

SUMMARY
Interleukin-23 (IL-23), an IL-12 family cytokine, plays pivotal roles in pro-inflammatory T helper 17 responses linked to autoimmune and inflammatory diseases. Despite intense therapeutic targeting, structural and mechanistic insights into receptor complexes mediated by IL-23, and IL-12 family members in general, have remained elusive. We determined a crystal structure of human IL-23 in complex with its cognate receptor, IL-23R, and revealed that IL-23R bound to IL-23 exclusively via its N-terminal immunoglobulin domain. The structural and functional hotspot of this interaction partially restructured the helical IL-23p19 subunit of IL-23, and restrained its IL-12p40 subunit to cooperatively bind the shared receptor IL-12Rβ1 with high affinity. Together with structural insights from the interaction of IL-23 with the inhibitory antibody briakinumab and by leveraging additional IL-23:antibody complexes, we propose a mechanistic paradigm for IL-23 and IL-12 whereby cognate receptor binding to the helical cytokine subunits primes recruitment of the shared receptors via the IL-12p40 subunit.
eTOC
IL-23, a human cytokine under intense clinical targeting, is pivotal to cellular responses underlying widespread inflammatory and autoimmune diseases, such as psoriasis and rheumatoid arthritis. Bloch et al. determine the structure of IL-23 bound by one its receptors, IL-23R, and reveal how IL-23R activates IL-23 for recruiting IL-12Rβ1 to the signalling assembly. Together with identifying an interaction hotspot such findings may contribute to additional approaches for the mechanistic and therapeutic interrogation of receptor complexes mediated by IL-12 family members.
INTRODUCTION
IL-12 family cytokines (IL-12, IL-23, IL-27, IL-35) are predominantly produced by activated antigen-presenting cells, such as dendritic cells and activated macrophages, and act as key immunological playmakers to coordinate innate and adaptive immune responses mainly via regulation of T-cell populations (Eberl, 2016; Hasegawa et al., 2016). Hallmarked by their heterodimeric nature and intriguing cross-utilization of cytokine subunits and sharing of signalling receptors, IL-12 family cytokines operate on opposing sides of the immunological balance. For instance, the archetypal IL-12 (Gubler et al., 1991; Wolf et al., 1991) and IL-23 (Oppmann et al., 2000) are charted as pro-inflammatory cytokines contrasting the rather protective roles of IL-27 and IL-35 (Vignali and Kuchroo, 2012).
IL-23 is the most extensively studied member of the IL-12 family of cytokines. Heterodimeric IL-23 comprises a p19 helical-bundle subunit (IL-23p19), which is disulphide-linked to a p40 subunit (IL-12p40) (Oppmann et al., 2000). The latter is shared with IL-12 thereby defining a key structural and functional divergence within the IL-12 family. For instance, IL-23 signals via its specific receptor interleukin-23 receptor (IL-23R) and interleukin-12 receptor subunit β1 (IL-12Rβ1), which is also utilized by IL-12 (Parham et al., 2002). Yet functionally, the two cytokines trigger diametrically opposite immunological pathways. IL-12 drives differentiation of naive T cells into interferon-γ (IFN-γ)-producing T helper 1 (Th1) cells in type 1 immunity, whereas IL-23 is synonymous with type 3 immune responses pivotal to the survival and expansion of CD4+ T helper 17 (Th17) cells (reviewed by (Eberl, 2016)).
It is precisely the potency by which IL-23 can drive production of interleukin-17 (IL-17) by Th17 cells and IL-17-producing γδ T (γδT17) cells, that has propelled IL-23 to a major therapeutic target (Gaffen et al., 2014). Indeed, IL-23R is required in vivo for effector Th17 cell responses (McGeachy et al., 2009), which are now closely associated with many autoimmune and chronic inflammatory disorders, including psoriasis, psoriatic arthritis, Crohn’s disease, rheumatoid arthritis, multiple sclerosis, inflammatory bowel disease, and uveitis (Duerr et al., 2006; Korn et al., 2009; Lowes et al., 2014; Lubberts, 2015; Murphy et al., 2003). Nevertheless, the IL-23-IL-17 axis appears to entail differential roles for the two cytokines in intestinal immunoregulation, as their inhibition in Crohn’s disease yields opposing effects, with IL-23 emerging as the therapeutically relevant target (Lee et al., 2015; Maxwell et al., 2015). Furthermore, elevated expression of IL-23 and IL-23R (and IL-17A) has been observed in several cancers, such as those of the skin, lung, breast, and colon (Grivennikov et al., 2012; Langowski et al., 2006; Zhang et al., 2014). The therapeutic context of IL-23 targeting continues to grow as recently evidenced by strategies to treat and manage pustular psoriasis (Arakawa et al., 2016), pityriasis rubra pilaris (Feldmeyer et al., 2017), and inflamed lesions in patients with leukocyte adhesion deficiency type 1 (Moutsopoulos et al., 2017).
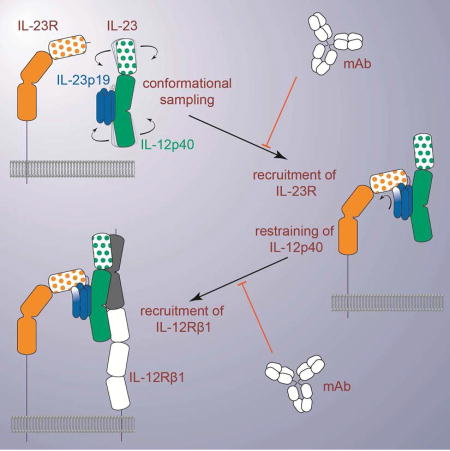
Despite the wealth of information on the immunoregulatory functions and disease-related context of IL-23 and IL-12 family cytokines, the field is characterized by a paucity of structural information. For instance, structural information of complexes of IL-12 family cytokines with cognate receptors is currently lacking. By elucidating the crystal structure of the IL-23:IL-23R complex and based on biochemical and biophysical undertakings we here provided insights into how IL-23 becomes structurally primed by IL-23R to mediate a tripartite complex with IL-12Rβ1. Supported by functional interrogation of key interactions in a mouse model of skin inflammation and by consolidating emerging structure-function considerations we propose a mechanism for the pro-inflammatory signaling complex mediated by IL-23. Restructuring of the helical IL-23p19 subunit of IL-23 as result of its interaction with the N-terminal immunoglobulin (Ig) domain of IL-23R constitutes a mechanistic prerequisite, which leads to receptor-mediated restraining of the p40 subunit of IL-23 to enable a high affinity interaction with the shared receptor IL-12Rβ1 in cooperative fashion. Thus, our mechanistic proposal segregates cognate and shared receptor binding to the helical (α) and non-helical (β) subunits of the heterodimeric cytokines in the IL-12 family, respectively.
RESULTS
Crystal structure of the human IL-23:IL-23R complex
IL-23 signals via its cognate receptor IL-23R and the shared receptor IL-12Rβ1, and is thought to mediate a stoichiometric ternary complex with the ectodomains of the two receptors (Figure 1A). While IL-12Rβ1 orthologues consistently display 5 extracellular domains (Figure S1A), mammalian IL-23R consist of just three extracellular domains compared to evolutionarily more distant verterbrates, which have 2–3 additional membrane-proximal fibronectin type III (FnIII) domains (Figure S1B,C). Current models for the assembly of cell-surface complexes mediated by IL-12 family cytokines have been largely based on the structural principles derived from the interleukin-6 (IL-6) complex with interleukin-6 receptor subunit alpha (IL-6Rα) and glycoprotein 130 (gp130) and other gp130 complexes (Boulanger et al., 2003; Skiniotis et al., 2005).
Figure 1. Structure of the IL-23:IL-23R complex.

A) Schematic representation of protein components participating in IL-23 and IL-12 signaling complexes. EC: Extracellular, PM: Plasma Membrane, CP: Cytoplasm, p19: IL-23p19, p40: IL-12p40. B) Cartoon representation of the IL-23:IL-23R crystal structure. Nanobody 22E11 used as a crystallization adjuvant is shown in gray surface representation. C) Close-up view of the IL-23R:IL-23p19 interface in the vicinity of W156 in IL-23p19. D) Restructuring of IL-23 upon binding to IL-23R. E) Close-up view of the interactions around the AB-loop of IL-23p19. F) Top-down view of the IL-23:IL-23R complex with IL-23R (surface). See also Figure S1, Figure S2, Figure S3, Table S1, and Table S2.
Given the paucity of structural information for any IL-12 family complex and the growing importance of specific therapeutic targeting of human IL-23 (Teng et al., 2015), we pursued structural studies of human IL-23 and its complexes with the ectodomains of its specific receptor IL-23R and the shared receptor IL-12Rβ1 (Figure 1A). We produced recombinant human IL-23, and the ectodomains of IL-23R (residues 1–317) and IL-12Rβ1 (residues 1–540) (Figure 1A), via inducible stable expression and secretion in HEK293S MGAT1−/− cells, which limit N-linked glycosylation of proteins to Man5GlcNAc2 glycan trees (Reeves et al., 2002; Verstraete et al., 2011). We biochemically reconstituted and purified stoichiometric IL-23:IL-23R binary and IL-23:IL-23R:IL-12Rβ1 ternary complexes to enable structural studies (Figure S2A–C). However, the IL-23:IL-23R:IL-12Rβ1 ternary complex displayed low stability and solubility, which greatly hampered efforts to propagate it for structural studies. On the other hand, the IL-23:IL-23R binary complex proved much more amenable to structural studies, and initially led to poorly diffracting crystals (~20 Å resolution). To improve crystal quality, we leveraged a collection of single domain VHH camelid antibodies targeting human IL-23 (Saunders et al., 2009) (PDB entry 3qwr). Introduction of Nb22E11 to the IL-23:IL-23R complex yielded much improved crystals, which could be further optimized via shaving of N-linked glycans in purified IL-23:IL-23R:Nb22E11 complex by Jack-bean α-mannosidase (Figure S2D,E). X-ray diffraction data from such crystals enabled structure determination of the complex to 2.8 Å resolution (Figure 1B–F, Table 1, Figure S2F–I), thereby setting the stage for accurate structural and functional insights.
Table 1.
Crystallographic data and refinement statistics
| Protein | IL-23:IL-23R:Nb22E11 | IL-23 | IL-23:BriakinumabFab | BriakinumabFab |
|---|---|---|---|---|
| PDB code | 5mzv | 5mxa | 5njd | 5n2k |
| Crystallization Condition | 0.2M MgCl2 0.1M Tris pH 6.6 11% (w/v) PEG8000 | 19.5% (w/v) PEG1000, 100 mM Citrate Phosphate pH 4.4, 200 mM Li2SO4 | 2M (NH4)2SO4, 100mM BisTris pH 6 | 200 mM NH4I, 20% (w/v) PEG3350 pH 6.2 |
| Cryo-protectant | 20% Ethylene Glycol | 5% PEG1000 | Saturated (NH4)2SO4 | 20% Ethylene Glycol |
| Data Collection† | ||||
| Beamline | PetraIII-P14 | PetraIII-P14 | PetraIII-P14 | PetraIII-P14 |
| Wavelength | 0.98 Å | 0.98 Å | 0.98 Å | 0.98 Å |
| Space Group | P 21 | P 61 | P 41212 | P 21 |
| a,b,c (Å) | 67.22 112.19 109.87 | 109.80 109.80 87.69 | 191.61 191.61 519.11 | 85.25 172.54 138.16 |
| α,β,γ (°) | 90.0 106.24 90.0 | 90 90 120 | 90 90 90 | 90.0 106. 16 90.0 |
| Resolution (Å) | 76.9−2.8 (2.9−2.8) | 87.7−2.5 (2.65−2.50) | 95.8−3.9 (4.14 − 3.90) | 81.88−2.22 (2.34−2.22) |
| Total reflections | 131247 (13351) | 213965 (33428) | 439822 (66604) | 732035 (110384) |
| Unique reflections | 38061 (3803) | 20825 (3297) | 86556 (13014) | 178979 (26837) |
| Multiplicity | 3.4 (3.5) | 10.3 (10.1) | 5.1 (5.1) | 4.1 (4.1) |
| Completeness (%) | 98.4 (99.2) | 99.7 (98.2) | 97.7 (92.5) | 94.7 (96.5) |
| Mean I/σ(I) | 12.4 (2.8) | 24.6 (2.2) | 4.2 (1.2) | 8.9 (2.2) |
| Wilson B-factor (Å2) | 72.2 | 69.4 | n.a. | 53.4 |
| R-meas (%) | 8.1 (52.8) | 6.6 (103.1) | 43.8 (137.8) | 11.0 (91.7) |
| CC1/2 (%) | 99.7 (63.4) | 100 (73.1) | 96.0 (43.0) | 99.5 (68.6) |
| Refinement‡ | ||||
| Resolution range (Å) | 76.9 − 2.8 (2.9 − 2.8) | 64.5 − 2.5 (2.59 − 2.50) | 95.8 − 3.9 (4.05 − 3.9) | 81.9 − 2.22 (2.30 − 2.22) |
| Reflections in refinement | 38058 (3805) | 20823 (2020) | 86486 (7722) | 178458 (17823) |
| Reflections used for R-free | 1594 (156) | 1716 (165) | 1835 (165) | 2153 (215) |
| R-work | 0.211 (0.319) | 0.187 (0.290) | 0.273 (0.358) | 0.203 (0.281) |
| R-free | 0.249 (0.321) | 0.226 (0.347) | 0.313 (0.354) | 0.233 (0.323) |
| Non-hydrogen atoms | 7116 | 3484 | 39446 | 26313 |
| Macromolecular atoms | 6803 | 3398 | 39015 | 25304 |
| Ligands | 238 | 50 | 431 | 4 |
| Protein residues | 856 | 441 | 5146 | 3382 |
| RMS(bonds) (Å) | 0.003 | 0.009 | 0.004 | 0.003 |
| RMS(angles) (°) | 0.65 | 0.91 | 0.57 | 0.61 |
| Ramachandran favored (%) | 94 | 97 | 94 | 97 |
| Ramachandran allowed (%) | 6.2 | 3.3 | 5.6 | 2.7 |
| Ramachandran outliers (%) | 0 | 0 | 0.4 | 0.09 |
| Rotamer outliers (%) | 0.5 | 1.1 | 0.3 | 0.5 |
| Clashscore | 1.1 | 1.2 | 1.2 | 1.1 |
| Average B-factor (Å2) | 84.5 | 79.5 | 120.1 | 63.80 |
| macromolecules | 83.4 | 79.6 | 120.2 | 64.4 |
| ligands | 123.8 | 88.2 | 106.7 | 52.2 |
| solvent | 57.7 | 57.1 | - | 49.1 |
| TLS groups | 14 | 10 | 24 | 33 |
|
|
||||
Values reported by XDS.
Values reported by Phenix.
The crystal structure of the IL-23:IL-23R:Nb22E11 complex provided structural insights into the IL-12 family of cytokine-receptor complexes. IL-23 deploys only its IL-23p19 subunit to contact IL-23R at its N-terminal Ig-like domain (D1) establishing a mixed interaction interface of ~900 Å2 mediated by a total of 51 residues (Figure 1B–F, Table S1, Figure S2I). The two domains forming the cytokine-binding homology region (CHR) of IL-23R (D2 and D3) stayed clear of IL-23 and extended away from the cytokine-receptor interaction site via a nearly linear arrangement of D1 and D2, followed by a sharp bend at the D2–D3 linker to orient D3 at right-angles to the plane formed by D1, D2 and IL-23p19 (Figure 1B,F). The CHR domains included the WSXWS motif in D3 (W304 Q305 P306 W307 S308), and adopted an inter-domain orientation reminiscent of CHR domains in the common gamma (γc) family of receptors (Verstraete et al., 2014; Wang et al., 2009). This motif formed a zipper-like arrangement of stacking interactions alternating between sidechains in the motif and sidechains on neighbouring β-strands along D3 (Figure S3A). W304 is also a putative C-mannosylation site, however no electron density was observed to support such post-translational modification. On the other hand, we observed clear electron density for six N-linked glycosylation sites in the ectodomain of human IL-23R at N47 and N81 in D1, two stacked glycans at N141 and N180 in D2, and at N262 and N273 in D3 (Figure 1B). Finally, previously reported glycosylation of N29 near the N-terminus N29 (Zhao et al., 2010) was not supported by our experimental electron density, and might be incompatible with the observed binding interface. Nanobody Nb22E11 binds at the junction of the two N-terminal domains of IL-12p40 (PDB code 3qwr), far away from the IL-23:IL-23R interaction site.
A limited interface (~290 Å2) between D1 and D2 of IL-23R and an interface covering 480 Å2 between D2 and D3 are in line with previously determined structures of type I cytokine receptors featuring an Ig-like domain linked to CHR domains, such as IL-12p40, gp130 and IL-6Rα. In IL-23R, D1 links to D2 by a nearly linear prolongation of the D1 G β-strand leading into the A β-strand in D2, consistent with proposals that such minimal interfaces between D1 and D2 may facilitate inter-domain plasticity (Skiniotis et al., 2005). The D2:D3 interface, however, is not only larger but also stabilized by several hydrogen bonds and salt bridges (Table S1). The relative orientation of D2 and D3 is rather well-conserved among type I cytokine receptors, consistent with the cytokine binding function of the elbow region between D2 and D3 in other receptors. Thus, the binary complex of IL-23 with IL-23R provided insights into the possible structural plasticity and role of the three extracellular domains of IL-23R in binding the cytokine, and the key conclusion that the N-terminal Ig domain of IL-23R serves as the sole cytokine-binding domain.
Recruitment of IL-23R restructures IL-23
To address possible conformational changes in human IL-23 upon binding to IL-23R receptor and to enable appropriate structural comparisons, we acquired an additional snapshot of the structure of unbound IL-23 by crystallizing glycosylated human IL-23 produced in HEK293S MGAT1−/− cells (Shirouzono et al., 2012) (Table 1, Figure 1D).
Arguably, the hallmarks of the IL-23:IL-23R interaction interface are threefold and feature conformational changes in both the cytokine and the receptor. First, IL-23 did not bind to the CHR domains of IL-23R, a mode of interaction commonly displayed by members of the cytokine superfamily activating type I cytokine receptors (Figure 1B). Second, the five N-terminal residues of D1 of IL-23R (G24–N29) that are not part of its canonical Ig fold, adopted a hook-like structure tethered at the disulfide bridge defined by C30–C115 and reached out into the loops atop helices B and D of IL-23p19 (Figure 1C,F). This led to partial ordering of the loop linking helices A and B (AB loop) in IL-23p19 (residues 54–60) (Figure 1E). The indole ring of W156 at the N-terminal end of helix D in IL-23p19 stacked tightly against the peptide plane of G116 in the G β-strand of IL-23RD1 (Figure 1D, Figure S3B,C). Interestingly, the IL-6:gp130 interface displays a very similar interaction based on conserved residues (Boulanger et al., 2003) (Figure S1D). The interaction in the IL-23:IL-23R complex featured additional stabilizing interactions via hydrogen bonds between the main chain amide nitrogen of W156 in IL-23p19 and residue D118 in IL-23R. Third, W156 marked the end point of a restructuring of the N-terminal end of helix D in IL-23p19, which transitioned from a canonical α-helix to a 310-helix (Figure 1D, Figure 2C, Figure S3B,C) upon binding IL-23R. This 310 helical conformation has also been observed in IL-23 in complex with a Fab fragment derived from the monoclonal antibody 7G10 targeting IL-23p19 (PDB entry 3d85; (Beyer et al., 2008)). In that case, W156 was located just at the edge of the Fab:IL-23 interaction epitope, highlighting the local conformational plasticity centered at W156 on helix D in IL-23p19.
Figure 2. Conformational selection in IL-23 upon IL-23R binding.

A) Structural superposition of IL12-p40 subunits as visualized in all available crystal structures (herein and PDB codes 1f42, 1f45, 3d85, 3d87, 3duh, 3hmx, 3qwr, 4grw, 5mj3 and 5mj4) with respect to the D2 domain. B) Schematic recapitulation of the IL-23:IL-23R binding event. C) Structural transition of the N-terminus of helix D in IL-23p19 from a canonical α-helix (grey, PDB 5mj3) to a 310 helix (orange) in the IL-23:IL-23R complex. See also
Even though the epicenter of the IL-23:IL-23R interaction was the interface between IL-23RD1 and IL-23p19, IL-23RD1 also contacted IL-12p40 close to the D1 D2 interface, albeit to a very limited extent (260 Å2). This interface was characterized by rather loose van der Waals contacts, and only a single well-defined residue-residue interaction, namely, the bifurcated interaction of the guanidine group of Arg62 in IL-23RD1 with the side- and main chain of Asp109 in IL-12p40 at the periphery of the interaction interface. A comprehensive listing of all the interactions observed at the IL-23:IL-23R interface can be found in Table S1.
Despite the limited footprint of IL-23R at the D1–D2 interface of IL-12p40, we wondered about the impact of IL-23R recruitment on the overall domain structure of IL-12p40. To this end we carried out structural superpositions with respect to D2 of all available structures of IL-12p40, including in the unbound form (PDB entries 3d87 and 3duh). We found that IL-12p40 displayed extensive domain flexibility (Figure 2A) manifested by a hinge-like motion of D1 or D3 of 5°–10° with respect to D2. IL-12p40 in the cont ext of its complex with IL-23R adopts the most extreme conformation, whereby D1 of IL-12p40 turns away from D2 and D3 swings towards IL-23p19 (Figure 2B). Together, the structural rearrangements at the IL-23:IL-23R interaction interface and the ensuing conformational restraining of the p40 subunit of IL-23 point to pronounced receptor-mediated restructuring events in IL-23 that may have mechanistic implications.
A functional hotspot in IL-23 underlies high affinity binding to IL-23R
We tapped into the structural insights from the IL-23:IL-23R complex to interrogate the possible functional relevance of interactions observed at the IL-23:IL-23R interface. In particular, we aimed to investigate whether mutagenesis of W156 in IL-23p19 at the heart of the interaction interface (Figure 1D, Figure 2C) would manifest a strong phenotype in pro-inflammatory Th17-relevant readouts in vitro and in skin inflammation in vivo. This is because the functional role of this particular position had been postulated based on structural considerations of the receptor-free cytokine (Lupardus and Garcia, 2008), and has only recently been probed via reporter cellular assays in vitro (Schroder et al., 2015). Furthermore, W156 participated in interaction interfaces between IL-23 and therapeutic biologics and designed protein scaffolds (Figure 2A) (Beyer et al., 2008; Desmet et al., 2014; Li et al., 2017). We further selected L56 and L160 in human IL-23p19, which become buried at the complex interface, as well as K164, which interacts with the main chain carbonyl oxygens of G24 and N27 of IL-23R and was further stabilized by E58 in IL-23p19 (Figure 1C,E).
Following structure-based sequence alignments to identify the equivalent amino acid positions in mouse IL-23 (Figure 3A,B), we produced and purified recombinant wildtype mouse IL-23 (mIL-23WT) and mutant variants mIL-23p19W157A, mIL-23p19L161E, mIL-23p19K165S and mIL-23p19L56E. The biochemical behavior and stability of all purified targeted mutant variants of mouse IL-23 were very similar to IL-23WT. To assess the affinity of the mutant variants to mouse IL-23R (mIL-23R) we employed bio-layer interferometry (BLI) and found that mIL-23p19W157A and mIL-23p19L161E were unable to engage into a measurable interaction with mIL-23R compared to the high affinity displayed by mouse IL-23WT (Figure 3C). The recombinant human IL-23 had similar binding affinity and kinetics to mIL-23R (Figure S5A). On the other hand, IL-23p19K165S showed moderately impaired affinity to the receptor, while binding of IL-23p19L56E was very comparable to IL-23WT (Figure 3C). We probed the cellular context of these findings by examining the secretion of IL-17A and interleukin-22 (IL-22), two key cytokines in skin inflammation, upon differentiation of conventional naïve T cells into Th17 cells following stimulation by wildtype and mutant IL-23 variants. In each case, we found that mIL-23p19W157A was unable to drive IL-17A and IL-22 secretion, with mIL-23p19L161E also showing impaired activity (Figure 3D,E). The behavior of mIL-23p19L161E in the cellular assays contrasts its apparent lack of binding to IL-23R in our BLI experiments. This suggests that the affinity of mIL-23p19L161E for IL-23R is likely well into the micromolar range, which would be unmeasurable within the range of concentrations employed in our BLI experiments (2–500 nM). However, in a cellular context, where the dimensionality of the cell membrane and interactions with the shared receptor IL-12Rβ1 could serve as compensating parameters, mIL-23p19L161E can exhibit measurable bioactivity. Thus, the lack of any measurable binding affinity and bioactivity for mIL-23p19W157A in vitro mounts a strong case for the centrality of W157 on mIL-23 in the assembly of signaling complexes.
Figure 3. W156 in mouse IL-23p19 is a functional hotspot.

A) Sequence alignment of human and mouse IL-23p19. Residues at the human IL-23:IL-23R interface and positions chosen for mutagenesis in mouse IL-23 are shown in blue and red, respectively. Predicted secretion signal sequences are underlined. B) Structural context of interrogated positions (violet) at the human IL-23:IL-23R interface. C) Experimental BLI setup and representative response curves fitted with a 1:1 binding model (red) to quantify the kinetics (ka, kd) and binding affinity (KD) of wildtype and mutant IL-23 to IL-23R. D, E) IL-17A and IL-22 secretion by Th17 cells upon differentiation from purified naive CD4+ T by the addition of mouse IL-23. Error bars represent the standard deviation calculated from a technical replicate. F) Representative H&E staining of cutaneous biopsies obtained from C57BL/6J mice treated with PBS, IL-23 or IL-23 mutants. Diffuse epidermal hyperplasia (acanthosis) with associated compact hyperkeratotic and parakeratosis of the stratum corneum caused by wildtype IL-23 is indicated with an asterisk. The vertical black bar in the mIL-23WT panel indicates epidermal thickness measurement. G) Quantification of epidermal thickness by microscopy. H–J) Expression of Krt16, S100a8, and S100a9 in the dorsal skin of C57BL/6J mice injected with PBS, IL-23, or IL-23 mutants. p-values were determined using one-way ANOVA followed by Holm-Sidak's multiple comparisons test (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). Data are presented as ± S.E.M. n=[3–6]. See also Figure S4 and Figure S5A.
We sought to investigate the functional relevance of our structure-guided mutants of mIL-23 in a mouse model for skin inflammation in C57BL/6J mice. We established the working dose of mIL-23 via intradermal administration of mIL-23WT at opposing sides of shaved dorsal skin for four consecutive days and by comparisons with topically applied imiquimod (IMQ), a known inducer of epidermal hyperplasia (Figure S4A–D). mIL-23 injections were sufficient to induce pathologic features, which included diffuse epidermal hyperplasia (acanthosis) with associated compact hyperkeratosis and parakeratosis of the stratum corneum, and the formation of Munro’s microabscesses (Figure S4B–D). These observations correlated with an upregulation of Krt16, and S100a8 mRNA expression levels on day 4 (Figure S4E,F).
Next, we investigated the capacity of the mIL-23 mutants to elicit such skin pathologies. Mice treated with mIL-23WT showed a marked thickening of epidermis and leukocyte infiltration compared to mice injected with PBS (Figure 3C). In contrast to mIL-23p19WT, which presented with a pronounced thickening of epidermis and leukocyte infiltration, mIL-23p19W157A exhibited a complete lack of pathologies and was indistinguishable from the control phenotype (Figure 3F,G). mIL-23p19L161E showed a statistically significant decrease (p<0.05) of epidermal hyperplasia with associated compact hyperkeratosis and parakeratosis of the stratum corneum when compared to IL-23p19W157A (Figure 3F,G). Mutants mIL-23p19K165S and mIL-23p19L56E caused only mild skin pathologies and epidermal thickness that was quantified as statistically insignificant (p>0.05) compared to mIL-23WT (Figure 3F,G). Quantification of epidermal thickness showed that these histological changes were not significantly different compared to mIL-23WT (p>0.05) (Figure 3F,G). Finally, we verified the histological differences observed between the different mutants using quantitative reverse transcription PCR analysis of dorsal skin tissue to quantify the expression of hyperproliferation markers, such as keratin-16, S100-A8, and S100-A9. Consistent with histology, Krt16, S100a8 and S100a9 transcriptional levels were significantly decreased in mIL-23p19W157A (p<0.01) and mIL-23p19L161E (p<0.05) compared to mIL-23WT (Figure 3H–J). A non-significant trend was observed with mIL-23p19K165S and mIL-23p19L56E.
Therefore, our findings identify position W157 in mouse IL-23, and by extrapolation W156 in human IL-23, as a bona fide functional and structural hotspot that underlies high affinity binding to IL-23R to trigger restructuring of IL-23 for signaling purposes.
IL-23R primes IL-23 for IL-12Rβ1 recruitment
To draw the binding blueprint for the assembly of the human IL-23:IL-23R:IL12Rβ1 complex we employed purified recombinant human IL-23 and cognate receptor constructs in binding studies by microcalorimetry, and characterized stoichiometric cytokine-receptor complexes by size exclusion chromatography coupled to inline multi-angle laser light scattering (SEC-MALLS). We first investigated the interaction capacity of the two receptors by titrating IL-23R into IL-12Rβ1, but were unable to measure any discernible binding (Figure S5B). We subsequently titrated IL-23 into either IL-12Rβ1 or IL-23R to establish the relative affinities of binary cytokine-receptor complexes. Whereas IL-23 binds to IL-12Rβ1 with an equilibrium dissociation constant (KD) of 2 µM, its affinity for IL-23R is ~50-fold higher (KD= 44 nM) (Figure 4A,B). The apparent binding in titrations with IL-23R does not obey a 1:1 stoichiometric ratio suggesting that a fraction of recombinant IL-23R is inactive. This receptor fraction might be carrying an additional N-linked glycan at N29 (Zhao et al., 2010), which might prevent binding to IL-23p19 due to its proximity to the IL-23:IL-23R interaction site (Figure 1C,1D).
Figure 4. The IL-23:IL-23R binary complex enables high affinity binding of IL-12Rβ1.

A) Titration of IL-23 (72 µM) into IL-12Rβ1 (6.8 µM) B) Titration of IL-23 (55 µM) into IL-23R (6.5 µM). The stoichiometry of this experiment was set to 1 and the concentration in the cell was fitted to account for inactive IL-23R. The stoichiometry before correction is provided in parentheses. C) Titration of IL-12Rβ1 (38.8 µM) into preformed IL-23:IL-23R (3.6 µM). Fitted values are provided with their fitting errors. See also Figure S5.
We took advantage of our experimental setup to verify that the single domain VHH antibody we had used as a crystallization adjuvant does not affect the IL-23:IL-23R interaction. Indeed, titration of IL-23R into IL-23 and into a preformed complex of IL-23:Nb22E11 resulted in nearly identical binding profiles (Figure S5C,D). Furthermore, we sought to provide orthogonal evidence for the unexpected observation that the IL-23:IL-23R epitope involves only the Ig domain of IL-23R, in contrast to the employment of the CHR region in all other type I cytokine receptors characterized to date. Upon titrating IL-23RD2D3 (residues G122–E317) into IL-23 we did not observe a binding event (Figure S5D), essentially confirming the delineation of the IL-23 binding epitope to the N-terminal Ig-domain of IL-23R. Despite multiple attempts we were unable to express a stand-alone IL-23RD1 for binding studies.
Having concluded that our structural view of the IL-23:IL-23R complex is the high affinity binary receptor complex of IL-23, we proceeded to titrate IL-12Rβ1 into preformed IL-23:IL-23R complex. IL-12Rβ1 could be recruited to this complex to form the ternary assembly with a KD=25 nM, i.e. a ~100-fold higher affinity compared to its binary interaction with IL-23 (Figure 4C). We corroborated this sequential assembly of a ternary complex by SEC-MALLS by supplementing purified IL-23:IL-23R binary complex with 1:1 stoichiometry with IL-12Rβ1, to show that IL-12Rβ1 can be recruited to a chromatographically distinct species bearing a molecular mass consistent with 1:1:1 stoichiometry (Figure S2B).
Together, our orthogonal experimental evidence for the assembly mechanism of the receptor complex mediated by IL-23 supports a sequential and cooperative process, whereby a stable binary IL-23:IL23R complex is the mechanistic prerequisite for the recruitment of the shared receptor IL-12Rβ1 to complete the ternary cytokine-receptor complex.
The p40 subunit of IL-23 mediates IL-12Rβ1 binding
In the absence of direct structural evidence about where IL-12Rβ1 might be recruited to the cooperative IL-23:receptor complex, we leveraged the diverse molecular tools we created for this study and available mutagenesis studies, and sought to expand and consolidate structural information from complexes of antibody fragments with IL-23. The IL-23:IL-23R:IL-12Rβ1 ternary complex has been hypothesized to follow the “site I–II–III” assembly paradigm inspired by seminal structural studies of the cytokine-receptor complex comprising IL-6, IL-6Rα, and gp130 (Boulanger et al., 2003). This model places the helical bundle of IL-23p19 at the cross-roads of the assembly providing interaction sites for all cytokine and receptor modules concerned: site I for p40, site II for IL-12Rβ1, and site III for IL-23R. Our structural elucidation of the human IL-23:IL-23R complex has shown how sites I and III are utilized (Figure 1B,F).
To provide insights into how the shared receptor IL-12Rβ1 might interact with IL-23, we investigated the binding profile of the p40 subunit of IL-23 to IL-12Rβ1 by ITC. This yielded a very similar binding isotherm, affinity, and thermodynamic parameters as for the interaction of heterodimeric IL-23 with IL-12Rβ1 (Figure S5F, Figure 4A). This provided direct evidence that the IL-23p19 is likely not involved in binding IL-12Rβ1, and that the low affinity interaction between IL-23 and IL-12Rβ1 is exclusively mediated by the p40 subunit.
In light of this emerging new mode of binding, we further hypothesized that a careful selection of antagonistic molecules against IL-23 activity (Figure S6A) might provide additional clues about the binding epitope for IL-12Rβ1. We focused on the single domain VHH camelid antibodies 124C4, 22E11 and 37D5 (Saunders et al., 2009), which bind to sizeable albeit non-overlapping sites on IL-23, the therapeutic monoclonal antibody ustekinumab (Benson et al., 2011) binding to the D1–D2 junction of IL-12p40 (Luo et al., 2010), and the monoclonal antibody briakinumab (ABT-874) (Reich et al., 2011). As structural information about briakinumab was not available, we produced and purified the Fab fragment of briakinumab and determined the crystal structure of its complex with IL-23 and in the unbound form (Figure 5A, Table 1). Briakinumab targetted, like 22E11 and ustekinumab, the D1–D2 junction of IL-12p40 but is also tilted towards the IL-23R binding epitope in a manner incompatible with IL-23R binding (Figure 5A, Figure S6B–D).
Figure 5. The IL-12Rβ1 binding epitope maps to IL-12p40.

A) Cartoon representation of IL-23 structures (vermillion) in complex with antagonists: IL-23:briakinumab-Fab (reported herein); IL-23 in complex with three nanobodies (PDB code: 4grw), IL-23:Ustekinumab-Fab (pdb code: 3hmx) B) Cropped Coomassie-stained reducing SDS-PAGE gel of purified IL-23:IL-12Rβ1 complex crosslinked in the absence (control) or presence of IL-23 antagonists. Protein bands corresponding to crosslinked species (arrow with star) and IL-12Rβ1 in the absence of IL-23 antagonists (arrow) are indicated. C) Previously proposed model for the IL-23:receptor complex. D) Herein proposed model of the IL-23:IL-23R:IL-12Rβ1 ternary complex. See also Figure S6.
Next, we evaluated the ability of the five antibodies to abrogate the interaction between IL-23 and IL-12Rβ1. As the intrinsic low affinity of the IL-23:IL-12Rβ1 complex (Figure 4A) imposed limitations in the employment of conventional biophysical methods, we resorted to chemical cross-linking of IL-23:IL-12Rβ1 in the presence of each antibody followed by analyses of the cross-linked species by SDS-PAGE and Western-blotting. In the absence of steric hindrance, IL-23 and IL-12Rβ1 would be expected to produce chemical cross-links corresponding to a molecular species of ~120 kDa. We found that compared to the control, all five antibodies efficiently inhibited IL-23:IL-12Rβ1 complexes, providing evidence that their interaction epitopes are overlapping with IL-12Rβ1 binding (Figure 5B).
Thus, the conformational restraining of IL-23 upon binding of IL-23R to enable co-operative recruitment of IL-12Rβ1 appears to be manifested in two main ways. First, binding of IL-23R selects for a specific IL-12p40 conformation characterized by marked displacement of D1 and D3 with respect to D2 (Figure 2B). Second, the wedging of D1 of IL-23R close to the D1–D2 interface of IL-12p40 is poised to restrict the conformational degrees of freedom of IL-12p40. Indeed, the rather limited and loose interaction interface between IL-23RD1 and IL-12p40 observed in our IL-23:IL-23R binary complex might undergo compaction and extension upon recruitment of IL-12Rβ1 to the ternary complex. IL-12p40D2D3 has been shown to be the minimal requirement for the interaction of IL-23 with IL-12Rβ1 (Schroder et al., 2015). Together, such structural and biochemical insights indicate that the p40 subunit of IL-23 is likely the major mediator of interactions with the shared receptor IL-12Rβ1.
Disease-protecting mutations due to SNP can be mapped to the IL-23:IL-23R complex
Our structural template for the IL-23:IL-23R complex provided a platform for rationalizing at the protein level the most frequent missense single nucleotide polymorphisms (SNP) identified in IL-23R and IL-23 via genome-wide association studies or targeted sequencing efforts. To this end we leveraged the curated browser of the Exome Aggregation Consortium (ExAC) (Lek et al., 2016). Nearly all most commonly occurring missense SNPs in the coding regions of the extracellular domain of IL-23R mapped to domains D2 and D3 far from the IL-23R:IL-23 interface (Table S2). A number of them have been linked to protective effects in IBD and/or Crohn’s disease. For instance, a G149R substitution in IL-23RD2 is thought to exert its protective behavior in IBD and Crohn’s disease via reduced expression at the cell surface (Momozawa et al., 2011; Onodera et al., 2015; Rivas et al., 2011; Sivanesan et al., 2016). Nevertheless, at least one substitution, R86Q, does localize in the cytokine-binding domain of IL-23R and associates with a protective effect in IBD (Momozawa et al., 2011) (Table S2). Furthermore, several SNPs correspond to amino acid substitutions within the region in IL-23p19 that interacts with IL-23R, but also throughout the IL-12p40 subunit of IL-23 (Table S2). However, only a V298F substitution in D3 of IL-12p40 has been linked to a protective effect in IBD (Prescott et al., 2015). Whereas several amino acid substitutions resulting from SNPs in coding regions of IL-23 and IL-23R have been linked to protective roles in IBD and Crohn’s Disease, their impact at the molecular level currently remains unclear in light of the structural information we reported here.
DISCUSSION
IL-23 constitutes a major clinical target against widespread autoimmune and inflammatory diseases (Teng et al., 2015), and remains under intense investigation for its role in immunoregulation while structural and mechanistic insights into cytokine-receptor complexes in the IL-12 cytokine family have eluded the field of decades.
The findings we presented here enable a proposal for the assembly mechanism of the receptor complex mediated by IL-23 (Figure 6). The cornerstone thereof is the binary high-affinity complex of IL-23 with IL-23R. Despite featuring CHR extracellular domains, which cognate type I cytokine receptors typically employ for binding cytokines, IL-23R exclusively uses its N-terminal Ig domain to dock onto IL-23p19 as a primary interaction interface and to loosely contact IL-12p40 via a secondary interaction site. This direct visualization of the cytokine binding domain of IL-23R contrasts the rationale provided for the ability of a soluble version of human IL-23RCHR (residues 124–313) produced in E. coli to inhibit the differentiation of Th17 cells and secretion of IL-17A in vitro and in vivo studies (Guo et al., 2012). The epicenter of the IL-23:IL-23R interaction localizes at the N-terminus of helix D in IL-23p19, which serves as a structural switch by undergoing restructuring to project the functional hotspot residue W156 to its active conformation. The IL-23:IL-23R binary complex is also an obligate mechanistic step for the recruitment of the shared receptor IL-12Rβ1, which otherwise has poor affinity to IL-23 and no measurable binding to IL-23R. The binding site for IL-12Rβ1 does not involve the IL-23p19 subunit and is thus distinct from the hitherto site II inspired by the IL-6:IL-6Rα:gp130 complex (Figure 6). We further note that IL-12p80, the dimeric disulfide-linked form of the IL-12p40 subunit, was shown to have both agonistic signaling properties as well as antagonistic behavior over a range of activities such as macrophage chemotaxis, inflammatory responses, and protective activity in a mycobacterial model, binds IL-12Rβ1 while lacking an alpha-helical subunit (Cooper and Khader, 2007). Together with recent studies that failed to identify mutations in IL-23p19 that abrogate interactions with IL-12Rβ1 (Schroder et al., 2015), our findings point to a ternary assembly model that does not feature a IL-23p19:IL-12Rβ1 interface (site II), but a IL-12p40:IL-12Rβ1 interaction instead. Thus, our proposed assembly mechanism essentially segregates cognate and shared receptor binding to the helical (α) and non-helical (β) subunits of the heterodimeric cytokines in the IL-12 family, respectively (Figure 6).
Figure 6. Assembly mechanism of the receptor complex mediated by IL-23.

The signaling complex mediated by IL-23 proceeds via the sequential recruitment of the two cognate receptors, and involves conformational selection, and restructuring of IL-23 by IL-23R to recruit IL-12Rβ1 with high affinity. Such cytokine-receptor ternary complex is poised to support phosphorylation of intracellular Jak2 and Tyk2 tyrosine kinases associated with the intracellular parts of the receptors, to initiate signaling cascades. Extracellular antagonism of such assemblies can be achieved at two different stages along the ternary complex itinerary depending on the specificity of IL-23 targeting. See also Figure S6A.
Whereas the apparent cooperativity in the assembly of the IL-23:IL-23R:IL-12Rβ1 ternary complex is pronounced, as evidenced by the increase in affinity of IL-12Rβ1 to the IL-23:IL-23R complex, it does not appear to involve extensive receptor-receptor interactions between the extracellular domains of the two receptors contrary to other cytokine receptors (Felix et al., 2015; Spangler et al., 2015; Verstraete et al., 2017). In what appears to be a reversal of roles as dictated by current paradigms for cytokine-driven receptor activation, IL-23 becomes structurally activated by its high affinity receptor, IL-23R, to enable recruitment of the shared receptor IL-12Rβ1 (Figure 6). Together, such insights may have implications as to what can be deduced for other members of the IL-12 family. Our present findings on IL-23 can be most readily extrapolated to the archetypal IL-12, given that the two cytokines share the IL-12p40 subunit, signal through IL-12Rβ1 as a common receptor, and exhibit pro-inflammatory activities. In particular, the conserved domain organization in IL-23R and IL-12Rβ2 in terms of an N-terminal Ig-domain preceding the CHR domains indicates that the two receptors will likely share common cytokine binding principles. Of note is the conservation of the Ile-Cys-Gly amino acid sequence cassette in the G β-strand of the Ig domains of IL-12Rβ2 and IL-23R, which in IL-23R accommodates the functionally critical W156 in IL-23p19. We would thus expect that Y189 in IL-12p35 would serve a similar role in IL-12, and that IL-12Rβ2 will be the high-affinity receptor for IL-12.
Over the last few years evidence has been mounting that the IL-12 cytokine family displays expanded mix-and-match scenarios of cytokine subunits and utilization of signaling receptors covering a broad spectrum of immune responses (reviewed by (Hasegawa et al., 2016)). For instance, IL-39 has been recently introduced as a pro-inflammatory cytokine comprising the helical subunit IL-23p19 coupled to Epstein-Barr virus-induced protein 3 (EBI3), and has been proposed to signal via IL-23R and gp130 (Wang et al., 2016). Furthermore, IL-35, which comprises the helical IL-12p35 subunit coupled to EBI3, has been reported to mediate immunosuppressive responses through heterodimeric assemblies of IL-12Rβ2 and gp130, but also through homodimers of each receptor chain (Collison et al., 2012). It is currently unclear how our findings could help rationalize such receptor complexes mediated by IL-35. However, it is likely that higher order assemblies might be necessary or that other co-receptors might be at play at the cell surface. Interestingly, IL-23 was recently shown to act on human CD16+MDL-1+DAP12+ osteoclast precursors to induce osteoclast differentiation in inflammatory arthritis. This pathway instigated by IL-23 in myeloid cells uses DNAX activating protein of 12 kDa (DAP12) to recruit Spleen Tyrosine Kinase (SYK) and induce calcium-dependent activation of NFAT cytoplasmic 1 (NFATc1) (Shin et al., 2015).
Lastly, the structural and mechanistic insights we have presented here may further fuel efforts to antagonize signaling receptor assemblies mediated by IL-23 for therapeutic purposes. This is an area that has been extensively developed over the last decade via antibody and non-antibody protein scaffolds alike (Desmet et al., 2014; Hawkes et al., 2017; Ramamurthy et al., 2012). The need to differentiate therapeutic targeting of IL-23 from IL-12 has been called upon (Benson et al., 2011; Kulig et al., 2016). Indeed, the recent clinical approvals of guselkumab and tildrakizumab, two monoclonal antibodies against IL-23p19 for the treatment of plaque psoriasis (Blauvelt et al., 2017; Kopp et al., 2015; Reich et al., 2017), reflects such efforts for therapeutic differentiation. Thus, strategies addressing specifically the obligate IL-23:IL-23R binary complex or the functional hotspot centered at W156 in IL-23p19 may offer additional avenues to address the broad spectrum of diseases linked to IL-23.
STAR ★ METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
- METHOD DETAILS
-
◦Plasmids, constructs, and cell lines for protein expression in mammalian cells
-
◦Protein expression in HEK293 and purification from conditioned media
-
◦Recombinant protein expression in E.coli
-
◦Crystal structure determination and refinement
-
◦Bio-layer Interferometry (BLI)
-
◦Isothermal Titration Calorimetry (ITC)
-
◦Multi-angle laser light scattering (MALLS)
-
◦In vitro differentiation of naïve T cells into Th17
-
◦RNA extraction and real-time quantitative PCR
-
◦Skin histology
-
◦Protein cross-linking
-
◦
SUPPLEMENTAL INFORMATION
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Savvas Savvides (savvas.savvides@ugent.be) The HEK293S MGAT−/− cell line and derivatives thereof cannot be freely distributed as some rights remain with the cell line developers.
EXPERIMENTAL MODEL DETAILS
Mouse model for skin inflammation
8 week old male C57BL/6J mice were purchased from Jackson Laboratories (Sacramento, CA). The University of California at Davis Institutional Animal Care and Use Committee approved all animal protocols. Mice were intradermally injected with PBS, or 10 µg of recombinant IL-23 or IL-23 mutants daily at opposing sides of shaved dorsal skin for 4 consecutive days. The IL-23 dose was empirically determined to cause optimal and reproducible skin inflammation in mice (Figure S4). Naïve mice and mice treated with a daily topical dose of 35 mg of IMQ cream (5%) (Aldara; Medicis Dermatologics) on the dorsal skin were used as negative and positive controls respectively.
METHOD DETAILS
Plasmids, constructs, and cell lines for protein expression in mammalian cells
For testing expression of protein constructs and transient expression in HEK293 cells, constructs were cloned into the pHLsec plasmid (Aricescu et al., 2006). For generating stable cell lines, selected constructs were cloned into the pCDNA4TO (Thermo Fisher scientific) plasmid. For ease of cloning an EcoRI-site was inserted 3’ of the AflII site in the multi cloning site (MCS) of the pCDNA4 plasmid to resemble the pHLsec MCS.
The reference sequences of the cDNA encoding for hIL-12p40 residues Met1-Ser328 (NM_002187.2), hIL-23p19 residues Met1-Pro189 (NM_016584.2), mIL-12p40 residues Met1-Ser335 (NM_001303244.1) and mIL-23p19 residues Met1-Ala196 (NM_031252.2) were synthesized by Geneart and were a kind gift from Complix NV. However, to allow for cloning into the pHLsec vector, the AAT codon for N248 in the hIL-12p40 sequence was replaced with AAC. Human and mouse IL-12p40 were cloned without any purification tags. Human and mouse IL-23p19 were cloned in frame with a C-terminal hexahistidine tag. Mutants of mIL-23p19 were generated using overlap extension PCR, employing a PCR protocol adapted for Q5 polymerase (NEB) and allowing the final PCR to first run for 5 extra cycles without primers.
Sequence optimized DNA encoding for the reference sequences of IL-23R residues Met1-Glu317 (full extra cellular domains (ECD)) or Gly122-Glu317 (ECD D2D3) (NP_653302.2) and IL-12Rβ1 (full ECD) residues Met1-Arg544 (NP_005526.1) were purchased from Genscript. IL-23R was cloned in frame with a C-terminal Factor-Xa site followed by a hexahistidine tag, the D2D3 construct was further cloned in frame with an N-terminal chicken RTPµ-like secretion signal. IL-12Rβ1 was cloned in frame with a C-terminal FactorXa site followed by an AviHis tag.
Sequence optimized DNA encoding the light and heavy chains of ustekinumab Fab and briakinumab Fab were purchased from IDT as GBlocks. The N-terminal signal peptide sequences were exchanged for a chicken RTPµ-like signal peptide sequence. The light chains were cloned without any purification tag. The heavy chains were cloned in frame with a C-terminal thrombin site followed by a hexahistidine tag.
For the generation of monoclonal cell lines the constructs in pCDNA4TO plasmids were linearized in the AMPR gene by digestion with either Pvu1 or Sca1. The linearized DNA was purified using ethanol precipitation. Prior to transfection the medium of the HEK293S MGAT1−/− TETR cells was exchanged for HEPES buffered MEM supplemented with 0.1 mM chloroquine. The cells were transfected according to the calcium phosphate method (Sambrook and Russell, 2006). For the heterodimeric hIL-23 plasmids encoding each chain were co-transfected in a 1:1 ratio. Transformants were selected by supplementing the growth medium with 200 mg/l Zeocin. Individual colonies were picked and tested for target protein expression upon tetracycline induction using SDS-PAGE and western blot.
Protein expression in HEK293 and purification from conditioned media
All protein production in mammalian cells was done via the adherent HEK293S MGAT1−/− TETR cell line with the exception of the antibody Fab fragments, murine IL-23 and mutants thereof, which were expressed in adherent HEK293T cells. HEK293 cells were maintained in DMEM + 10 % FCS and were supplemented with 50 mg/l Zeocin for stable cell lines. The growth medium was exchanged to DMEM supplemented with 3.6 mM valproic acid upon protein expression. Expression in stable cell lines 2 mg/l tetracycline was additionally added to the medium.
Transient expression in HEK293 cells was achieved using polyethyleneimine as transfection reagent (Aricescu et al., 2006). For the heterodimeric Fab fragments and mIL-23, plasmids encoding for each chain were co-transfected in a 1:1 ratio. Conditioned medium was harvested from the HEK293 cultures once the cell morphology and integrity deteriorated assessed by light microscopy. Medium was clarified via centrifugation and filtered through a 0.22 µm filter prior to chromatographic steps.
The standard chromatography running buffer used during protein purification and SEC-MALLS experiments was HBS (20 mM HEPES pH 7.4, 150 mM NaCl). IMAC elution buffer was HBS supplemented with 250 mM imidazole, 5% (v/v) glycerol and 0.05% (v/v) Tween20. His-tagged proteins were captured using Immobilized Metal Affinity Chromatography IMAC (cOmplete His-tag purification resin, Roche) and polished via size exclusion chromatography (SEC) (HiLoad 16/600 superdex 75 or 200, GE Lifesciences). The AviHis tag of IL-12Rβ1 was removed by overnight digestion of the purified protein with Bovine Factor Xa (Prozyme) at 287 K in HBS supplemented with 2mM CaCl2. To remove any undigested protein and the enzyme, the digestion mixture was loaded onto a HiTrap benzamidine FFcolumn (GE lifesciences) in series with a HisTrap FF column (GE Lifesciences). The flowthrough containing tagless IL-12Rβ1 was polished on SEC. The AviHis tag of the heavy chain of the ustekinumab and briakinumab Fabs were removed by overnight digestion of the purified protein with human Thrombin (Novagen) at 287 K in HBS supplemented with 2 mM CaCl2. Undigested protein was removed via IMAC. The flow-through containing tag-free Fab fragments was polished on SEC. For the murine IL-23 and mutants thereof used for animal studies, endotoxin levels were measured using a Endosafe PTS limulus amebocyte lysate assay (Charles river) and were below 5 EU/mg.
Recombinant protein expression in E.coli
DNA encoding nanobodies were sequence-optimized for expression in E. coli and were purchased from IDT as GBlocks. The sequences were cloned in a variant of the pMECS vector whereby the nanobody construct was in frame with a N-terminal PelB signal peptide sequence and a C-terminal StrepTagII tag. Single domain VHH camelid antibodies were expressed as described by (Pardon et al., 2014) in E. coli WK6. The capture step using IMAC was replaced by a Streptactin based affinity-purification according to manufacturer’s protocol (IBA Lifesciences) and the protein was polished using SEC (HiLoad 16/600 Superdex 75).
Crystal structure determination and refinement
For crystals of the IL23:IL23R:Nb22E11 complex, the complex was reconstituted and purified via SEC. Glycans were trimmed by overnight enzymatic digestion with Jack bean α-mannosidase (Prozyme) in HBS supplemented with 2 mM ZnCl2 and 50 mM Bis-Tris pH 6.5. After overnight digestion, the complex was further purified via SEC and concentrated to 7 mg/ml. Commercial sparse matrix sitting drop crystallization screens were set up using a Mosquito liquid handling robot (TTP Labtech) using a 100 nl protein mixed with 100 nl mother liquor geometry in SwissSci 96-well triple drop plates. Plates were incubated at 287 K. Hits were optimized in both sitting and hanging drop format. An original hit in the JCSG+ screen (0.2M MgCl2 0.1M Tris pH 7 10% PEG8000) was optimized to 0.2M MgCl2 0.1M Tris pH 6.6 11% (w/v) PEG8000 in hanging drop format. Crystals were cryoprotected in mother liquor supplemented with 20 % ethylene glycol prior to being cryo-cooled in liquid nitrogen. Diffraction data was collected at 100 K at the P14 microfocus beam line at PETRA III, Hamburg.
Diffraction data was integrated and using XDS (Kabsch, 2010). The anisotropic datasets were truncated and rescaled using the STARANISO server. Initial phases were obtained using maximum likelihood molecular replacement in Phaser (McCoy et al., 2007) using a composite model for IL-23 (pdb 4oe8) 22E11 nanobody (pdb 4grw) complex. Structure building and refinement was performed iteratively in COOT (Emsley et al., 2010) and Phenix.refine (Adams et al., 2010) or BUSTER (Bricogne et al., 2016). The IL-23R structure was built from scratch into the electron density. Two isotropic atomic displacement parameters (ADP, one for mainchain atoms and one for sidechain atoms) were refined per residue together with rigid body harmonic displacement parameters described by a translation libration screw-rotation (TLS) model. TLS groups were determined in Phenix and glycans were assigned to their closest protein group. Higher resolution local torsion-angle restraints were imposed for IL-23 (pdb 4oe8) and the nanobody 22E11 (pdb 4grw).
For IL-23:briakinumab-Fab crystals the complex was purified via SEC and concentrated to 9 mg/ml. Screens were set up and optimized as described above. An original hit in the JCSG+ and Index screen (2 M (NH4)2SO4, 100 mM BisTris pH 5.5) was optimized to 2 M (NH4)2SO4, 100 mM BisTris pH 6 in sitting drop format. Crystals were cryoprotected in 90% (v/v) saturated (NH4)2SO4, 10% (v/v) 1 M BisTris pH 6 prior to being cryocooled in liquid nitrogen. Diffraction data was collected at 100 K at the P14 microfocus beam line at PETRA III, Hamburg. The data were processed as described above with the exception that the “BEAM_DIVERGENCE” parameter had to be doubled in XDS, greatly improving the dataset consistency. Initial phases were obtained from a dataset of an isomorphous crystal diffracting to 4.8Å using maximum likelihood molecular replacement in Phaser. Search models were IL-23 (pdb 4oe8) and a briakinumab homology model created in Modeller (Eswar et al., 2006) split into constant and variable domains without CDR loops. Structure building and refinement was performed iteratively in COOT and Phenix.refine or BUSTER. One overall isotropic ADP parameter was refined together with a TLS model. One TLS group was assigned per chain. Higher resolution local torsion-angle restraints were imposed for IL23 (pdb 4oe8) and the briakinumab Fab (structure reported in this paper) as well as local torsion angle non-crystallographic symmetry (NCS) restraints.
For briakinumab Fab crystals the protein was concentrated to 19 mg/ml and a pH versus PEG3350 concentration or (NH4)2SO4 concentration screen was set up in hanging drop format. A single crystal from an optimized crystallization hit (21.6% (w/v) PEG 3350 40 mM Glycine pH 9.5) was used to prepare a seed stock (D'Arcy et al., 2007) at a nominal 1/1000 dilution using a PTFE seed bead (Hampton Research). This seed stock was used to set up micro-seeded matrix screens of the PEG-ION HT sparse matrix screen and an optimization screen of the original hit with 600 nl drops (200 nl protein, 100 nl seed stock and 300 nl mother liquor). Crystals developed faster and in more drops of the optimization screen of the original hit compared to the non-seeded screen. An isomorphous crystal also grew in condition A12 (200 mM NH4I, 20% (w/v) PEG 3350 pH6.2) of the PEGION HT screen (Hampton Research). This crystal was cryoprotected in 80% (v/v) mother-liquor supplemented with 20% (v/v) ethylene glycol prior to being cryo-cooled in liquid nitrogen.
Diffraction data were collected at 100 K at the P14 microfocus beam line at PETRA III, Hamburg. The data was processed as described above with the difference that the dataset was isotropic and wN’t processed with the STARANISO server. Initial phases were obtained using maximum likelihood molecular replacement in Phaser. The briakinumab homology model, split in constant and variable domains without CDR loops as described above, was used as search model. Structure building and refinement was performed as described above. Individual isotropic ADP parameters were refined together with a TLS model. The TLS groups were assigned to each domain. Local torsion angle non-crystallographic symmetry (NCS) restraints were imposed.
Crystals of glycosylated human IL-23 produced in HEK293S MGAT1−/− TETR cells were grown as previously described (Shirouzono et al., 2012). IL-23 was concentrated to 6 mg/ml and a narrow pH versus PEG1000 concentration screen was set up around the published condition (18% (w/v) PEG 1000, 200 mM Li2SO4, 100 mM sodium citrate, 100 mM sodium phosphate pH 4.2) in both hanging- and sitting-drop geometry using 200 nl drops (100 nl protein mixed with 100 nl mother liquor). A crystal grown in hanging drop with 19.5% (w/v) PEG1000, 100 mM sodium citrate, 100 mM sodium phosphate pH 4.4, 200 mM Li2SO4 was cryoprotected in mother liquor supplemented up to 25 % (w/v) PEG1000 prior to being cryocooled in liquid nitrogen. Diffraction data was collected at 100 K at the P14 microfocus beam line at PETRA III, Hamburg.
The data was processed as described above with the difference that the dataset was isotropic and wN’t processed with the STARANISO server. Initial phases were obtained using maximum likelihood molecular replacement in Phaser using the IL-23 model from PDB entry 4oe8. Structure building and refinement was performed as described above. Individual isotropic ADP parameters were refined together with a TLS model. TLS groups were assigned by Phenix.
Bio-layer Interferometry
The binding kinetics and dissociation constant of IL-23, and mutants thereof, towards murine IL-23R were characterized by biolayer interferometry (BLI). Binding assays were performed using an Octet Red 96 machine (FortéBIO) in assay buffer (20 mM HEPES pH 7.4, 300 mM NaCl, 0.05 % (w/v) BSA, 0.05% (v/v) Tween 20) at 303 K. Lyophilized mouse IL-23R Fc fusion protein (R&D Systems) was reconstituted in PBS to 0.1 mg/ml as recommended by the manufacturer. mIL-23R Fc was immobilized at 5 µg/ml in PBS onto anti-human IgG Fc capture biosensors (FortéBIO) until an optical shift of ~0.5 nm was achieved. To correct for system artifacts, system drift, and non-specific binding to the biosensors (the latter was apparent at higher analyte concentrations) the experiments were set up in a double referenced geometry. Additional control biosensors were therefore not coated with mIL-23R-Fc. After equilibrating the biosensors in assay buffer, the biosensors were dipped in a 3-fold dilution series (517 nM, 172 nM, 57 nM, 19 nM, 6 nM, 2 nM, and blanc control) of cytokine for 300 s during the association phase followed by a 600 s dissociation phase in assay buffer. All biosensors were regenerated after each cycle in 10 mM glycine pH 1.75. After subtraction of the control sensorgrams, a 1:1 binding model was fitted to each dilution series. Assay design and data acquisition were performed using the BLI Data acquisition software 9.0.0.49 (FortéBIO) and data analysis was performed using the Data Analysis software 9.0.0.14 (FortéBIO).
Isothermal Titration Calorimetry
Prior to all measurements all protein pairs were buffer matched on SEC in standard SEC running buffer (20 mM HEPES pH 7.4, 150 mM NaCl). Experiments were carried out using a VP-ITC MicroCalorimeter except for the IL-23 in IL-23RCHR and the IL-12Rβ1 in IL-23R titrations, both of which were carried out using a MicroCal PEAQ-ITC instrument. The experiments were conducted at 310 K. Injection spacing was chosen to allow for the signal to get back to a stable baseline. Data were analyzed using the PEAQ-ITC analysis software (version 1.1.0.1262, Malvern) and fit using a “one set of sites” model. In case of experiments involving free IL-23R in the cell or syringe, the concentration of IL-23R was corrected to allow for a 1:1 stoichiometry.
Multi-angle Laser Light Scattering (MALLS)
Protein samples of 100 µl at approximately 1mg/ml were injected onto a Superdex increase 10/300 GL column (GE Lifesciences) connected in line to a UV-detector (Shimadzu), a miniDawnTReos (Wyatt) multi-angle laser light scattering detector and an optilab T-Rex (Wyatt) refractometer thermostated at 298 K. The refractive increment value (dn/dc) was adapted to reflect the expected glycosylation of the protein using the formula
, whereby the MWglycan is estimated at 1.2 kDa per N-linked glycan. IL-23 has one clear N-linked glycan in available crystal structures, IL-23R has 7 visible N-linked glycans in our crystal structure, IL-12Rβ1 has 6 predicted N-linked glycosylation sites and the Fab fragments have none.
Band broadening was corrected for using reference measurements of BSA (Pierce). Data analysis was carried out using the Astra 6.1.6 software and standard deviations were calculated in the Excel software.
In vitro differentiation of naïve T cells into Th17
Conventional naive CD4+ T cells obtained from spleens of C57BL/6J 8-week old male mice were FACS sorted to over 99% purity after being stained with PE-Cy7-conjugated anti-CD62L (Tonbo Biosciences), FITC-conjugated anti-CD4 (BD Biosciences), V500-conjugated anti-CD3ε (BD Biosciences), violetFluor 450-conjugated anti-CD19 (Tonbo Biosciences), PE-conjugated anti-CD8α (eBioscience), APC-Cy7-conjugated anti-CD44 (eBioscience) and APC-conjugated anti-CD1d tetramer (Sidobre and Kronenberg, 2002). After purification, cells were cultured in RPMI containing 10% fetal calf serum, glutamine, non-essential amino acids, sodium pyruvate, 2-mercaptoethanol, penicillin and streptomycin. Naive CD4+ T cells were seeded in a 96-well U-bottom plate (1 × 105 cells/well) coated with anti-CD3ε mAb (3 µg/ml; eBioscience) and then further activated with soluble anti-CD28 mAb (5 µg/ml; eBioscience). Culture medium was supplemented with increasing concentrations (0, 1.5625, 6.25, 25, 100 ng/ml or 0–1.7 nM) of recombinant mouse IL-23 or IL-23 mutants to induce Th17 differentiation. After 4 days of stimulation, concentrations of IL-17A and IL-22 in culture supernatants were detected by ELISA (eBioscience).
RNA Extraction and Real-Time Quantitative PCR
Total RNA was extracted using the RNeasy mini kit (Qiagen), following the manufacturer’s instructions. The purity of the RNA was assessed by the ratio of absorbance at 260 and 280 nm. RNA preparation was reverse-transcribed using the Omniscript RT Kit (Qiagen). The resultant cDNA were amplified by qPCR using Brilliant III Ultra-Fast SYBR QPCR (Agilent technologies) with the following primers: : Mouse B2m, 5’-CTGCTACGTAACACAGTTCCACCC-3’ and 5’-CATGATGCTTGATCACATGTCTCG-3’; mouse Krt16, 5’- TATCCACTCCTCCTCACAGC -3’ and 5’- GCTGGTTGAACCTTGCTCCT-3’; mouse S100a8, 5’- TGCGATGGTGATAAAAGTGG-3’ and 5’- GGCCAGAAGCTCTGCTACTC -3’; mouse S100a9, 5’- CACAGTTGGCAACCTTTATG-3’ and 5’- CAGCTGATTGTCCTGGTTTG-3’. Experiments were performed in triplicate and gene expression levels were normalized to the Beta-2-Microglobulin housekeeping gene.
Skin Histology
Dorsal skin was collected and fixed in 4% formaldehyde (Fisher Chemical), embedded in paraffin, cut longitudinally into 5-µm sections and stained with haematoxylin and eosin (Sigma-Aldrich). Images were acquired using a Keyence BZ 9000 microscope. Thickness of epidermis was measured by using the BZ-Analyzer software (Keyence) and data shown represents the average of 10 measurements per section, with 2 sections per mouse.
Protein crosslinking
The ability of IL-23 antagonists to disrupt the IL-23:IL-12Rβ1 complex was assessed using chemical crosslinking. The crosslinking experiment was performed in HBS at RT. 4 µl (40 pmol) of purified IL-23:IL-12Rβ1 complex was crosslinked with 1µl (500 pmol) freshly dissolved BS3 (Thermo Scientific) for 30 min in the absence (control) or presence of 2 µl (80 pmol) of IL-23 antagonists. The reaction was quenched by adding 1 µl 1M TRIS pH 9. The resulting sample was analyzed on SDS-PAGE under reducing conditions and stained with Coomassie blue. The presence of both IL-12p40 and IL-12Rβ1 in the crosslinked species was ascertained by western blot using polyclonal anti human IL-12p40 and anti-human IL-12 Rβ1 primary antibodies (R&D).
QUANTIFICATION AND STATISTICAL ANALYSIS
In-vivo experiments
Significance of observed differences was determined by one-way ANOVA followed by Holm-Sidak's multiple comparisons test. Analysis was performed with the GraphPad Prism Software, version 6.
ITC analysis
Fitted values are reported with their fitting errors as reported by the MicroCal PEAQ-ITC Analysis software version 1.1.0.1262. The errors (also called standard errors) in the fit parameters are calculated as square root of respective diagonal elements of variance-covariance matrix (calculated from partial derivatives of the fitted function with respect to the fitted parameters). The errors calculated with default fitting procedure are then scaled (multiplied) with the square root of the reduced chi-square.
DATA AVAILABILITY
Protein Data Bank accession codes
Crystallographic coordinates and structure factors have been deposited to the Protein Data Bank (www.rcsb.org) as follows: human IL-23:IL-23R:Nb22E11 complex (5mzv), unbound human IL-23 (5mxa), IL-23:BriakinumabFab complex (5njd), and BriakinumabFab (5n2k).
COMPETING FINANCIAL INTERESTS
none
Supplementary Material
HIGHLIGHTS.
Determined crystal structure of human IL-23 in complex with cognate IL-23R.
IL-23 is restrained upon IL-23R binding to enable recruitment of IL-12Rβ1.
A single residue in IL-23 is crucial for its pro-inflammatory activity.
Receptor binding sites on IL-23 segregate to the p19 and IL-12p40 subunits.
Acknowledgments
We thank the staff of beam lines P14 (PETRAIII) and Proxima2A (SOLEIL) for beam time allocation and excellent technical support. We further thank Veronique De Backer (Ghent University Hospital) for technical support. YB and SD are supported by pre-doctoral fellowships from the Flanders Agency for Innovation and Entrepreneurship (VLAIO-Flanders, Belgium). SG is supported by a postdoctoral fellowship from Research Foundation Flanders (FWO, Belgium). LVDB, FH, and MD are supported by the Ghent University Hospital Spearhead Initiative for Immunology Research. FH is supported by the Jeffrey Modell Foundation. IEA acknowledges support from the NIAMS/NIH (Grant AR62173) and a National Psoriasis Foundation Translational Research Grant. SNS acknowledges research support from Research Foundation Flanders (FWO grant G0C2214N), the Hercules Foundation (no. AUGE-11-029), and the VIB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
YB designed and performed recombinant protein production, biochemical, biophysical, and structural studies. YB and SNS analyzed data. RM, SD, and KS contributed to molecular tools and biochemical studies. LB and IEA designed and performed studies employing the animal model of skin inflammation. LVdB, MD, FH, SG, and DE designed and performed in vitro cellular studies. YB and SNS wrote the manuscript with contributions from all authors. SNS conceived and supervised the project.
References
- Adams PD, Afonine PV, Bunkczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D: Biological Crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa A, Ruzicka T, Prinz JC. Therapeutic Efficacy of Interleukin 12/Interleukin 23 Blockade in Generalized Pustular Psoriasis Regardless of IL36RN Mutation Status. JAMA Dermatol. 2016;152:825–828. doi: 10.1001/jamadermatol.2016.0751. [DOI] [PubMed] [Google Scholar]
- Aricescu aR, Lu W, Jones EY. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta crystallographica. Section D, Biological crystallography. 2006;62:12431250. doi: 10.1107/S0907444906029799. [DOI] [PubMed] [Google Scholar]
- Benson JM, Sachs CW, Treacy G, Zhou H, Pendley CE, Brodmerkel CM, Shankar G, Mascelli MA. Therapeutic targeting of the IL-12/23 pathways: generation and characterization of ustekinumab. Nat Biotechnol. 2011;29:615–624. doi: 10.1038/nbt.1903. [DOI] [PubMed] [Google Scholar]
- Beyer BM, Ingram R, Ramanathan L, Reichert P, Le HV, Madison V, Orth P. Crystal structures of the pro-inflammatory cytokine interleukin-23 and its complex with a high-affinity neutralizing antibody. Journal of molecular biology. 2008;382:942–955. doi: 10.1016/j.jmb.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Blauvelt A, Papp KA, Griffiths CE, Randazzo B, Wasfi Y, Shen YK, Li S, Kimball AB. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: Results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. 2017;76:405–417. doi: 10.1016/j.jaad.2016.11.041. [DOI] [PubMed] [Google Scholar]
- Boulanger MJ, Chow D-c, Brevnova EE, Garcia KC. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science. 2003;300:2101–2104. doi: 10.1126/science.1083901. [DOI] [PubMed] [Google Scholar]
- Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C, Womack TO. Buster 2016 [Google Scholar]
- Collison LW, Delgoffe GM, Guy CS, Vignali KM, Chaturvedi V, Fairweather D, Satoskar AR, Garcia KC, Hunter Ca, Drake CG, et al. The composition and signaling of the IL-35 receptor are unconventional. Nature immunology. 2012;13:290–299. doi: 10.1038/ni.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AM, Khader Sa. IL-12p40: an inherently agonistic cytokine. Trends in Immunology. 2007;28:33–38. doi: 10.1016/j.it.2006.11.002. [DOI] [PubMed] [Google Scholar]
- D'Arcy A, Villard F, Marsh M. An automated microseed matrix-screening method for protein crystallization. Acta Crystallographica Section D: Biological Crystallography. 2007;63:550–554. doi: 10.1107/S0907444907007652. [DOI] [PubMed] [Google Scholar]
- Desmet J, Verstraete K, Bloch Y, Lorent E, Wen Y, Devreese B, Vandenbroucke K, Loverix S, Hettmann T, Deroo S, et al. Structural basis of IL-23 antagonism by an Alphabody protein scaffold. Nature communications. 2014;5:5237. doi: 10.1038/ncomms6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Hillary SA, Abraham C, Regueiro M, Griffiths A, et al. A Genome-Wide Association Study Identifies IL23R as an Inflammatory Bowel Disease Gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberl G. Immunity by equilibrium. Nature Reviews Immunology. 2016;16:524–532. doi: 10.1038/nri.2016.75. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallographica Section D: Biological Crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics. 2006;Chapter 5(Unit-5):6. doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmeyer L, Mylonas A, Demaria O, Mennella A, Yawalkar N, Laffitte E, Hohl D, Gilliet M, Conrad C. Interleukin 23–Helper T Cell 17 Axis as a Treatment Target for Pityriasis Rubra Pilaris. JAMA Dermatology. 2017;153:304. doi: 10.1001/jamadermatol.2016.5384. [DOI] [PubMed] [Google Scholar]
- Felix J, De Munck S, Verstraete K, Meuris L, Callewaert N, Elegheert J, Savvides SN. Structure and Assembly Mechanism of the Signaling Complex Mediated by Human CSF-1. Structure. 2015;23:1621–1631. doi: 10.1016/j.str.2015.06.019. [DOI] [PubMed] [Google Scholar]
- Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23–IL-17 immune axis: from mechanisms to therapeutic testing. Nature Reviews Immunology. 2014;14:585–600. doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491:254–258. doi: 10.1038/nature11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubler U, Chua AO, Schoenhaut DS, Dwyer CM, McComas W, Motyka R, Nabavi N, Wolitzky AG, Quinn PM, Familletti PC. Coexpression of two distinct genes is required to generate secreted bioactive cytotoxic lymphocyte maturation factor. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:4143–4147. doi: 10.1073/pnas.88.10.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Luo C, Wang C, Zhu Y, Wang X, Gao X, Yao W. Protection against Th17 cells differentiation by an interleukin-23 receptor cytokine-binding homology region. PloS one. 2012;7:e45625. doi: 10.1371/journal.pone.0045625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa H, Mizoguchi I, Chiba Y, Ohashi M, Xu M, Yoshimoto T. Expanding diversity in molecular structures and functions of the IL-6/IL-12 heterodimeric cytokine family. Frontiers in Immunology. 2016;7:1–10. doi: 10.3389/fimmu.2016.00479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol. 2017;140:645–653. doi: 10.1016/j.jaci.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta crystallographica. Section D, Biological crystallography. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp T, Riedl E, Bangert C, Bowman EP, Greisenegger E, Horowitz A, Kittler H, Blumenschein WM, McClanahan TK, Marbury T, et al. Clinical improvement in psoriasis with specific targeting of interleukin-23. Nature. 2015;521:222–226. doi: 10.1038/nature14175. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annual Review of Immunology. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- Kulig P, Musiol S, Freiberger SN, Schreiner B, Gylveszi G, Russo G, Pantelyushin S, Kishihara K, Alessandrini F, Kndig T, et al. IL-12 protects from psoriasiform skin inflammation. Nature Communications. 2016;7:13466. doi: 10.1038/ncomms13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, Basham B, McClanahan T, Kastelein RA, Oft M. IL-23 promotes tumour incidence and growth. Nature. 2006;442:461–465. doi: 10.1038/nature04808. [DOI] [PubMed] [Google Scholar]
- Lee Jacob S, Tato Cristina M, Joyce-Shaikh B, Gulan F, Cayatte C, Chen Y, Blumenschein Wendy M, Judo M, Ayanoglu G, McClanahan Terrill K, et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity. 2015;43:727–738. doi: 10.1016/j.immuni.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wei H, Krystek SR, Jr, Bond D, Brender TM, Cohen D, Feiner J, Hamacher N, Harshman J, Huang RY, et al. Mapping the Energetic Epitope of an Antibody/Interleukin-23 Interaction with Hydrogen/Deuterium Exchange, Fast Photochemical Oxidation of Proteins Mass Spectrometry, and Alanine Shave Mutagenesis. Anal Chem. 2017;89:2250–2258. doi: 10.1021/acs.analchem.6b03058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowes MA, Surez-Farinas M, Krueger JG. Immunology of psoriasis. Annual review of immunology. 2014:227–255. doi: 10.1146/annurev-immunol-032713-120225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubberts E. The IL-23-IL-17 axis in inflammatory arthritis. Nat Rev Rheumatol. 2015;11:415–429. doi: 10.1038/nrrheum.2015.53. [DOI] [PubMed] [Google Scholar]
- Luo J, Wu S-J, Lacy ER, Orlovsky Y, Baker A, Teplyakov A, Obmolova G, Heavner Ga, Richter H-T, Benson J. Structural basis for the dual recognition of IL-12 and IL-23 by ustekinumab. Journal of molecular biology. 2010;402:797–812. doi: 10.1016/j.jmb.2010.07.046. [DOI] [PubMed] [Google Scholar]
- Lupardus PJ, Garcia KC. The structure of interleukin-23 reveals the molecular basis of p40 subunit sharing with interleukin-12. Journal of molecular biology. 2008;382:931–941. doi: 10.1016/j.jmb.2008.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell Joseph R, Zhang Y, Brown William A, Smith Carole L, Byrne Fergus R, Fiorino M, Stevens E, Bigler J, Davis John A, Rottman James B, et al. Differential Roles for Interleukin-23 and Interleukin-17 in Intestinal Immunoregulation. Immunity. 2015;43:739–750. doi: 10.1016/j.immuni.2015.08.019. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of Applied Crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, McClanahan TK, O'Shea JJ, Cua DJ. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17–producing effector T helper cells in vivo. Nature Immunology. 2009;10:314–324. doi: 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momozawa Y, Mni M, Nakamura K, Coppieters W, Almer S, Amininejad L, Cleynen I, Colombel JF, de Rijk P, Dewit O, et al. Resequencing of positional candidates identifies low frequency IL23R coding variants protecting against inflammatory bowel disease. Nat Genet. 2011;43:43–47. doi: 10.1038/ng.733. [DOI] [PubMed] [Google Scholar]
- Moutsopoulos NM, Zerbe CS, Wild T, Dutzan N, Brenchley L, DiPasquale G, Uzel G, Axelrod KC, Lisco A, Notarangelo LD, et al. Interleukin-12 and Interleukin-23 Blockade in Leukocyte Adhesion Deficiency Type 1. New England Journal of Medicine. 2017;376:1141–1146. doi: 10.1056/NEJMoa1612197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent Pro- and Antiinflammatory Roles for IL-23 and IL-12 in Joint Autoimmune Inflammation. The Journal of Experimental Medicine. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera K, Arimura Y, Isshiki H, Kawakami K, Nagaishi K, Yamashita K, Yamamoto E, Niinuma T, Naishiro Y, Suzuki H, et al. Low-Frequency IL23R Coding Variant Associated with Crohn's Disease Susceptibility in Japanese Subjects Identified by Personal Genomics Analysis. PLoS One. 2015;10:e0137801. doi: 10.1371/journal.pone.0137801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- Pardon E, Laeremans T, Triest S, Rasmussen SGF, Wohlknig A, Ruf A, Muyldermans S, Hol WGJ, Kobilka BK, Steyaert J. A general protocol for the generation of Nanobodies for structural biology. Nature Protocols. 2014;9:674–693. doi: 10.1038/nprot.2014.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. Journal of immunology. 2002;168:5699–5708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- Prescott NJ, Lehne B, Stone K, Lee JC, Taylor K, Knight J, Papouli E, Mirza MM, Simpson MA, Spain SL, et al. Pooled sequencing of 531 genes in inflammatory bowel disease identifies an associated rare variant in BTNL2 and implicates other immune related genes. PLoS Genet. 2015;11:e1004955. doi: 10.1371/journal.pgen.1004955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamurthy V, Krystek SR, Bush A, Wei A, Emanuel SLa. Structures of adnectin/protein complexes reveal an expanded binding footprint. Structure. 2012;20:259–269. doi: 10.1016/j.str.2011.11.016. [DOI] [PubMed] [Google Scholar]
- Reeves PJ, Callewaert N, Contreras R, Khorana HG. Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc Natl Acad Sci U S A. 2002;99:13419–13424. doi: 10.1073/pnas.212519299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich K, Armstrong AW, Foley P, Song M, Wasfi Y, Randazzo B, Li S, Shen YK, Gordon KB. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: Results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. J Am Acad Dermatol. 2017;76:418–431. doi: 10.1016/j.jaad.2016.11.042. [DOI] [PubMed] [Google Scholar]
- Reich K, Langley RG, Papp KA, Ortonne J-P, Unnebrink K, Kaul M, Valdes JM. A 52-Week Trial Comparing Briakinumab with Methotrexate in Patients with Psoriasis. New England Journal of Medicine. 2011;365:1586–1596. doi: 10.1056/NEJMoa1010858. [DOI] [PubMed] [Google Scholar]
- Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, Boucher G, Ripke S, Ellinghaus D, Burtt N, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43:1066–1073. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Calcium-phosphate-mediated Transfection of Eukaryotic Cells with Plasmid DNAs. Cold Spring Harbor Protocols. 2006;2006 doi: 10.1101/pdb.prot095430. pdb.prot3871. [DOI] [PubMed] [Google Scholar]
- Saunders MJS, Blanchetot C, Rommelaere H, Vercammen J, De HJJW. Amino acid sequences directed against heterodimeric cytokines and/or their receptors and polypeptides comprising the same. WO2009068627 A3 Patent. 2009
- Schroder J, Moll JM, Baran P, Grotzinger J, Scheller J, Floss DM. Non-canonical interleukin 23 receptor complex assembly: p40 protein recruits interleukin 12 receptor beta1 via site II and induces p19/interleukin 23 receptor interaction via site III. J Biol Chem. 2015;290:359–370. doi: 10.1074/jbc.M114.617597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H-S, Sarin R, Dixit N, Wu J, Gershwin E, Bowman EP, Adamopoulos IE. Crosstalk among IL-23 and DNAX Activating Protein of 12 kDa-Dependent Pathways Promotes Osteoclastogenesis. Journal of immunology. 2015;194:316–324. doi: 10.4049/jimmunol.1401013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirouzono T, Chirifu M, Nakamura C, Yamagata Y, Ikemizu S. Preparation, crystallization and preliminary X-ray diffraction studies of the glycosylated form of human interleukin-23. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012;68:432–435. doi: 10.1107/S1744309112005295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidobre S, Kronenberg M. CD1 tetramers: a powerful tool for the analysis of glycolipid-reactive T cells. J Immunol Methods. 2002;268:107–121. doi: 10.1016/s0022-1759(02)00204-1. [DOI] [PubMed] [Google Scholar]
- Sivanesan D, Beauchamp C, Quinou C, Lee J, Lesage S, Chemtob S, Rioux JD, Michnick SW. IL23R (Interleukin 23 Receptor) variants protective against inflammatory bowel diseases (IBD) display loss of function due to impaired protein stability and intracellular trafficking. Journal of Biological Chemistry. 2016;291:8673–8685. doi: 10.1074/jbc.M116.715870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skiniotis G, Boulanger MJ, Garcia KC, Walz T. Signaling conformations of the tall cytokine receptor gp130 when in complex with IL-6 and IL-6 receptor. Nature Structural \& Molecular Biology. 2005;12:545–551. doi: 10.1038/nsmb941. [DOI] [PubMed] [Google Scholar]
- Spangler JB, Moraga I, Mendoza JL, Garcia KC. Insights into cytokine-receptor interactions from cytokine engineering. Annu Rev Immunol. 2015;33:139–167. doi: 10.1146/annurev-immunol-032713-120211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng MWL, Bowman EP, McElwee JJ, Smyth MJ, Casanova J-L, Cooper AM, Cua DJ. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nature Medicine. 2015;21:719–729. doi: 10.1038/nm.3895. [DOI] [PubMed] [Google Scholar]
- Verstraete K, Peelman F, Braun H, Lopez J, Van Rompaey D, Dansercoer A, Vandenberghe I, Pauwels K, Tavernier J, Lambrecht BN, et al. Structure and antagonism of the receptor complex mediated by human TSLP in allergy and asthma. Nat Commun. 2017;8:14937. doi: 10.1038/ncomms14937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstraete K, Remmerie B, Elegheert J, Lintermans B, Haegeman G, Vanhoenacker Pa. Inducible production of recombinant human Flt3 ectodomain variants in mammalian cells and preliminary crystallographic analysis of Flt3 ligand-receptor complexes. Acta crystallographica. Section F, Structural biology and crystallization communications. 2011;67:325–331. doi: 10.1107/S1744309111003319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstraete K, van Schie L, Vyncke L, Bloch Y, Tavernier J, Pauwels E, Peelman F, Savvides SN. Structural basis of the proinflammatory signaling complex mediated by TSLP. Nat Struct Mol Biol. 2014;21:375–382. doi: 10.1038/nsmb.2794. [DOI] [PubMed] [Google Scholar]
- Vignali DAA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nature Immunology. 2012;13:722–728. doi: 10.1038/ni.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lupardus P, Laporte SL, Garcia KC. Structural biology of shared cytokine receptors. Annual review of immunology. 2009;27:29–60. doi: 10.1146/annurev.immunol.24.021605.090616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wei Y, Xiao H, Liu X, Zhang Y, Han G, Chen G, Hou C, Ma N, Shen B, et al. A novel IL-23p19/Ebi3 (IL-39) cytokine mediates inflammation in Lupus-like mice. European Journal of Immunology. 2016;46:1343–1350. doi: 10.1002/eji.201546095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf SF, Temple PA, Kobayashi M, Young D, Dicig M, Lowe L, Dzialo R, Fitz L, Ferenz C, Hewick RM, et al. Cloning of Cdna for Natural-Killer-Cell Stimulatory Factor, a Heterodimeric Cytokine With Multiple Biologic Effects on T-Cells and Natural-Killer-Cells. Journal of Immunology. 1991;146:3074–3081. [PubMed] [Google Scholar]
- Zhang L, Li J, Li L, Zhang J, Wang X, Yang C, Li Y, Lan F, Lin P. IL-23 selectively promotes the metastasis of colorectal carcinoma cells with impaired Socs3 expression via the STAT5 pathway. Carcinogenesis. 2014;35:1330–1340. doi: 10.1093/carcin/bgu017. [DOI] [PubMed] [Google Scholar]
- Zhao J, Liu Y-H, Reichert P, Pflanz S, Pramanik B. Glycosylation analysis of interleukin-23 receptor: elucidation of glycosylation sites and characterization of attached glycan structures. Journal of mass spectrometry. 2010;45:1416–1425. doi: 10.1002/jms.1858. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Protein Data Bank accession codes
Crystallographic coordinates and structure factors have been deposited to the Protein Data Bank (www.rcsb.org) as follows: human IL-23:IL-23R:Nb22E11 complex (5mzv), unbound human IL-23 (5mxa), IL-23:BriakinumabFab complex (5njd), and BriakinumabFab (5n2k).