

Abstract
Background
Infectious disease is the single greatest threat to taxa such as amphibians (chytrid fungus), bats (white nose syndrome), Tasmanian devils (devil facial tumor disease), and black-footed ferrets (canine distemper virus, plague). Although understanding the genetic basis to disease susceptibility is important for the long-term persistence of these groups, most research has been limited to major-histocompatibility and Toll-like receptor genes. To better understand the genetic basis of infectious disease susceptibility in a species of conservation concern, we sequenced all known/predicted immune response genes (i.e., the immunomes) in 16 Florida gopher tortoises, Gopherus polyphemus. All tortoises produced antibodies against Mycoplasma agassizii (an etiologic agent of infectious upper respiratory tract disease; URTD) and, at the time of sampling, either had (n = 10) or lacked (n = 6) clinical signs.
Results
We found several variants associated with URTD clinical status in complement and lectin genes, which may play a role in Mycoplasma immunity. Thirty-five genes deviated from neutrality according to Tajima’s D. These genes were enriched in functions relating to macromolecule and protein modifications, which are vital to immune system functioning.
Conclusions
These results are suggestive of genetic differences that might contribute to disease severity, a finding that is consistent with other mycoplasmal diseases. This has implications for management because tortoises across their range may possess genetic variation associated with a more severe response to URTD. More generally: 1) this approach demonstrates that a broader consideration of immune genes is better able to identify important variants, and; 2) this data pipeline can be adopted to identify alleles associated with disease susceptibility or resistance in other taxa, and therefore provide information on a population’s risk of succumbing to disease, inform translocations to increase genetic variation for disease resistance, and help to identify potential treatments.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4452-0) contains supplementary material, which is available to authorized users.
Keywords: Genome-wide association study, Target enrichment, Sequence capture, Next-generation sequencing, Immunogenetics, Gopherus polyphemus, Immunomes
Background
Several species of conservation concern are in direct danger of extinction from disease. For example, devil facial tumor disease, a contagious cancer, is driving Tasmanian devils (Sarcophilus harrisii) to extinction at an alarming rate [1]. Similarly, the last native black-footed ferret (Mustela nigripes) population crashed as a result of canine distemper disease. Disease and reintroduced populations are susceptible to plague [2]. Further millions of bats and amphibians have died as a result of white-nose syndrome and chytrid fungus, respectively [3, 4].
Some diseases are projected to spread as a result of changing land use and climate as well as through anthropogenic spread [5]. For example, ticks (Ixodes scapularis), and the associated pathogens they vector, are expected to spread north as a consequence of climate change [6]. Other examples of diseases predicted to spread include West Nile virus, sarcoptic mange, and cholera ([7–9], see [10] for review). Regardless of the driver, infectious diseases in plant and animal populations are spreading and threatening many species of conservation concern.
Infectious disease susceptibility in plants and animals may have a genetic basis, as changes in the coding portions of immune response genes can affect protein conformation and alter pathogen recognition and binding [11]. Most studies have focused on a handful of immune genes (e.g. MHC (major histocompatibility complex), TLR (Toll-like receptors)) although several hundred genes are involved in the immune response. Genetic variants underlying disease susceptibility throughout the genome may be identified by associating immunogenetic variation and disease clinical status in a genome-wide association study (GWAS). GWASs are commonly conducted with agricultural crops (e.g., [12]), model organisms (e.g., [13]), and humans (e.g., [14]) to look for genetic variation associated with disease susceptibility. For example, researchers found 18 candidate genes associated with resistance to head smut disease in corn, leading to improved corn cultivar selection in an important agricultural crop [12]. Similarly, GWAS work in humans has helped identify the mechanisms of hepatitis C infection [14]. In contrast, GWAS in wildlife populations has been very limited despite its tremendous potential to identify the genetic basis of disease resistance and susceptibility [15–20]. For example, investigators used restriction-site associated DNA sequencing (RADseq) to find associations between candidate loci and bacterial cold water disease resistance in rainbow and steelhead trout [17]. Seven single nucleotide polymorphisms (SNPs) were associated with survival status and/or time, which could presumably be used to increase fitness in aquaculture or wild stocks.
The gopher tortoise, Gopherus polyphemus, is a threatened tortoise that can contract a chronic, and occasionally fatal, infectious upper respiratory tract disease (URTD) caused by Mycoplasma agassizii [21]. There may be a genetic component to URTD susceptibility as mycoplasmas are widespread, but gopher tortoise die-offs attributable to URTD have been documented only in Georgia and Florida [22, 23]. Tortoise populations show genetic structure at microsatellite loci between URTD-positive and -negative regions, thus there may also be genetic differences in immune response genes [24].
The gopher tortoise is federally listed as threatened in the portion of its range west of the Mobile and Tombigbee Rivers in Alabama, and the eastern tortoises are candidates for listing on the Endangered Species Act [25]. As such, the tortoise has been the subject of many genetic studies using neutral genetic markers such as microsatellites to inform management (e.g., [24]), but no research has identified genetic variation associated with susceptibility or resistance to URTD. This is crucial because tortoise populations may have adequate levels of genetic variation based on neutral microsatellites, but may be depauperate in adaptive genetic variation that influences immune responses to infections such as URTD, and therefore, population viability. Herein, we identify synonymous and non-synonymous variants in hundreds of genes that influence immune responses. We examined which variants associate with URTD non-clinical and clinical gopher tortoises to better understand the genetic basis for susceptibility to URTD. We also evaluate population genetic diversity, differentiation, and admixture among individuals from three populations in Florida. Finally, we assess which SNPs may be under selection, which genes may deviate from neutrality, and the specific functions of these non-neutral loci. We undertake this work using a data pipeline that can be used to identify important immune gene variants in a wide range of wildlife species in danger of extinction from disease.
Methods
Samples
Between 2002 and 2006, 11 populations in Florida were sampled annually as part of a long-term field study of gopher tortoise health [26]. Serum and erythrocytes were collected from all animals, and red blood cells were stored at − 80 °C until deoxyribonucleic acid (DNA) extraction. We selected 16 gopher tortoises representing two clinical states (tortoises with and without clinical signs of URTD; Table 1) from 3 populations (Cecil Field/Branan Field Wildlife and Environmental Area (CF), Fort Cooper State Park (FC), and Perry Oldenburg Wildlife and Environmental Area (OLD); Fig. 1) that had documented URTD. The sites chosen represented populations at different transition stages of URTD. The FC site has been studied since 2003, and from 2003 to 2005, seroprevalence to M. agassizii was extremely low, and mortality events and observed clinical disease were rare. However, in 2006, increases in seroprevalence, clinical signs, and mortality events were observed, indicating that the FC site had transitioned from an M. agassizii URTD-negative site to an acute epizootic site. Prior to 2003, the seroprevalence of M. agassizii at the CF site ranged from 0 to 20%. In 2003, dramatic increases were seen in seroprevalence (77% positive), tortoises exhibited a nasal discharge (33%), and tortoises (58%) positive by PCR for M. agassizii. Mortality events were observed, seroprevalence remained high, but clinical disease expression and severity decreased over time, suggesting that the CF site had transitioned from an acute epizootic site to an endemically stable site. The OLD site was endemically stable but had experienced a large-scale die-off circa 1996–1998 attributed to URTD [26]. In 2006, a sharp increase in seroconversion was observed, with an increase in clinical signs and slight increases in mortality events, supporting the concept that the OLD population was undergoing recrudescence of mycoplasmal URTD. All 16 tortoises in the 2 clinical states were observed more than once between 2002 and 2006 and had a least one positive enzyme-linked immunosorbent assay indicating active production of antibodies against M. agassizii [27]. Clinical tortoises had at least one incident of mild to severe nasal discharge. Because tortoises with URTD have intermittent expression of clinical signs, an important caveat is that tortoises were observed only once for several hours each year at time of capture, and therefore we cannot rule out the possibility that animals without clinical signs had previously been clinically ill. In experimental infections an individual can show clinical signs one day, be subclinical, and then present again with clinical signs [21]. However, at each time of capture, non-clinical tortoises were never observed with nasal discharge, an important diagnostic clinical sign of URTD.
Table 1.
Description of Gopherus polyphemus samples by clinical status and diploid genotypes at several bases along the gene LOC101950941 on NCBI contig NW_007281632.1
| Sample | Clinical status | NW_007281632.1 Bases | ||||
|---|---|---|---|---|---|---|
| 53,575 | 53,576 | 53,577 | 53,578 | 53,579 | ||
| FC19 | Non-clin | A,A | A,A | T,T | A,A | AG,AG |
| OLD77 | Non-clin | A,A | A,A | T,T | A,G | AG,AG |
| CF53 | Non-clin | A,A | A,A | T,T | A,A | AG,AG |
| CF69 | Non-clin | A,A | AT,AT | G,G | A,A | A,A |
| FC58 | Non-clin | A,A | A,AT | G,T | A,G | A,AG |
| OLD106 | Non-clin | A,A | AT,AT | G,G | A,A | A,A |
| CF72 | Clin | A,A | AT,AT | G,T | A,A | A,A |
| CF80 | Clin | A,A | AT,AT | G,G | A,A | A,A |
| CF90 | Clin | A,A | AT,AT | G,G | A,A | A,A |
| CF219 | Clin | A,A | AT,AT | G,G | A,A | A,A |
| FC13 | Clin | A,A | AT,AT | G,G | A,A | A,A |
| FC15 | Clin | A,A | AT,AT | G,G | A,A | A,A |
| FC47 | Clin | A,A | AT,AT | G,G | A,A | A,A |
| OLD65 | Clin | A,A | AT,AT | G,G | A,A | A,A |
| OLD92 | Clin | A,A | AT,AT | G,G | A,A | A,A |
| OLD107 | Clin | A,A | AT,AT | G,G | A,A | A,A |
Non-clin for tortoises that were never observed with nasal discharge during the duration of the field study and Clin for tortoises that had at least one incident of mild to severe nasal discharge
CF Cecil Field, FC Fort Cooper, and OLD Oldenburg
Fig. 1.
Location of Florida study populations. CF for Cecil Field/Branan Field Wildlife and Environmental Area, FC for Fort Cooper State Park, and OLD for Perry Oldenburg Wildlife and Environmental Area
Target enrichment
We used a next-generation sequencing approach and previously developed ribonucleic acid (RNA) baits [28, 29] from MYcroarray Inc. (Ann Arbor, MI, USA) in an in-solution hybridization experiment to capture the immunomes of 16 gopher tortoises. Briefly, we used the GO2TR workflow [24] to design a target region of exons from Chrysemys picta bellii (western painted turtle) genome [25] relating to the gene ontology term “immune response” to create 120-bp RNA baits. We followed the steps of a previous immunome study [28], except we created libraries using Bioo Scientific Nextflex Pre-and Post-Capture Combo Kit-Set A (catalog no. 5144–51, Bioo Scientific Corp., Austin, TX, USA) and used Bioo Scientific reagents and protocols for target enrichment.
Data analysis
We used the same analysis pipeline of [28] to call SNPs and indels and used those previously identified variants for variant quality score filtering. Briefly, we performed adapter and quality trimming with TRIMMOMATIC v0.32 [30], merged overlapping paired-end reads with BBMerge v5.4 (https://sourceforge.net/projects/bbmap/), aligned merged and unpaired reads and paired reads separately to the C. p. bellii 3.0.3 genome [31] using BWA-MEM v0.7.12 [32], then less stringently with STAMPY v1.0.23 [33], and followed the Genome Analysis Toolkit’s v3.3.0 [34] best practices for variant calling using the Haplotype Caller. For population genomic analyses, we used the same programs and steps as [28] except we included di- (n = 16,169), tri- (n = 651), and tetra-allelic (n = 1) SNP loci. To determine what polymorphic variants were under selection, we used BayeScan v2.1 [35], an FST outlier test, and used a false discovery rate [36] of 0.1.
We calculated Tajima’s D [37] for each population separately using VariScan [38] for all gene regions (i.e., loci) that were at least 60 bp and had reads at ≥20X coverage. We used a variable cutoff for significant negative and positive values of Tajima’s D based on the empirical P value calculated for each population and locus combination as follows: first we simulated extreme values of Tajima’s D using 1000 simulations for a population of five individuals with a single locus containing 1–138 segregating sites using the program ms [39]. Then we used custom R [40] code to calculate the P value of the empirical Tajima’s D given the simulated extreme values for a particular number of segregating sites and retained only loci with a false discovery rate less than 0.05. We performed functional enrichment analysis using FatiGO [41] implemented in Blast2GO v3.1 [42] to determine if non-neutral genes were enriched for specific biological functions relative to rest of genome.
We labeled alternate SNPs and indels as synonymous or non-synonymous using snpEff v4.0e [43]. We performed case/control association tests with PLINK v1.07 [44], analyzing SNPs and indels separately. We repeated association test using ROADTRIPS v2.0 [45], which controls for unknown population structure and individual relatedness.
Results
From two Illumina MiSeq runs using 75-bp paired end reads, we obtained 21.5 million reads that passed quality control, and each tortoise had 1.3 ± 0.2 (mean ± SD) million reads. Mean coverage was 47.5 ± 7.6 reads (Additional file 1: Figure S1 and Additional file 2: Figure S2). We targeted 632 immune genes and 5425 exons, and each tortoise had reads for 593.9 ± 3.5 genes and 4198 ± 82.3 exons. After variant filtering, imputation, and removing non-polymorphic loci, we identified 16,821 SNPs and 3226 indels.
SNP allelic richness was lowest in CF, intermediate in OLD, and highest in FC. Observed and expected heterozygosity was lowest in CF but equivalent in FC and OLD (Table 2). Pairwise FST values were 0.027 (CFxFC), 0.060 (CFxOLD), and − 0.003 (FCxOLD): low values were expected based on geographic proximities of the populations.
Table 2.
Estimates of genetic diversity for each tortoise population
| Site | AR | HO | HE |
|---|---|---|---|
| CF | 1.75a | 0.32a | 0.29a |
| FC | 1.78c | 0.33b | 0.30b |
| OLD | 1.77b | 0.34b | 0.30b |
AR allelic richness, HO and HE for observed and expected heterozygosity, respectively, CF Cecil Field, FC Fort Cooper, and OLD Oldenburg
Superscript letters for each column indicate differences at the 0.05 level using a post-hoc Tukey test. Shared letters within a column indicate no difference while unique letters indicate differences in levels
Population admixture inferred with SNPs using principle component analysis (PCA, Fig. 2) and STRUCTURE ([46], Fig. 3a) suggested two clusters: the first with CF and the second with FC and OLD mixed together. These results agreed with the optimal K value of two based on STRUCTURE HARVESTER [47]. There was no meaningful grouping of samples when we used clinical status to cluster individuals in STRUCTURE (Fig. 3b).
Fig. 2.
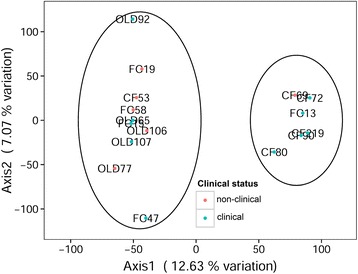
Principle component analysis for 16,821 all polymorphic SNPs. Circles indicate optimum clusters identified using STRUCTURE and STRUCTURE HARVESTER
Fig. 3.
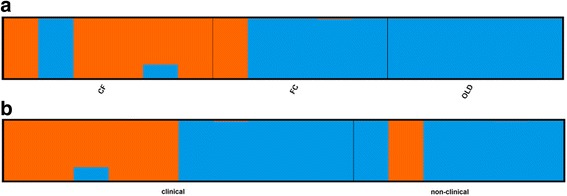
STRUCTURE plots using (a) populations and (b) clinical status with optimum number of clusters K = 2 determined by STRUCTURE HARVESTER. CF for Cecil Field/Branan Field Wildlife and Environmental Area, FC for Fort Cooper State Park, and OLD for Perry Oldenburg Wildlife and Environmental Area. Non-clinical tortoises were never observed with nasal discharge during the duration of the field study. Clinical tortoises had at least one incident of mild to severe nasal discharge
There were two SNPs but no indel loci putatively under selection. The first SNP, NW_007281406.1:2,298,828 was non-synonymous and present in the 1st codon position of the 354th amino acid in the gene IFIT1-like. The second SNP, NW_007322484.1:105, was synonymous and found in the gene IFI44-like.
There were 1556 polymorphic gene regions of which 70 deviated from neutrality. Tajima’s D was negative (i.e., ≤ − 1.56) for 27 regions suggesting either population expansion or positive selection but positive (≥ 2.06) for 43 regions suggesting either population bottlenecks, balancing selection, or strong population structure. These regions represented 35 genes (Additional file 3: Table S1). Thirteen genes had positive, 19 genes had negative, and 3 genes (IPO11, SIN3A, and TRAF6) had both positive and negative (i.e., multiple regions occurred in the same gene) Tajima’s D values.
Functional enrichment analysis of the 35 genes that contained regions deviating from neutrality showed overrepresentation of several gene functions, most notably the gene ontology terms: endoplasmic reticulum, cellular protein modification process, protein modification process, macromolecule modification, and transferase activity (Fig. 4).
Fig. 4.
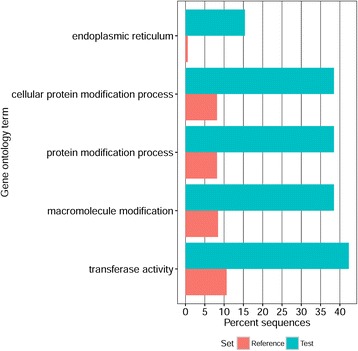
Functional enrichment analysis of gopher tortoise genes deviating from neutrality (i.e., Test) compared to the rest of the genome (i.e., Reference) based on percent of sequences with particular gene ontology terms
Association tests with PLINK revealed several SNPs and indels (Table 3, Additional file 4: Table S2) that were associated with clinical status but which were not significantly associated after correction for multiple tests. Variants that lack statistical significance are typical of GWAS studies, especially when variant effect sizes are small, unless hundreds or thousands of subjects are examined [48], therefore it is important to use less strict significance criteria and report the top ranking variants (e.g., [12]). Association tests with ROADTRIPS produced the same list of SNPs and indels as PLINK (data not shown). SNPs with the strongest associations were contained in genes such as Austrelaps superbus venom factor 1-like and LOC101950941. Indels with the strongest associations were in genes such as LOC101950941 and TNFRSF5-like.
Table 3.
Top SNPs and indels from PLINK GWAS association analysis between affected (clinical) and unaffected (non-clinical) tortoises
| Position | Gene | Type | Unadjusted P |
|---|---|---|---|
| SNPsa | |||
| NC_024232.1:1,105,959 | A. superbus venom factor 1-like | non-syn | 0.0001388 |
| NC_024232.1:1,105,940 | A. superbus venom factor 1-like | syn | 0.0001388 |
| NC_024232.1:1,105,284 | A. superbus venom factor 1-like | syn | 0.0001388 |
| NW_007281632.1:53,578 | Uncharacterized LOC101950941 | non-syn | 0.0001725 |
| NW_007281632.1:53,577 | Uncharacterized LOC101950941 | non-syn | 0.0001725 |
| Indelsb | |||
| NW_007281632.1:53,576 | Uncharacterized LOC101950941 | syn | 0.0001725 |
| NW_007281632.1:53,579 | Uncharacterized LOC101950941 | non-syn | 0.0001725 |
| NC_024234.1:2,112,869 | TNFRSF5-likec | syn | 0.0007433 |
Syn synonymous and non-syn non-synonymous
aAll of these SNPs had a false discovery rate of 0.4164 after correction for multiple tests
bAll of these indels had a false discovery rate of 0.2193 after correction for multiple tests
cTumor necrosis factor receptor superfamily member 5-like
We found alleles at two non-synonymous SNPs and one non-synonymous indel shared by all but one URTD clinical tortoise (CF72, observed twice, once with URTD signs and again without) but also shared by two non-clinical animals (CF69 and OLD77). The SNPs were found on the following National Center for Biotechnology Information (NCBI) contig NW_007281632.1 at positions 53,577 and 53,578, and the indel was found at position 53,579 (Table 1). All were located in the gene LOC101950941.
Discussion
We sequenced the immunomes of 10 tortoises with clinical signs of URTD and 6 tortoises without clinical signs of URTD to: 1) estimate genetic diversity, differentiation, and admixture in each sampling location, and; 2) to identify SNPs putatively under selection, genes deviating from neutrality, and to better understand the genetic basis of URTD susceptibility, and; 3) to outline an approach that could be used to identify important genetic variants associated with diseases in other threatened species. We discuss each of these below.
Genetic differentiation and admixture were in line with expectations. First, pairwise FST values showed genetic differentiation corresponded to geographic distance. Second, genetic admixture inferred by PCA and STRUCTURE methods was congruent and reasonable as FC and OLD are geographically close and cluster together whereas CF is farther away and clusters separately. Because the populations represented three transition states in URTD epidemiology, differences also could be reflective of changes in the population after transition from initial URTD acute outbreaks to endemically stable and/or recrudescent outbreaks rather than geography alone. Clinically ill tortoises at CF could represent a subpopulation of more susceptible animals, which might be predicted to be among those impacted most during an initial disease outbreak. The historic OLD die-off [26] could have resulted in loss of the most susceptible animals from the population during the acute disease outbreak. Long term surveillance of populations would be needed to address this possibility.
Two SNPs appeared to be under selection. The first SNP, NW_007281406.1:2,298,828, was in the gene IFIT1-like; IFIT1 can bind to viral RNA with a triphosphate group on the 5′ end (i.e., PPP-RNA producing viruses, [49]). Specifically, the absence of IFIT1 leads to increased growth and pathogenicity of PPP-RNA producing viruses. The second SNP, NW_007322484.1:105, was synonymous and found in the gene IFI44-like; IFI44 may function in reducing viral transcription by suppressing long terminal repeat promoter activity of viruses such as HIV-1 [50]. Both of these SNPs are involved with response to viruses, and there are many viruses that affect turtles and tortoises (reviewed in [51]). For example, the genus Ranavirus as well as Herpesviruses affect G. polyphemus [52]. In fact, the G. polyphemus with Ranavirus examined by AJ Johnson, AP Pessier, JFX Wellehan, A Childress, TM Norton, NL Stedman, DC Bloom, W Belzer, VR Titus, R Wagner, et al. [52] was found only 50 km from the CF population. It is not clear if immunity to Ranaviruses or other viruses is influenced by variation in IFIT1 and IFI44.
There were 27 gene regions that had negative Tajima’s D values. We do not think this was due to population expansion, as G. polyphemus populations have declined across their range, especially during the past century [53]. Therefore, it is more likely that these genes are experiencing the effects of positive selection [54]. Forty-three regions had positive Tajima’s D values, which is unlikely to be due to population bottlenecks as estimates of genetic diversity are very high for the populations assessed in this study relative to other gopher tortoise populations [28]. These positive values may be due to balancing selection as we did not detect strong population structure between OLD and FC samples [54]. It is interesting that three genes (IPO11, SIN3A, and TRAF6) had regions that were potentially under positive and balancing selection. This finding may be because we only analyzed gene regions that were at least 60 bp and had 20× coverage resulting in partial gene sequences as opposed to the full gene sequences analyzed by other studies (e.g., [55]).
Functional enrichment analysis of the 35 gene regions that deviated from neutrality (Additional file 3: Table S1) suggested several things. First, these genes were enriched relative to the rest of the genome for gene ontology functions relating to the modification of macromolecules and proteins, which usually fold and mature in the lumen of the endoplasmic reticulum [56]. Second, these genes were enriched with transferase activity, which includes glycosylation of glycoproteins (i.e., adding sugars to glycoproteins) that can take place after glycoproteins leave the endoplasmic reticulum and travel to the Golgi apparatus (reviewed in [57]). Third, both macromolecule and protein modifications can also take place in Golgi apparatus, especially modifications for proteins destined to be secreted outside of cells [57], such as cytokines, which are vital to immune system functioning [58].
Genes that contained SNPs and indels with the strongest associations to clinical status may influence key biological functions in Mycoplasma immunity and are useful as hypotheses to test whether specific alleles may make tortoises more likely to present URTD signs. These variants were found in the A. superbus venom factor 1-like and LOC101950941 genes. A. superbus venom factor 1 (and possibly A. superbus venom factor 1-like) encodes a protein component of Austrelaps superbus (lowland copperhead snake) venom that is structurally and functionally similar to the complement protein C3 [59], which initiates the alternative pathway of the complement cascade, an important innate immune system process that results in the opsonization or lysis of pathogens and initiation of inflammatory responses [58]. A. superbus venom factor 1 may function like its analog C3 in mycoplasmal infections. Specifically, mycoplasmas such as M. pulmonis activate C3 [60], which can be broken down into C3a and C3b. C3b coats the surfaces of microbes, leading to opsonization or lysis [58].
LOC101950941 likely encodes a C-type lectin-like receptor found on natural killer cells (NK) based on a Coding Domain Search [61]. The full protein is predicted to be 405 amino acids in length, and the C-type lectin-like receptor domains are found between amino acids 70–154 and 283–400. The region between amino acids 70–154 contains the NW_007281632.1:53,577 and NW_007281632.1:53,578 SNPs and the NW_007281632.1:53,576 and NW_007281632.1:53,579 indels identified as important in this study. Because these SNPs and indels are non-synonymous (except NW_007281632.1:53,576), they may influence the functionality of the lectin receptors. Natural killer cells (NK) are part of the innate branch of the immune system and can detect unusual carbohydrates using lectin receptors to probe host cells for foreign or naturally occurring carbohydrates [58]. NK have a complicated role in mycoplasma immunity. First, NK are attracted to the site of mycoplasma infection in response to cytokines secreted by macrophages. Then NK produce INF-γ, an important cytokine that activates macrophages, in response to mycoplasma infection [62]. Interestingly, NK can be detrimental to clearing mycoplasma by negatively affecting the innate immune response [62]. In particular, mice depleted of NK and then infected with M. pulmonis have lower mycoplasmal load than mice with intact NK [63]. Thus, NK can have a negative influence on clearing of mycoplasmas by innate immune responses.
The strongest associated indels were present in the genes LOC101950941 and TNFRSF5-like. The indel at NW_007281632.1:53,579, which was contained within LOC101950941 was also found in our analysis of indels with non-synonymous alleles shared by tortoises with clinical signs. The TNFRSF5 gene (also referred to as CD40) encodes a receptor on antigen-presenting cells of the immune system such as dendritic and B cells as well as macrophages. TNFRSF5 functions in a variety of immune and inflammatory responses including the survival and proliferation of B cells [64] and induces the production of cytokines in dendritic cells and macrophages [58]. Mycoplasma arthritidis and M. fermentans have membrane-associated lipoproteins that upregulate CD40 expression by dendritic cells and macrophages [65–67], and it is likely that surface lipoproteins of M. agassizii could similarly impact CD40 upregulation. Therefore it is reasonable to suggest that there may be interactions between variation at the TNFRSF5-like gene and mycoplasma immunity in G. polyphemus.
There are several conservation implications based on the associations found between genetic variation and URTD clinical status. First, previous immunome work by JP Elbers, RW Clostio and SS Taylor [28] showed that 16 tortoises with unknown URTD clinical status from Louisiana, Alabama, Georgia, and Florida (4 per population) possessed the same alleles at SNPs NW_007281632.1:53,578 and NW_007281632.1:53,577 as the URTD clinical tortoises sequenced in this work. If the strong associations observed between genetic variation and URTD clinical status carry over to other tortoises, and these associations prove to be causal rather than correlated, then it is possible that tortoises across the range of the species possess genetic variation that makes them more likely to present URTD clinical signs. In particular, the Georgia population surveyed by JP Elbers, RW Clostio and SS Taylor [28] had the same variants as the URTD clinical tortoises and evidence of Mycoplasmal-URTD. Most tortoises from Green Grove, an area of Jones Ecological Research Center in Southwest Georgia, had been exposed to URTD since 125 out of 136 (92%) produced antibodies against M. agassizii; 44 out of 191 (23%) presented mild to severe clinical signs of URTD; and 101 out of 366 (28%) had evidence of chronic URTD lesions [22]. This suggests some correlation between the genetic variation associated with disease susceptibility and actual presentation of disease. If further experimental studies and more careful examination of these genes show a causal relationship, managers may wish to translocate tortoises from appropriate donor to at risk populations to bolster genetic variation at these loci. Future work might also examine the severity of Mycoplasmal-URTD infections, which may be influenced by particular immunome variants. In particular, some gopher tortoises may be susceptible to Mycoplasmal-URTD but possess immunome variants such that clinical signs are not severe, resulting in little to no impediment to normal function.
More generally, understanding the genetic basis of disease susceptibility is critical for a variety of reasons. First it can provide information on populations’ risk to succumbing to disease. For example, mice with the H-2Dk allele can properly recognize cytomegalovirus-infected cells [68], thus mice populations with low frequencies of this allele may be at increased risk of developing chronic infections, especially in the salivary glands. Second, identifying genetic mechanisms of disease resilience can inform management, in particular if managers are able to spread “beneficial” variation to populations that are depauperate in genetic variation at susceptibility loci by translocating genetically variable individuals. Finally, genetic understanding of disease predisposition can lead to potential treatments. For example, tumor cells in devil facial tumor disease (DFTD) are not recognized by the adaptive immune system of Tasmanian devils because the tumors do not express major histocompatibility complex molecules, thus introducing MHC-expressing DFTD cells could be a potential treatment for DFTD [1].
Conclusions
Previous studies examining the genetic basis of disease resistance focus on a limited number and type of genes (e.g., MHC and TLR genes, [69, 70]). Here we provide evidence that a framework to examine several hundred genes involved in the immune response is better able to identify important genetic variants that confer resistance to disease. This framework can be adopted as an initial step to find solutions for a wide-range of species now in danger of extinction from disease.
Additional files
Coverage plots for first eight samples showing number of sequencing reads at or above specified proportions. A value at 50 Depth and 0.5 fraction means 50% of bases were at or above 50X coverage. (TIFF 149 kb)
Coverage plots for last eight samples showing number of sequencing reads at or above specified proportions. (TIFF 124 kb)
Genes with regions deviating from neutrality. (DOCX 17 kb)
Description of Gopherus polyphemus samples by clinical status and diploid genotypes at several bases along the A. superbus venom factor 1-like and TNFRSF5-like genes for NCBI contigs NC_024232.1 and NC_024234.1 respectively. Non-clin for tortoises that were never observed with nasal discharge during the duration of the field study and Clin for tortoises that had at least one incident of mild to severe nasal discharge. CF for Cecil Field, FC for Fort Cooper, and OLD for Oldenburg. (DOCX 17 kb)
Acknowledgements
We are grateful to R. Carmouche of Pennington Biomedical Research Center’s Genomic Core Facility for performing next-generation sequencing laboratory work. We used Louisiana State University High Performance Computing resources to analyze next-generation sequencing data.
Funding
This material is based upon work that is supported by the National Institute of Food and Agriculture, United States Department of Agriculture, McIntire Stennis project LAB04066 and LAB94169. The Lucius Gilbert Foundation provided support for J.P.E. This project/work used genomics core facilities that are supported in part by COBRE (NIH 8 P20 GM103528) and NORC (NIH 2P30DK072476) center grants from the National Institutes of Health. Gopher tortoise sample collection was supported by grants from the National Science Foundation, Ecology of Infectious Diseases program (DEB-0224953; to M. B. Brown, M. K. Oli, and P. A. Klein) and the National Institutes of Health (5K08AI57722; to L. D. Wendland).
Availability of data and materials
Raw sequencing data are available from the Sequence Read Archive (accession: SRP069350). Detailed analytical methods and scripts to create tables and figures are available from https://github.com/jelber2/immunome_urtd
Abbreviations
- 5′
Five prime
- AR
Allelic richness
- bp
Base pair
- CF
Cecil Field/Branan Field Wildlife and Environmental Area
- Clin
Clinical
- DFTD
Devil facial tumor disease
- DNA
Deoxyribonucleic acid
- FC
Fort Cooper State Park
- FST
Fixation index for population subdivision
- GWAS
Genome-wide association study
- HE
Expected heterozygosity
- HIV
Human immunodeficiency virus
- HO
Observed heterozygosity
- Indels
Insertions/deletions
- INF-γ
Interferon gamma
- km
Kilometers
- MHC
Major histocompatibility complex
- NCBI
National Center for Biotechnology Information
- NK
Natural killer cells
- Non-clin
Non-clinical
- non-syn
Non-synonymous
- OLD
Perry Oldenburg Wildlife and Environmental Area
- PCA
Principle component analysis
- PPP-RNA
Triphosphate ribonucleic acid
- RADseq
Restriction-site associated DNA sequencing
- RNA
Ribonucleic acid
- SNPs
Single nucleotide polymorphisms
- syn
Synonymous
- TLR
Toll-like receptors
- URTD
Upper respiratory tract disease
Authors’ contributions
JPE, MBB, and SST designed the study, JPE performed SNP analyses, and MBB provided tortoise samples. JPE, MBB, and SST wrote the paper. All authors read and approved the final manuscript.
Ethics approval
The University of Florida Institutional Animal Care and Use Committee, and Florida Fish and Wildlife Conservation Commission approved all protocols for animal use (research permit #WX02160d).
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4452-0) contains supplementary material, which is available to authorized users.
Contributor Information
Jean P. Elbers, Email: jean.elbers@gmail.com
Mary B. Brown, Email: mbbrown@ufl.edu
Sabrina S. Taylor, Email: staylor@lsu.edu
References
- 1.Siddle HV, Kreiss A, Tovar C, Yuen CK, Cheng Y, Belov K, Swift K, Pearse AM, Hamede R, Jones ME, et al. Reversible epigenetic down-regulation of MHC molecules by devil facial tumour disease illustrates immune escape by a contagious cancer. P Natl Acad Sci USA. 2013;110:5103–5108. doi: 10.1073/pnas.1219920110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams ES, Mills K, Kwiatkowski DR, Thome ET, Boerger-Fields A. Plague in a black-footed ferret (Mustela nigripes) J Wildlife Dis. 1994;30:581–585. doi: 10.7589/0090-3558-30.4.581. [DOI] [PubMed] [Google Scholar]
- 3.Berger L, Roberts AA, Voyles J, Longcore JE, Murray KA, Skerratt LF. History and recent progress on chytridiomycosis in amphibians. Fungal Ecol. 2016;19:89–99. doi: 10.1016/j.funeco.2015.09.007. [DOI] [Google Scholar]
- 4.Thogmartin WE, Sanders-Reed CA, Szymanski JA, McKann PC, Pruitt L, King RA, Runge MC, Russell RE. White-nose syndrome is likely to extirpate the endangered Indiana bat over large parts of its range. Biol Conserv. 2013;160:162–172. doi: 10.1016/j.biocon.2013.01.010. [DOI] [Google Scholar]
- 5.Daszak P, Cunningham AA, Hyatt AD. Anthropogenic environmental change and the emergence of infectious diseases in wildlife. Acta Trop. 2001;78:103–116. doi: 10.1016/S0001-706X(00)00179-0. [DOI] [PubMed] [Google Scholar]
- 6.Ogden NH, Bigras-Poulin M, O'Callaghan CJ, Barker IK, Lindsay LR, Maarouf A, Smoyer-Tomic KE, Waltner-Toews D, Charron D. A dynamic population model to investigate effects of climate on geographic range and seasonality of the tick Ixodes scapularis. In J Parasitol. 2005;35:375–389. doi: 10.1016/j.ijpara.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 7.Reed KD, Meece JK, Henkel JS, Shukla SK. Birds, migration and emerging zoonoses: West Nile virus, Lyme disease, influenza a and enteropathogens. Clin Med Res. 2003;1:5–12. doi: 10.3121/cmr.1.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wachsmuth IK, Evins GM, Fields PI, Olsvik x, rjan PT, Bopp CA, Wells JG, Carrillo C, et al. The molecular epidemiology of cholera in Latin America. J Infect Dis. 1993;167:621–626. doi: 10.1093/infdis/167.3.621. [DOI] [PubMed] [Google Scholar]
- 9.Skerratt L, Martin R, Handasyde K. Sarcoptic mange in wombats. Aust Vet J. 1998;76:408–410. doi: 10.1111/j.1751-0813.1998.tb12389.x. [DOI] [PubMed] [Google Scholar]
- 10.Daszak P, Cunningham AA, Hyatt AD. Emerging infectious diseases of wildlife--threats to biodiversity and human health. Science. 2000;287:443–449. doi: 10.1126/science.287.5452.443. [DOI] [PubMed] [Google Scholar]
- 11.Fremont DH, Matsumura M, Stura EA, Peterson PA, Wilson IA. Crystal structures of two viral peptides in complex with murine MHC class I H-2Kb. Science. 1992;257:919–927. doi: 10.1126/science.1323877. [DOI] [PubMed] [Google Scholar]
- 12.Wang M, Yan J, Zhao J, Song W, Zhang X, Xiao Y, Zheng Y. Genome-wide association study (GWAS) of resistance to head smut in maize. Plant Sci. 2012;196:125–131. doi: 10.1016/j.plantsci.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 13.Magwire MM, Fabian DK, Schweyen H, Cao C, Longdon B, Bayer F, Jiggins FM. Genome-wide association studies reveal a simple genetic basis of resistance to naturally coevolving viruses in Drosophila melanogaster. PLoS Genet. 2012;8:e1003057. doi: 10.1371/journal.pgen.1003057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rauch A, Kutalik Z, Descombes P, Cai T, Di Iulio J, Mueller T, Bochud M, Battegay M, Bernasconi E, Borovicka J, et al. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology. 2010;138:1338–1345. doi: 10.1053/j.gastro.2009.12.056. [DOI] [PubMed] [Google Scholar]
- 15.Donaldson ME, Davy CM, Willis CKR, McBurney S, Park A, Kyle CJ. Profiling the immunome of little brown myotis provides a yardstick for measuring the genetic response to white-nose syndrome. Evol Appl. 2017;00:1–15. doi: 10.1111/eva.12514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morris KM, Wright B, Grueber CE, Hogg C, Belov K. Lack of genetic diversity across diverse immune genes in an endangered mammal, the Tasmanian devil (Sarcophilus harrisii) Mol Ecol. 2015;24:3860–3872. doi: 10.1111/mec.13291. [DOI] [PubMed] [Google Scholar]
- 17.Palti Y, Vallejo RL, Gao G, Liu S, Hernandez AG, Rexroad CE, 3rd, Wiens GD. Detection and validation of QTL affecting bacterial cold water disease resistance in rainbow trout using restriction-site associated DNA sequencing. PLoS One 2015;10:e0138435. [DOI] [PMC free article] [PubMed]
- 18.Reed KM, Mendoza KM, Settlage RE. Targeted capture enrichment and sequencing identifies extensive nucleotide variation in the Turkey MHC-B. Immunogenetics. 2016;68:219–229. doi: 10.1007/s00251-015-0893-7. [DOI] [PubMed] [Google Scholar]
- 19.Roffler GH, Amish SJ, Smith S, Cosart T, Kardos M, Schwartz MK, Luikart G. SNP discovery in candidate adaptive genes using exon capture in a free-ranging alpine ungulate. Mol Ecol Resour. 2016;16:1147–1164. doi: 10.1111/1755-0998.12560. [DOI] [PubMed] [Google Scholar]
- 20.Schweizer RM, Robinson J, Harrigan R, Silva P, Galverni M, Musiani M, Green RE, Novembre J, Wayne RK. Targeted capture and resequencing of 1040 genes reveal environmentally driven functional variation in grey wolves. Mol Ecol. 2016;25:357–379. doi: 10.1111/mec.13467. [DOI] [PubMed] [Google Scholar]
- 21.Brown MB, GS ML, Klein PA, Crenshaw BC, Schumacher IM, Brown DR, Jacobson ER. Upper respiratory tract disease in the gopher tortoise is caused by Mycoplasma agassizii. J Clin Microbiol. 1999;37:2262–2269. doi: 10.1128/jcm.37.7.2262-2269.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGuire JL, Smith LL, Guyer C, Lockhart JM, Lee GW, Yabsley MJ. Surveillance for upper respiratory tract disease and mycoplasma in free-ranging gopher tortoises (Gopherus polyphemus) in Georgia, USA. J Wildlife Dis. 2014;50:733–744. doi: 10.7589/2013-11-300. [DOI] [PubMed] [Google Scholar]
- 23.Gates CA, Allen MJ, JED B, Stillwaugh DM, Shattler SR. Characterization of a gopher tortoise mortality event in west-central Florida. Fla Scientist. 2002;65:185–197. [Google Scholar]
- 24.Clostio RW, Martinez AM, LeBlanc KE, Anthony NM. Population genetic structure of a threatened tortoise across the south-eastern United States: implications for conservation management. Anim Conserv. 2012;15:613–625. doi: 10.1111/j.1469-1795.2012.00557.x. [DOI] [Google Scholar]
- 25.USFWS Endangered and threatened wildlife and plants; 12-month finding on a petition to list the gopher tortoise (Gopherus polyphemus) as threatened in the eastern portion of its range. Fed Regist. 2011;76:45130–45162. [Google Scholar]
- 26.Wendland LD, Wooding J, White CL, Demcovitz D, Littell R, Berish JED, Ozgul A, Oli MK, Klein PA, Christman MC, et al. Social behavior drives the dynamics of respiratory disease in threatened tortoises. Ecology. 2010;91:1257–1262. doi: 10.1890/09-1414.1. [DOI] [PubMed] [Google Scholar]
- 27.Wendland LD, Zacher LA, Klein PA, Brown DR, Demcovitz D, Littell R, Brown MB. Improved enzyme-linked immunosorbent assay to reveal Mycoplasma agassizii exposure: a valuable tool in the management of environmentally sensitive tortoise populations. Clin Vaccine Immunol. 2007;14:1190–1195. doi: 10.1128/CVI.00108-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elbers JP, Clostio RW, Taylor SS. Population genetic inferences using immune gene SNPs mirror patterns inferred by microsatellites. Mol Ecol Resour. 2017;17:481–491. doi: 10.1111/1755-0998.12591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elbers JP, Taylor SS. GO2TR: a gene ontology-based workflow to generate target regions for target enrichment experiments. Conserv Genet Resour. 2015;7:851–857. doi: 10.1007/s12686-015-0487-6. [DOI] [Google Scholar]
- 30.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaffer HB, Minx P, Warren DE, Shedlock AM, Thomson RC, Valenzuela N, Abramyan J, Amemiya CT, Badenhorst D, Biggar KK, et al. The western painted turtle genome, a model for the evolution of extreme physiological adaptations in a slowly evolving lineage. Genome Biol. 2013;14:R28. doi: 10.1186/gb-2013-14-3-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 1303:2013, 3997.
- 33.Lunter G, Goodson M. STAMPY: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011;21:936–939. doi: 10.1101/gr.111120.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Foll M, Gaggiotti O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics. 2008;180:977–993. doi: 10.1534/genetics.108.092221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289–300. [Google Scholar]
- 37.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vilella AJ, Blanco-Garcia A, Hutter S, Rozas J. VariScan: analysis of evolutionary patterns from large-scale DNA sequence polymorphism data. Bioinformatics. 2005;21:2791–2793. doi: 10.1093/bioinformatics/bti403. [DOI] [PubMed] [Google Scholar]
- 39.Hudson RR. Generating samples under a Wright-Fisher neutral model. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- 40.R Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2016. Available from: http://www.R-project.org.
- 41.Al-Shahrour F, Diaz-Uriarte R, Dopazo J. FatiGO: a web tool for finding significant associations of gene ontology terms with groups of genes. Bioinformatics. 2004;20:578–580. doi: 10.1093/bioinformatics/btg455. [DOI] [PubMed] [Google Scholar]
- 42.Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 43.De Baets G, Van Durme J, Reumers J, Maurer-Stroh S, Vanhee P, Dopazo J, Schymkowitz J, Rousseau F. SNPeffect 4.0: on-line prediction of molecular and structural effects of protein-coding variants. Nucleic Acids Res. 2012;40:D935–D939. doi: 10.1093/nar/gkr996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thornton T, McPeek MS. ROADTRIPS: case-control association testing with partially or completely unknown population and pedigree structure. Am J Hum Genet. 2010;86:172–184. doi: 10.1016/j.ajhg.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pritchard JK, Stephens M, Donnelly P. Inference Of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Earl DA, vonHoldt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4:359–361. doi: 10.1007/s12686-011-9548-7. [DOI] [Google Scholar]
- 48.Spencer CC, Su Z, Donnelly P, Marchini J. Designing genome-wide association studies: sample size, power, imputation, and the choice of genotyping chip. PLoS Genet. 2009;5:e1000477. doi: 10.1371/journal.pgen.1000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pichlmair A, Lassnig C, Eberle C-A, Gorna MW, Baumann CL, Burkard TR, Burckstummer T, Stefanovic A, Krieger S, Bennett KL, et al. IFIT1 is an antiviral protein that recognizes 5[prime]-triphosphate RNA. Nat Immunol. 2011;12:624–630. doi: 10.1038/ni.2048. [DOI] [PubMed] [Google Scholar]
- 50.Power D, Santoso N, Dieringer M, Yu J, Huang H, Simpson S, Seth I, Miao H, Zhu J. IFI44 suppresses HIV-1 LTR promoter activity and facilitates its latency. Virology. 2015;481:142–150. doi: 10.1016/j.virol.2015.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marschang RE. Viruses infecting reptiles. Viruses. 2011;3:2087f2126. doi: 10.3390/v3112087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson AJ, Pessier AP, Wellehan JFX, Childress A, Norton TM, Stedman NL, Bloom DC, Belzer W, Titus VR, Wagner R, et al. Ranavirus infection of free-ranging and captive box turtles and tortoises in the United States. J Wildlife Dis. 2008;44:851–863. doi: 10.7589/0090-3558-44.4.851. [DOI] [PubMed] [Google Scholar]
- 53.Auffenberg W, Franz R. 1982. The status and distribution of the gopher tortoise (Gopherus polyphemus). In: Bury RB, editor. North American Tortoises: Conservation and Ecology. Wildlife Research Report 12. Washington DC: US Fish and Wildlife Service; 1982. p. 95–126.
- 54.Biswas S, Akey JM. Genomic insights into positive selection. Trends Genet. 2006;22:437–446. doi: 10.1016/j.tig.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 55.Ferrer-Admetlla A, Bosch E, Sikora M, Marquès-Bonet T, Ramírez-Soriano A, Muntasell A, Navarro A, Lazarus R, Calafell F, Bertranpetit J, et al. Balancing selection is the main force shaping the evolution of innate immunity genes. J Immunol. 2008;181:1315–1322. doi: 10.4049/jimmunol.181.2.1315. [DOI] [PubMed] [Google Scholar]
- 56.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Bio. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 57.Farquhar MG, Palade GE. The Golgi apparatus (complex)-(1954-1981)-from artifact to center stage. J Cell Biol. 1981;91:77s–103s. doi: 10.1083/jcb.91.3.77s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Janeway CA, Travers P, Walport M, Shlomchik MJ. Immunobiology: the immune system in health and disease. 5. New York: Garland Science; 2001. [Google Scholar]
- 59.Rehana S, Manjunatha Kini R. Molecular isoforms of cobra venom factor-like proteins in the venom of Austrelaps superbus. Toxicon. 2007;50:32–52. doi: 10.1016/j.toxicon.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 60.Simmons WL, Denison AM, Dybvig K. Resistance of Mycoplasma pulmonis to complement lysis is dependent on the number of Vsa tandem repeats: shield hypothesis. Infect Immun. 2004;72:6846–6851. doi: 10.1128/IAI.72.12.6846-6851.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marchler-Bauer A, Bryant SH. CD-search: protein domain annotations on the fly. Nucleic Acids Res. 2004;32:W327–W331. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Woolard MD, Hudig D, Tabor L, Ivey JA, Simecka JW. NK cells in gamma-interferon-deficient mice suppress lung innate immunity against Mycoplasma spp. Infect Immun. 2005;73:6742–6751. doi: 10.1128/IAI.73.10.6742-6751.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bodhankar S, Woolard MD, Sun X, Simecka JW. NK cells interfere with the generation of resistance against mycoplasma respiratory infection following nasal-pulmonary immunization. J Immunol. 2009;183:2622–2631. doi: 10.4049/jimmunol.0802180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schattner EJ, Elkon KB, Yoo D-H, Tumang J, Krammer PH, Crow MK, Friedman SM. CD40 ligation induces Apo-1/Fas expression on human B lymphocytes and facilitates apoptosis through the Apo-1/Fas pathway. J Exp Med. 1995;182:1557–1565. doi: 10.1084/jem.182.5.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cole BC, Mu H-H, Pennock ND, Hasebe A, Chan FV, Washburn LR, Peltier MR. Isolation and partial purification of macrophage- and dendritic cell-activating components from Mycoplasma arthritidis: association with organism virulence and involvement with toll-like receptor 2. Infect Immun. 2005;73:6039–6047. doi: 10.1128/IAI.73.9.6039-6047.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weigt H, Mühlradt PF, Emmendörffer A, Krug N, Braun A. Synthetic mycoplasma-derived lipopeptide MALP-2 induces maturation and function of dendritic cells. Immunobiology. 2003;207:223–233. doi: 10.1078/0171-2985-00234. [DOI] [PubMed] [Google Scholar]
- 67.Link C, Gavioli R, Ebensen T, Canella A, Reinhard E, Guzmán CA. The toll-like receptor ligand MALP-2 stimulates dendritic cell maturation and modulates proteasome composition and activity. Eur J Immunol. 2004;34:899–907. doi: 10.1002/eji.200324511. [DOI] [PubMed] [Google Scholar]
- 68.Desrosiers M-P, Kielczewska A, Loredo-Osti JC, Adam SG, Makrigiannis AP, Lemieux S, Pham T, Lodoen MB, Morgan K, Lanier LL, et al. Epistasis between mouse KLRa and major histocompatibility complex class I loci is associated with a new mechanism of natural killer cell–mediated innate resistance to cytomegalovirus infection. Nat Genet. 2005;37:593–599. doi: 10.1038/ng1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Savage AE, Zamudio KR. MHC genotypes associate with resistance to a frog-killing fungus. P Natl Acad Sci USA. 2011;108:16705–16710. doi: 10.1073/pnas.1106893108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zekarias B, Ter Huurne AA, Landman WJ, Rebel JM, Pol JM, Gruys E. Immunological basis of differences in disease resistance in the chicken. Vet Res. 2002;33:109–125. doi: 10.1051/vetres:2002001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Coverage plots for first eight samples showing number of sequencing reads at or above specified proportions. A value at 50 Depth and 0.5 fraction means 50% of bases were at or above 50X coverage. (TIFF 149 kb)
Coverage plots for last eight samples showing number of sequencing reads at or above specified proportions. (TIFF 124 kb)
Genes with regions deviating from neutrality. (DOCX 17 kb)
Description of Gopherus polyphemus samples by clinical status and diploid genotypes at several bases along the A. superbus venom factor 1-like and TNFRSF5-like genes for NCBI contigs NC_024232.1 and NC_024234.1 respectively. Non-clin for tortoises that were never observed with nasal discharge during the duration of the field study and Clin for tortoises that had at least one incident of mild to severe nasal discharge. CF for Cecil Field, FC for Fort Cooper, and OLD for Oldenburg. (DOCX 17 kb)
Data Availability Statement
Raw sequencing data are available from the Sequence Read Archive (accession: SRP069350). Detailed analytical methods and scripts to create tables and figures are available from https://github.com/jelber2/immunome_urtd