

Many enzyme activity and metabolite quantitative trait loci (QTL) colocalize, including a pleiotropic QTL at ACCELERATED CELL DEATH6 that points to a trade-off between defense and central metabolism.
Abstract
Central metabolism is a coordinated network that is regulated at multiple levels by resource availability and by environmental and developmental cues. Its genetic architecture has been investigated by mapping metabolite quantitative trait loci (QTL). A more direct approach is to identify enzyme activity QTL, which distinguishes between cis-QTL in structural genes encoding enzymes and regulatory trans-QTL. Using genome-wide association studies, we mapped QTL for 24 enzyme activities, nine metabolites, three structural components, and biomass in Arabidopsis thaliana. We detected strong cis-QTL for five enzyme activities. A cis-QTL for UDP-glucose pyrophosphorylase activity in the UGP1 promoter is maintained through balancing selection. Variation in acid invertase activity reflects multiple evolutionary events in the promoter and coding region of VAC-INV. cis-QTL were also detected for ADP-glucose pyrophosphorylase, fumarase, and phosphoglucose isomerase activity. We detected many trans-QTL, including transcription factors, E3 ligases, protein targeting components, and protein kinases, and validated some by knockout analysis. trans-QTL are more frequent but tend to have smaller individual effects than cis-QTL. We detected many colocalized QTL, including a multitrait QTL on chromosome 4 that affects six enzyme activities, three metabolites, protein, and biomass. These traits are coordinately modified by different ACCELERATED CELL DEATH6 alleles, revealing a trade-off between metabolism and defense against biotic stress.
INTRODUCTION
Primary metabolism underpins plant growth by providing the building blocks and energy required for cell division, expansion, and maintenance and for the production of stress and defense metabolites. Plant growth is regulated by interconnected transcriptional and metabolic networks that integrate information about resource availability, hormones, the clock, and environmental and developmental cues (Kooke and Keurentjes, 2012; Chaiwanon et al., 2016). Coordinated and multilevel studies of the individual components of central metabolism are required to disentangle this complex polygenic regulatory mechanism and to improve our understanding of plant growth. This includes a deeper understanding of the genetic control of enzyme abundance. Enzymes catalyze the interconversion of metabolites and play a key role in the control of metabolic flux and, hence, growth. In particular, it is important to know to what extent enzyme abundance is regulated by polymorphisms in the structural genes that encode individual enzymes or by regulatory genes that have the potential to exert coordinated control on large numbers of enzymes and to integrate the control of the abundance of cohorts of enzymes during development or environmental responses.
Genotype-phenotype relationships can be deciphered using natural genetic variation. Arabidopsis thaliana is an outstanding model due to its genetic adaptation to different natural habitats and its extensive variation in morphology, metabolism, and growth (Alonso-Blanco et al., 2009). Natural variation in Arabidopsis for many traits in primary and secondary metabolism has been reported (Causse et al., 1995a; Mitchell-Olds and Pedersen, 1998; Sergeeva et al., 2004; Cross et al., 2006; Keurentjes et al., 2006, 2008; Meyer et al., 2007; Sulpice et al., 2009, 2010).
Correlation analyses across Arabidopsis and maize (Zea mays) populations have uncovered coordinated changes in metabolite levels and enzyme activities and strong links between the primary metabolic network and growth (Sulpice et al., 2010; Zhang et al., 2010; Toubiana et al., 2016). Multivariate analysis of metabolite profiles across panels of genotypes provides predictions of biomass (Meyer et al., 2007; Riedelsheimer et al., 2012) and has delivered first insights into which features promote rapid growth. For example, a negative correlation between starch turnover and biomass formation indicates that fast-growing Arabidopsis accessions use their carbohydrates more efficiently than slower-growing lines (Cross et al., 2006; Sulpice et al., 2009, 2013). This was recently shown to be partly due to fast-growing accessions having a lower rate of protein turnover (Ishihara et al., 2017).
A mechanistic understanding of genetic regulation requires the identification of quantitative trait loci (QTL; loci that contribute to inherited variance of a quantitative trait). Many studies have mapped QTL for metabolite levels (Causse et al., 1995b, 2002, 2004; Fridman et al., 2002; Baxter et al., 2005; Fu et al., 2009). These QTL colocalize with biomass QTL more frequently than expected by chance, pointing again to a close link between metabolism and growth (Lisec et al., 2008).
A more direct approach to study the genetic regulation of metabolism would be to determine QTL for enzyme abundance. Metabolite levels are regulated by interactions between enzymes at the single cell level and are modulated by the concerted action of enzymes and transporters in multicellular organisms. Depending on pathway topology, an increase in pathway flux can lead to an increase or decrease in the level of a particular metabolite (Fernie and Stitt, 2012). Enzyme abundance QTL could be located in cis, i.e., in the structural gene encoding the enzyme, or in trans, i.e., in regulatory genes affecting transcription, translation, activity, or stability of the enzyme. Understanding the relative contribution of cis- and trans-QTL to variation in enzyme activity is of major importance. For instance, analyses of glucosinolate metabolism have highlighted the impact of polymorphisms in individual enzymes and in regulatory genes (Kliebenstein et al., 2001b; Wentzell et al., 2007; Burow et al., 2010). The colocalization of trans-QTL for different enzymes might explain why metabolic traits vary in a coordinated manner.
Recent advances in proteomics make it possible to quantify many proteins in large numbers of samples in model organisms with small genomes (Picotti et al., 2013; Schubert et al., 2015). However, these technologies are not yet applicable to plants. Proxies for enzyme abundance can be obtained by determining in vitro enzyme activity, using assay conditions that detect maximum activity (Stitt and Gibon, 2014). While conventional enzyme assay methods are slow, robotized platforms have been established that provide accurate measurements of many enzymes in a large number of samples (Gibon et al., 2004b; Keurentjes et al., 2008; Steinhauser et al., 2011; Biais et al., 2014; Toubiana et al., 2016).
Enzyme activity QTL for individual enzymes and panels of enzymes have been mapped using inbred populations (Causse et al., 1995b; Mitchell-Olds and Pedersen, 1998; Prioul et al., 1999; Limami et al., 2002; Sergeeva et al., 2004, 2006; Thévenot et al., 2005; Keurentjes et al., 2008; Zhang et al., 2010; Steinhauser et al., 2011). However, these studies were limited by the restricted genetic variation in biparental populations and the relatively low mapping resolution, which can lead to incorrect calls for colocalization and incorrect assignments of cis-QTL (Zhang et al., 2010). Genome-wide association studies (GWAS) profit from the long evolutionary history of Arabidopsis. Through outcrossing and self-fertilization, this species combines a high allelic diversity with a relatively rapid decay of linkage disequilibrium (LD; ∼10 kb) over the entire genome. LD, a key-concept in association studies, describes the degree to which an allele of one single nucleotide polymorphism (SNP) co-occurs with an allele of another SNP within a population. For Arabidopsis, a 250,000 SNP array with a marker density higher than the extent of LD supports a mapping resolution close to the gene or even nucleotide level (Bergelson and Roux, 2010; Horton et al., 2012; Korte and Farlow, 2013; Alonso-Blanco et al., 2016). However, several factors complicate the selection and validation of candidate genes: the high gene density in the Arabidopsis genome, the presence of extended islands of high LD due to either a recent selective sweep or low recombination, and the occurrence of genetic and/or allelic heterogeneity (Brachi et al., 2011). Furthermore, the presence of LD can reflect two different scenarios if an association is detected between a SNP and a trait: (1) The SNP influencing the phenotype has been genotyped and is statistically associated with the trait, or (2) genotyping has not captured the causal SNP but has captured a tag SNP that is in high LD with the causal SNP and statistically but indirectly associated to the phenotype (Bush and Moore, 2012).
GWAS have already been employed to detect QTL for primary and secondary metabolites (Chan et al., 2010a, 2011; Riedelsheimer et al., 2012; Verslues et al., 2014; Wu et al., 2016). These studies have confirmed the involvement of genes previously identified by reverse genetics and QTL mapping with biparental populations and have also revealed novel genes. However, they did not directly address the genetic regulation of enzyme activity and the contribution of cis- and trans-QTL. Furthermore, pleiotropy and QTL colocalization have been poorly analyzed, and it remains unclear what generates the strongly connected metabolite networks observed in mapping populations.
We performed GWAS on 349 diverse Arabidopsis accessions for 24 enzyme activities, nine related metabolites, three structural components, and plant biomass in two independent experiments, with three main aims: first, to identify novel genes involved in the regulatory mechanisms of plant primary metabolism and biomass; second and more specifically, to provide insights into the genetic architecture underlying the regulation of enzyme activities; and, third, to search for regulatory or pleiotropic hubs that generate coordinated changes in many metabolic traits.
RESULTS
Natural Variation in Primary Metabolism
Using a mapping population of 349 diverse Arabidopsis accessions selected previously from the HapMap Panel (Horton et al., 2012), we performed two experiments (Exp1 and Exp2) under slightly different growth conditions. In Exp1, plants were grown in a 10-h photoperiod and harvested 37 d after sowing (DAS), and in Exp2, plants were grown in a 12-h photoperiod and harvested 28 DAS. We did this to increase robustness and to break pleiotropic correlations between traits, which might lead to false secondary associations. We investigated 24 enzyme activities, nine metabolites, three structural traits, and rosette fresh weight, with the majority of traits analyzed in both experiments (Table 1; Supplemental Data Set 1). Enzyme assays were optimized to measure maximum velocity (Vmax) activity, which should be proportional to protein abundance (Piques et al., 2009).
Table 1. Primary Metabolic Traits, Experiments Performed, and Heritability.
| Targeted Metabolic Pathway | Trait Acronym | Complete Trait Name | Exp | Broad-Sense H2 |
Marker-Based h2 |
||
|---|---|---|---|---|---|---|---|
| Exp1 | Exp2 | Exp1 | Exp2 | ||||
| Biomass | FW | Fresh weight | 1 | 45.2 | – | 44.0 | – |
| Structural components | ChlA | Chlorophyll A | 1,2, KO | 40.0 | 50.0 | 36.0 | 52.0 |
| ChlB | Chlorophyll B | 1,2, KO | 14.3 | 21.4 | 13.2 | 12.8 | |
| Prot | Protein | 1,2, KO | 30.9 | 39.3 | 31.6 | 39.4 | |
| AA | NO3− | Nitrate | 1,2, KO | 46.3 | 50.4 | 46.3 | 50.7 |
| AA | Amino acids | 1,2, KO | 50.6 | 47.8 | 49.0 | 47.9 | |
| Sugars | Fru | Fructose | 1,2, KO | 14.9 | 32.8 | 14.6 | 32.9 |
| Glc | Glucose | 1,2, KO | 28.7 | 55.4 | 27.1 | 55.5 | |
| G6P | Glucose-6-phosphate | 1,2, KO | 19.4 | 33.4 | 18.0 | 34.1 | |
| Suc | Sucrose | 1,2, KO | 12. 9 | 31.2 | 10.2 | 31.9 | |
| Starch | Starch | 1,2, KO | 39.9 | 46.1 | 37.0 | 46.5 | |
| Organic acids | Fum | Fumarate | 1,2, KO | 63.7 | 57.5 | 63.9 | 58.0 |
| Mal | Malate | 1,2, KO | 73.8 | 71.6 | 74.8 | 71.0 | |
| Sucrose breakdown | FK | Fructokinase | 1,2 | 8.9 | 0.0 | 6.5 | 0.96 |
| GK | Glucokinase | 1,2 | 8.7 | 3.1 | 4.2 | 3.2 | |
| aINV | Acid invertase | 1,2, KO | 50.8 | 54.2 | 50.4 | 54.9 | |
| nINV | Neutral invertase | 1,2, KO | 26.5 | 33.0 | 26.1 | 31.1 | |
| Glycolysis and respiration | G6PDH | Glucose-6-phosphate-dehydrogenase | 1,2 | 2.3 | 6.1 | 0.3 | 1.3 |
| FBP | Fructose-1,6-bisphosphatase | 1,2 | 9.5 | 0.0 | 8.9 | 0.6 | |
| Fumarase | Fumarate hydratase | 1,2, KO | 9.2 | 13.1 | 10.1 | 16.7 | |
| MDH | Malate dehydrogenase (NAD) | 2, KO | – | 15.9 | – | 18.6 | |
| TPI | Triose-phosphate isomerase | 2, KO | – | 16.2 | – | 18.2 | |
| PEPC | Phosphoenolpyruvate carboxylase | 2, KO | – | 25.7 | – | 25.7 | |
| TK | Transketolase | 2 | – | 0.0 | 6.1 | ||
| Calvin-Benson cycle | iRUB | Rubisco (initial) | 1,2 | 2.2 | 13.8 | 6.4 | 13.3 |
| mRUB | Rubisco (maximal) | 1,2 | 3.2 | 23.4 | 2.3 | 24.0 | |
| Sucrose synthesis | SPS | Sucrose phosphate synthase | 1,2 | 58.3 | 8.8 | 55.7 | 10.9 |
| UGP | UDP-glucose pyrophosphorylase | 1,2, KO | 36.5 | 37.4 | 34.4 | 37.7 | |
| PGM | Phosphoglucomutase | 1,2 | 0.0 | 5.3 | 2.7 | 9.4 | |
| cPGI | Phosphoglucoisomerase (cytosolic) | 1,2, KO | 9.9 | 18.2 | 12.1 | 19.3 | |
| tPGI | Phosphoglucoisomerase (total) | 1,2, KO | 2.8 | 3.6 | 7.6 | 10.9 | |
| Starch synthesis | pPGI | Phosphoglucoisomerase (plastidial) | 1,2, KO | 0.5 | 0.0 | 6.3 | 1.7 |
| AGP | ADP-glucose pyrophosphorylase | 1,2, KO | 34.6 | 19.5 | 33.1 | 19.8 | |
| Nitrogen metabolism | NRVm | Nitrate reductase (maximal velocity) | 1,2, KO | 12.7 | 30.2 | 14.2 | 31.1 |
| NRVs | Nitrate reductase (selective velocity) | 1,2, KO | 24.2 | 34.8 | 23.5 | 35.0 | |
| GDH | Glutamate dehydrogenase | 2, KO | – | 34.4 | – | 33.7 | |
| SKDH | Shikimate dehydrogenase | 2 | – | 7.4 | – | 6.0 | |
Targeted metabolic pathway, complete trait names, trait acronyms, and experiments (Exp) performed for each trait: GWAS (1, 2) and/or with KO lines (KO). Broad-sense heritability (H2) and marker-based heritability (h2) obtained using replicates for model GAPIT are displayed for each experiment.
Most traits exhibited a high level of variation. The variation usually approximated a normal-shaped distribution, although only 13 traits passed the normality test in one (ChlA [chlorophyll A], ChlB, nitrate [NO3−], Suc, Fum [fumarate], NRVm, NRVs, GDH, MDH [malate dehydrogenase], and PEPC [phosphoenolpyruvate carboxylase]) or both experiments (Prot [total protein], ADP-glucose pyrophosphorylase [AGP], and UDP-glucose pyrophosphorylase [UGP]) (Supplemental Figure 1A; P > 0.01, modified Shapiro-Wilks). For most traits, the values are positively correlated between the two experiments, and the plasticity parameters follow normal distributions (Supplemental Figure 1B). For sugars and organic acids, the plasticity distribution plots are skewed to the left (G6P [glucose-6-phosphate], Suc, Fum, and Mal [malate]) or right (Fru and Glc), indicating that trait values are higher in the second or first experiment, respectively. However, the moderate to strong positive correlations (r = 0.26–0.6, PB < 0.01) between the two experiments for these traits indicate that the genotypes are robust in their response to the two experimental conditions. The plasticity distribution plots of FK (fructokinase), GK (glucokinase), G6PDH, FBP, fumarase, and SPS (sucrose phosphate synthase) are not normally distributed, which indicates that for these traits a large number of genotypes are plastic in their response to different conditions. This is also demonstrated by the very low correlations between the two experiments for these six traits (r < 0.1). Overall, these results illustrate that for most traits the genotypes are robust in their response to the different environmental conditions of the two experiments and that it is appropriate to perform GWAS and analyze both experiments together. However, for the six enzymes that show genotype by environment (GxE) interactions, poor overlap in GWAS might reflect genuine differences under different conditions.
Trait variation was quantified by calculating the coefficient of variation (CV; Supplemental Data Set 2). Low CV (<10%) was observed for structural components (ChlA and ChlB in both experiments and Prot in Exp2) and some enzyme activities in individual experiments. For the remaining traits, CV was >20% in both experiments. In general, enzyme activity CVs were between 20 and 60%, with G6PDH displaying the highest CV (50–61%). Among the metabolites, Glc had the highest CV (43–63%). Previous but more limited studies in Arabidopsis also detected an overall normal distribution of trait values, suggesting quantitative regulation, moderate CVs, and larger variation for metabolites and enzymes than for structural components.
To identify which factors contribute to trait variation, we calculated both broad-sense heritability (H2) and marker-based heritability (h2) using individual replicates (Kruijer et al., 2015) and averages (i.e., line-based heritability) for each trait and experiment (Table 1; Supplemental Data Set 2 and Supplemental Figure 1C). For estimates of marker-based heritability, we compared six models with different kinship matrices (EMMAX, GAPIT, PK1, PK2, PK3, and PK4). We chose GAPIT as the default model in the subsequent analyses because it controls for both population structure and cryptic relations between individuals (i.e., covariates and kinship matrix). For biomass, structural components, AA (total amino acids), sugars, starch, and organic acids, H2 is moderate to large (13–74%), with good replication between the two experiments (Table 1). In most cases, H2 exceeds h2, suggesting that phenotypic variation is the result of heritable additive variation (h2) and nonheritable variation (H2 includes epistasis). However, the estimated values for h2 are very close to the H2 values, indicating that epistasis does not play a major role in the genetic regulation of these metabolic traits (Table 1). For enzymes, the estimates of H2 and h2 are low to moderate (0–58%) (Supplemental Data Set 2). For acid invertase activity (aINV), nINV, UGP, AGP, NRVm, NRVs, PEPC, GDH, and SPS, H2 is relatively high in one or both experiments but for other enzymes (G6PDH, fumarase, MDH, PGM, phosphoglucose isomerase [PGI], iRUB, SKDH, TPI, and TK), H2 is low (<20%) in both experiments and sometimes extremely low. Low H2 values can reflect low between-line variation and/or high within-line variation, and indeed, within-line variation is very large for these enzymes (Supplemental Figure 1D). Nevertheless, the heritability values detected for the majority of traits suggest that natural genetic variation is a major causal factor for the observed phenotypic variation and that GWAS can be employed to identify candidate genes underlying this variance.
Primary Metabolism Is a Highly Coordinated Network
To identify traits that vary in a concerted manner, we performed Spearman rank correlation tests (Figure 1; Supplemental Data Set 3). There were many highly significant correlations within a given experiment (Figures 1A and 1B). Most correlations between the different enzyme activities were positive, as previously reported for a smaller accession panel (Sulpice et al., 2010). This included enzymes involved in the Calvin-Benson cycle, sucrose and starch synthesis, and nitrogen metabolism (0.32 < r < 0.85, PB < 0.001), pointing to coordinated regulation of enzyme abundance in these pathways. Enzyme activities correlated strongly and positively with major end products of primary metabolism (starch, Suc, AA, and Prot; Figures 1A and 1B; Supplemental Data Set 3). Correlations between metabolites were strong in Exp1 but weaker in Exp2 (Figures 1A and 1B). Biomass was determined only in Exp1 and correlated negatively with protein and starch (r = −0.64, r = −0.49; PB < 0.01), as previously reported (Sulpice et al., 2009, 2010), and with other metabolites including Suc, G6P, and AA (all PB < 0.01), and positively with NO3− (r = 0.24, PB < 0.05) and Fum (r = 0.3, PB < 0.01). Biomass correlated negatively with enzyme activities in contrast to earlier reports (Sulpice et al., 2010). The strongest negative correlations between enzyme activities and biomass were for NRVs (r = −0.51, PB < 0.001; NRVm, r = −0.48, PB < 0.001), tPGI (r = −0.5, PB < 0.001), and UGP (r = −0.5, PB < 0.001) activities (Figure 1A).
Figure 1.

Spearman Rank Correlation Matrices for Primary Metabolic Traits.
Two independent experiments (Exp1 and Exp2) were performed, differing in photoperiod (10 h versus 12 h) and the age of harvested plants (37 versus 28 DAS). This was done to increase robustness and to break pleiotropic correlations between traits, which might lead to false secondary associations. More traits were investigated in Exp2 than in Exp1; these traits are omitted here. A correlation scale is included to the right of (C): positive correlations are shown in shades of blue (1 to 0) and negative correlations in shades of red (−1 to 0). Coefficients are filtered according to Bonferroni correction (PB < 0.1). Correlations within the same trait category are highlighted with black frames (triangles in [A] and [B] and squares in [C]). All traits are expressed on a FW basis. The traits are arranged along the same axes and in the same order as they are listed in Supplemental Data Set 3. Names and abbreviations can be found in Table 1.
(A) Correlations between traits measured within Exp1.
(B) Correlations between traits measured within Exp2.
(C) Cross-correlations between Exp1 and Exp2.
Interexperiment correlations (Figure 1C; i.e., correlation for metabolic traits between the two experiments) were much weaker than intraexperiment correlations. Most of the significant correlations in the interexperiment regression were for comparisons of the same trait, rather than of different traits between the two experiments. These were again stronger for structural components and metabolites than for enzymes (r = 0.2–0.6, PB < 0.01).
Coordinated changes in trait values may be due to indirect genetic relationships, for example, pleiotropy. However, the observation that correlations are weakened or lost when experiments performed under slightly different conditions are compared indicates that environmental factors exert a strong influence on the network structure of primary metabolism.
Genome-Wide Association Analysis of Primary Metabolism
We performed genome-wide association (GWA) mapping using six models that differed in the approaches used to control for confounding effects (see Methods for a more detailed description). For most traits, model EMMAX gave the highest LOD scores [logarithm of the odds, −log10 (P value)] (Supplemental Data Set 4), probably because it is the only model that does not include cofactors to control for population structure (i.e., Q matrix). However, in general, the six models performed similarly. Subsequent analyses used GAPIT as the representative model.
We performed GWAS independently for Exp1 and Exp2 and compared the results to identify associations that are robust across conditions. The separate analyses of Exp1 and Exp2 detected 80 and 134 SNPs at a significance threshold of LOD = 5.5 (false discovery rate [FDR] < 0.05), and 7100 and 8117 SNPs with LOD ≥ 3, respectively (Supplemental Data Set 4). Although more false positive associations are expected at the lower threshold, earlier studies have often reported large-effect loci that are weakly but causally associated with quantitative trait variation (Atwell et al., 2010; Kooke et al., 2016).
We therefore chose LOD ≥ 3 as a threshold to search for shared associations. This threshold was passed in both experiments by 126 SNPs corresponding to 131 SNP-trait associations (SNPt) (Supplemental Data Set 5). The LOD values for these SNPt were strongly correlated between the two experiments (r2 = 0.6, P < 0.001; Supplemental Figure 2). The high correlation was mostly driven by significant SNPs (LOD > 6.5) associated with UGP and aINV activities. The cross-validated SNPs were associated with two structural components (ChlA and Prot), nine metabolites (NO3−, AA, G6P, Glc, Fru, Suc, starch, fumarase, and Mal), and seven enzyme activities (aINV, nINV, GK, Fum, tPGI, UPG, and NRVs) (Supplemental Figure 2). In 16 cases, we identified multiple contiguous SNPs for the same trait (Supplemental Data Set 5A). LD analysis revealed that these SNPs are linked, allowing them to be assigned to unique genomic regions (i.e., QTL; Supplemental Data Set 6). This analysis also uncovered four QTL where different but linked SNPs were associated with the same trait in Exp1 and Exp2 (Supplemental Data Set 5B). In total, we detected 70 cross-validated QTL (LOD ≥ 3 in both experiments; Supplemental Data Set 5). Their LOD scores were significantly correlated between Exp1 and Exp2 (r2 = 0.368, P < 0.001; Figure 2). These included two cases where different traits were associated with the same QTL.
Figure 2.

QTL Cross-Validation between Exp1 and Exp2.
For the 19 traits where robust QTL were identified (Supplemental Data Set 5), the highest-LOD SNP in Exp1 was plotted against the highest-LOD SNP in Exp2. Only QTL with LOD ≥ 3 in at least one experiment are included. LOD = 3 thresholds (black lines) and 10% FDR (LOD = 4.8, dashed lines) are shown. QTL are shown in different colored and shaped symbols according to trait and class. For cPGI, a cross-validated QTL was identified considering QTL-ID rather than SNP-ID between experiments. Linear regression for the validated QTL data set was r2 = 0.368, P < 0.001. The SNP-to-SNP cross-validation is presented in Supplemental Figure 2. Names and abbreviations for all traits can be found in Table 1.
Fourteen SNPs were retained that did not meet our criteria but had a high-LOD score in one experiment and colocated with other cross-validated SNPt. Eight significant SNPs at FDR < 0.1 (LOD > 4.8) for FK, nINV, FBP, fumarase, SPS, pPGI, and AGP were retained because these traits showed high plasticity between conditions, and for five of them, we were unable to identify cross-validated SNPs when analyzing both experiments together (Supplemental Data Set 5). We also retained some interesting suggestive associations, including one SNP for AA, four SNPs for AGP, and two SNPs for GDH. In total, we captured 165 unique SNPs, corresponding to 191 SNPt and 88 QTL (Supplemental Data Set 5). Six of the resulting QTL support intervals were large, ranging from 19 to 68.2 kb, and some included more than one gene.
We chose candidate genes based on the position of the SNP(s) with the highest LOD score, complemented by manual inspection for obvious functional annotations. This procedure short-listed 116 candidate genes (Supplemental Data Set 5; small selection in Table 2). The candidates were distributed across 21 functional categories (MapMan v3.5.1R2; Thimm et al., 2004; http://mapman.gabipd.org; Figure 3A), including metabolic enzymes (13 genes), regulation of transcription (10 genes), ubiquitin-dependent protein degradation (11 genes), RNA processing and splicing (6 genes), protein processing, secretion and posttranslational modification (8 genes), and transporters (7 genes).
Table 2. Overview of QTL Identified by GWAS for Enzyme Activity, Metabolites, and Colocalizing QTL.
| Trait | QTL ID | QTL Size | Gene (AGI) | Gene ID | Function | LOD1 | LOD2 | Var1 | Var2 |
|---|---|---|---|---|---|---|---|---|---|
| Enzyme cis-QTL | |||||||||
| AGP | 73 | 1 | AT5G19220 | APL1 | ADP-Glc PPase large subunit 1 | 1.8 | 4.3 | 1.8 | 4.9 |
| aINV | 3 | 24 | AT1G12240 | VAC-INV | β-Fructofuranosidase | 7.7 | 6.8 | 10.2 | 8.4 |
| Fumarase | 30 | 5 | AT2G47510 | FUM1 | Fumarase 1 (mitochondrial) | 3.5 | 3.0 | 4.0 | 3.2 |
| UGP | 32 | 19 | AT3G03250 | UGP1 | UDP-Glc PPase 1 | 8.6 | 12.5 | 11.2 | 16.7 |
| Enzyme trans-QTL | |||||||||
| aINV | 19 | 1 | AT1G62710 | BETA-VPE | Vacuolar-processing enzyme | 7.2 | 4.7 | 9.5 | 5.4 |
| Fumarase | 79 | 1 | AT5G46110 | TPT | Triose-P/phosphate translocator | – | 5.7 | – | 7.0 |
| nINV | 6 | 1 | AT1G27720 | TAF4B | TBP-associated factor 4B | 4.9 | – | 6.0 | – |
| nINV | 44 | 1 | AT3G49430 | SR34A | Pre-mRNA splicing factor SF2 | 6.5 | 3.1 | 8.5 | 2.8 |
| nINV | 78 | 1 | AT5G44560 | VPS2.2 | Vacuolar protein sorting 2-2 | 3.8 | 6.2 | 4.5 | 8.0 |
| NRVs | 67 | 1 | AT5G01550 AT5G01560 | LECRKA4.2 LECRKA4.3 | Lectin receptor kinase A4.2/4.3 | 5.3 | 3.3 | 6.4 | 3.5 |
| tPGI | 46 | 28 | AT3G58560 | CCR4A | Carbon catabolite repressor 4 | 4.1 | 4.2 | 5.2 | 4.4 |
| Metabolite level QTL | |||||||||
| Fum | 21 | 1 | AT1G68600 | – | Malate transporter | 3.9 | 4.9 | 4.1 | 5.5 |
| Mal | 42 | 1 | AT3G47520 | pMDH | MDH, plastidial | 4.3 | 4.5 | 4.9 | 4.9 |
| AA | 31 | 1 | AT2G47570 | RPL18e | Ribosomal protein L18e | 3.8 | 2.4 | 4.1 | 2.4 |
| Fru | 63 | 4 | AT4G23060 | IQD22 | Protein IQ-domain 22 | 4.6 | 5.7 | 5.7 | 6.3 |
| G6P | 27 | 2.5 | AT2G33150 | KAT2 | 3-Ketoacyl-CoA thiolase 2 | 5.8 | 4.0 | 7.3 | 4.4 |
| Colocalized QTL | |||||||||
| Fum, Mal | 83 | 12 | AT5G50950 | FUM2 | Fumarase 2 (cytosolic) | 4.1-2.4 | 3.2-3.6 | 4.6-2.4 | 3.4-3.7 |
| Glc, Suc, Starch | 72 | 1 | AT5G12080 | MSL10 | Osmolyte channel 10 | 2.9-NA-4.5 | NA-2.9-2.6 | 3.2-NA-5.2 | NA-2.4-2.6 |
| Mal, MDH, GDH | 33 | 1 | AT3G07220 | FHA2 | SMAD/FHA domain protein | – | 4.4-3.9-3.1 | – | 4.7-4.3-3.2 |
| FW, Prot, AGP, UGP, tPGI | 48 | 1 | AT4G02500 | XXT2 | Xyloglucan 6-xylosyltransferase | 3.3 to 5.2 | – | 3.6 to 6.5 | – |
| FW, Prot, AA, Fum, G6P, | 56 | 68.2 | AT4G14400 | ACD6 | Ankyrin repeat-containing protein | 3.3 to 7.8 | 3.1 to 6.6 | 3.7 to 10.5 | 2.8 to 8.1 |
| aINV, nINV | |||||||||
| Fumarase, cPGI, tPGI, GDH |
Trait, QTL ID, QTL size in kilobases, candidate gene AGI code, gene ID, and function are shown. LOD = log10(P value) for Exp1 (LOD1) and Exp2 (LOD2) are included. Percentage of variation explained by the highest-LOD SNP is mentioned for each experiment separately (Var1 and Var2). A full description of gene function is available in Supplemental Data Set 5.
Figure 3.

Assignment of Candidate Genes to Functional Categories and Analysis of QTL Colocalization.
Candidate genes were selected based on the position of the SNP with the highest LOD score. QTL and QTL colocalization were determined by assessing LD between contiguous SNPs (within and between traits), with LOD ≥ 3.
(A) Number of genes falling in different ontology categories according to trait classes: structural components (black), metabolites (white), and enzyme activities (gray) (MapMan v3.5.1, http://mapman.gabipd.org; Thimm et al., 2004).
(B) Specific and colocalized QTL obtained for each trait class: structural components, metabolites, and enzyme activities. Colocalized QTL within a trait category are specified in smaller circles.
The number of robust QTL detected for a given metabolic trait varied greatly. The largest number was detected for aINV activity (16) and Mal (15), followed by Fum (9), Prot (5), NO3− (5), and G6P (5) (Supplemental Data Set 7). The proportion of total amount of variation explained by the summed QTL depended on the trait. This value was highest for aINV, Mal, and Fum (>30%), between 10 and 30% for a further 17 traits, and <10% for 10 traits (Supplemental Data Set 7). It should be noted, however, that summing values might lead to a bias since, for example, epistatic effects are not taken into account.
Genetic Regulation of Enzyme Activity in cis and in trans
Five enzyme activity QTL were detected in cis (i.e., mapping to structural genes encoding that enzyme). This represents 12.5% of all enzyme activity QTL (Table 2; Supplemental Data Set 5). A strong QTL for UGP activity (LOD1 = 8.6, LOD2 = 12.5) was detected with the most significantly associated SNPs located 914 bp (Exp1) and 2877 bp (Exp2) downstream of the start codon of UDP-GLUCOSE PYROPHOSPHORYLASE1 (UGP1), one of two genes encoding UGP in Arabidopsis. aINV activity was significantly associated (LOD1 = 7.7, LOD2 = 6.8) with VACUOLAR INVERTASE (VAC-INV), with the peak SNP located 1122 bp upstream of the start codon. AGP activity (LOD1 = 1.8, LOD2 = 4.3) was associated with SNPs in the coding region of the starch biosynthesis gene ADP-GLUCOSE PYROPHOSPHORYLASE LARGE SUBUNIT1 (APL1). A QTL for cPGI activity was robustly associated with the sugar isomerase gene SIS (LOD1 = 4.5, LOD2 = 3.0), with the highest-LOD SNPs located 14,988 and 20,188 bp downstream of the start codon. Fumarase activity associated with FUMARASE1 (FUM1; mitochondrial) with the strongest SNPs (LOD1 = 4.5, LOD2 = 3.8) located 10 and 14 kb upstream of the coding region and lower scoring SNPs within the coding region (LOD1 = 3.5, LOD2 = 3.0). The percentage of variation explained by these cis-QTL ranged from 4% for fumarase up to 10.2 and 16.7% for aINV and UGP, respectively (Table 2).
Thirty-five trans-QTL were detected for enzyme activity. In addition to its cis-QTL, aINV activity associated in trans with BETA-VPE (LOD1 = 7.2, LOD2 = 4.7). BETA-VPE is a vacuolar-processing enzyme that is likely involved in protein degradation and turnover. This trans-QTL explained 9.5% of variation. We identified two candidate genes for nINV variation, the protein involved in splicing SRp34a (LOD1 = 6.5, LOD2 = 3.1) and the vacuole-localized protein VPS2.2 (LOD1 = 3.8, LOD2 = 6.2). These QTL explained 8.5% and 8% of trait variation, respectively (Table 2). NRVs activity associated with a QTL containing two lectin receptor protein kinases (LECRKA4.2 and LECRKA4.3, LOD1 = 5.3, LOD2 = 3.3). These have previously been implicated as negative regulators of abscisic acid responses (Xin et al., 2009). Total PGI activity associated with a QTL in the coding region of ATCCR4a (LOD1 = 5.2, LOD2 = 4.4). This gene and its neighbor, ATCCR4b, are orthologs to yeast CARBON CATABOLITE REPRESSOR4 (CCR4), which is involved in RNA degradation (Dupressoir et al., 2001). Recent analyses revealed higher levels of starch and lower levels of sucrose in Arabidopsis ccr4a and ccr4b knockout (KO) lines compared with wild-type controls, although no changes in tPGI nor evidence of posttranscriptional regulation of genes encoding PGI enzymes were observed (Suzuki et al., 2015). All of these QTL were significantly associated in at least one experiment with an FDR control between 1 and 10% (LOD = 4.8–6.3). (Supplemental Data Set 5).
For enzymes showing great plasticity between conditions, we often identified significantly associated QTL in only one experiment. For example, in Exp1, nINV associated with a transcription factor (TAF4B, LOD1 = 4.9), FK associated with an E3 ubiquitin-protein ligase (LOD1 = 5.1) and a TRAF-like protein involved in signaling (LOD1 = 4.9), and pPGI associated with a ubiquitin-like protease (LOD1 = 5.0). In Exp2, FBP associated with an anion transporter (LOD2 = 6.1), SPS with a forkhead-associated domain-containing protein (LOD = 5.8), and fumarase activity with the TRIOSE-PHOSPHATE TRANSLOCATOR (TPT; LOD2 = 5.7).
Altogether, these findings indicate that the genetic regulation of the activity of enzymes occurs through natural variation in their structural genes (in cis) and excessive modulation through modifiers in trans, with the latter being far more numerous but some of the former showing the largest effect sizes.
Metabolite QTL Mapping to Biosynthesis Pathways and Growth Metabolism
Two metabolite QTL mapped to structural genes for an enzyme that uses the metabolite as a substrate (Table 2). We classify these as “putative cis-QTL.” The first was a QTL for Mal content (LOD1 = 4.3, LOD2 = 4.5) that mapped to the PLASTIDIC MALATE DEHYDROGENASE (pMDH) (Scheibe, 1987). The most significant SNP is located 11,983 bp upstream of the start codon (Supplemental Data Set 5). This SNP did not show significant LD with other SNPs in the pMDH genomic region, which might explain why GWAS did not detect SNP associations closer to or within pMDH. On average, accessions carrying the major “C” allele (frequency = 0.80) show significantly higher levels of Mal than accessions carrying the minor “T” allele. MDH activity was not significantly different between the two haplotypes, but this might be explained because MDH activity was assayed using NAD+ as cofactor, while the plastid form uses NADP.
The second putative cis-QTL was for Fum (LOD1 = 4.1, LOD2 = 3.2) and Mal content (LOD1 = 2.4, LOD2 = 3.6). It mapped to FUM2, which encodes the cytosolic fumarase (Pracharoenwattana et al., 2010). Fumarase catalyzes the reversible hydration/dehydration of Fum to Mal. The most strongly associated SNPs are located upstream of the start codon (21,513 and 9751 bp for Fum and Mal, respectively). Both SNPs are polymorphic between Col-0 and C24, the parental lines of an inbred population in which corresponding metabolite QTL were previously detected (Lisec et al., 2008). The allelic effect for these SNPs predicts higher fumarase activity, higher levels of Fum, and lower levels of Mal for accessions carrying the Col-0 allele. In agreement, the Col-0 haplotype (i.e., AC, allele frequency = 0.11) displayed significantly different trait values than the C24 haplotype (i.e., GG, allele frequency = 0.30) in the association mapping population in both experiments. However, these two haplotypes are not as abundant as the intermediate haplotype (i.e., GC, allele frequency = 0.59), which might explain the relatively low power to detect this QTL in the association mapping population compared with the biparental inbred population. Interestingly, another QTL for Fum content mapped to a malate transporter gene (LOD1 = 3.9, LOD2 = 4.9), again pointing to close links between Mal and Fum (Supplemental Data Set 5).
Several metabolite QTL mapped to genes involved in signaling or growth. A QTL for AA mapped in the coding region for a ribosomal protein L18e (RPL18e, LOD1 = 3.8, LOD2 = 2.4), a QTL for Fru content mapped to IQ-DOMAIN22 (IQD22; LOD1 = 4.6, LOD2 = 5.7), which belongs to a multigene family of calmodulin binding proteins (Abel et al., 2005), and a QTL for NO3− content mapped to LOB DOMAIN-CONTAINING PROTEIN37 (LBD37; LOD1 = 4.1, LOD2 = 3.2), which is involved in nitrogen signaling and the regulation of central metabolism (Rubin et al., 2009). Finally, a QTL for G6P mapped to KAT2 (LOD1 = 5.8, LOD2 = 4.0), a 3-ketoacyl-CoA thiolase involved in peroxisomal β-oxidation.
Putative cis-QTL and trans-QTL both contribute to variation in metabolite levels. Although the former represent only 6.7% of all metabolite QTL, they make a slightly stronger contribution to metabolite variation (4.7% versus 4.4% of total variation, respectively). A higher number of trans-QTL was detected for metabolite traits than for enzyme activities (4.7 and 1.5 per trait, respectively), suggesting that many genes have an indirect effect on metabolite levels. For both metabolite and enzyme activity QTL, although far fewer cis-QTL (8) were detected than trans-QTL (80), the individual cis-QTL effects were on average stronger than the trans-QTL effects (7.2% and 5.1%, respectively; Supplemental Data Set 7).
Colocalized QTL Suggest a High Degree of Pleiotropy
The high connectivity between metabolic traits in individual experiments (Figures 1A and 1B; see also Introduction) led us to search for colocalizing QTL. Fourteen QTL associated with two or more metabolic traits (Supplemental Data Set 5; small selection listed in Table 2). The most abundant colocalizations were between structural component QTL and metabolite QTL (4) and between different metabolite QTL (4) (Figure 3B). Starch (LOD1 = 4.5, LOD2 = 2.6), and (although weakly in only one experiment) Glc (LOD1 = 2.9) and Suc (LOD2 = 2.9) associated with MECHANOSENSITIVE CHANNEL OF SMALL CONDUCTANCE-LIKE10 (MSL10) (Haswell et al., 2008). Mal content (LOD2 = 4.4) and MDH (LOD2 = 3.9) and GDH (LOD2 = 3.1) activities associated in Exp2 to FHA2, which encodes a protein involved in protein-protein interactions (Ahn et al., 2013). In Exp1, a QTL associated with fresh weight (FW), Prot, AGP, UGP, and tPGI activity (maximum LOD1 = 5.2 for AGP activity) was identified within XXT2 (Supplemental Data Set 5; Table 2). XXT2 encodes a xyloglucan xylosyltransferase that is important for xyloglucan synthesis (Cavalier et al., 2008) and loosening of the cell wall during expansion growth (Park and Cosgrove, 2012).
The most striking multitrait QTL extended over a 68.2-kb region on chromosome 4 (Chr4) (Figure 3B; Supplemental Data Set 5; QTL ID = 56). This QTL associated with FW (LOD1 = 3.9, LOD2 = NA), Prot (LOD1 = 3.3; not detected in Exp2), AA (LOD1 = 5.0, LOD2 = 3.5), G6P (LOD1 = 3.8, LOD2 = 2.1), Fum (LOD1 = 4.5, LOD2 = 4.4), and six enzyme activities: aINV (LOD1 = 7.8, LOD2 = 6.8), nINV (LOD1 = 4.1, LOD2 = 3.1), cPGI (LOD1 = 3.2; not detected in Exp2), tPGI (LOD1 = 3.5; not detected in Exp2), fumarase (LOD1 = 4.6; not detected in Exp2), and GDH (LOD1 = NA, LOD2 = 5.0). The trait variation explained by this QTL ranged from moderate (3.7% for Prot) to high (10.5% for aINV) (Table 2; see below for more analysis).
These results point to a high degree of pleiotropy in the regulation of central metabolism, resource allocation, and growth. This could contribute to the high connectivity in the metabolic traits observed in this and previous studies (see Discussion).
Analysis of KO Lines for Selected Candidate Genes
We next analyzed the metabolic phenotypes of homozygous T-DNA insertion lines (KO lines) for 35 of the candidate genes. Analysis of KO lines is often used to confirm QTL-trait associations by identifying the gene-to-phenotype link, although it is only useful if the causal allele has a similar phenotype to the null allele. We included multiple lines for a given candidate that targeted different parts of the gene. In total, 74 KO lines were grown in parallel with Col-0 (wild type) in conditions resembling Exp2 and analyzed for 26 metabolic traits (Supplemental Data Set 8).
Each KO line and metabolic trait was tested for deviation from the population mean (i.e., the average level of the trait across all 74 KO lines plus the wild-type value). This approach assumes that for most traits, the KO lines would present wild-type-like values and that the large number of KOs (74 lines) would dilute the impact of the few lines with an altered trait value. The wild type displayed trait levels around the population mean, except for significantly higher NO3− content and TPI activity (Supplemental Figure 3). For 21 of the 26 analyzed traits, we found one to 12 lines that differed significantly from the population mean (Supplemental Data Set 8 and Supplemental Figure 3).
These analyses supported the choice of candidate genes for several cis-QTL (Figure 4). A significant decrease (P < 0.05) was observed in the targeted enzyme activity in KO lines of VAC-INV (86–50% lower), UGP1 (75–42% lower), and APL1 (85–82% lower) compared with the population average (Figure 4; Supplemental Figure 4 and Supplemental Data Set 8). An earlier coarse mapping study with a Ler x Cvi recombinant inbred line (RIL) population provided further validation for the cis-QTL at VAC-INV, UGP1, and SIS but did not detect the QTL for AGP activity at APL1 (Keurentjes et al., 2008). The impact of a fum2 KO on Mal and Fum content has also been previously documented (Pracharoenwattana et al., 2010; Brotman et al., 2011). Attempts to obtain homozygous KO lines for pMDH/AT3G47520 have failed (Selinski et al., 2014), possibly due to the essential function of this enzyme.
Figure 4.

Analysis of KO Metabolic Profiles for Selected Candidate Genes.
Wild-type plants (Col-0 N6673, six biological replicates, each of three plants) and 74 KO lines (three biological replicates per line, each of three plants) were grown in a complete randomized design in the same conditions as in Exp2. Full rosettes were harvested 28 DAS at the end of the light period. Trait values were z-score normalized across the values for all genotypes in the KO panel and plotted as a heat map. KOs are ordered vertically according to chromosome location and T-DNA insertion position in the gene. Measured traits are ordered according to the three categories: structural components (black), metabolites (white), and enzyme activities (gray). The trait(s) for which KO lines were selected and the locus targeted are stated at the left and right side of the heat map, respectively. Each KO line was compared with the average behavior of wild-type and all 74 KO lines, and significant differences (P < 0.05) are indicated with an asterisk. This figure shows selected KO lines. A complete overview of all KO lines is provided in Supplemental Figure 3, and all data are provided in Supplemental Data Set 8. Further experiments to validate cis-QTL for enzyme activities are provided in Supplemental Figure 4, for the colocalized QTL for MDH, GDH, and Mal levels at FHA2 in Supplemental Figure 5, for the QTL for Fru at IQD22 in Supplemental Figure 6, and for the QTL for starch and sugars at MSL10 in Supplemental Figure 7.
These analyses also supported candidate genes selected for several trans-QTL. One was the association between NO3− content and LBD37 that, as already mentioned, is involved in the regulation of nitrate metabolism (Rubin et al., 2009). The QTL for MDH and GDH activities and Mal content on Chr3 was narrowed down to a SMAD/FHA domain-containing protein (FHA2/ AT3G07220). The most significantly associated SNP was located between AT3G07220 and AT3G07230. In an initial experiment using lines with T-DNA insertions in AT3G07220 and the intergenic region, the latter displayed significantly higher values for all three traits (Figure 4). In the second experiment, the wild type and lines with T-DNA insertions in the intergenic region and an intron of AT3G07220 and in the 5′UTR (untranslated region) of AT3G07230 were harvested at dawn (EN) and dusk (ED) to obtain information for diurnal changes in Mal levels (Supplemental Figure 5A). As enzyme activities show little diurnal change (Gibon et al., 2004b), GDH and MDH activities were evaluated for the combined time points. Significant changes (P < 0.05) were detected for Mal levels and MDH activity for the KO line targeting the intron of AT3G07220, but not for the line targeting the 5′UTR of AT3G07230 (Supplemental Figure 5B). The intergenic T-DNA insertion line confirmed the higher Mal level detected in the first experiment but did not show significant changes for MDH activity, possibly because fewer biological replicates were analyzed. GDH activity was not significantly altered (Supplemental Figure 5B). Together, these results suggest that Mal levels and MDH and GDH activities might be direct or indirect targets of FHA2.
The phenotypes of KO lines targeting IQD22 supported the contribution of this calmodulin binding family member to variation in Fru levels. In the initial experiment, one of two KO lines displayed a significant increase in Fru and decrease in Suc levels (Figure 4) and, while starch levels were not changed, the starch:Suc ratio was higher than in the wild type. In the second experiment, we analyzed three additional homozygous iqd22 lines together with the previously identified functional KO and the wild type. Almost all iqd22 lines displayed a significantly higher starch:Suc ratio and a trend toward lower Suc than the wild type, although no significant differences could be detected (Supplemental Figures 6A and 6B). Fru contents were at the limit of detection in this experiment, preventing testing for line-specific differences. In addition to the metabolic phenotype, we noted that the leaf initiation rate was increased and flowering time (expressed as total leaf number) was significantly earlier in iqd22 lines compared with the wild type (Supplemental Figures 6C and 6D).
The analyses of KO lines also supported the association of MSL10 with starch, Suc, and Glc content. In the KO panel, msl10 displayed significantly higher levels of Glc and Fru and lower levels of Suc (Figure 4). To further test whether MSL10 has a role in starch regulation, Col-0, msl10, and an overexpressor line (Pro35S:MSL10, termed OEX) were grown in a 12-h photoperiod and harvested every four hours over the 24-h cycle (Supplemental Figure 7). In the wild type, starch accumulated in a linear manner in the light period, degraded in a linear manner at night, and was almost (but not completely) exhausted at dawn, as typically observed (Stitt and Zeeman, 2012). The OEX line synthesized starch more quickly than Col-0, reaching a maximum at ZT = 8 (P < 0.05), and msl10 exhibited slower starch degradation at the beginning of the night (P < 0.05; Supplemental Figure 7).
KO Lines ugp1, vac-inv, and apl1 Exhibit Seed Abortion Phenotypes
The ugp1, vac-inv, and apl1 KO lines did not exhibit any obvious change in rosette biomass or morphology during vegetative growth compared with the wild type (see also Meng et al., 2009; Leskow et al., 2016). Seed set is known to be especially dependent on metabolism (Lauxmann et al., 2016), and this trait was previously reported to be impaired in the double ugp1 ugp2 mutant (Meng et al., 2009; Park et al., 2010). The ugp1, vac-inv, and apl1 KO lines displayed shorter and aborted siliques (Figure 5A) and a significant increase in seed abortion (P < 0.05; Figure 5B) in long-day conditions (16-h photoperiod) compared with wild-type plants. This phenotype was significant in all three sectors of the stem for apl1 and ugp1, but only in the rosette leaf branches for vac-inv.
Figure 5.

Importance of cis-QTL in Reproductive Growth.
(A) Silique abortion phenotype observed for apl1, ugp1, and vac-inv grown in a 16-h photoperiod. Silique abortion was scored in main stem (red arrow), cauline-leaf branches (yellow arrows), and rosette branches (blue arrows) separately. Bar = 1 cm.
(B) Number of nonaborted (black) and aborted (gray) siliques on the main stem, cauline-leaf branches, and rosette branches (mean ± sd, n = 3 plants). ANOVA analysis was performed separately for nonaborted (lowercase letters) and aborted (capital letters) siliques. Means with a common letter are not significantly different (P < 0.05).
ACCELERATED CELL DEATH6 Modulates Central Metabolism in a Pleiotropic Manner
As noted, many QTL explained variation in multiple traits. The most extreme case was a multitrait QTL for six enzyme activities (aINV, nINV, cPGI, tPGI, fumarase, and GDH), three metabolites (G6P, AA, and Fum), Prot, and FW detected in Chr4 (Table 2). This locus covered 68.2 kb and contained 24 genes in strong LD (Figure 6), 10 of which showed associations in GWAS. We assessed seven of these 10 genes using 14 homozygous KO lines (Figure 4). Significant changes in individual traits were found for KO lines targeting AT4G14368/SALK_006734, AT4G14420/SALK_018752, and AT4G14440/SALK_012852 (Figure 4). The most striking and significant changes in multiple traits were found for SALK_059132, which targets ACCELERATED CELL DEATH6 (ACD6)/AT4G14400.
Figure 6.

Analysis of the Multitrait QTL on Chr4.
(A) Haplotypes for Col-0 and Est-1 obtained with the five SNPs in high LD on Chr4 (QTL 56; Table 2; Supplemental Data Set 5). The eight SNPs significantly associated (FDR < 0.1) with QTL 56 are shown. SNPs close to ACD6 are highlighted in red and marked along the figure with vertical black dashed lines.
(B) Manhattan plot for the 68.2 kb where the QTL 56 was detected for 11 traits. LOD ≥ 3 for different traits and different experiments are color- and symbol-coded (legend at right). The horizontal line shows FDR = 0.1 at LOD = 4.8.
(C) Genomic structure in the region covered by Chr4-QTL. Genes are scale-depicted with gray arrows indicating the orientation. AGI codes for candidate genes selected for validation are shown in black and ACD6 = AT4G14400 is shown in red.
(D) Heat map of LD between the eight SNPs called in (A) measured as the partial coefficient of correlation (R2, scale included in the panel).
A previous QTL mapping study in a Col-0 x Est-1 RIL population identified the Est-1 allele of ACD6 as causal for slower leaf initiation, necrosis and decreased biomass (Todesco et al., 2010). Est-1 and Est-1-like accessions carry a “hyperactive” ACD6 allele, while Col-0 and Col-0-like accessions carry a so-called “reference” allele. These studies also showed that three nonsynonymous changes in the C-terminal transmembrane domain of ACD6 are responsible for the phenotypic differences between alleles. The insertion in the SALK_059132 KO line interrupts the last exon of ACD6, which encodes this transmembrane domain.
Our GWAS detected eight highly associated SNPs nearACD6. Five of them showed significant LD (r2 > 0.2, P < 0.01) and were associated with more than one metabolic trait (Figure 6A; Supplemental Data Set 5). These five SNPs define 13 haplotypes, only two of which occur at a frequency above the detection threshold for GWAS (minor allele frequency ≥0.05). The CTTTT haplotype is found in Col-0 and occurs more frequently (0.80, 280 accessions), while the ACGAG haplotype is found in Est-1 and occurs at a moderate frequency (0.05, 19 accessions). According to the allelic effect for these SNPs, the metabolic trait values are predicted to be lower in accessions with the CTTTT haplotype than in accessions with the ACGAG haplotype, except for Fum and FW, which should be higher. In agreement with these predictions, across our panel of 349 accessions, the mean trait values for accessions with the CTTTT and accessions with the ACGAG haplotype differed significantly in the expected direction (Supplemental Figure 8A).
Published sequence information (Todesco et al., 2010) for 46 of the accessions in our panel allowed us to classify 15 of them as Est-1-like and 31 as Col-0-like accessions. The mean trait values for these sequence-verified classes showed similar directional variation to that found when the complete population was classified based on the CTTTT and ACGAG haplotypes (Supplemental Figure 8B). The only exception was fumarase activity, which showed a similar trait mean in both groups of accessions. While this might indicate that fumarase activity is regulated by a different gene in the support region, it is also possible that in this small set of accessions, the effect of ACD6 on Fum activity is masked by the strong cis-QTL at FUM1/AT2G47510.
As the hyperactive ACD6 allele is carried by Est-1, we investigated the metabolic phenotypes of two knockdown lines in the Est-1 background (Est-1amiR.19 and Est-1amiR.20). At both dusk (ED) and dawn (EN), these lines showed a significant decrease in all six enzyme activities (aINV, nINV, cPGI, tPGI, fumarase, and GDH), AA, G6P, and Prot, and a significant increase in Fum and biomass (FW and dry weight) compared with the Est-1 wild type (Supplemental Figure 9). We also examined a dominant gain-of-function mutant for ACD6 in the Col-0 background (acd6-1). This mutant was obtained by EMS mutagenesis (Rate et al., 1999) and exhibits elevated defense responses, patches of dead cells, and reduced growth, resembling, although stronger, the phenotype of the Est-1 ACD6 allele. The metabolic phenotype of acd6-1 showed many similarities to accessions with the ACD6 Est-1 like allele, including significantly higher enzyme activities (nINV, cPGI, tPGI, and GDH) at ED and a significant decrease in biomass (FW and dry weight) (Supplemental Figure 10). In contrast, AA and Prot did not change consistently, and Fum increased rather than decreased.
In summary, the allelic effect of the minor Chr4 haplotype (ACGAG) matches the effect of the Est-1 ACD6 hyperactive allele. Furthermore, the metabolic phenotype of Est-1 knockdown lines and of the acd6-1 mutant are in line with the idea that the Chr4-QTL is driven by a change from an ACGAG to a CTTTT haplotype. These observations support the hypothesis that genetic variation at ACD6 is the causal factor underlying the pleiotropic metabolic QTL on Chr4 and indicate that ACD6 is a major modulator of plant growth, central metabolism, and plant defense.
Dissection of the cis-QTL at UGP1
We analyzed the strong cis-QTL at UGP1 to gain insights into gene evolution. UGP1 accounts for ∼75% of total UGP activity (Supplemental Figure 4). Earlier studies using the Ler x Cvi RIL population detected a UGP activity QTL in a genomic region containing UGP1 and an expression QTL (eQTL) for UGP1 (Keurentjes et al., 2008). In our GWAS, the most strongly associated SNPs were located ∼1 kb (m81737, TAIR10 position 748476, LOD1 = 8.6) and 3 kb (m81730, TAIR10 position 746513, LOD2 = 12.5) downstream of the UGP1/AT3G03250 coding sequence. LD analysis over a 26.8-kb region surrounding the UGP1 gene revealed an 11-kb LD block containing three genes, AT3G03240, AT3G03250/UGP1, and AT3G03260 (Figure 7A), and revealed that m81737 and m81730 are in strong LD with two SNPs located 206 and 1272 bp upstream of the ATG start codon of UGP1 (m81749, m81750; r2 > 0.8, P < 0.001; Figure 7A). Using these four SNPs, we defined 11 haplotypes (Figure 7B).
Figure 7.

cis-QTL for UGP Activity on Chr3.
(A) Manhattan plot for cis-QTL detected for UGP activity on Chr3. LOD for Exp1 (red) and Exp2 (black) are shown against the genome position of the SNP. FDR = 0.1 threshold (LOD = 4.8) is indicated with a horizontal dashed line. LD is depicted as a heat map of the coefficient of correlation (R2, scale included in the panel). An LD block (0.8 > R2 > 0.2, P < 0.001) was identified including the highest LOD SNPs (m81737 and m81730 for Exp1 and Exp2, respectively) and extending over three genes (AT3G03240, UGP1/AT3G03250, and AT3G03260). Two SNPs (m81749 and 81750) upstream of the ATG of UGP1 are in high LD with m81730 and m81737. Main haplotypes for the four polymorphisms are depicted at the top of the panel.
(B) UGP average activity from Exp1 and Exp2 plotted against the haplotypes defined by the four SNPs mentioned in (A). The frequency of each haplotype is indicated below the panel. The major haplotypes ACAA (low activity, red) and CTCG (high activity, gray) are significantly different (***P < 0.001) and are mainly responsible for the trait variation observed in GWA and in Ler x Cvi RIL population (Keurentjes et al., 2008). Four minor haplotypes defined by more than two accessions were also identified.
(C) Phylogenetic analysis of UGP1 LD block using 164 sequenced accessions. The major haplotypes defined in (B) are separated in the neighbor-joining tree using a shared allele distance matrix. Every accession is depicted as a small colored circle according to the haplotype; accessions with four different unique haplotypes are in black. The most common accessions are listed: Col-0, Cvi, Ler, and C24. Percentages for bootstrap values (1000 pseudo-replicates) are shown only for the groups discussed in the text.
(D) Tajima’s D value in sliding windows for the UGP1 LD block. The UGP1 gene location and orientation is depicted. Significantly high Tajima’s D values are marked with asterisks (*P < 0.05).
Most of the trait variation was accounted for by a major high-activity haplotype (CTCG, frequency = 0.66, carried by Cvi) and a major low-activity haplotype (ACAA, frequency = 0.28, carried by Ler). Two minor haplotypes (CTCA and CTAA, frequency = 0.006–0.023) showed slightly but not significantly higher UGP activity than the major high-activity haplotype, and two minor haplotypes (ACAG and CCCG, frequency = 0.020–0.006) showed low activity, although still higher than the major low-activity haplotype (Figure 7B). Five other haplotypes were restricted to single accessions. Full sequence information for 164 accessions (http://signal.salk.edu/atg1001/3.0/gebrowser.php) was used to investigate if lines carrying the minor high-activity haplotypes (CTCA and CTAA) are more similar to the major high- or the major low-activity haplotypes (Supplemental File 1). A neighbor-joining tree obtained from shared allele distance was generated using all polymorphic information (i.e., all SNPs and indels; Figure 7C). All lines carrying the major high-activity CTCG haplotype grouped in a unique branch (I; Figure 7C). The major low-activity ACAA haplotype was distributed between two distinct and well supported groups (II and III). Group II also contained members of a very similar minor low-activity haplotype (ACAG). A sub-branch of Group III contained the minor high-activity haplotypes CTCA and CTAA. This analysis indicates that the minor high-activity haplotypes derive from the major low-activity haplotype.
We computed Tajima’s D (Tajima, 1989) in sliding windows for the 164 sequenced accessions to assess the neutrality of mutations along the LD block (Figure 7D). A high Tajima’s D indicates that polymorphisms are evolving in a non-neutral manner. Tajima’s D was significantly positive (D = 2.06, P < 0.05) in two regions: 1000 to 1500 bp upstream of the start codon and downstream of AT3G03250, around m81730 and m81737. This suggests coevolution of the upstream and downstream regions, with a probable hitchhiking effect on the entire AT3G03250 gene.
Dissection of the cis-QTL at VAC-INV
VAC-INV accounts for up to 86% of total aINV activity (Supplemental Figure 4). The most significantly associated SNP (m7011) was located 1122 bp upstream of the start codon (Figure 8A). The two alleles present at m7011 are evenly distributed in the population (C frequency = 0.51, T frequency = 0.49), and C displays higher activity than T (P < 0.001; Figure 8B). Curiously, aINV activity coarse-mapped to VAC-INV in the Ler x Cvi biparental RIL population (Keurentjes et al., 2008), even though these two accessions are not polymorphic for m7011. This led us to search for further causal polymorphisms. Another SNP in the promoter region, m7010, showed moderate to high association with aINV and is in significant LD with m7011 (Figure 8A). This SNP is polymorphic between Cvi (A) and Ler (G) with A allele in higher frequency (0.65) and having higher aINV activity (Figure 8C). Three additional SNPs in exon 3 (m7017, m7018, and m7019) and a SNP in intron 3 (m7020) showed moderate to high association with aINV activity. SNPs m7017-20 are in extreme LD to each other (LD > 0.8, P < 0.001). Only two haplotypes are represented by more than three accessions, with the TCTT haplotype (frequency = 0.64, Cvi) showing higher activity than the CTCG haplotype (frequency = 0.34, Ler) (Figure 8D). Based on SNP data alone, there was no strong LD between m7011 or m7010 and the m7017-20 block. However, when LD analysis was repeated using full-genome sequence data from 164 accessions, we uncovered LD between m7010, the m7017-20 block, and another three nonsynonymous SNPs (C/G, C/A, and A/C) in the fourth and the fifth exons of VAC-INV (Figure 8E). Using these nine SNPs, we defined 18 haplotypes, including six that occur at a frequency between 0.06 and 0.29, three that are very rare (frequency 0.01–0.03), and nine that are unique. When we inspected the haplotypes with frequency ≥0.05 (Figure 8F), haplotypes carrying opposite alleles for all nine SNPs (ACTCTTCCA versus GTCTCGGAC) showed aINV activity differing significantly for Exp1 (P < 0.001) and marginally for Exp2 (P = 0.06), suggesting that these haplotypes drive GWAS. The other haplotypes with mixed effect alleles showed, as expected, intermediate values. The observation that the SNPs in the haplotype block are not homogeneously linked indicates that variation in VAC-INV might be a result of multiple evolutionary events, each associated to one or a few SNPs.
Figure 8.

cis-QTL for aINV Activity on Chr1.
(A) Manhattan plot for the genomic region on Chr1 where the cis-QTL was detected for aINV activity. LOD for Exp1 (red) and Exp2 (black) are shown against the genome position of the SNP. FDR = 0.1 at LOD = 4.8 is shown with a horizontal dashed line. High-LOD SNPs discussed in the text are indicated. Gene arrangements including VAC-INV are depicted with arrows. LD is shown as a heat map of the coefficient of correlation (R2, scale included in the panel).
(B) to (D) Average aINV activity from Exp1 and Exp2 plotted against the alleles at SNPs m7011 (B), m7010 (C), and m7017-20 (D). Significant differences in aINV activity were checked using t test (***P < 0.001).
(E) VAC-INV gene structure and LD heat map for the LD block marked in (A), using the complete sequence information for 164 accessions. Three nonsynonymous SNPs in VAC-INV coding sequence that are in LD with the associated SNPs highlighted in (A) are displayed. Significant values of LD are shown, and values between the nine SNPs of interest are given in red.
(F) The nine SNPs displayed in (E) generate six major haplotypes (frequency > 0.05). The average aINV activity from Exp1 and Exp2 for a given haplotype is plotted, showing opposing haplotypes from left to right.
DISCUSSION
GWAS has become a standard procedure for dissecting complex genetic traits, with proven success in plants (Meijón et al., 2014; Dubin et al., 2015), but it has only occasionally been applied to plant primary metabolism (Zhang et al., 2015; Wu et al., 2016). Here, we produced a comprehensive, highly defined QTL data set for plant central metabolism. We used this data set to explore the importance of cis- and trans-regulation of enzyme activity, to investigate whether genetic regulation contributes to the highly coordinated network structure of primary metabolism and to dissect genetic factors that underlie trait variation at the major pleiotropic locus ACD6 and the structural genes UGP1 and VAC-INV.
The heritability values obtained in our analysis resemble those in previous studies of metabolic traits in Arabidopsis, tomato (Solanum lycopersicum), and wheat (Triticum aestivum) (Kliebenstein et al., 2001a; Keurentjes et al., 2006; Toubiana et al., 2012; Alseekh et al., 2015; Matros et al., 2017). Broad-sense heritability (H2) for biomass, structural components, metabolites, and several enzyme activities (aINV, nINV, PEPC, AGP, UGP, NRVs, and GDH) was moderate to high (20–74%), with marker-based h2 (Kruijer et al., 2015) very close to H2 values. This indicates that additive genetic variation is the main source of phenotypic variation and that epistasis does not play a substantial role in the variation of these traits. The other enzyme activities showed low (<20%) or very low (∼0%) H2. Low heritability can be due to complexity of the trait, environmental variation, experimental error, and/or substantial GxE (Flint-Garcia et al., 2005). For enzymes with low to very low H2, within-line variation was very high in one or both experiments (Supplemental Figure 1D). For some enzymes, like FBP, GK, fumarase, and G6PDH, this was partly due to technical noise, but for others (e.g., FK, nINV, mRUB, and TK), it may reflect between-plant variation. Although QTL detection for these enzymes might be improved by including more biological replicates, this would substantially increase the requirements for growth space and analytic time.
The proportion of traits for which QTL were detected resembles those of previous studies. In two independent experiments, 12 (37%) and 24 (67%) of the 36 scored traits showed statistically significant associations (LOD ≥ 5.5, FDR control of 5%), while in earlier GWAS on primary metabolism, associations were detected for 38% of all annotated metabolites (Wu et al., 2016).
Environmental variation can be controlled in GWAS by phenotyping the population in multiple trials, as has been done in metabolite-GWAS for maize (Riedelsheimer et al., 2012; Wen et al., 2014; Liu et al., 2016) and Arabidopsis (Chan et al., 2010a, 2010b). We performed two experiments with plants at different ages under slightly different photoperiods. Plant metabolism is subject to age- or plant-size-dependent effects (Geronimo and Beevers, 1964; Meyer et al., 2007) and is modified in response to environmental changes such as photoperiod (Graf et al., 2010; Sulpice et al., 2014; Yang et al., 2016). These factors modified the metabolic network, as shown by the differing correlation networks in the two experiments (Figure 1; see also Sulpice et al., 2013) and a weakening of correlations in the cross-experiment comparison. Furthermore, the activities of some enzymes, such as FK, GK, G6PDH, FBP, fumarase, and SPS, showed high plasticity. These results indicate that the coordinated response of metabolic trait values across accessions is partly driven by coordinated responses to environmental or developmental cues (see below for further discussion). Nevertheless, we were able to identify 70 QTL shared between the two experiments. These QTL presumably reflect genotype-phenotype relationships that are relatively robust against small changes in photoperiod or plant age.
We identified these QTL using a threshold of LOD ≥ 3 in both experiments. Even though the incidence of false positives may increase at this low threshold, previous findings indicate that many causal loci are excluded by the stringent Bonferroni-corrected thresholds (Kooke et al., 2016). We detected robust QTL for 19 (63%) of the 30 scored traits. Our failure to detect QTL for the remaining traits is in line with earlier studies, which have shown that proven causal allelic variation sometimes associates only weakly with variation in quantitative traits. The FLOWERING LOCUS C (FLC) illustrates this scenario. Although this locus is strongly involved in the regulation of flowering time, Atwell et al. (2010) found only weak associations to a few of the surrounding SNPs, and no significantly associated SNPs within the FLC coding region.
The individual cis-QTL explained on average 7.2% of the total inherited variation for the linked trait, with the highest value for the cis-QTL at UGP1 (16.7%). These values are lower than in an earlier QTL analysis in a biparental Ler x Cvi population (Keurentjes et al., 2008), in which a putative cis-QTL for UDPG content explained 65%, and tentative cis-QTL at a PGM structural gene and at UGP1 explained 56% and 37% of total variation, respectively. This difference may partly reflect the increased genetic diversity in a GWA panel, which dilutes the effect of any one single polymorphism. In our GWAS, individual trans-QTL explained 5.1% of total variation and, although individually low, the summed trans-QTL for Fum, Mal, and aINV activity explained more than 30% of the total variation in these traits (Supplemental Data Set 7). The failure to detect QTL for some traits and the low explanatory power of individual QTL may be due to additional factors such as pleiotropy, epistatic interactions, trait complexity, and the quality of the phenotypic data (Oraguzie, 2007). In addition, some of the missing heritability may be due to allelic heterogeneity (i.e., the presence of multiple alleles at one locus influencing a trait) (Wood et al., 2011).
cis-QTL in Structural Genes Encoding for Enzymes
We applied GWAS to a large set of enzymes involved in central metabolism, extending earlier studies that used biparental populations or addressed single individuals or a small number of enzymes (Keurentjes et al., 2008; Brotman et al., 2011). Our analysis provides strong evidence that natural variation in central metabolism is partly due to cis-variation in enzyme structural genes. These represented the strongest individual QTL detected in our diverse panel (see above).
The mode of regulation varies from gene to gene. For instance, it is likely that the strong cis-QTL at UGP1 (LOD = 12.5, effect size 16.7%; Figure 7) is the result of variation in UGP1 expression, driven by different UGP1 alleles. This conclusion is supported by the following: the identification of SNPs in the UGP1 promoter (m81749 and m81750) associated with high and low activity, the strong correlation between the two major haplotypes and UGP activity, and the results of a previous study in a biparental population that indicated that a cis-eQTL for UGP1 affects UGP activity (Keurentjes et al., 2008).
We detected a strong cis-QTL for aINV activity at VAC-INV (LOD = 7.7, effect size 10.2%; Figure 8). Earlier studies using a Ler x Cvi population coarse-mapped an INV activity QTL (Sergeeva et al., 2006) and a weak cis-eQTL to this genomic region (Keurentjes et al., 2008). However, subsequent studies on isogenic lines derived from a Ler x Cvi cross did not detect differences in expression between the two putatively causal alleles, and the authors proposed that differences in the VAC-INV coding region are responsible for the activity QTL (Leskow et al., 2016). Our analyses show that aINV activity variation is not only due to Ler and Cvi alleles, but also to other polymorphisms in the promoter and the coding region. These findings point to a combined contribution of multiple evolutionary events, with up to nine SNP haplotypes explaining the allelic effects in our GWA population (Figure 8).
The cis-QTL for AGP activity is probably the result of nonsynonymous SNPs in the gene encoding the catalytic APL1 subunit. APL1 is the most highly expressed member of a small gene family that encodes the regulatory subunit of AGP (Ballicora et al., 2004).
Plastid and cytosolic PGI activity are controlled by different genes (Kunz et al., 2014). We detected a cis-QTL for cPGI activity at SIS/AT5G42740. A QTL for PGI activity and a weak eQTL were previously coarse-mapped to this genomic region in a Ler x Cvi population (Keurentjes et al., 2008). Although not validated across experiments, pPGI activity was associated very weakly with a SNP located in the genomic region of PGI1/AT4G24620.
A cis-QTL for fumarase activity associated with FUM1/AT2G47510, which encodes mitochondrial fumarase (Pracharoenwattana et al., 2010). While the strongest SNPs were located upstream of the coding region, SNPs were also detected in the coding region. The Arabidopsis genome also contains a gene encoding a cytosolic fumarase (FUM2). FUM2 is associated with Fum and Mal content. This finding is in agreement with earlier coarse-mapping in a biparental population (Lisec et al., 2008) and analyses of metabolite levels in fum2 mutants (Pracharoenwattana et al., 2010; Brotman et al., 2011). Recently, a QTL for FUM2 was linked to a large insertion in the promoter of this gene in C24-like accessions (Riewe et al., 2016). The SNPs associated with FUM2 in our GWA population are in high LD with the indel (insertion/deletion) identified by Riewe et al. (2016). We did not detect fumarase activity QTL at FUM2. This might be masked by the cis-QTL at FUM1, which encodes the majority of the fumarase activity in Arabidopsis (Pracharoenwattana et al., 2010). Alternatively, fumarase activity might be modified in a manner that our standardized assays do not detect (e.g., via a change in substrate affinity). The vast majority of the Mal and Fum in plant cells is located in the vacuole (Krueger et al., 2011; Arrivault et al., 2014). The lack of effect of the FUM1 enzyme activity QTL on the levels of these metabolites, and the association of Mal and Fum metabolite QTL with FUM2, indicate that cytosolic fumarase determines the vacuolar levels of Mal and Fum.
Evolutionary Implications of Variations in Structural Genes for Enzymes
Some enzymes are encoded by single genes and others by small gene families. Some of the cis-QTL detected in our study mapped to single genes, for example, mitochondrial fumarase, cytosolic fumarase, and cytosolic PGI (FUM1, FUM2, and SIS). On the other hand, UGP is encoded by two genes (UGP1 and UGP2) (Meng et al., 2008), APL1 is one of four members of a gene family for the regulatory subunit of AGP (Ballicora et al., 2004), and VAC-INV is one of four annotated acid invertases (Tymowska-Lalanne and Kreis, 1998). This raises the question of why multiple genes are retained; do they confer an advantage or are they just remnants of earlier whole-genome or local duplications? Moderate changes in enzyme abundance usually have little effect on pathway flux (Kacser and Burns, 1973; Fell and Thomas, 1995; Schuster et al., 1999), including flux in plant central metabolism (Stitt and Sonnewald, 1995; Stitt et al., 2010). This indicates that multiple gene copies are sometimes superfluous, at least under the conditions used in laboratory experiments. On the other hand, in some cases, mutations in individual members of enzyme gene families result in clear phenotypes, including VAC-INV in Arabidopsis (Sergeeva et al., 2006). We therefore combined information about enzyme activity variation in our large association panel with public domain information on whole-genome sequences to explore recent evolution at two loci where we detected strong cis-QTL.
The variation in UGP activity is mainly due to the presence of a high activity (CTCG) and a low activity (ACAA) haplotype (Figure 7). One explanation for the maintenance of the latter at a quite high frequency would be that overall UGP activity is in excess. However, our analyses indicate that balancing selection is acting to maintain both the high and low activity haplotypes (Figure 7D). This effect might be partly a consequence of selection on UGP2, which also contributes to total UGP activity (Meng et al., 2009). In the Ler x Cvi population, an UGP2 eQTL with an opposite effect to UGP1 was identified, suggesting that UGP2 is upregulated in accessions in which UGP1 expression is reduced. Our GWAS did not detect a QTL at UGP2, probably because the effect was much smaller than the QTL at UGP1. Earlier studies in a biparental population indicated that the effect of a QTL of UGP2 is only 20% that of UGP1 (Keurentjes et al., 2008). The idea of balancing selection is also supported by the detection of two very low frequency haplotypes (<<0.05, CTCA and CTAA) that have slightly higher activity than the major high-activity haplotype (CTCG) but are phylogenetically more similar to the major low-activity haplotype (ACAA). The rare high activity haplotypes presumably result from allele variation that compensates for the low activity in accessions carrying ACAA. Interestingly, these haplotypes are present in geographically distant accessions and did not propagate further in the population, supporting the idea that evolutionary forces are maintaining UGP activity within a certain range.
Invertase plays a key role in sucrose import and allocation. Although there are several acid invertase genes, two biparental studies (Sergeeva et al., 2006; Keurentjes et al., 2008) and our GWAS detected only one cis-QTL for aINV activity, which is always associated with VAC-INV. We might have missed other cis-QTL because VAC-INV encodes much of the total aINV activity and because some aINV genes are expressed in specific tissues or developmental stages (Tymowska-Lalanne and Kreis, 1998). Nevertheless, the strong cis-QTL, together with the finding that the variance in aINV activity is probably due to many independent events, point to the importance of the genetic regulation of VAC-INV. Furthermore, we detected many trans-QTL for aINV activity. Irrespective of whether they act on VAC-INV or on other members of the family, their frequency further underlines the importance of the genetic regulation of acid invertase.
Sergeeva et al. (2006) reported a root extension phenotype in vac-inv, and Leskow et al. (2016) reported reduced inflorescence biomass for several VAC-INV alleles, pointing to its general role in vegetative and reproductive growth. No marked phenotypes have been reported for ugp1 or for apl1 in long days. This raised the question of whether these cis-QTL carry any disadvantages. A short period of darkness leads to a shortfall in carbon supply, seed abortion, and shorter siliques in Arabidopsis (Lauxmann et al., 2016), indicating that seed set is sensitive to perturbations in central metabolism. We found seed abortion phenotypes not only for vac-inv but also for ugp1 and apl1 (Figure 5). Defective seed development was also reported in Arabidopsis pMDH/pmdh heterozygotes (Selinski et al., 2014), which in our GWAS associated with Mal content. Mal content and MDH and GDH activity associated with FHA2, and fha2 mutants exhibit an altered silique phenotype (Ahn et al., 2013). These various studies all point to seed set being especially sensitive to minor lesions in metabolism that have little effect at other stages in the plant’s life history.
Metabolic Regulation by trans-Regulation
Our GWAS detected many more trans-QTL (80) than cis-QTL (8). This resembles earlier findings from studies using biparental populations (Keurentjes et al., 2008). Enzymes, like other proteins, are subject to transcription, translation, processing, transport, posttranslational modification, and degradation. These processes were well represented in the detected trans-QTL (Figure 3), supporting the idea that many processes in addition to transcriptional regulation contribute to the regulation of protein level. The high number of trans-QTL and large range of functional categories was even more striking for metabolite QTL. Only a low number of metabolite-level QTL mapped to structural genes for related enzymes (Table 2). This may reflect the finding that moderate changes in individual enzyme abundance usually have little effect on metabolic flux (see above). Interestingly, some metabolite QTL associated with genes for transporters, highlighting their importance in determining metabolite levels. The rather high proportion of trans-QTL compared with cis-QTL in our study of enzyme activity QTL contrasts with a recent GWAS of eQTL in poplar (Mähler et al., 2017) in which the number of cis-QTL exceeded trans-QTL by a factor of four. This difference might reflect a difference in detection thresholds, affecting the cis/trans ratio due to dissimilarity in effect sizes of the two classes, or alternatively, the strong contribution of posttranscriptional events to the regulation of protein abundance.
In addition to confirming known links (e.g., LBD37 for NO3− levels; Rubin et al., 2009), we validated several novel trans-QTL. We assigned a QTL in MDH and GDH activities and Mal content to a SMAD/FHA domain-containing protein (FHA2). Members of this family recognize a phosphothreonine epitope and are involved in protein-protein interactions (Ahn et al., 2013). We detected a QTL for Fru at IQD22, and experiments using KO mutants demonstrated that IQD22 also affects Suc and the starch:Suc ratio (Figure 4; Supplemental Figures 6A and 6B). IQD22 belongs to a large family of IQ67-Domain (IQD) proteins, which are targets of plant-specific CaM/CaML (calmodulin/calmodulin-like) proteins (Abel et al., 2005). IQD22 is repressed by gibberellic acid (GA) and induced by the DELLA protein RGA (Zentella et al., 2007), which inhibits GA signaling and restricts plant growth (Silverstone et al., 1998). Our results indicate that IQD22 regulates carbohydrate allocation during GA-induced expansion growth, possibly to provide osmolytes or energy to support growth.
We detected and validated a QTL colocalized for starch, Suc, and Glc content at MSL10 (Figure 4; Supplemental Figure 7). MSL family members act as osmotic safety valves to release osmolytes under increased membrane tension (Maksaev and Haswell, 2012). These proteins have been implicated in pollen development (Hamilton et al., 2015), osmotic regulation in plastids (Wilson et al., 2016), redox homeostasis in mitochondria (Lee et al., 2016), and cell death (Veley et al., 2014). The link between MSL10, sugars, and starch metabolism might be related to osmotic and turgor regulation.
Pleiotropic Regulation of Primary Metabolism
Metabolic traits show strong connectivity in biparental mapping lines and GWA panels (see Introduction). As mentioned above, network structure is altered by relatively small changes in growth conditions (Figure 1; see also Sulpice et al., 2013), indicating that connectivity is partly due to the coordinated regulation of metabolism by metabolic, environmental, or developmental cues. To determine if genetic regulation also contributes to the connectivity in central metabolism, we searched for pleiotropic loci at which several metabolic QTL colocalize.
We detected 14 genomic regions with colocalized QTL for enzyme activities, metabolite levels, structural components, or biomass. This result supports the hypothesis that coordinated genetic regulation contributes to the strong correlation network revealed by cross-genotype comparisons of primary metabolic trait values (Figure 1). Colocalization of metabolic QTL is a widespread phenomenon within both biparental populations and GWA panels. In a biparental Col-0 x C24 population, more metabolic QTL colocated with biomass QTL than expected by chance (Lisec et al., 2008). Transcript, protein, metabolite, and plant phenotype variation in a Ler x Cvi RIL population could be reduced to six QTL hot spots that collectively explained much of the variation at each phenotypic level (Fu et al., 2009), and two of these hot spots (VAC-INV and MAM) were related to plant metabolism, pointing to an important role for metabolism in defining the overall plant phenotype.
Nonetheless, many metabolic traits did not share a QTL with other traits. This might indicate that they are regulated in a less concerted manner. For example, it is possible that some traits escape integrated regulation when large-effect factors segregate. However, this may also reflect the low detection power of GWAS, as illustrated by the irregular detection of QTL in the two replicate experiments. The colocalized QTL identified in our GWAS also differed from those reported in a biparental population (Keurentjes et al., 2008). Such discrepancies may reflect differences in allele balance between GWA and biparental populations as well as their differing power to detect QTL. It is also possible that some colocations reported using biparental mapping are spurious because they are defined by large genome intervals in which independent QTL may colocalize by chance (Zhang et al., 2010). Specific analyses that are beyond the scope of this study might help to improve the statistical power to detect pleiotropic variants (Tyler et al., 2013; Tyler and Carter, 2017).
We validated three QTL at which several traits colocalized (Table 2): FHA2, MSL10 (see above), and a large genomic interval of 68.2 kb on Chr4 that contains 24 genes in strong LD and associates with six enzyme activities (aINV, nINV, cPGI, tPGI, fumarase, and GDH), three metabolites (G6P, AA, and Fum), Prot, and FW. Analyses of haplotype effects and KO lines showed that many of these associations are due to the presence of different ACD6 alleles (Supplemental Figures 8 and 9).
ACD6 has previously been linked to plant defense, spontaneous necrosis, and plant biomass (Rate et al., 1999; Lu et al., 2005; Atwell et al., 2010; Todesco et al., 2010). Arabidopsis accessions with the hyperactive Est-1-like allele of ACD6 show preactivated defense pathways, higher pathogen resistance, and reduced biomass compared with Col-0-like accessions (Todesco et al., 2010). ACD6 was not previously implicated in the regulation of central metabolism, and analyses of two biparental populations did not identify any metabolic QTL at ACD6 (Keurentjes et al., 2008; Lisec et al., 2008). This is probably because accessions Ler, Cvi, C24, and Col-0 all carry the reference Col-0-like allele (Todesco et al., 2010). The multitrait metabolic QTL detected in our study correlated strongly with the presence of the Est-1-like allele. Investment in defense might require modification of metabolism at the expense of rapid growth. Alternatively, higher enzyme activities might be maintained in smaller plants to sustain higher levels of sugars, proteins, and amino acids and provide robustness against a fluctuating or adverse environment. The reduced Fum levels might be a result of the higher levels of AA and proteins in accessions carrying an ACD6 hyperactive allele; cytosolic fumarase has been shown to play a key role in amino acid synthesis (Pracharoenwattana et al., 2010).
The effect of the ACD6 hyperactive allele reflects the correlation network seen for metabolic traits, with most enzyme activities, protein, G6P, and AA being negatively correlated with biomass, while Fum is positively correlated with biomass. This indicates that the major multitrait QTL at ACD6 makes a substantial contribution to natural genetic variation in central metabolism in Arabidopsis. Interestingly, of the accessions that have the highest protein levels and lowest biomass in our study, many derive from locations with strong seasonal differences (Sweden and Finland), where they may experience stressful situations in which slow growth and increased defense might be preferred, hinting at a trade-off between plant defense and central metabolism and growth.
In conclusion, first, we identified large numbers of enzyme activity and metabolite QTL, cross-validated them in two independent experiments, and validated many using a large KO panel. The strongest enzyme activity QTL mapped to structural genes for the enzymes, but we also found many trans-QTL that cumulatively had a larger effect than the cis-QTL and mapped to a wide range of gene categories. Second, analysis of the variation in structural genes for enzymes uncovered complex and dynamic evolution at these loci. Third, we found many colocalized QTL for different traits, pointing to coordinated genetic regulation of central metabolism. The QTL with the most marked effect was located at ACD6, pointing to a trade-off between defense and central metabolism. Finally, the coordinated changes in metabolic traits across mapping populations observed in this and previous studies are partly due to coordinated genetic regulation and partly due to coordinated responses of central metabolism to environmental and developmental cues.
METHODS
Mapping Population
The GWA mapping population comprised 349 Arabidopsis thaliana natural accessions belonging to the HapMap panel (Horton et al., 2012). This panel was selected because the accessions maximize diversity and minimize redundancy and close family relatedness (Baxter et al., 2010).
Plant Growth Conditions and Experimental Design for GWAS
For GWAS, two independent experiments (Exp1 and Exp2) were designed and performed at an interval of 12 months to ensure replication and the robustness of QTL identification. For each experiment, seeds were sown on filter paper with demineralized water, stratified at 4°C in dark conditions for 5 d, and transferred to a culture room (16-h photoperiod, 24°C) for 42 h to induce seed germination. Six plants per accession were transplanted to wet 4 × 4-cm Rockwool blocks in a climate chamber. They served as two biological replicates (three pooled plants each). Plants from the same biological replicate were planted in consecutive (Exp1) or random (Exp2) order, and biological replicates were located in different sides of the growth chamber. Chamber climate conditions were as follows: 10-h photoperiod for Exp1 and 12-h photoperiod for Exp2, fluorescent light intensity 125 μmol m−2 s−1 with a wavelength from 380 to 800 nm (Master TL5; Philips), temperature 20°C day/18°C night, and relative humidity 70%. All plants were watered daily for 5 min with 1/1000 Hyponex solution. Full rosettes were harvested after 37 DAS for Exp1 and 28 DAS for Exp2. The harvest time was set to the end of the light period.
KO Lines and Mutants
Homozygous T-DNA insertion lines from public collections (SALK, SAIL, GABI-KAT, and JIC) for 45 candidate genes were obtained from the NASC (European Arabidopsis Stock Centre). In total, 102 KO lines were ordered. Of these, three KO lines did not germinate, and other lines were discarded because they were heterozygous (20) or could not be confirmed by PCR amplification (5). Seed of MSL10 OEX (Pro35S:MSL10) were kindly provided by Elizabeth Haswell (Washington University in St. Louis). Knockdown lines for ACD6 in the Est-1 background (Est.amiR.19 and Est.amiR.20) were kindly provided by Detlef Weigel (Max Planck Institute for Developmental Biology), and the gain-of-function mutant acd6-1 was kindly provided in-house by Roosa Laitinen (Max Planck Institute of Molecular Plant Physiology). Additional homozygous KO lines for IQD22 analyses were obtained in-house from heterozygous KO lines ordered from NASC.
Plant Growth Conditions and Experimental Design for KO Panel Validation
For KO analysis, a complete randomized design in a chamber with the same conditions as described for Exp2 was used. Seeds and plants were treated as stated above. Nine plants were collected in three biological replicates for KO lines and 18 plants in six biological replicates for wild-type plants. Col-0 (N6673 and N1093, two different seed stocks) and Col-8 (N60000, formerly background of SALK lines) were included as wild-type controls. Full rosettes were harvested 28 DAS at the end of the light period.
For further experiments with the fha2, iqd22, msl10, and acd6 mutants, seeds were germinated and grown for the first week in a 16-h photoperiod, a temperature of 20°C day/6°C night, humidity of 75%, and a light intensity of 145 μmol m−2 s−1. The seedlings were transferred to a Phytotron where the growth was continued in an 8-h photoperiod with temperature 20°C day/16°C night, humidity of 75%, and light intensity of 145 μmol m−2 s−1. After the second week, plants of average sizes were transferred to 10-cm-diameter pots (five plants per pot) filled with the same soil as for germination and grown for 4 weeks in a controlled environment chamber (Percival Scientific) with a relative humidity 70% and a fluorescent light intensity of 125 μmol m−2 s-1with a light spectrum as described by Annunziata et al. (2017). The temperature for fha2, iqd22, and msl10 was set to 20°C day/18°C night and for the acd6 mutant to a constant temperature of 22°C, since acd6-1 growth is extremely impaired at lower temperatures (Todesco et al., 2010). Three to five pots were grown per mutant/wild type and time point. All full rosettes from each pot were harvested and pooled together as one biological replicate. For the msl10 experiment, plants were harvested every 4 h starting at the end of the previous night (ZT = 0). For the fha2 and acd6 experiments, plants were harvested at dawn (ZT = 0, end of the previous night) and dusk (ZT = 12, end of the light period). For iqd22, harvest was only done at dawn (ZT = 0, end of the previous night).
For silique abortion and flowering time experiments with the apl1, vac-inv, ugp1, and iqd22 mutants, seeds were germinated and grown for the first 2 weeks as described above. After the second week, plants were transferred to 5-cm-diameter pots (one plant per pot) filled with the same soil as for germination and grown in a 16 h-photoperiod in a greenhouse with temperature between 20°C and 18°C and 60 to 80% humidity. For each line and wild type, 25 to 35 pots of plants were grown. Flowering time was scored as the total leaf number on the day of bolting (flower bud ∼0.5 cm) and as the leaf initiation rate (days to flower/total leaf number). Days to flower was counted from sowing until the day of bolting. Silique formation was followed until maturation on at least 10 plants, and the number of siliques was quantified on three plants.
Phenotyping
The traits analyzed in Exp1 and Exp2 included ChlA and ChlB, total protein (Prot), total amino acids (AA), nitrate (NO3−), Glc, Fru, Suc, starch, G6P, Mal, Fum, and 24 enzyme activities targeting sucrose synthesis and degradation, starch synthesis, glycolysis, the tricarboxylic acid cycle, Calvin-Benson cycle, pentose phosphate pathway, and NO3− conversion pathway. Accessions were weighed before freezing only for Exp1 (FW), and six enzymes were further included for Exp2 to cover nitrogen metabolism and the Calvin-Benson cycle in more detail. In the KO panel and in further experiments with the fha2, iqd22, msl10, acd6, apl1, vac-inv, and ugp1 mutants, only 26 traits were measured: all structural components and metabolites but only 14 enzyme activities. For a full description of the traits measured in each experiment, see Table 1.
Enzyme and Metabolite Assays
Biological replicates (three to six Arabidopsis rosettes each) were used for metabolic and enzymatic assays. For each biological replicate or sample, a technical replicate (two aliquots from the same sample extract) was analyzed. Samples from the same experiment were measured within 2 weeks in batches of 80 or 40 per plate for metabolites and enzymes, respectively. They were randomized within and between plates, and reference material was included in each plate to control for plate differences. However, FBP, GK, fumarase, and G6PDH activity measurements presented high technical noise. Chemicals and enzymes for metabolite and enzymatic assays were purchased as described by Gibon et al. (2004a). Metabolites were extracted from 20 mg of ground frozen material. Total protein was assayed using the Bradford method (Bradford, 1976). ChlA, ChlB, starch, Glc, Fru, Suc, and AA were determined by enzymatic assays in ethanolic extracts of 20 mg of frozen plant material as described by Cross et al. (2006). Mal and Fum were measured as described by Nunes-Nesi et al. (2007). Assays were performed in 96-well microplates using a Janus pipetting robot (Perkin-Elmer). Absorbance was determined using a Synergy or ELX-800-UV microplate reader (Bio-Tek Friedrichshall). For enzyme measurements, 20 mg of powdered frozen material was extracted as described by Gibon et al. (2004b) with the modifications introduced by Nunes-Nesi et al. (2007). AGP, SPS, fumarase, acidic and neutral INV, GK, FK, FBP, G6PDH, and NR were determined as described by Gibon et al. (2004b). NR activity was measured in two assay conditions, one (Vm) to detect total activity and one (Vs) that contained Mg2+ to allow binding of an inhibitory 14-3-3 protein to the phosphorylated form, allowing detection of the active dephosphorylated NR. Rubisco was measured according to Sulpice et al. (2007). Cytosolic and plastidic PGI were quantified following Weeden and Gottlieb (1982). PGM was assayed as published by Manjunath et al. (1998) and UGP as stated by Keurentjes et al. (2008). Enzyme activities were expressed on a fresh weight basis (nmol g−1 FW min−1). Assays were prepared in 96-well microplates using a Multiprobe II pipetting robot equipped with a cooling block, an incubation block set to 25°C, a shaker, and a gripper (Perkin-Elmer).
Descriptive Statistics
For GWAS, the technical replicates were used to obtain the biological replicate values for each trait. Trait means for each accession 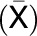 were obtained by averaging the two biological replicates. No additional transformation was applied. Histograms for trait values and Spearman’s rho correlation coefficients were determined using Infostat (Di Rienzo et al., 2011). Coefficient of variation (CV) was calculated as
were obtained by averaging the two biological replicates. No additional transformation was applied. Histograms for trait values and Spearman’s rho correlation coefficients were determined using Infostat (Di Rienzo et al., 2011). Coefficient of variation (CV) was calculated as 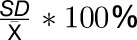 . The phenotypic plasticity index was calculated as (maximum − minimum)/maximum for each averaged genotype in the two environments; if the genotypic value of Exp2 was larger than Exp1, the phenotypic plasticity index was multiplied by −1.
. The phenotypic plasticity index was calculated as (maximum − minimum)/maximum for each averaged genotype in the two environments; if the genotypic value of Exp2 was larger than Exp1, the phenotypic plasticity index was multiplied by −1.
For KO analysis, trait means for each KO line and the wild type were obtained by averaging between two to six biological replicates. KO/wild-type comparisons were made using one-way ANOVA after correcting for batch noise (Lisec et al., 2008). For all traits, Col-8 and Col-0 N1093 always showed a skewed behavior compared with the entire population. Thus, only Col-0 N6673 was used as a wild-type control. To increase robustness, significant differences were determined by comparing each individual line with the population mean (all KO lines plus Col-0 N6673 wild type) as a reference value. This approach was based on the assumption that for most traits, the KO lines would present wild-type-like values and that the large size of the panel (74 lines) would dilute the impact of the very small number of lines in which the trait value was altered. Correction for multiple comparisons was done using a procedure based on clustering (DGC) implemented in Infostat. This test well controlled the type I error rate by maintaining an acceptable power in well-conducted experiments (low CV for the mean difference) and improves its general performance when increasing the number of means to compare.
For additional experiments using fha2, iqd22, msl10, acd6, apl1, vac-inv, and ugp1 mutants, Col-0 N6673 and an in-house Col-0 were included as the wild type. Both wild-type seed stocks showed similar values for all traits and were used as one wild-type control. ANOVA followed by multiple test correction (DGC) was applied to detect significant differences in fha2, iqd22, msl10, apl1, vac-inv, and ugp1 experiments. For acd6 experiments, a t test comparing the natural accession (Col-0 or Est-1) to the gain-of-function or knockdown mutants was performed.
GWA Mapping
All phenotyped accessions were genotyped for 214,051 SNPs previously (Horton et al., 2012). Two statistical packages were used in R to perform GWA analysis: EMMAX (Efficient Mixed-Model Association eXpedited) and GAPIT (Genomic Association and Prediction Integrated Tool) (Kang et al., 2010; Lipka et al., 2012).
Both packages use a mixed linear model (MLM) containing fixed and random effects. Including individuals as random effects gives MLM the ability to incorporate information about relationships among individuals. This information about relationships is conveyed through the kinship (K) matrix and the population structure or the so-called Q matrix. The Q+K approach improves statistical power compared with Q only, and K and Q matrixes can be obtained using different approaches. Thus, we evaluate the performance of GWA including K and/or Q factors obtained using different approaches: (1) K was obtained according to Kang et al. (2008) and Q was omitted (Model EMMAX); (2) K was obtained according to VanRaden (2008) and Q was accounted for by including the first three principal components from principal component analysis (Model GAPIT); and (3) K was obtained by SPAGeDi (Hardy and Vekemans, 2002) according to Loiselle et al. (1995) and Q was calculated using Discriminant Analysis of Principal Components (Jombart et al., 2010). For the last approach, four groups of SNPs were used for K+Q calculation with the aim of testing the effect of different set of markers on the control of confounding effects (Models PK1 to PK4). Each group of SNPs consisted of ∼1300 markers selected from groups of 5000 SNPs and discarding the ones showing linkage disequilibrium, to ensure a set of independent markers. In total, six different GWA models were run for Exp1 and Exp2.
The MLM can be described as:
| (1) |
where Y is the vector of observed phenotypes; β is an unknown vector containing fixed effects, including the genetic marker, population structure (Q), and the intercept; u is an unknown vector of random additive genetic effects from multiple background QTL for individuals/lines; X and Z are the known design matrices; and e is the unobserved vector of random residual. The u and e vectors are assumed to be normally distributed with a null mean and a variance of:
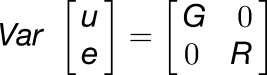 |
(2) |
where 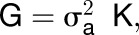 with
with 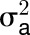 as the additive genetic variance and K as the kinship matrix. Homogeneous variance is assumed for the residual effect; i.e.,
as the additive genetic variance and K as the kinship matrix. Homogeneous variance is assumed for the residual effect; i.e., 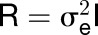 , where
, where 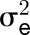 is the residual variance. The proportion of the total variance explained by the genetic variance is defined as heritability (h2).
is the residual variance. The proportion of the total variance explained by the genetic variance is defined as heritability (h2).
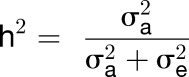 |
(3) |
In GWA, marker-based h2 is obtained by fitting a mixed model with random genetic effects. The covariances between these effects are modeled by the kinship matrix estimated from DNA markers. Moreover, h2 can be obtained using either one-step procedure including phenotypic repetitions for the same individual in the calculation (Kruijer et al., 2015), or two-step procedure, where the phenotypic mean is first estimated and then included in the mixed model to obtain the genetic variance (also called line-based h2) (Lipka et al., 2012). The h2 calculations according to Kruijer et al. (2015) and using the two-step procedure are stated in Supplemental Data Set 2.
REML-based estimations of broad-sense heritability (H2) were done using lme4 package in R, the following model was adjusted to the phenotypic traits per experiment:
| (4) |
where µ is the common mean, g is the random effect of the genotype ith and e is the typical residual error with normal distribution.
GWA Analysis, Candidate Gene Selection, and Validation
GWA was run using the R packages EMMA for Model EMMAX and GAPITfor the remaining models (Kang et al., 2008; Lipka et al., 2012). P values of association were log-transformed to LOD values [–log10(P value)] and regarded as significant after FDR correction (Benjamini and Hochberg, 1995) if ≤0.1∼LOD = 4.8, ≤0.05∼LOD = 5.5, and ≤0.01∼LOD = 6.3 for a 10, 5, or 1% control of false discovery. However, the initial threshold of comparison was set to LOD ≥ 3. First, we preselected those SNPs with LOD ≥ 3 in all models for each experiment. Then, these SNPs were compared between experiments and those with LOD ≥ 3 in both experiments were retained as cross-validated SNPs. The cross-validated SNPs showed robustness across conditions. For traits showing high plasticity between experiments (i.e., FK, nINV, FBP, fumarase, SPS, pPGI, and AGP), SNPs with LOD ≥ 4.8 in either experiment were retained as plastic associations. We also selected all SNPs that colocalized for several traits despite cross-validation between experiments. To determine the genes involved in the association, a general examination of the region with the candidate SNP was done looking for gene functional annotations that would further support a selection. When not possible, the gene or genes where the high-LOD SNPs were located were selected as putative candidates. When the SNPs were placed between two genes, both genes, upstream and downstream of the SNP, were selected. To determine QTL size and trait colocalization, LD analysis was performed on contiguous SNPs with LOD ≥ 3 in either experiment or trait. SNPs were considered linked and belonging to the same QTL when the squared coefficient of correlation (R2) was ≥0.2.
Nucleotide Diversity Analysis and Genetic Distances
Sequences for UGP1 and VAC-INV analyses were obtained from the Arabidopsis 1001 genome project (http://signal.salk.edu/atg1001/3.0/gebrowser.php; 164 accessions). For deviation from neutrality, Tajima’s D statistic (Tajima, 1989) was computed in sliding windows (window length = 100 sites, step size = 25 sites) using DnaSP version 4.0 (Rozas et al., 2003). Linkage disequilibrium analysis was performed with DnaSP version 4.0 (Rozas et al., 2003) for complete sequence data. For SNP data and LD figures, the LDheatmap package (Shin et al., 2006) was used in R (R Core Team, 2016). UGP1 sequences were aligned with MAFFT software using the G-INS-1 method (Katoh et al., 2002). Sequence alignment for the UGP1 LD-block is provided in Supplemental File 1. The neighbor joining tree was obtained by calculating shared allele distance (Jin and Chakraborty, 1994) from the sequence alignment using PowerMarker (Liu and Muse, 2005). Branch support was obtained by bootstrap method (1000 pseudo-replicates) implemented in the same software. Only bootstrap values (>50%) supporting the large groups discussed in the text are shown.
Accession Numbers
Sequence data from this article can be found in the GenBank/EMBL libraries under the following accession numbers: AT3G03250 (UGP1), AT1G12240 (VAC-INV), AT5G19220 (APL1), AT5G42740 (SIS), AT2G47510 (FUM1), AT1G62710 (BETA-VPE), AT3G49430 (SRp34a), AT5G44560 (VPS2.2), AT5G01550 (LECRKA4.2), AT5G01560 (LECRKA4.3), AT3G58560 (ATCCR4a), AT3G58580 (ATCCR4b), AT1G27720 (TAF4B), AT1G49210, AT2G15710, AT1G10570, AT5G44370, AT3G54350, AT5G46110 (TPT), AT3G47520 (pMDH), AT5G50950 (FUM2), AT1G68600, AT2G47570 (RPL18e), AT4G23060 (IQD22), AT5G67420 (LBD37), AT2G33150 (KAT2), AT5G12080 (MSL10), AT3G07220 (FHA2), AT4G02500 (XXT2), and AT5G10140 (FLC).
Supplemental Data
Supplemental Figure 1. Phenotypic variation, phenotypic plasticity, and heritability for metabolic traits.
Supplemental Figure 2. SNP-to-SNP cross-validation between Exp1 and Exp2.
Supplemental Figure 3. Metabolic profiles for the KO panel.
Supplemental Figure 4. Validation of cis-QTL identified for enzyme activity variation.
Supplemental Figure 5. Further experiments to confirm the QTL associated with Mal, MDH, and GDH activity.
Supplemental Figure 6. Metabolic profiles and developmental parameters of iqd22 KO lines.
Supplemental Figure 7. Diurnal changes in starch for msl10 mutants.
Supplemental Figure 8. Trait means for the major haplotypes defined by the high-LOD SNPs associated in Chr4-QTL and trait means for accessions carrying Col-0 and Est-1 ACD6 alleles.
Supplemental Figure 9. Metabolic effect of knocking down the ACD6 hyperactive allele.
Supplemental Figure 10. Metabolic profile for the gain-of-function acd6 mutant in the Col-0 background.
Supplemental Data Set 1. Phenotypic values for the 349 accessions used in GWAS.
Supplemental Data Set 2. Descriptive statistics.
Supplemental Data Set 3. Spearman rank correlation matrix between traits for the two different experiments.
Supplemental Data Set 4. Numbers of SNPs detected for the six models tested.
Supplemental Data Set 5. Candidate SNPs identified in the regulation of 30 out of 37 traits analyzed by GWA in two independent experiments.
Supplemental Data Set 6. LD analysis between contiguous SNPs with LOD ≥ 3.
Supplemental Data Set 7. Summarized information for GWA results.
Supplemental Data Set 8. Metabolic phenotype for the KO panel.
Supplemental File 1. Alignment used to produce the phylogenetic tree in Figure 7.
Acknowledgments
This research was supported by the Max Planck Society (Germany) and the Centre for Biosystems Genomics (The Netherlands). C.M.F. was supported by an Alexander von Humboldt Fellowship. We thank Roosa Laitinen, Detlef Weigel, and Elizabeth Haswell for kindly providing material for follow-up experiments on QTL validation. Ursula Krause, Marina Salles, and Daria Nitarska helped with KO growth and sample preparation at MPI, and Padraic Flood, Johanna Molenaar, and Natalia Carreño helped during plant harvest at Wageningen University. We thank Willem Kruijer for helpful discussion and comments on heritability.
AUTHOR CONTRIBUTIONS
J.J.B.K., M.S., and R.S. conceived the project. C.M.F. and R.K. performed GWAS analysis and candidate gene selection. C.M.F. performed experiments with additional KO lines to support initial validations from KO panel, executed further analyses for determining causal mutations on cis-QTL, and analyzed all data. C.M.F. and M.A.L. performed KO screening for flowering time and seed abortion phenotypes. R.K. and F.F.M.B. grew and harvested samples from GWA population and KO panel. M.G.A. helped to obtained homozygous KO lines for follow-up experiments and performed starch and sugar analysis on MSL10 KO and OEX lines. B.E., M.H., and N.K. performed metabolite and enzyme activity assays. A.S. gave technical support. C.M.F., R.K., M.S., and J.J.B.K. wrote the manuscript. All authors drafted the manuscript and agreed with the last version of the manuscript.
Footnotes
Articles can be viewed without a subscription.
References
- Abel S., Savchenko T., Levy M. (2005). Genome-wide comparative analysis of the IQD gene families in Arabidopsis thaliana and Oryza sativa. BMC Evol. Biol. 5: 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn E.R., Cho H.K., Pai H.S. (2013). The forkhead-associated domain 2 (FHA2) in Arabidopsis plays a role in plant fertility by regulating stamen development. Planta 237: 1015–1023. [DOI] [PubMed] [Google Scholar]
- Alonso-Blanco C., Aarts M.G.M., Bentsink L., Keurentjes J.J.B., Reymond M., Vreugdenhil D., Koornneef M. (2009). What has natural variation taught us about plant development, physiology, and adaptation? Plant Cell 21: 1877–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Blanco C., et al. ; 1001 Genomes Consortium (2016). 1,135 Genomes reveal the global pattern of polymorphism in Arabidopsis thaliana. Cell 166: 481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alseekh S., et al. (2015). Identification and mode of inheritance of quantitative trait loci for secondary metabolite abundance in tomato. Plant Cell 27: 485–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziata M.G., Apelt F., Carillo P., Krause U., Feil R., Mengin V., Lauxmann M.A., Köhl K., Nikoloski Z., Stitt M., Lunn J.E. (2017). Getting back to nature: a reality check for experiments in controlled environments. J. Exp. Bot. 68: 4463–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrivault S., Guenther M., Florian A., Encke B., Feil R., Vosloh D., Lunn J.E., Sulpice R., Fernie A.R., Stitt M., Schulze W.X. (2014). Dissecting the subcellular compartmentation of proteins and metabolites in arabidopsis leaves using non-aqueous fractionation. Mol. Cell. Proteomics 13: 2246–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwell S., et al. (2010). Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465: 627–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballicora M.A., Iglesias A.A., Preiss J. (2004). ADP-glucose pyrophosphorylase: A regulatory enzyme for plant starch synthesis. Photosynth. Res. 79: 1–24. [DOI] [PubMed] [Google Scholar]
- Baxter C.J., Sabar M., Quick W.P., Sweetlove L.J. (2005). Comparison of changes in fruit gene expression in tomato introgression lines provides evidence of genome-wide transcriptional changes and reveals links to mapped QTLs and described traits. J. Exp. Bot. 56: 1591–1604. [DOI] [PubMed] [Google Scholar]
- Baxter I., Brazelton J.N., Yu D., Huang Y.S., Lahner B., Yakubova E., Li Y., Bergelson J., Borevitz J.O., Nordborg M., Vitek O., Salt D.E. (2010). A coastal cline in sodium accumulation in Arabidopsis thaliana is driven by natural variation of the sodium transporter AtHKT1;1. PLoS Genet. 6: e1001193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57: 289–300. [Google Scholar]
- Bergelson J., Roux F. (2010). Towards identifying genes underlying ecologically relevant traits in Arabidopsis thaliana. Nat. Rev. Genet. 11: 867–879. [DOI] [PubMed] [Google Scholar]
- Biais B., et al. (2014). Remarkable reproducibility of enzyme activity profiles in tomato fruits grown under contrasting environments provides a roadmap for studies of fruit metabolism. Plant Physiol. 164: 1204–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachi B., Morris G.P., Borevitz J.O. (2011). Genome-wide association studies in plants: the missing heritability is in the field. Genome Biol. 12: 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M.M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248–254. [DOI] [PubMed] [Google Scholar]
- Brotman Y., Riewe D., Lisec J., Meyer R.C., Willmitzer L., Altmann T. (2011). Identification of enzymatic and regulatory genes of plant metabolism through QTL analysis in Arabidopsis. J. Plant Physiol. 168: 1387–1394. [DOI] [PubMed] [Google Scholar]
- Burow M., Halkier B.A., Kliebenstein D.J. (2010). Regulatory networks of glucosinolates shape Arabidopsis thaliana fitness. Curr. Opin. Plant Biol. 13: 348–353. [DOI] [PubMed] [Google Scholar]
- Bush W.S., Moore J.H. (2012). Chapter 11: Genome-wide association studies. PLOS Comput. Biol. 8: e1002822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causse M., Rocher J.P., Henry A.M., Charcosset A., Prioul J.L., Devienne D. (1995a). Genetic dissection of the relationship between carbon metabolism and early growth in maize with emphasis on key enzyme loci. Mol. Breed. 1: 259–272. [Google Scholar]
- Causse M., Rocher J.P., Henry A.M., Charcosset A., Prioul J.L., Devienne D. (1995b). Genetic dissection of the relationship between carbon metabolism and early growth in maize, with emphasis on key-enzyme loci. Mol. Breed. 1: 259–272. [Google Scholar]
- Causse M., Saliba-Colombani V., Lecomte L., Duffé P., Rousselle P., Buret M. (2002). QTL analysis of fruit quality in fresh market tomato: a few chromosome regions control the variation of sensory and instrumental traits. J. Exp. Bot. 53: 2089–2098. [DOI] [PubMed] [Google Scholar]
- Causse M., Duffe P., Gomez M.C., Buret M., Damidaux R., Zamir D., Gur A., Chevalier C., Lemaire-Chamley M., Rothan C. (2004). A genetic map of candidate genes and QTLs involved in tomato fruit size and composition. J. Exp. Bot. 55: 1671–1685. [DOI] [PubMed] [Google Scholar]
- Cavalier D.M., Lerouxel O., Neumetzler L., Yamauchi K., Reinecke A., Freshour G., Zabotina O.A., Hahn M.G., Burgert I., Pauly M., Raikhel N.V., Keegstra K. (2008). Disrupting two Arabidopsis thaliana xylosyltransferase genes results in plants deficient in xyloglucan, a major primary cell wall component. Plant Cell 20: 1519–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaiwanon J., Wang W., Zhu J.Y., Oh E., Wang Z.Y. (2016). Information integration and communication in plant growth regulation. Cell 164: 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan E.K., Rowe H.C., Kliebenstein D.J. (2010a). Understanding the evolution of defense metabolites in Arabidopsis thaliana using genome-wide association mapping. Genetics 185: 991–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan E.K., Rowe H.C., Hansen B.G., Kliebenstein D.J. (2010b). The complex genetic architecture of the metabolome. PLoS Genet. 6: e1001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan E.K., Rowe H.C., Corwin J.A., Joseph B., Kliebenstein D.J. (2011). Combining genome-wide association mapping and transcriptional networks to identify novel genes controlling glucosinolates in Arabidopsis thaliana. PLoS Biol. 9: e1001125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross J.M., von Korff M., Altmann T., Bartzetko L., Sulpice R., Gibon Y., Palacios N., Stitt M. (2006). Variation of enzyme activities and metabolite levels in 24 Arabidopsis accessions growing in carbon-limited conditions. Plant Physiol. 142: 1574–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rienzo J.A., Casanoves F., Balzarini M.G., Gonzalez L., Tablada M., Robledo C.W. (2011). InfoStat v2014. http://www.infostat.com.ar.
- Dubin M.J., et al. (2015). DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. eLife 4: e05255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupressoir A., Morel A.P., Barbot W., Loireau M.P., Corbo L., Heidmann T. (2001). Identification of four families of yCCR4- and Mg2+-dependent endonuclease-related proteins in higher eukaryotes, and characterization of orthologs of yCCR4 with a conserved leucine-rich repeat essential for hCAF1/hPOP2 binding. BMC Genomics 2: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell D.A., Thomas S. (1995). Physiological control of metabolic flux: the requirement for multisite modulation. Biochem. J. 311: 35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernie A.R., Stitt M. (2012). On the discordance of metabolomics with proteomics and transcriptomics: coping with increasing complexity in logic, chemistry, and network interactions scientific correspondence. Plant Physiol. 158: 1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint-Garcia S.A., Thuillet A.C., Yu J., Pressoir G., Romero S.M., Mitchell S.E., Doebley J., Kresovich S., Goodman M.M., Buckler E.S. (2005). Maize association population: a high-resolution platform for quantitative trait locus dissection. Plant J. 44: 1054–1064. [DOI] [PubMed] [Google Scholar]
- Fridman E., Liu Y.S., Carmel-Goren L., Gur A., Shoresh M., Pleban T., Eshed Y., Zamir D. (2002). Two tightly linked QTLs modify tomato sugar content via different physiological pathways. Mol. Genet. Genomics 266: 821–826. [DOI] [PubMed] [Google Scholar]
- Fu J., et al. (2009). System-wide molecular evidence for phenotypic buffering in Arabidopsis. Nat. Genet. 41: 166–167. [DOI] [PubMed] [Google Scholar]
- Geronimo J., Beevers H. (1964). Effects of aging and temperature on respiratory metabolism of green leaves. Plant Physiol. 39: 786–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibon Y., Blasing O.E., Palacios-Rojas N., Pankovic D., Hendriks J.H., Fisahn J., Hohne M., Gunther M., Stitt M. (2004a). Adjustment of diurnal starch turnover to short days: depletion of sugar during the night leads to a temporary inhibition of carbohydrate utilization, accumulation of sugars and post-translational activation of ADP-glucose pyrophosphorylase in the following light period. Plant J. 39: 847–862. [DOI] [PubMed] [Google Scholar]
- Gibon Y., Blaesing O.E., Hannemann J., Carillo P., Höhne M., Hendriks J.H., Palacios N., Cross J., Selbig J., Stitt M. (2004b). A Robot-based platform to measure multiple enzyme activities in Arabidopsis using a set of cycling assays: comparison of changes of enzyme activities and transcript levels during diurnal cycles and in prolonged darkness. Plant Cell 16: 3304–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf A., Schlereth A., Stitt M., Smith A.M. (2010). Circadian control of carbohydrate availability for growth in Arabidopsis plants at night. Proc. Natl. Acad. Sci. USA 107: 9458–9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton E.S., Jensen G.S., Maksaev G., Katims A., Sherp A.M., Haswell E.S. (2015). Mechanosensitive channel MSL8 regulates osmotic forces during pollen hydration and germination. Science 350: 438–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy O.J., Vekemans X. (2002). SPAGEDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2: 618–620. [Google Scholar]
- Haswell E.S., Peyronnet R., Barbier-Brygoo H., Meyerowitz E.M., Frachisse J.M. (2008). Two MscS homologs provide mechanosensitive channel activities in the Arabidopsis root. Curr. Biol. 18: 730–734. [DOI] [PubMed] [Google Scholar]
- Horton M.W., et al. (2012). Genome-wide patterns of genetic variation in worldwide Arabidopsis thaliana accessions from the RegMap panel. Nat. Genet. 44: 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara H., Moraes T.A., Pyl E.T., Schulze W.X., Obata T., Scheffel A., Fernie A.R., Sulpice R., Stitt M. (2017). Growth rate correlates negatively with protein turnover in Arabidopsis accessions. Plant J. 91: 416–429. [DOI] [PubMed] [Google Scholar]
- Jin L., Chakraborty R. (1994). Estimation of genetic distance and coefficient of gene diversity from single-probe multilocus DNA fingerprinting data. Mol. Biol. Evol. 11: 120–127. [DOI] [PubMed] [Google Scholar]
- Jombart T., Devillard S., Balloux F. (2010). Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacser H., Burns J.A. (1973). The control of flux. Symp. Soc. Exp. Biol. 27: 65–104. [PubMed] [Google Scholar]
- Kang H.M., Zaitlen N.A., Wade C.M., Kirby A., Heckerman D., Daly M.J., Eskin E. (2008). Efficient control of population structure in model organism association mapping. Genetics 178: 1709–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H.M., Sul J.H., Service S.K., Zaitlen N.A., Kong S.Y., Freimer N.B., Sabatti C., Eskin E. (2010). Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42: 348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Misawa K., Kuma K., Miyata T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30: 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keurentjes J.J., Sulpice R., Gibon Y., Steinhauser M.C., Fu J., Koornneef M., Stitt M., Vreugdenhil D. (2008). Integrative analyses of genetic variation in enzyme activities of primary carbohydrate metabolism reveal distinct modes of regulation in Arabidopsis thaliana. Genome Biol. 9: R129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keurentjes J.J., Fu J., de Vos C.H., Lommen A., Hall R.D., Bino R.J., van der Plas L.H., Jansen R.C., Vreugdenhil D., Koornneef M. (2006). The genetics of plant metabolism. Nat. Genet. 38: 842–849. [DOI] [PubMed] [Google Scholar]
- Kliebenstein D.J., Gershenzon J., Mitchell-Olds T. (2001a). Comparative quantitative trait loci mapping of aliphatic, indolic and benzylic glucosinolate production in Arabidopsis thaliana leaves and seeds. Genetics 159: 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliebenstein D.J., Lambrix V.M., Reichelt M., Gershenzon J., Mitchell-Olds T. (2001b). Gene duplication in the diversification of secondary metabolism: tandem 2-oxoglutarate-dependent dioxygenases control glucosinolate biosynthesis in Arabidopsis. Plant Cell 13: 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooke R., Keurentjes J.J.B. (2012). Multi-dimensional regulation of metabolic networks shaping plant development and performance. J. Exp. Bot. 63: 3353–3365. [DOI] [PubMed] [Google Scholar]
- Kooke R., Kruijer W., Bours R., Becker F., Kuhn A., van de Geest H., Buntjer J., Doeswijk T., Guerra J., Bouwmeester H., Vreugdenhil D., Keurentjes J.J. (2016). Genome-wide association mapping and genomic prediction elucidate the genetic architecture of morphological traits in Arabidopsis. Plant Physiol. 170: 2187–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte A., Farlow A. (2013). The advantages and limitations of trait analysis with GWAS: a review. Plant Methods 9: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger S., Giavalisco P., Krall L., Steinhauser M.C., Büssis D., Usadel B., Flügge U.I., Fernie A.R., Willmitzer L., Steinhauser D. (2011). A topological map of the compartmentalized Arabidopsis thaliana leaf metabolome. PLoS One 6: e17806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruijer W., Boer M.P., Malosetti M., Flood P.J., Engel B., Kooke R., Keurentjes J.J., van Eeuwijk F.A. (2015). Marker-based estimation of heritability in immortal populations. Genetics 199: 379–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz H.H., Zamani-Nour S., Häusler R.E., Ludewig K., Schroeder J.I., Malinova I., Fettke J., Flügge U.I., Gierth M. (2014). Loss of cytosolic phosphoglucose isomerase affects carbohydrate metabolism in leaves and is essential for fertility of Arabidopsis. Plant Physiol. 166: 753–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauxmann M.A., et al. (2016). Reproductive failure in Arabidopsis thaliana under transient carbohydrate limitation: flowers and very young siliques are jettisoned and the meristem is maintained to allow successful resumption of reproductive growth. Plant Cell Environ. 39: 745–767. [DOI] [PubMed] [Google Scholar]
- Lee C.P., Maksaev G., Jensen G.S., Murcha M.W., Wilson M.E., Fricker M., Hell R., Haswell E.S., Millar A.H., Sweetlove L.J. (2016). MSL1 is a mechanosensitive ion channel that dissipates mitochondrial membrane potential and maintains redox homeostasis in mitochondria during abiotic stress. Plant J. 88: 809–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leskow C.C., et al. (2016). Allelic differences in a vacuolar invertase affect Arabidopsis growth at early plant development. J. Exp. Bot. 67: 4091–4103. [DOI] [PubMed] [Google Scholar]
- Limami A.M., Rouillon C., Glevarec G., Gallais A., Hirel B. (2002). Genetic and physiological analysis of germination efficiency in maize in relation to nitrogen metabolism reveals the importance of cytosolic glutamine synthetase. Plant Physiol. 130: 1860–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipka A.E., Tian F., Wang Q., Peiffer J., Li M., Bradbury P.J., Gore M.A., Buckler E.S., Zhang Z. (2012). GAPIT: genome association and prediction integrated tool. Bioinformatics 28: 2397–2399. [DOI] [PubMed] [Google Scholar]
- Lisec J., Meyer R.C., Steinfath M., Redestig H., Becher M., Witucka-Wall H., Fiehn O., Torjek O., Selbig J., Altmann T., Willmitzer L. (2008). Identification of metabolic and biomass QTL in Arabidopsis thaliana in a parallel analysis of RIL and IL populations. Plant J. 53: 960–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K., Muse S.V. (2005). PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21: 2128–2129. [DOI] [PubMed] [Google Scholar]
- Liu N., Xue Y., Guo Z., Li W., Tang J. (2016). Genome-wide association study identifies candidate genes for starch content regulation in maize kernels. Front. Plant Sci. 7: 1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loiselle B.A., Sork V.L., Nason J., Graham C. (1995). Spatial genetic-structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am. J. Bot. 82: 1420–1425. [Google Scholar]
- Lu H., Liu Y., Greenberg J.T. (2005). Structure-function analysis of the plasma membrane- localized Arabidopsis defense component ACD6. Plant J. 44: 798–809. [DOI] [PubMed] [Google Scholar]
- Mähler N., Wang J., Terebieniec B.K., Ingvarsson P.K., Street N.R., Hvidsten T.R. (2017). Gene co-expression network connectivity is an important determinant of selective constraint. PLoS Genet. 13: e1006402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksaev G., Haswell E.S. (2012). MscS-Like10 is a stretch-activated ion channel from Arabidopsis thaliana with a preference for anions. Proc. Natl. Acad. Sci. USA 109: 19015–19020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunath S., Lee C.-H.K., VanWinkle P., Bailey-Serres J. (1998). Molecular and biochemical characterization of cytosolic phosphoglucomutase in maize. Expression during development and in response to oxygen deprivation. Plant Physiol. 117: 997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matros A., et al. (2017). Genome-metabolite associations revealed low heritability, high genetic complexity, and causal relations for leaf metabolites in winter wheat (Triticum aestivum). J. Exp. Bot. 68: 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijón M., Satbhai S.B., Tsuchimatsu T., Busch W. (2014). Genome-wide association study using cellular traits identifies a new regulator of root development in Arabidopsis. Nat. Genet. 46: 77–81. [DOI] [PubMed] [Google Scholar]
- Meng M., Wilczynska M., Kleczkowski L.A. (2008). Molecular and kinetic characterization of two UDP-glucose pyrophosphorylases, products of distinct genes, from Arabidopsis. Biochim. Biophys. Acta 1784: 967–972. [DOI] [PubMed] [Google Scholar]
- Meng M., Geisler M., Johansson H., Harholt J., Scheller H.V., Mellerowicz E.J., Kleczkowski L.A. (2009). UDP-glucose pyrophosphorylase is not rate limiting, but is essential in Arabidopsis. Plant Cell Physiol. 50: 998–1011. [DOI] [PubMed] [Google Scholar]
- Meyer R.C., Steinfath M., Lisec J., Becher M., Witucka-Wall H., Törjék O., Fiehn O., Eckardt A., Willmitzer L., Selbig J., Altmann T. (2007). The metabolic signature related to high plant growth rate in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 104: 4759–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell-Olds T., Pedersen D. (1998). The molecular basis of quantitative genetic variation in central and secondary metabolism in Arabidopsis. Genetics 149: 739–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes-Nesi A., Carrari F., Gibon Y., Sulpice R., Lytovchenko A., Fisahn J., Graham J., Ratcliffe R.G., Sweetlove L.J., Fernie A.R. (2007). Deficiency of mitochondrial fumarase activity in tomato plants impairs photosynthesis via an effect on stomatal function. Plant J. 50: 1093–1106. [DOI] [PubMed] [Google Scholar]
- Oraguzie N.C. (2007). Association Mapping in Plants. (New York: Springer; ). [Google Scholar]
- Park J.I., Ishimizu T., Suwabe K., Sudo K., Masuko H., Hakozaki H., Nou I.S., Suzuki G., Watanabe M. (2010). UDP-glucose pyrophosphorylase is rate limiting in vegetative and reproductive phases in Arabidopsis thaliana. Plant Cell Physiol. 51: 981–996. [DOI] [PubMed] [Google Scholar]
- Park Y.B., Cosgrove D.J. (2012). Changes in cell wall biomechanical properties in the xyloglucan-deficient xxt1/xxt2 mutant of Arabidopsis. Plant Physiol. 158: 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P., et al. (2013). A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature 494: 266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piques M., Schulze W.X., Höhne M., Usadel B., Gibon Y., Rohwer J., Stitt M. (2009). Ribosome and transcript copy numbers, polysome occupancy and enzyme dynamics in Arabidopsis. Mol. Syst. Biol. 5: 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pracharoenwattana I., Zhou W., Keech O., Francisco P.B., Udomchalothorn T., Tschoep H., Stitt M., Gibon Y., Smith S.M. (2010). Arabidopsis has a cytosolic fumarase required for the massive allocation of photosynthate into fumaric acid and for rapid plant growth on high nitrogen. Plant J. 62: 785–795. [DOI] [PubMed] [Google Scholar]
- Prioul J.L., Pelleschi S., Sene M., Thevenot C., Causse M., de Vienne D., Leonardi A. (1999). From QTLs for enzyme activity to candidate genes in maize. J. Exp. Bot. 50: 1281–1288. [Google Scholar]
- R Core Team (2016). R: A Language and Environment for Statistical Computing. (Vienna, Austria: R Foundation for Statistical Computing; ). [Google Scholar]
- Rate D.N., Cuenca J.V., Bowman G.R., Guttman D.S., Greenberg J.T. (1999). The gain-of-function Arabidopsis acd6 mutant reveals novel regulation and function of the salicylic acid signaling pathway in controlling cell death, defenses, and cell growth. Plant Cell 11: 1695–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedelsheimer C., Lisec J., Czedik-Eysenberg A., Sulpice R., Flis A., Grieder C., Altmann T., Stitt M., Willmitzer L., Melchinger A.E. (2012). Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize. Proc. Natl. Acad. Sci. USA 109: 8872–8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riewe D., Jeon H.J., Lisec J., Heuermann M.C., Schmeichel J., Seyfarth M., Meyer R.C., Willmitzer L., Altmann T. (2016). A naturally occurring promoter polymorphism of the Arabidopsis FUM2 gene causes expression variation and is associated with metabolic and growth traits. Plant J. 88: 826–838. [DOI] [PubMed] [Google Scholar]
- Rozas J., Sánchez-DelBarrio J.C., Messeguer X., Rozas R. (2003). DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496–2497. [DOI] [PubMed] [Google Scholar]
- Rubin G., Tohge T., Matsuda F., Saito K., Scheible W.R. (2009). Members of the LBD family of transcription factors repress anthocyanin synthesis and affect additional nitrogen responses in Arabidopsis. Plant Cell 21: 3567–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibe R. (1987). NADP+-malate dehydrogenase in C-3-plants - Regulation and role of a light-activated enzyme. Physiol. Plant. 71: 393–400. [Google Scholar]
- Schubert O.T., Gillet L.C., Collins B.C., Navarro P., Rosenberger G., Wolski W.E., Lam H., Amodei D., Mallick P., MacLean B., Aebersold R. (2015). Building high-quality assay libraries for targeted analysis of SWATH MS data. Nat. Protoc. 10: 426–441. [DOI] [PubMed] [Google Scholar]
- Schuster S., Dandekar T., Fell D.A. (1999). Detection of elementary flux modes in biochemical networks: a promising tool for pathway analysis and metabolic engineering. Trends Biotechnol. 17: 53–60. [DOI] [PubMed] [Google Scholar]
- Selinski J., König N., Wellmeyer B., Hanke G.T., Linke V., Neuhaus H.E., Scheibe R. (2014). The plastid-localized NAD-dependent malate dehydrogenase is crucial for energy homeostasis in developing Arabidopsis thaliana seeds. Mol. Plant 7: 170–186. [DOI] [PubMed] [Google Scholar]
- Sergeeva L.I., Vonk J., Keurentjes J.J., van der Plas L.H., Koornneef M., Vreugdenhil D. (2004). Histochemical analysis reveals organ-specific quantitative trait loci for enzyme activities in Arabidopsis. Plant Physiol. 134: 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeeva L.I., Keurentjes J.J., Bentsink L., Vonk J., van der Plas L.H., Koornneef M., Vreugdenhil D. (2006). Vacuolar invertase regulates elongation of Arabidopsis thaliana roots as revealed by QTL and mutant analysis. Proc. Natl. Acad. Sci. USA 103: 2994–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J.-H., Blay S., McNeney B., Graham J. (2006). LDheatmap: An R function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. J. Stat. Softw. 16: Code Snippet 3. [Google Scholar]
- Silverstone A.L., Ciampaglio C.N., Sun T. (1998). The Arabidopsis RGA gene encodes a transcriptional regulator repressing the gibberellin signal transduction pathway. Plant Cell 10: 155–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhauser M.C., Steinhauser D., Gibon Y., Bolger M., Arrivault S., Usadel B., Zamir D., Fernie A.R., Stitt M. (2011). Identification of enzyme activity quantitative trait loci in a Solanum lycopersicum x Solanum pennellii introgression line population. Plant Physiol. 157: 998–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt M., Sonnewald U. (1995). Regulation of metabolism in transgenic plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 46: 341–368. [Google Scholar]
- Stitt M., Zeeman S.C. (2012). Starch turnover: pathways, regulation and role in growth. Curr. Opin. Plant Biol. 15: 282–292. [DOI] [PubMed] [Google Scholar]
- Stitt M., Gibon Y. (2014). Why measure enzyme activities in the era of systems biology? Trends Plant Sci. 19: 256–265. [DOI] [PubMed] [Google Scholar]
- Stitt M., Lunn J., Usadel B. (2010). Arabidopsis and primary photosynthetic metabolism - more than the icing on the cake. Plant J. 61: 1067–1091. [DOI] [PubMed] [Google Scholar]
- Sulpice R., Tschoep H., VON Korff M., Büssis D., Usadel B., Höhne M., Witucka-Wall H., Altmann T., Stitt M., Gibon Y. (2007). Description and applications of a rapid and sensitive non-radioactive microplate-based assay for maximum and initial activity of D-ribulose-1,5-bisphosphate carboxylase/oxygenase. Plant Cell Environ. 30: 1163–1175. [DOI] [PubMed] [Google Scholar]
- Sulpice R., Flis A., Ivakov A.A., Apelt F., Krohn N., Encke B., Abel C., Feil R., Lunn J.E., Stitt M. (2014). Arabidopsis coordinates the diurnal regulation of carbon allocation and growth across a wide range of photoperiods. Mol. Plant 7: 137–155. [DOI] [PubMed] [Google Scholar]
- Sulpice R., Nikoloski Z., Tschoep H., Antonio C., Kleessen S., Larhlimi A., Selbig J., Ishihara H., Gibon Y., Fernie A.R., Stitt M. (2013). Impact of the carbon and nitrogen supply on relationships and connectivity between metabolism and biomass in a broad panel of Arabidopsis accessions. Plant Physiol. 162: 347–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulpice R., et al. (2010). Network analysis of enzyme activities and metabolite levels and their relationship to biomass in a large panel of Arabidopsis accessions. Plant Cell 22: 2872–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulpice R., et al. (2009). Starch as a major integrator in the regulation of plant growth. Proc. Natl. Acad. Sci. USA 106: 10348–10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y., Arae T., Green P.J., Yamaguchi J., Chiba Y. (2015). AtCCR4a and AtCCR4b are involved in determining the poly(A) length of granule-bound starch synthase 1 transcript and modulating sucrose and starch metabolism in Arabidopsis thaliana. Plant Cell Physiol. 56: 863–874. [DOI] [PubMed] [Google Scholar]
- Tajima F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenot C., Simond-Côte E., Reyss A., Manicacci D., Trouverie J., Le Guilloux M., Ginhoux V., Sidicina F., Prioul J.L. (2005). QTLs for enzyme activities and soluble carbohydrates involved in starch accumulation during grain filling in maize. J. Exp. Bot. 56: 945–958. [DOI] [PubMed] [Google Scholar]
- Thimm O., Blasing O., Gibon Y., Nagel A., Meyer S., Kruger P., Selbig J., Muller L.A., Rhee S.Y., Stitt M. (2004). MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37: 914–939. [DOI] [PubMed] [Google Scholar]
- Todesco M., et al. (2010). Natural allelic variation underlying a major fitness trade-off in Arabidopsis thaliana. Nature 465: 632–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toubiana D., Semel Y., Tohge T., Beleggia R., Cattivelli L., Rosental L., Nikoloski Z., Zamir D., Fernie A.R., Fait A. (2012). Metabolic profiling of a mapping population exposes new insights in the regulation of seed metabolism and seed, fruit, and plant relations. PLoS Genet. 8: e1002612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toubiana D., Xue W., Zhang N., Kremling K., Gur A., Pilosof S., Gibon Y., Stitt M., Buckler E.S., Fernie A.R., Fait A. (2016). Correlation-based network analysis of metabolite and enzyme profiles reveals a role of citrate biosynthesis in modulating N and C metabolism in Zea mays. Front. Plant Sci. 7: 1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler A.L., Carter G.W. (2017). Genetic interactions improve models of quantitative traits. Nat. Genet. 49: 486–488. [DOI] [PubMed] [Google Scholar]
- Tyler A.L., Lu W., Hendrick J.J., Philip V.M., Carter G.W. (2013). CAPE: an R package for combined analysis of pleiotropy and epistasis. PLOS Comput. Biol. 9: e1003270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymowska-Lalanne Z., Kreis M. (1998). Expression of the Arabidopsis thaliana invertase gene family. Planta 207: 259–265. [DOI] [PubMed] [Google Scholar]
- VanRaden P.M. (2008). Efficient methods to compute genomic predictions. J. Dairy Sci. 91: 4414–4423. [DOI] [PubMed] [Google Scholar]
- Veley K.M., Maksaev G., Frick E.M., January E., Kloepper S.C., Haswell E.S. (2014). Arabidopsis MSL10 has a regulated cell death signaling activity that is separable from its mechanosensitive ion channel activity. Plant Cell 26: 3115–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verslues P.E., Lasky J.R., Juenger T.E., Liu T.W., Kumar M.N. (2014). Genome-wide association mapping combined with reverse genetics identifies new effectors of low water potential-induced proline accumulation in Arabidopsis. Plant Physiol. 164: 144–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeden N.F., Gottlieb L.D. (1982). Dissociation, reassociation, and purification of plastid and cytosolic phosphoglucose isomerase isozymes. Plant Physiol. 69: 717–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen W., Li D., Li X., Gao Y., Li W., Li H., Liu J., Liu H., Chen W., Luo J., Yan J. (2014). Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat. Commun. 5: 3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzell A.M., Rowe H.C., Hansen B.G., Ticconi C., Halkier B.A., Kliebenstein D.J. (2007). Linking metabolic QTLs with network and cis-eQTLs controlling biosynthetic pathways. PLoS Genet. 3: 1687–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M.E., Mixdorf M., Berg R.H., Haswell E.S. (2016). Plastid osmotic stress influences cell differentiation at the plant shoot apex. Development 143: 3382–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood A.R., et al. (2011). Allelic heterogeneity and more detailed analyses of known loci explain additional phenotypic variation and reveal complex patterns of association. Hum. Mol. Genet. 20: 4082–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S., Alseekh S., Cuadros-Inostroza A., Fusari C.M., Mutwil M., Kooke R., Keurentjes J., Fernie A.R., Willmitzer L., Brotman Y. (2016). Combined use of genome-wide association data and correlation networks unravels key regulators of primary metabolism in Arabidopsis thaliana. PLoS Genet. 12: e1006363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Z., Wang A., Yang G., Gao P., Zheng Z.L. (2009). The Arabidopsis A4 subfamily of lectin receptor kinases negatively regulates abscisic acid response in seed germination. Plant Physiol. 149: 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., Seaton D.D., Krahmer J., Halliday K.J. (2016). Photoreceptor effects on plant biomass, resource allocation, and metabolic state. Proc. Natl. Acad. Sci. USA 113: 7667–7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentella R., Zhang Z.L., Park M., Thomas S.G., Endo A., Murase K., Fleet C.M., Jikumaru Y., Nambara E., Kamiya Y., Sun T.P. (2007). Global analysis of della direct targets in early gibberellin signaling in Arabidopsis. Plant Cell 19: 3037–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N., Gibon Y., Gur A., Chen C., Lepak N., Höhne M., Zhang Z., Kroon D., Tschoep H., Stitt M., Buckler E. (2010). Fine quantitative trait loci mapping of carbon and nitrogen metabolism enzyme activities and seedling biomass in the maize IBM mapping population. Plant Physiol. 154: 1753–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N., et al. (2015). Genome-wide association of carbon and nitrogen metabolism in the maize nested association mapping population. Plant Physiol. 168: 575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]