; node V in Figure 4) and previously studied woody plants (c. 4.04 Ma, Juglans cathayensis, Bai, Wang, & Zhang, 2015; c. 3.31 Ma, Taiwania cryptomerioides, Chou, Thomas, Ge, LePage, & Wang, 2011; c. 3.29 Ma, Taxus wallichiana, Gao et al., 2007) was congruently inferred to be mid‐Pliocene, which was shortly after Taiwan attained its modern form. The lowest differentiation index between CC and TW (Tables S5 and S6) and the more similar leaf blades (Figure 4) further support the idea that TW populations are descendants of CC.
Our SDM analysis provides a detailed picture of the last glacial cycle (Figure 5a,b) and glimpses of the preceding cycles. Based on the paleoclimate record, temperature in each glacial period was similar (Bloom, 2010). Potential suitable habitat distribution of Triplostegia in Mainland China at c. 2.15 Ma might be similar to that during the LGM (Figure 5b): Triplostegia expanded to the coastal regions of East China and reached TW via land bridges in the Taiwan Strait during the glacial period. Because of repeated glacial–interglacial cycles, CC and TW populations may have experienced frequent gene flow via land bridges during the Quaternary. However, genetic exchange between SW and CC might have been obstructed by altitudinal gradients. This may explain why CC haplotypes are phylogenetically closer to those of distant TW rather than the nearby SW. Nie et al. (2007) and Xiang et al. (2017) also support our scenario that migration existed from CC to TW during the Early Pleistocene by either land bridges or long‐distance dispersal. Neither pollen grains nor seeds of Triplostegia have strong dispersal abilities. The long‐distance dispersal hypothesis is thus not likely to be the cause of the disjunct Triplostegia distributions. Therefore, current distribution pattern in Triplostegia might be the consequence of topographic isolation and postglacial contraction.
4.3. Conservation implications
Identifying areas with stable niches and understanding their importance in determining the current distribution of species represent a pivotal task for biodiversity conservation (Marta, Lacasella, Gratton, Cesaroni, & Sbordoni, 2016). The predicted high climatic suitability areas of Triplostegia in 2070s are more restricted than that of the present, especially in southern HHM (Figure 5c,d). Additionally, areas with high climatic suitability in Mainland China are predicted to show a slightly northward migration, whereas climatic suitability in Taiwan may suffer from declining (Figure 5d). The potential reason may be that Mainland China has very northern mountain habitats that are suitable for Triplostegia; however, Taiwan has neither northern cold areas nor higher mountains for Triplostegia dispersal with continued global warming (Figure 5d). Moreover, global mean annual temperatures are likely to rise by an additional 1–4°C by the year 2100 (Stocker et al., 2013). It is undoubtedly a big challenge for the survival of many organisms, especially cold‐adapted taxa that inhabit high mountains, such as Triplostegia, which may experience local extinction at the warm edge, and their suitable habitat distribution in the near future will be as limited as or more restricted than during the LIG (Beever, Ray, Wilkening, Brussard, & Mote, 2011; Wiens, 2016).
Species distribution modeling only uses species occurrence data and environmental variables to predict the potential geographical distribution of species (Guisan & Thuiller, 2005). However, climate change is not the sole factor affecting species' habitat suitability. Human activities (e.g., overharvesting, urbanization, fire suppression, and nitrogen pollution) also have profound effects on the distribution of organisms (MacDougall, McCann, Gellner, & Turkington, 2013). Although we predicted a northward range shift under continuing global warming, the potential habitats may be no longer suitable for survival because of human activities. Indeed, we failed to discover Triplostegia in numerous places with detailed specimen records. Triplostegia represents series of organisms that are already confined to mountaintops (QTP) and/or islands (TW). Therefore, conservation actions are needed to protect these taxa, which are sensitive to climate change and habitat disturbance.
5. CONCLUSION
In this study, we explored how geology and climate together influenced the evolutionary history of Triplostegia, which is an ecologically sensitive and cold‐adapted herbaceous genus. High genetic divergences were detected among Triplostegia populations in SW, CC, and TW, and several interglacial refugia were recognized in the central Hengduan Mts. (particularly Muli County, Sichuan Province). Ancestral area reconstruction revealed that Triplostegia originated from SW, and diversification began during the late Miocene as a result of global cooling and uplift of the Hengduan Mts., then colonized CC in the mid‐Pliocene when global temperature further decreased, and finally dispersed to TW by land bridge at c. 2.15 Ma. Based on phylogeographic and SDM analyses, frequent gene flow might have existed between CC and TW populations with repeated continued distribution between two regions during the Pleistocene glacial cycles. Nevertheless, biotic exchange between SW and CC might have been isolated by altitudinal gradients triggered by dramatic physiographic changes in HHM. Our research represents the first comprehensive phylogeographic study on herbaceous plants to investigate the HHM‐TW disjunction and supports that postglacial range contraction together with topographic heterogeneity resulted in the HHM‐TW disjunction.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
L.‐M.L., J.‐F.Y., and Z.‐D.C. planned and designed the research. Y.‐T.N. performed experiments. Y.‐T.N., J.‐Z.W., and T.Y. analyzed the data. Z.‐D.C., J.‐F.Y., L.‐M.L., and T.Y. conducted the field work. L.‐M.L., Z.‐D.C., Y.‐T.N., J.‐L.Z., X.‐X.W., and J.‐H.L. wrote the manuscript.
DATA ACCESSIBILITY
All haplotype sequences identified in this study were deposited in GenBank (accession numbers MF737219–MF737340).
Supporting information
ACKNOWLEDGMENTS
The authors are grateful to Jingbo Zhang, Honglei Li, Miao Sun, Shih‐Wen Chung, and Shengxiang Yu for field assistance and sample collection. We thank Ching‐I Peng and Hang Sun for their jointly organizing collaborative project on biodiversity studies of disjunct vascular plants distributions between the East Himalayas, Hengduan Mts., and Taiwan. The research was supported by grants from The National Natural Science Foundation of China (NNSF 31590822, 31270268, and 31061160184) and The National Basic Research Program of China (2014CB954101). Field work was partially supported by Sino‐African Joint Research Center, Chinese Academy of Sciences (SAJC201613), and Science and Technology Basic Work (2013FY112100).
Niu Y‐T, Ye J‐F, Zhang J‐L, et al. Long‐distance dispersal or postglacial contraction? Insights into disjunction between Himalaya–Hengduan Mountains and Taiwan in a cold‐adapted herbaceous genus, Triplostegia . Ecol Evol. 2018;8:1131–1146. https://doi.org/10.1002/ece3.3719
REFERENCES
- Bai, W. N. , Wang, W. T. , & Zhang, D. Y. (2015). Phylogeographic breaks within Asian butternuts indicate the existence of a phytogeographic divide in East Asia. New Phytologist, 209, 1757–1772. [DOI] [PubMed] [Google Scholar]
- Baldwin, B. G. (1993). Molecular phylogenetics of Calycadenia (Compositae) based on ITS sequences of nuclear ribosomal DNA: Chromosomal and morphological evolution reexamined. American Journal of Botany, 80, 222–238. https://doi.org/10.2307/2445043 [Google Scholar]
- Bandelt, H. J. , Forster, P. , & Röhl, A. (1999). Median‐joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution, 16, 37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036 [DOI] [PubMed] [Google Scholar]
- Baskin, J. M. , Hidayati, S. N. , Baskin, C. C. , Walck, J. L. , Huang, Z. Y. , & Chien, C. T. (2006). Evolutionary considerations of the presence of both morphophysiological and physiological seed dormancy in the highly advanced euasterids II order Dipsacales. Seed Science Research, 16, 233–242. https://doi.org/10.1017/SSR2006256 [Google Scholar]
- Beever, E. A. , Ray, C. , Wilkening, J. L. , Brussard, P. F. , & Mote, P. W. (2011). Contemporary climate change alters the pace and drivers of extinction. Global Change Biology, 17, 2054–2070. https://doi.org/10.1111/j.1365-2486.2010.02389.x [Google Scholar]
- Bell, C. D. , & Donoghue, M. J. (2005). Dating the Dipsacales: Comparing models, genes, and evolutionary implications. American Journal of Botany, 92, 284–296. https://doi.org/10.3732/ajb.92.2.284 [DOI] [PubMed] [Google Scholar]
- Bloom, A. J. (2010). Global climate change: Convergence of disciplines. Davis, CA: University of California Sinauer Associates Press. [Google Scholar]
- Bueno, M. L. , Pennington, R. T. , Dexter, K. G. , Kamino, L. H. Y. , Pontara, V. , Neves, D. R. M. , … de Oliveira‐Filho, A. T. (2016). Effects of Quaternary climatic fluctuations on the distribution of Neotropical savanna tree species. Ecography, 39, 1–12. [Google Scholar]
- Camargo, A. , Werneck, F. P. , Morando, M. , Sites, J. W. Jr , & Avila, L. J. (2013). Quaternary range and demographic expansion of Liolaemus darwinii (Squamata: Liolaemidae) in the Monte Desert of Central Argentina using Bayesian phylogeography and ecological niche modelling. Molecular Ecology, 22, 4038–4054. https://doi.org/10.1111/mec.12369 [DOI] [PubMed] [Google Scholar]
- Carlson, S. E. , Linder, H. P. , & Donoghue, M. J. (2012). The historical biogeography of Scabiosa (Dipsacaceae): Implications for Old World plant disjunctions. Journal of Biogeography, 39, 1086–1100. https://doi.org/10.1111/j.1365-2699.2011.02669.x [Google Scholar]
- Chen, Z. D. , Ying, J. S. , & Lu, A. M. (2012). Disjunct distribution of seed plants between southwestern China and Taiwan island of China. Chinese Bulletin of Botany, 47, 551–570. [Google Scholar]
- Chou, Y. W. , Thomas, P. I. , Ge, X. J. , LePage, B. A. , & Wang, C. N. (2011). Refugia and phylogeography of Taiwania in East Asia. Journal of Biogeography, 38, 1992–2005. https://doi.org/10.1111/j.1365-2699.2011.02537.x [Google Scholar]
- Clark, M. K. , House, M. A. , Royden, L. H. , Whipple, K. X. , Burchfiel, B. C. , Zhang, X. , & Tang, W. (2005). Late Cenozoic uplift of southeastern Tibet. Geology, 33, 525–528. https://doi.org/10.1130/G21265.1 [Google Scholar]
- Comes, H. P. , & Kadereit, J. W. (1998). The effect of Quaternary climatic changes on plant distribution and evolution. Trends in Plant Science, 3, 432–438. https://doi.org/10.1016/S1360-1385(98)01327-2 [Google Scholar]
- Crandall, K. A. , & Templeton, A. R. (1993). Empirical tests of some predictions from coalescent theory with applications to intraspecific phylogeny reconstruction. Genetics, 134, 959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba, D. , Taboada, G. L. , Doallo, R. , & Posada, D. (2012). jModelTest 2: More models, new heuristics and parallel computing. Nature Methods, 9, 772 https://doi.org/10.1038/nmeth.2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorofeev, P. (1963). Tertiary floras of western Siberia. Moscow‐Leningrad, Russia: Akademia Nauk SSSR; (In Russian). [Google Scholar]
- Drummond, A. J. , Nicholls, G. K. , Rodrigo, A. G. , & Solomon, W. (2002). Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics, 161, 1307–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond, A. J. , & Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology, 7, 214 https://doi.org/10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond, A. J. , Suchard, M. A. , Xie, D. , & Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution, 29, 1969–1973. https://doi.org/10.1093/molbev/mss075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupanloup, I. , Schneider, S. , & Excoffier, L. (2002). A simulated annealing approach to define the genetic structure of populations. Molecular Ecology, 11, 2571–2581. https://doi.org/10.1046/j.1365-294X.2002.01650.x [DOI] [PubMed] [Google Scholar]
- Excoffier, L. , & Lischer, H. E. L. (2010). Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10, 564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x [DOI] [PubMed] [Google Scholar]
- Favre, A. , Päckert, M. , Pauls, S. U. , Jähnig, S. C. , Uhl, D. , Michalak, I. , & Muellner‐Riehl, A. N. (2015). The role of the uplift of the Qinghai‐Tibetan Plateau for the evolution of Tibetan biotas. Biological Reviews, 90, 236–253. https://doi.org/10.1111/brv.12107 [DOI] [PubMed] [Google Scholar]
- Fawcett, T. (2006). An introduction to ROC analysis. Pattern Recognition Letters, 27, 861–874. https://doi.org/10.1016/j.patrec.2005.10.010 [Google Scholar]
- Faye, A. , Deblauwe, V. , Mariac, C. , Richard, D. , Sonké, B. , Vigouroux, Y. , & Couvreur, T. L. P. (2016). Phylogeography of the genus Podococcus (Palmae/Arecaceae) in Central African rain forests: Climate stability predicts unique genetic diversity. Molecular Phylogenetics and Evolution, 105, 126–138. https://doi.org/10.1016/j.ympev.2016.08.005 [DOI] [PubMed] [Google Scholar]
- Friis, E. M. (1985). Angiosperm fruits and seeds from the Middle Miocene of Jutland (Denmark). Biologiske Skrifter, Der Kongelige Danske Videnskabernes Selskab, 24, 1–165. [Google Scholar]
- Fu, Y. X. (1997). Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics, 147, 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, L. M. , Moller, M. , Zhang, X. M. , Hollingsworth, M. L. , Liu, J. , Mill, R. R. , … Li, D. Z. (2007). High variation and strong phylogeographic pattern among cpDNA haplotypes in Taxus wallichiana (Taxaceae) in China and North Vietnam. Molecular Ecology, 16, 4684–4698. https://doi.org/10.1111/j.1365-294X.2007.03537.x [DOI] [PubMed] [Google Scholar]
- Ge, J. Y. , Dai, Y. , Zhang, Z. S. , Zhao, D. A. , Li, Q. , Zhang, Y. , … Guo, Z. T. (2013). Major changes in East Asian climate in the mid‐Pliocene: Triggered by the uplift of the Tibetan Plateau or global cooling? Journal of Asian Earth Sciences, 69, 48–59. https://doi.org/10.1016/j.jseaes.2012.10.009 [Google Scholar]
- Gueta, T. , & Carmel, Y. (2016). Quantifying the value of user‐level data cleaning for big data: A case study using mammal distribution models. Ecological Informatics, 34, 139–145. https://doi.org/10.1016/j.ecoinf.2016.06.001 [Google Scholar]
- Guisan, A. , & Thuiller, W. (2005). Predicting species distribution: Offering more than simple habitat models. Ecology Letters, 8, 993–1009. https://doi.org/10.1111/j.1461-0248.2005.00792.x [DOI] [PubMed] [Google Scholar]
- Halvorsen, R. , Mazzoni, S. , Dirksen, J. W. , Næsset, E. , Gobakken, T. , & Ohlson, M. (2016). How important are choice of model selection method and spatial autocorrelation of presence data for distribution modelling by MaxEnt? Ecological Modelling, 328, 108–118. https://doi.org/10.1016/j.ecolmodel.2016.02.021 [Google Scholar]
- Hamilton, M. B. (1999). Four primer pairs for the amplification of chloroplast intergenic regions with intraspecific variation. Molecular Ecology, 8, 521–523. [PubMed] [Google Scholar]
- Hewitt, G. M. (1996). Some genetic consequences of ice ages, and their role in divergence and speciation. Biological Journal of the Linnean Society, 58, 247–276. https://doi.org/10.1111/j.1095-8312.1996.tb01434.x [Google Scholar]
- Hewitt, G. M. (1999). Post‐glacial re‐colonization of European biota. Biological Journal of the Linnean Society, 68, 87–112. https://doi.org/10.1111/j.1095-8312.1999.tb01160.x [Google Scholar]
- Hewitt, G. (2000). The genetic legacy of the Quaternary ice ages. Nature, 405, 907–913. https://doi.org/10.1038/35016000 [DOI] [PubMed] [Google Scholar]
- Hijmans, R. J. , Cameron, S. E. , Parra, J. L. , Jones, P. G. , & Jarvis, A. (2005). Very high resolution interpolated climate surfaces for global land areas. International Journal of Climatology, 25, 1965–1978. https://doi.org/10.1002/(ISSN)1097-0088 [Google Scholar]
- Hong, D. Y. , Ma, L. M. , & Barrie, F. R. (2011). Dipsacaceae In Wu Z. Y., Hong D. Y., & Raven P. H. (Eds.), Flora of China (pp. 654–660). Beijing, China and St. Louis, MO: Science Press and Missouri Botanical Garden Press. [Google Scholar]
- Hoorn, C. , Mosbrugger, V. , Mulch, A. , & Antonelli, A. (2013). Biodiversity from mountain building. Nature Geoscience, 6, 154 https://doi.org/10.1038/ngeo1742 [Google Scholar]
- Jakob, S. S. , Ihlow, A. , & Blattner, F. R. (2007). Combined ecological niche modelling and molecular phylogeography revealed the evolutionary history of Hordeum marinum (Poaceae)‐niche differentiation, loss of genetic diversity, and speciation in Mediterranean Quaternary refugia. Molecular Ecology, 16, 1713–1727. https://doi.org/10.1111/j.1365-294X.2007.03228.x [DOI] [PubMed] [Google Scholar]
- Katoh, K. , Misawa, K. , Kuma, K. I. , & Miyata, T. (2002). MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research, 30, 3059–3066. https://doi.org/10.1093/nar/gkf436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse, M. , Moir, R. , Wilson, A. , Stones‐Havas, S. , Cheung, M. , Sturrock, S. , … Duran, C. (2012). Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics, 28, 1647–1649. https://doi.org/10.1093/bioinformatics/bts199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby, E. , Reiners, P. W. , Krol, M. A. , Whipple, K. X. , Hodges, K. V. , Farley, K. A. , … Chen, Z. L. (2002). Late Cenozoic evolution of the eastern margin of the Tibetan Plateau: Inferences from 40Ar/39Ar and (U‐Th)/He thermochronology. Tectonics, 21, 1–20. [Google Scholar]
- Lahaye, R. , Savolainen, V. , Duthoit, S. , Maurin, O. , & Van der Bank, M. (2008). A test of psbK–psbI and atpF–atpH as potential plant DNA barcodes using the flora of the Kruger National Park as a model system (South Africa). [Nature Precedings document]. Retrieved from http://precedings.nature.com/documents/1896/version/1
- Łańcucka‐Środoniowa, M. (1967). Two new genera: Hemiptelea Planch. and Weigela Thunb. in the younger Tertiary of Poland. Acta Palaeobotanica, 8, 1–19. [Google Scholar]
- Li, W. J. , Sui, X. L. , Kuss, P. , Liu, Y. Y. , Li, A. R. , & Guan, K. Y. (2016). Long‐distance dispersal after the Last Glacial Maximum (LGM) led to the disjunctive distribution of Pedicularis kansuensis (Orobanchaceae) between the Qinghai‐Tibetan Plateau and Tianshan region. PLoS ONE, 11, e0165700 https://doi.org/10.1371/journal.pone.0165700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado, P. , & Rozas, J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25, 1451–1452. https://doi.org/10.1093/bioinformatics/btp187 [DOI] [PubMed] [Google Scholar]
- Liu, L. , Hao, Z. Z. , Liu, Y. Y. , Wei, X. X. , Cun, Y. Z. , & Wang, X. Q. (2014). Phylogeography of Pinus armandii and its relatives: Heterogeneous contributions of geography and climate changes to the genetic differentiation and diversification of Chinese white pines. PLoS ONE, 9, e85920 https://doi.org/10.1371/journal.pone.0085920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , Möller, M. , Provan, J. , Gao, L. M. , Poudel, R. C. , & Li, D. Z. (2013). Geological and ecological factors drive cryptic speciation of yews in a biodiversity hotspot. New Phytologist, 199, 1093–1108. https://doi.org/10.1111/nph.12336 [DOI] [PubMed] [Google Scholar]
- Lomolino, M. , Riddle, B. , Whittaker, R. , & Brown, J. (2010). Biogeography. Sunderland, MA: Sinauer Associates Press. [Google Scholar]
- Lu, S. Y. , Peng, C. I. , Cheng, Y. P. , Hong, K. H. , & Chiang, T. Y. (2001). Chloroplast DNA phylogeography of Cunninghamia konishii (Cupressaceae), an endemic conifer of Taiwan. Genome, 44, 797–807. https://doi.org/10.1139/g01-074 [PubMed] [Google Scholar]
- Luo, D. , Yue, J. P. , Sun, W. G. , Xu, B. , Li, Z. M. , Comes, H. P. , & Sun, H. (2016). Evolutionary history of the subnival flora of the Himalaya‐Hengduan Mountains: First insights from comparative phylogeography of four perennial herbs. Journal of Biogeography, 43, 31–43. https://doi.org/10.1111/jbi.12610 [Google Scholar]
- MacDougall, A. S. , McCann, K. S. , Gellner, G. , & Turkington, R. (2013). Diversity loss with persistent human disturbance increases vulnerability to ecosystem collapse. Nature, 494, 86–89. https://doi.org/10.1038/nature11869 [DOI] [PubMed] [Google Scholar]
- Magallón, S. , Gómez‐Acevedo, S. , Sánchez‐Reyes, L. L. , & Hernández‐Hernández, T. (2015). A metacalibrated time‐tree documents the early rise of flowering plant phylogenetic diversity. New Phytologist, 207, 437–453. https://doi.org/10.1111/nph.13264 [DOI] [PubMed] [Google Scholar]
- Maldonado, C. , Molina, C. I. , Zizka, A. , Persson, C. , Taylor, C. M. , Alban, J. , … Antonelli, A. (2015). Estimating species diversity and distribution in the era of Big Data: To what extent can we trust public databases? Global Ecology and Biogeography, 24, 973–984. https://doi.org/10.1111/geb.12326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchester, S. R. , & Donoghue, M. J. (1995). Winged fruits of Linnaeeae (Caprifoliaceae) in the Tertiary of Western North America: Diplodipelta gen. nov. International Journal of Plant Sciences, 156, 709–722. https://doi.org/10.1086/297293 [Google Scholar]
- Marta, S. , Lacasella, F. , Gratton, P. , Cesaroni, D. , & Sbordoni, V. (2016). Deciphering range dynamics: Effects of niche stability areas and post‐glacial colonization on alpine species distribution. Journal of Biogeography, 43, 2186–2198. https://doi.org/10.1111/jbi.12771 [Google Scholar]
- Martinez‐Freiria, F. , Velo‐Antón, G. , & Brito, J. C. (2015). Trapped by climate: Interglacial refuge and recent population expansion in the endemic Iberian adder Vipera seoanei. Diversity and Distributions, 21, 331–344. https://doi.org/10.1111/ddi.12265 [Google Scholar]
- Meng, H. H. , Su, T. , Gao, X. Y. , Li, J. , Jiang, X. L. , Sun, H. , & Zhou, Z. K. (2017). Warm‐Cold colonization: Response of oaks to uplift of the Himalaya‐Hengduan Mountains. Molecular Ecology, 26, 3276–3294. https://doi.org/10.1111/mec.14092 [DOI] [PubMed] [Google Scholar]
- Merow, C. , Smith, M. J. , & Silander, J. A. (2013). A practical guide to MaxEnt for modeling species distributions: What it does, and why inputs and settings matter. Ecography, 36, 1058–1069. https://doi.org/10.1111/j.1600-0587.2013.07872.x [Google Scholar]
- Miller, M. , Pfeiffer, W. , & Schwartz, T. (2010). Creating the CIPRES Science Gateway for inference of large phylogenetic trees, New Orleans, LA: Gateway Computing Environments Workshop (GCE) https://doi.org/10.1109/GCE.2010.5676129 [Google Scholar]
- Moore, B. R. , & Donoghue, M. J. (2007). Correlates of diversification in the plant clade Dipsacales: Geographic movement and evolutionary innovations. The American Naturalist, 170, S28–S55. https://doi.org/10.1086/519460 [DOI] [PubMed] [Google Scholar]
- Mulch, A. , & Chamberlain, C. P. (2006). Earth science: The rise and growth of Tibet. Nature, 439, 670–671. https://doi.org/10.1038/439670a [DOI] [PubMed] [Google Scholar]
- Myers, N. , Mittermeier, R. A. , Mittermeier, C. G. , Da Fonseca, G. A. B. , & Kent, J. (2000). Biodiversity hotspots for conservation priorities. Nature, 403, 853–858. https://doi.org/10.1038/35002501 [DOI] [PubMed] [Google Scholar]
- Nagalingum, N. S. , Marshall, C. R. , Quental, T. B. , Rai, H. S. , Little, D. P. , & Mathews, S. (2011). Recent synchronous radiation of a living fossil. Science, 334, 796–799. https://doi.org/10.1126/science.1209926 [DOI] [PubMed] [Google Scholar]
- Nathan, R. (2006). Long‐distance dispersal of plants. Science, 313, 786–788. https://doi.org/10.1126/science.1124975 [DOI] [PubMed] [Google Scholar]
- Nie, Z. L. , Wen, J. , & Sun, H. (2007). Phylogeny and biogeography of Sassafras (Lauraceae) disjunct between eastern Asia and eastern North America. Plant Systematics and Evolution, 267, 191–203. https://doi.org/10.1007/s00606-007-0550-1 [Google Scholar]
- Peng, C. I. , Tobe, H. , & Takahashi, M. (1995). Reproductive morphology and relationships of Tripostegia (Dipsacales). Botanische Jahrbuecher Fuer Systematik Pflanzengeschichte und Pflanzengeographie, 116, 505–516. [Google Scholar]
- Peng, Y. , Wan, Y. , & Luo, L. S. (2000). The study on the diversity and sustainable development in Hengduanshan Mountain of Yunnan. Human Geography, 15, 50–53. [Google Scholar]
- Peterson, A. T. , Ammann, C. M. , & Diniz‐Filho, J. A. F. (2013). Global patterns of connectivity and isolation of populations of forest bird species in the late Pleistocene. Global Ecology and Biogeography, 22, 596–606. https://doi.org/10.1111/geb.12010 [Google Scholar]
- Phillips, S. J. , Anderson, R. P. , Dudík, M. , Schapire, R. E. , & Blair, M. E. (2017). Opening the black box: An open‐source release of Maxent. Ecography, 40, 887–893. https://doi.org/10.1111/ecog.03049 [Google Scholar]
- Phillips, S. J. , Anderson, R. P. , & Schapire, R. E. (2006). Maximum entropy modeling of species geographic distributions. Ecological Modelling, 190, 231–259. https://doi.org/10.1016/j.ecolmodel.2005.03.026 [Google Scholar]
- Phillips, S. J. , & Dudík, M. (2008). Modeling of species distributions with Maxent: New extensions and a comprehensive evaluation. Ecography, 31, 161–175. https://doi.org/10.1111/j.0906-7590.2008.5203.x [Google Scholar]
- Pinot, S. , Ramstein, G. , Harrison, S. P. , Prentice, I. C. , Guiot, J. , Stute, M. , & Joussaume, S. (1999). Tropical paleoclimates at the Last Glacial Maximum: Comparison of Paleoclimate Modeling Intercomparison Project (PMIP) simulations and paleodata. Climate Dynamics, 15, 857–874. https://doi.org/10.1007/s003820050318 [Google Scholar]
- Pons, O. , & Petit, R. J. (1996). Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics Society of America, 144, 1237–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada, D. , & Crandall, K. A. (2001). Intraspecific gene genealogies: Trees grafting into networks. Trends in Ecology & Evolution, 16, 37–45. https://doi.org/10.1016/S0169-5347(00)02026-7 [DOI] [PubMed] [Google Scholar]
- Pyck, N. , & Smets, E. (2004). On the systematic position of Triplostegia (Dipsacales): A combined molecular and morphological approach. Belgian Journal of Botany, 137, 125–139. [Google Scholar]
- Qi, X. S. , Chen, C. , Comes, H. P. , Sakaguchi, S. , Liu, Y. H. , Tanaka, N. , … Qiu, Y. X. (2012). Molecular data and ecological niche modelling reveal a highly dynamic evolutionary history of the East Asian Tertiary relict Cercidiphyllum (Cercidiphyllaceae). New Phytologist, 196, 617–630. https://doi.org/10.1111/j.1469-8137.2012.04242.x [DOI] [PubMed] [Google Scholar]
- Qu, Y. H. , Song, G. , Gao, B. , Quan, Q. , Ericson, P. G. , & Lei, F. M. (2015). The influence of geological events on the endemism of East Asian birds studied through comparative phylogeography. Journal of Biogeography, 42, 179–192. https://doi.org/10.1111/jbi.12407 [Google Scholar]
- Radosavljevic, A. , Anderson, R. P. , & Araújo, M. (2014). Making better Maxent models of species distributions: Complexity, overfitting and evaluation. Journal of Biogeography, 41, 629–643. https://doi.org/10.1111/jbi.12227 [Google Scholar]
- Rambaut, A. (2009). FigTree v.1.3.1. Retrieved from http://tree.bio.ed.ac.uk/software/figtree/
- Raymond, M. , & Rousset, F. (2009). Tracer v.1.5. Retrieved from http://tree.bio.ed.ac,uk/software/tracer
- Rogers, A. R. , & Harpending, H. (1992). Population growth makes waves in the distribution of pairwise genetic differences. Molecular Biology and Evolution, 9, 552–569. [DOI] [PubMed] [Google Scholar]
- Ronquist, F. , & Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics, 19, 1572–1574. https://doi.org/10.1093/bioinformatics/btg180 [DOI] [PubMed] [Google Scholar]
- Ronquist, F. , Teslenko, M. , Van Der Mark, P. , Ayres, D. L. , Darling, A. , Höhna, S. , … Huelsenbeck, J. P. (2012). MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology, 61, 539–542. https://doi.org/10.1093/sysbio/sys029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandel, B. , Arge, L. , Dalsgaard, B. , Davies, R. G. , Gaston, K. J. , Sutherland, W. J. , & Svenning, J. C. (2011). The influence of late Quaternary climate‐change velocity on species endemism. Science, 334, 660–664. https://doi.org/10.1126/science.1210173 [DOI] [PubMed] [Google Scholar]
- Sang, T. , Crawford, D. J. , & Stuessy, T. F. (1997). Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). American Journal of Botany, 84, 1120 https://doi.org/10.2307/2446155 [PubMed] [Google Scholar]
- Shao, Y. Z. , & Xiang, Q. P. (2015). Species delimitation and phylogeography of the Abies chensiensis complex inferred from morphological and molecular data. Botanical Journal of the Linnean Society, 177, 175–188. https://doi.org/10.1111/boj.12235 [Google Scholar]
- Shi, Y. F. , & Yao, T. D. (2002). MIS 3b (54–44 ka BP) cold period and glacial advance in middle and low latitudes. Journal of Glaciology and Geocryology, 24, 1–9. [Google Scholar]
- Sibuet, J. C. , & Hsu, S. K. (1997). Geodynamics of the Taiwan arc‐arc collision. Tectonophysics, 274, 221–251. https://doi.org/10.1016/S0040-1951(96)00305-8 [Google Scholar]
- Sibuet, J. C. , & Hsu, S. K. (2004). How was Taiwan created? Tectonophysics, 379, 159–181. https://doi.org/10.1016/j.tecto.2003.10.022 [Google Scholar]
- Soejima, A. , & Wen, J. (2006). Phylogenetic analysis of the grape family (Vitaceae) based on three chloroplast markers. American Journal of Botany, 93, 278–287. https://doi.org/10.3732/ajb.93.2.278 [DOI] [PubMed] [Google Scholar]
- Stamatakis, A. (2006). RAxML‐VI‐HPC: Maximum likelihood‐based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics, 22, 2688–2690. https://doi.org/10.1093/bioinformatics/btl446 [DOI] [PubMed] [Google Scholar]
- Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. https://doi.org/10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis, A. , Hoover, P. , & Rougemont, J. (2008). A rapid bootstrap algorithm for the RAxML web servers. Systematic Biology, 57, 758–771. https://doi.org/10.1080/10635150802429642 [DOI] [PubMed] [Google Scholar]
- Stocker T. F., Qin D., Plattner G. K., Tignor M., Allen S. K., Boschung J., … Midgley B. M. (Eds.) (2013). Climate change 2013: The physical science basis. Contribution of Working Group I to the fifth assessment report of the Intergovernmental Panel on Climate Change. Cambridge, UK & New York NY: Cambridge University Press. [Google Scholar]
- Sun, Y. X. , Moore, M. J. , Yue, L. L. , Feng, T. , Chu, H. J. , Chen, S. T. , … Li, J. Q. (2014). Chloroplast phylogeography of the East Asian Arcto‐Tertiary relict Tetracentron sinense (Trochodendraceae). Journal of Biogeography, 41, 1721–1732. https://doi.org/10.1111/jbi.12323 [Google Scholar]
- Taberlet, P. , Gielly, L. , Pautou, G. , & Bouvet, J. (1991). Universal primers for amplification of three non‐coding regions of chloroplast DNA. Plant Molecular Biology, 17, 1105–1109. https://doi.org/10.1007/BF00037152 [DOI] [PubMed] [Google Scholar]
- Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics, 123, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Stecher, G. , Peterson, D. , Filipski, A. , & Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 30, 2725–2729. https://doi.org/10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Z. Y. , Wang, Z. H. , Zheng, C. Y. , & Fang, J. Y. (2006). Biodiversity in China's mountains. Frontiers in Ecology and the Environment, 4, 347–352. https://doi.org/10.1890/1540-9295(2006)004[0347:BICM]2.0.CO;2 [Google Scholar]
- The Angiosperm Phylogeny Group (2016). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Botanical Journal of the Linnean Society, 181, 1–20. [Google Scholar]
- Tian, S. , López‐Pujol, J. , Wang, H. W. , Ge, S. , & Zhang, Z. Y. (2010). Molecular evidence for glacial expansion and interglacial retreat during Quaternary climatic changes in a montane temperate pine (Pinus kwangtungensis Chun ex Tsiang) in southern China. Plant Systematics and Evolution, 284, 219–229. https://doi.org/10.1007/s00606-009-0246-9 [Google Scholar]
- Verboom, G. A. , Bergh, N. G. , Haiden, S. A. , Hoffmann, V. , & Britton, M. N. (2015). Topography as a driver of diversification in the Cape Floristic Region of South Africa. New Phytologist, 207, 368–376. https://doi.org/10.1111/nph.13342 [DOI] [PubMed] [Google Scholar]
- Wahlberg, N. , Wheat, C. W. , & Peña, C. (2013). Timing and patterns in the taxonomic diversification of Lepidoptera (butterflies and moths). PLoS ONE, 8, e80875 https://doi.org/10.1371/journal.pone.0080875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. C. (1989). Notes on the disjunction in the flora of China. Bulletin of Botanical Research, 9, 1–16. [Google Scholar]
- Wang, Y. H. , Comes, H. P. , Cao, Y. N. , Guo, R. , Mao, Y. R. , & Qiu, Y. X. (2017). Quaternary climate change drives allo‐peripatric speciation and refugial divergence in the Dysosma versipellis‐pleiantha complex from different forest types in China. Scientific Report, 7, 40261 https://doi.org/10.1038/srep40261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. H. , Jiang, W. M. , Comes, H. P. , Hu, F. S. , Qiu, Y. X. , & Fu, C. X. (2015). Molecular phylogeography and ecological niche modelling of a widespread herbaceous climber, Tetrastigma hemsleyanum (Vitaceae): Insights into Plio‐Pleistocene range dynamics of evergreen forest in subtropical China. New Phytologist, 206, 852–867. https://doi.org/10.1111/nph.13261 [DOI] [PubMed] [Google Scholar]
- Wang, H. F. , Landrein, S. , Dong, W. P. , Nie, Z. L. , Kondo, K. , Funamoto, T. , … Zhou, S. L. (2015). Molecular phylogeny and biogeographic diversification of Linnaeoideae (Caprifoliaceae s.l.) disjunctly distributed in Eurasia, North America and Mexico. PLoS ONE, 10, e0116485 https://doi.org/10.1371/journal.pone.0116485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. J. , McKay, B. D. , Dai, C. Y. , Zhao, N. , Zhang, R. Y. , Qu, Y. H. , … Yang, X. J. (2013). Glacial expansion and diversification of an East Asian montane bird, the green‐backed tit (Parus monticolus). Journal of Biogeography, 40, 1156–1169. https://doi.org/10.1111/jbi.12055 [Google Scholar]
- Wei, X. X. , Yang, Z. Y. , Li, Y. , & Wang, X. Q. (2010). Molecular phylogeny and biogeography of Pseudotsuga (Pinaceae): Insights into the floristic relationship between Taiwan and its adjacent areas. Molecular Phylogenetics and Evolution, 55, 776–785. https://doi.org/10.1016/j.ympev.2010.03.007 [DOI] [PubMed] [Google Scholar]
- Wen, J. , Zhang, J. Q. , Nie, Z. L. , Zhong, Y. , & Sun, H. (2014). Evolutionary diversifications of plants on the Qinghai‐Tibetan Plateau. Frontiers in Genetics, 5, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, T. J. , Bruns, T. , Lee, S. , & Taylor, J. W. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics In Innis M. A., Gelfand D. H., Sninsky J. J., & White T. J. (Eds.), PCR protocols: A guide to methods and applications (pp. 315–322). San Diego, CA: Academic Press. [Google Scholar]
- Wiens, J. J. (2016). Climate‐related local extinctions are already widespread among plant and animal species. PLoS Biology, 14, e2001104 https://doi.org/10.1371/journal.pbio.2001104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing, S. L. , Alroy, J. , & Hickey, L. J. (1995). Plant and mammal diversity in the Paleocene to early Eocene of the Bighorn Basin. Palaeogeography, Palaeoclimatology, Palaeoecology, 115, 117–155. https://doi.org/10.1016/0031-0182(94)00109-L [Google Scholar]
- Wu, P. , & Wang, M. Z. (2001). Relationship of the tropical elements of the bryophytes between Hengduan Mts., southwest China and Taiwan province, southeast China. Guizhou Science, 19, 6–9. [Google Scholar]
- Xiang, K. L. , Zhao, L. , Erst, A. S. , Yu, S. X. , Jabbour, F. , & Wang, W. (2017). A molecular phylogeny of Dichocarpum (Ranunculaceae): Implications for eastern Asian biogeography. Molecular Phylogenetics and Evolution, 107, 594–604. https://doi.org/10.1016/j.ympev.2016.12.026 [DOI] [PubMed] [Google Scholar]
- Xing, Y. W. , & Ree, R. H. (2017). Uplift‐driven diversification in the Hengduan Mountains, a temperate biodiversity hotspot. Proceedings of the National Academy of Sciences of the United States of America, 114, E3444–E3451. https://doi.org/10.1073/pnas.1616063114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, J. F. , Chen, Z. D. , Liu, B. , Qin, H. N. , & Yang, Y. (2012). Disjunct distribution of vascular plants between southwestern area and Taiwan area in China. Biodiversity Science, 20, 482–494. [Google Scholar]
- Ye, Z. , Zhu, G. , Chen, P. , Zhang, D. , & Bu, W. (2014). Molecular data and ecological niche modelling reveal the Pleistocene history of a semi‐aquatic bug (Microvelia douglasi douglasi) in East Asia. Molecular Ecology, 23, 3080–3096. https://doi.org/10.1111/mec.12797 [DOI] [PubMed] [Google Scholar]
- Yu, Y. , Harris, A. J. , Blair, C. , & He, X. J. (2015). RASP (Reconstruct Ancestral State in Phylogenies): A tool for historical biogeography. Molecular Phylogenetics and Evolution, 87, 46–49. https://doi.org/10.1016/j.ympev.2015.03.008 [DOI] [PubMed] [Google Scholar]
- Zachos, J. C. , Dickens, G. R. , & Zeebe, R. E. (2008). An early Cenozoic perspective on greenhouse warming and carbon‐cycle dynamics. Nature, 451, 279–283. https://doi.org/10.1038/nature06588 [DOI] [PubMed] [Google Scholar]
- Zanne, A. E. , Tank, D. C. , Cornwell, W. K. , Eastman, J. M. , Smith, S. A. , FitzJohn, R. G. , … Reich, P. B. (2014). Three keys to the radiation of angiosperms into freezing environments. Nature, 506, 89–92. [DOI] [PubMed] [Google Scholar]
- Zhang, W. H. , Chen, Z. D. , Li, J. H. , Chen, H. B. , & Tang, Y. C. (2003). Phylogeny of the Dipsacales s.l. based on chloroplast trnL–F and ndhF sequences. Molecular Phylogenetics and Evolution, 26, 176–189. https://doi.org/10.1016/S1055-7903(02)00303-2 [DOI] [PubMed] [Google Scholar]
- Zhang, J. J. , Li, Z. Z. , Fritsch, P. W. , Tian, H. , Yang, A. H. , & Yao, X. H. (2015). Phylogeography and genetic structure of a Tertiary relict tree species, Tapiscia sinensis (Tapisciaceae): Implications for conservation. Annals of Botany, 116, 727–737. https://doi.org/10.1093/aob/mcv112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. L. , Li, B. Y. , & Zheng, D. (2002). A discussion on the boundary and area of the Tibetan Plateau in China. Geographical Research, 21, 1–8. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All haplotype sequences identified in this study were deposited in GenBank (accession numbers MF737219–MF737340).