


Abstract
Opioids are among the most effective pain relievers; however, their abuse has been on the rise worldwide evident from an alarming increase in accidental opioid overdoses. This demands for an urgent increase in scientific endeavors for better understanding of main cellular mechanisms and circuits involved in opiate addiction. Preclinical studies strongly suggest that memories associated with positive and negative opioid experiences are critical in promoting compulsive opiate-seeking and opiate-taking behaviors, and relapse. Particular focus on synaptic plasticity as the cellular correlate of learning and memory has rapidly evolved in drug addiction field over the past two decades. Several critical addiction-related brain areas are identified, one of which is the ventral tegmental area (VTA), an area intensively studied as the initial locus for drug reward. Here, we provide an update to our previous review on “Opiates and Plasticity” highlighting the most recent discoveries of synaptic plasticity associated with opiates in the VTA. Electrophysiological studies of plasticity of addiction to date have been invaluable in addressing learning processes and mechanisms that underlie motivated and addictive behaviors, and now with the availability of powerful technologies of transgenic approaches and optogenetics, circuit-based studies hold high promise in fostering synaptic studies of opiate addiction.
Keywords: Synaptic plasticity, LTP, LTD, opiates, addiction, VTA
Graphical abstract
INTRODUCTION
Opiates are among the most-prescribed drugs in the United States, in part because they are particularly powerful analgesics and pain relieving agents. However, their ability to induce feeling of euphoria (intense pleasant sensations) makes them extremely addictive.1 Approximately 3 million people in the United states and 16 million worldwide have a current or past opioid use disorder (OUD).2 According to the Centers for Disease and Control Prevention (CDC), opioid overdose has become the leading cause of death of Americans under 50. The current “Opioid Epidemic” or “Opioid Crisis” has also urged the National Institutes of Health (NIH) to initiate partnership with private sectors to support collaborative research efforts on pain and opioid abuse.3 Chronic opiate intake induces tolerance, resulting in the need to increase doses to obtain the desired effects, which contributes to accidental deaths by overdose. Not surprisingly, approximately 80% of heroin users report the prior use of opioid pain relievers,4 demonstrating that previous exposure to opiates prompts long lasting changes in the brain circuits important in the development of OUD including opioid addiction, further contributing to the troubling rise in related overdoses. In fact, strong and durable memories of the opioid experience promotes compulsive opiate taking, craving, and relapse, highlighting the necessity for a better understanding of learning mechanisms that underlie addictive behaviors and vulnerability to relapse. Since the discovery of synaptic plasticity as the cellular correlate of learning and memory, strong overlaps between neural and cellular substrates of learning, and drug addiction have been recognized. The current perspective of a synaptic basis for drug addiction was a paradigm shift in the field of addiction introducing a pathological learning model of addiction where addictive drugs usurp synaptic mechanisms that underlie reward-based learning and motivated behaviors.5,6 With this perspective, neuroscientists have made significant progress towards understanding the molecular mechanisms underpinning the reinforcing, aversive ,and addictive properties of drugs of abuse through their interaction with learning mechanisms.5,7–11 Here, we provide an update on “Opiates and Plasticity”12 highlighting the most recent discoveries of synaptic plasticity associated with opiates. The main focus of this review is on the acute and chronic effects of opiates on synaptic plasticity of the mesocorticolimbic pathway originating from the ventral tegmental area (VTA), as this key brain area is intimately involved in early stages of opioid abuse. We refer readers to ref 12 for discussion on current views of drug addiction including dopamine hypothesis valid for different aspects of addictive behaviors which will not be covered here. In addition, see more excellent reviews for comprehensive discussion of neurocircuitry of drug addiction and drug-induced plasticity in brain areas mediating reward and motivated behavior such as nucleus accumbens (NAc), prefrontal cortex, hippocampus, and others: refs 1, 5, 6, and 13–25 We believe that a better understanding of synaptic basis of reward/motivated behaviors and opiate addiction will help identify novel pharmacological and nonpharmacological approaches targeting opiate-induced synaptic plasticity and provide a step forward in resolving the current serious public health crisis of opioid abuse.
MESOCORTICOLIMBIC PATHWAY AND DRUG ADDICTION
Early neuroadaptations associated with development of drug addiction are believed to occur in the mesocorticolimbic dopamine pathway originating from the VTA dopamine neurons. The VTA is a key brain structure involved in reward processing and motivation. Its role in drug addiction has been well-established and intensively studied.26,27 The increased release of dopamine in the VTA projection areas (specifically, the nucleus accumbens, NAc, also known as ventral striatum, and the prefrontal cortex, PFC) triggered in response to addictive drugs including opiates is believed to mediate positive reinforcing effects of the drugs and highlight motivational values of drugs leading to compulsive drug seeking, drug taking, craving, and relapse.28,29 In addition, diminished dopamine levels in the NAc are associated with dysphoric and negative feelings of drug withdrawal that also play a key role in compulsive craving and relapse.19 However, it is becoming clearer that increased dopamine neuronal activity participates not only in reward signaling and salience, but also in aversion. These contrasting effects are mediated by distinct anatomical and functional subsets of VTA dopamine neurons with differential axonal projections and receiving alternative afferents. For example, dopamine neurons projecting to shell of NAc that receive afferents from laterodorsal tegmentum (LDTg) and rostromedial tegmental area (RMTg) mediate reward, while dopamine neurons projecting to PFC receive excitatory inputs from the lateral habenula (LHb) and mediate aversion.23,30 Therefore, drug-induced concurrent increased dopamine release in VTA projection areas such as NAc and PFC may mediate different aspects of rewarding and aversive experiences associated with drugs of abuse. It is worth mentioning that in spite of the dominant dopamine hypothesis of drug addiction focusing on the mesolimbic dopamine pathway, human studies only support this hypothesis for stimulant and alcohol addiction, as demonstrated by the strong correlation between drug seeking behaviors and striatal dopamine receptor availability and dopamine release. The evidence for involvement of the dopamine system in individuals with opiate abuse is limited,31,32 although higher activity of VTA neurons is shown in response to heroin-associated cues in abstinent heroin users,33 highlighting the necessity of investigating dopamine and nondopamine mechanisms of the VTA in opiate addiction. Specifically investigation of glutamatergic and GABAergic synaptic plasticity in VTA neurons, in addition to studies of other critical brain areas involved in motivation and goal-directed behaviors, is needed. In this review, we will only focus on the VTA and comprehensively discuss most recent findings on the actions of opiates and opiate-induced plasticity in the VTA.
ACUTE ACTIONS OF OPIATES IN THE VTA
It is well-established that there is a considerable variability in neuronal populations within the VTA with dopamine neurons of the VTA comprising ~60–65% of this midbrain area. The nondopamine neurons of the VTA include different sub-populations of GABAergic (30%) and glutamatergic (5%) neurons. In addition to dopamine neurons, GABAergic and glutamatergic neurons of the VTA may play important roles in non-dopamine related aspects of addictive behavior34–36 although the major focus of the addiction field has been on drug-induced changes in dopamine neuron activity. Since different populations of dopamine neurons in the VTA project to different target structures including NAc, amygdala and PFC, and also receive different afferents, dopamine release from these distinct subsets of dopamine neurons is likely to be implicated in positive or negative motivational effects of opiates.23,30
All drugs of abuse are able to trigger synaptic plasticity in the VTA, posing the idea that drug-induced plasticity may be the critical common cellular substrate for the establishment of drug addiction for all drug classes.37–50 The predominant mechanism of acute opiate action in increasing dopamine cell activity and dopamine release is through disinhibition (i.e., inhibition of GABAergic interneurons of the VTA making GABAA synapses onto VTA dopamine neurons).51 Whereas the reinforcing actions of acute opiates are mediated through MORs, leading to an increase in dopamine release in the NAc, KOR activation seems to decrease dopamine release in this region which mediates dysphoric effects of opioids and opiates (Box 1). The addictive effects of opiates are believed to be mainly through their action on MORs located on VTA GABAergic interneurons, but opiates can also act upon other neurons within and outside of the VTA to mediate these effects, suggesting the presence of distinct neural circuits for acute opiate action. For example, it is now known that acute morphine can increase VTA dopamine neuronal activity through the activation of MORs located in GABAergic neurons in the tail of the VTA, a region also called the RMTg.52 Interestingly, the excitatory action of morphine on VTA dopamine neuron through RMTg (which make GABAA synapses onto dopamine neurons) requires VTA glutamatergic tone since blocking VTA NMDA and AMPA receptors completely prevents morphine-induced excitation of VTA dopamine neurons.52,53
Box 1. Opioid Receptors.
In general, opiate drugs and endogenous opioids act through Ithree main G protein coupled receptors: mu- (MOR), delta-(DOR), and kappa-(KOR) opioid receptors. Activation of opioid receptors typically results in opening of potassium channels such as G protein inwardly rectifying K+ channels (GIRKs), inhibition of calcium channels, inhibition of adenylyl cyclase, and inhibition of neurotransmitter release. The effects of opiates are mediated through the GTP-bound form of the alpha-subunit as well as free beta/gamma-subunits of these G protein coupled receptors.
RMTg GABAergic neurons also relay the inhibitory effects of LHb (an exclusively glutamatergic structure that mediate aversion and negative reward) on midbrain dopamine neurons.54 Interestingly, LHb neurons not only synapse on GABAergic RMTg neurons that project to dopamine neurons innervating NAc but also make direct connections on dopamine neurons projecting to PFC.55 In addition to activation of MORs located on RMTg and GABAergic interneurons of the VTA by opioids, MOR activation within the LHb inhibits a subset of LHb neurons both directly and indirectly by inhibiting the presynaptic release of glutamate onto these LHb neurons. Surprisingly, MOR activation also decreases presynaptic GABA release onto these LHb neurons.56 This may be explained by the fact that main GABAergic structures projecting to the LHb such as the entopeduncular nucleus and the VTA corelease GABA and glutamate onto LHb neurons;57,58 therefore, activation of MORs on such inputs might decrease both glutamate and GABA release onto LHb neurons although the balance may be shifted by opiates toward greater reduction of glutamate rather than GABA release hence less excitatory drive onto LHb neurons.
In addition to GABAA synapses arising from RMTg and VTA GABAergic interneuron inputs to the VTA, NAc medium spiny neurons (MSNs) also synapse onto both VTA dopamine neurons and GABAergic interneurons and have differential sensitivity to stimulant and opioids.59,60 NAc MSNs are the main projection neurons of this area and express either D1 or D2 dopamine receptors. D1MSNs selectively express the neuropeptide dynorphin as well as GABA and send direct projections to midbrain dopamine neurons. A recent study by Bonci’s team elegantly demonstrates the bidirectional modes of inhibition by NAc D1MSN projections to the VTA.60 Optical stimulation of NAc projections inhibits both dopamine (preferentially via GABAB synapses) and GABAergic interneurons (via activating GABAA synapses) of the VTA but the net effect of this optical stimulation is inhibition of dopamine neurons. More interestingly, selective deletion of GABAB receptors from dopamine neurons affects behavioral response to cocaine but not morphine.60 In spite of insensitivity of GABAB inhibition to acute morphine, these two inhibitory NAc projections to the VTA (either disinhibiting dopamine neurons via GABAA receptors on VTA GABAergic neurons or directly inhibiting dopamine neurons via GABAB receptors) may be differentially targeted by chronic morphine. For example, it is possible that opiate withdrawal selectively potentiates direct GABAB inhibition of dopamine neurons from this pathway, then contributing to hypoactivity of VTA dopamine neurons during aversive withdrawal states. On the other hand, if disinhibition of dopamine neurons via GABAA receptors is favored by opiates, this would promote bursts of dopamine release in response to opiates or opioid-associated cues, possibly triggering craving and relapse. Adding to this complexity of VTA microcircuits is the differential opioid receptor expression within the VTA which results in independent opioid-modulation of dopamine release in NAc and PFC that is regulated through MOR and KOR activation on different subpopulation of VTA dopamine and GABAergic neurons.59–63 See ref 22 for further comprehensive discussion on opioid regulation of dopamine release.
ACUTE OPIATE-INDUCED PLASTICITY IN THE VTA
Glutamatatergic and GABAergic synaptic transmissions are key components of the regulation of dopamine cell activity in the VTA and are known to play important roles in pathological drug-seeking behavior. Not surprisingly, acute morphine administration increases dopamine cell activity by triggering changes in synaptic strengths (i.e., synaptic plasticity) of glutamatergic and GABAergic synapses onto VTA dopamine neurons (Box 2).
Box 2. Synaptic Plasticity and Dopamine Cell Activity.
Synaptic plasticity as the cellular substrate of learning can be experimentally triggered using traditional induction paradigms (e.g., high or low frequency stimulation) or in response to near-coincident pairing of pre- and postsynaptic cell activities (i.e., spike timing dependent plasticity, STDP). Two best studied forms of synaptic plasticity are long-term potentiation (strengthening of synapses, LTP) and long-term depression (weakening of synapses, LTD). Almost every synapse in the brain including glutamatergic and GABAergic synapses are capable of exhibiting LTP and LTD. In general, expression of synaptic plasticity can be at presynaptic or postsynaptic loci. This means that plasticity can be expressed presynaptically by changes in the release of neurotransmitter from presynaptic terminals (e.g., increased GABA release in LTPGABA) or postsynaptically through changes in the number or conductance of postsynaptic receptors (e.g., an increased AMPA/ NMDA ratio as glutamatergic LTP). An induction of glutamatergic LTP and/or GABAergic LTD in VTA dopamine neurons will promote dopamine cell activity and increase dopamine release in VTA projection areas. On the other hand, an induction of glutamatergic LTD and/or GABAergic LTP in VTA dopamine neurons will dampen dopamine cell excitability and decrease dopamine release from the VTA Drugs of abuse including opiates can modulate dopamine release through induction of these different forms of plasticity.
Opiates and Glutamatergic Synaptic Plasticity in the VTA
Activation of glutamatergic input via AMPA and NMDA receptors expressed on dopamine neurons increases dopamine release. In fact, activation of pyramidal neurons of the PFC in vivo has been shown to promote bursting of VTA dopamine neurons.64 Other glutamatergic inputs to VTA dopamine neurons emanate from LDTg, lateral hypothalamus, and bed nucleus of stria terminalis (BNST).30 It is well documented that a single in vivo injection of morphine in rats increases the AMPA/NMDA ratio in VTA dopamine neurons (reflecting an induction of glutamatergic long-term potentiation, LTP) and promotes the insertion of GluA2 subunit-lacking calcium-permeable AMPA receptors at this synapse 24h after the treatment.45,50,65 Several forms of glutamatergic long-term depression (LTD) have also been described in the VTA,37,66–70 but it is yet to be determined whether these forms of activity-dependent glutamatergic plasticity are also targeted by opiates.
Opiates and GABAergic Synaptic Plasticity in the VTA
Regarding inhibitory plasticity, acute morphine acts on MOR to inhibit a form of presynaptic NMDA receptor-dependent GABAergic LTP (LTPGABA) by perturbing nitric oxide (NO)-guanylate cyclase signaling 24 h after treatment.40,71 Similar to addictive drugs, acute stress induces a similar neuroplasticity in the VTA (induction of glutamatergic LTP and inhibition of LTPGABA in VTA dopamine neurons),72,73 highlighting the role of stress in facilitation of drug reinstatement and relapse through priming synapses for further long-lasting drug-induced plasticity. Intriguingly, exposure to stress seems to inhibit LTPGABA through a MOR-independent mechanism that involves KORs. In vivo administration of a KOR inhibitor rescues LTPGABA in stressed animals.72 Moreover, constitutive activation of KOR following stress is required for maintaining the block of LTPGABA and for stress-induced reinstatement of cocaine-seeking.74 These studies emphasize the need for future investigations on the role of endogenous dynorphin (from NAc MSNs to the VTA) and KORs in opioid abuse and opiate-induced plasticity. Morphine is also able to modulate a form of postsynaptic LTD (LTDGABA) at GABAergic synapses onto VTA dopamine neurons. LTDGABA is expressed postsynaptically, dependent on D2 dopamine receptor activation, and requires the postsynaptic scaffolding A-kinase anchoring protein 79/150 (AKAP79/150) signaling complex which selectively regulates GABAergic synaptic transmission and strength through interaction with protein kinase A (PKA), and calcineurin.49,75 Generally, AKAP binding to PKA promotes insertion and stabilization of receptors at the synapse while calcineurin-induced dephosphorylation of receptors increases endocytosis and removal of receptors from the synapse. Similar to LTPGABA, morphine attenuates LTDGABA 24 h following a single injection.49 Our most recent data now suggest that morphine per se depresses GABAergic synaptic transmission onto VTA dopamine neurons both pre and post-synaptically.50 While the absence of LTDGABA that we observed after morphine administration may be related to an occlusion of this form of plasticity (due to prior morphine-induced reduction in postsynaptic GABAergic transmission), endocannabinoid (eCB) signaling seems to be involved in the decreased GABA release onto VTA dopamine neurons after morphine.50
There is also a strong possibility for a morphine-induced metaplasticity in which the morphine experience primes and modifies the subsequent induction of activity-dependent synaptic plasticity at both glutamatergic and GABAergic synapses onto VTA dopamine neurons. The concept of metaplasticity refers to plasticity of synaptic plasticity where capability of synapses to express subsequent LTP or LTD in response to induction protocols will be altered after exposure to a prior experience such as stress or drug experience. We have been able to demonstrate GABAergic metaplasticity in VTA dopamine neurons triggered by a severe early life stress event. GABAergic synapses onto VTA dopamine neurons are able to exhibit a physiological Hebbian form of plasticity, spike-timing dependent plasticity (STDP, including both LTP and LTD, Box 2, Figure 1) in VTA dopamine neurons. STDP requires NMDA receptor activation and is dependent on the AKAP150 signaling complex similar to LTDGABA.76,77 Interestingly, a single 24 h episode of maternal deprivation as a severe early life stress (which is known to significantly increase vulnerability to drug addiction) induces LTD and shifts STDP toward LTD at GABAergic synapses onto VTA dopamine neurons through epigenetic modifications of the AKAP scaffolding protein.77 Long-term changes in brain structure and plasticity that accompany exposure to drugs of abuse and severe stress require alterations in gene regulation. Both stress and addictive drugs are most likely enacting such transcriptional changes via epigenetic mechanisms. Such mechanisms including histone lysine-tail acetylation alter gene expression in neurons and are required for neuroplasticity underlying memory formation.78,79 We also found that histone deacetylase (HDAC) inhibition rescues GABAergic metaplasticity and restores normal AKAP signaling in maternally deprived rats, suggesting that early life stress-induced neuroplasticity in the VTA involves HDACs in the triggering of long-lasting GABAergic synaptic abnormalities and dopamine dysfunction that underlie susceptibility to addiction.77 Interestingly, acute morphine also increases HDAC2 expression and activity in VTA dopamine neurons and reduces histone H3 acetylation at lysine 9 (Ac-H3K9) in the VTA. Moreover, acute morphine-induced plasticity at both gluta-matergic and GABAergic synapses can be reversed by HDAC inhibition with an associated increase in acetylation of histone H3K9.50 Our results suggest that acute morphine- and early life stress-induced changes in VTA dopamine activity and synaptic plasticity engage HDAC2 activity locally in the VTA to maintain synaptic modifications through histone hypoacetylation (Figure 2). Whether morphine acting similarly to early life stress affects GABAergic STDP and induces metaplasticity in VTA synapses through changes in AKAP150 expression and signaling has yet to be determined. Moreover, transcriptional modulation of mechanisms governing AMPA and GABAA receptor trafficking in VTA dopamine neurons for maintenance of morphine- and stress-induced plasticities is completely unknown and merits further investigation. Orexin/hypocretin signaling in the VTA is also required for acute morphine-induced plasticity of dopamine neurons, representing another novel mechanism that may be subjected to epigenetic modifications by opioids.65
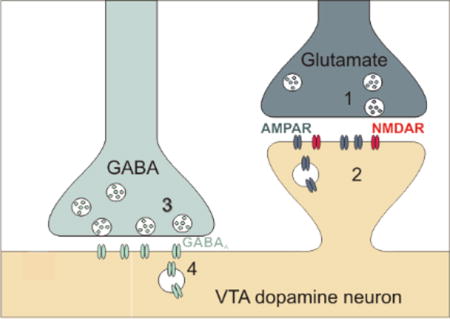
Figure 1.

Expression sites of plasticity at glutamatergic and GABAergic synapses onto VTA dopamine neurons indicated as 1–4.
Figure 2.

Epigenetic mechanisms supporting morphine-induced synaptic modifications in the VTA. Acute morphine increases HDAC2 activity in VTA dopamine neurons and reduces histone H3 acetylation at lysine 9 (Ac-H3K9) in the VTA 24 h following the injection. Morphine-induced synaptic changes at glutamatergic synapses involves eCB signaling to reduce GABAergic synaptic strength onto VTA dopamine neurons. Both plasticities can be recovered by a class I specific HDAC inhibitor (HDACi), through an increase in acetylation of histone H3K9. Epigenetic and synaptic modifications induced by morphine can promote dopamine neuron hyperexcitability, thereby increasing dopamine release in NAc. AKAP150 signaling controls the opposing effects of PKA and calcineurin (CaN) on GABAA receptor trafficking in the VTA. We assume that AKAP is a target for HDAC2-mediated transcriptional changes. Tilted ⊥ means inhibition. DA, dopamine; Ac, acetyl group; HDAC, histone deacetylase; HAT, histone acetyltransferase.
Taken together, it is clear that opioids and opiates target synaptic strength of glutamatergic and GABAergic synapses in the VTA and even their short-term actions result in long-lasting synaptic changes in this brain area that might significantly affect the susceptibility of synapses to further drug-induced plasticity with repeated exposures to drugs and stress. This in turn could result in persistent dysfunction of dopamine signaling from the VTA underlying many of the features of opiate addiction (Box 2).
CHRONIC OPIATE-INDUCED PLASTICITY IN THE VTA
Unfortunately, since our last review on the topic of opiate-induced plasticity in 2011,12 there is a serious lack of experimental data on the chronic effects of opiates on activity-dependent forms of synaptic plasticity (LTP, LTD, or STDP) at glutamatergic or GABAergic synapses in the VTA and other motivational brain circuits. Nevertheless, many similarities have been recognized between neuroadaptations in opioid dependence and withdrawal and the neural correlates of opiate-seeking and relapse.80–87 In this section, we will integrate and analyze some of the most recent data on opiate dependence, withdrawal, and addiction from a synaptic perspective to highlight how these opiate-induced synaptic modifications may underlie opiate addiction.
Based on the extensive literature on opiate dependence and withdrawal, it seems that chronic opiates potently influence glutamatergic and GABAergic function, and structural plasticity in the VTA through myriad interconnected processes, with the final outcome of decreased excitability of dopamine neurons underpinning aversive opiate withdrawal.
Opiates and Glutamatergic Synaptic Plasticity in the VTA
Withdrawal from a repeated morphine exposure results in increased AMPA receptor GluA1 subunit expression as well as increased NMDA receptor expression in the VTA, although it is assumed that depolarization-induced blockade following an excessive excitation of VTA dopamine neurons would decrease the activity of dopamine neurons and consequently diminish dopamine release.88 Morphine withdrawal also reduces presynaptic glutamate release onto VTA dopamine neurons through a metabotropic glutamate receptor, mGluR II-mediated mechanism.89
Opiates and GABAergic Synaptic Plasticity in the VTA
One of the most common forms of plasticity associated with chronic opiates is an increase in presynaptic GABA release on VTA dopamine neurons.12 Recent advances in optogenetic interrogation of neuronal circuitries and the availability of promoter driven Cre mouse and rat lines have begun to help addiction researchers to investigate the specific contributions of different synaptic inputs to the VTA and further delineate mechanisms underlying drug-induced plasticity in distinct VTA neural circuits. Importantly, engineering of new variants of channelrodopsins with the ability to activate individual synaptic inputs at higher frequencies is under way and will be instrumental for optical induction of synaptic plasticity for addiction-related studies. In fact, a recent study by Lüscher’s laboratory used retrograde tracing and optogenetics in transgenic mice to demonstrate the induction of an inhibitory plasticity at a distinct NAc inhibitory synapse onto VTA dopamine neurons following repeated exposure to cocaine in vivo. In this study, the authors have been able to induce a presynaptic LTP in response to high frequency optical stimulation of NAc D1-MSN GABAergic terminals onto VTA GABA neurons (iLTP at GABAA synapses).90 This plasticity is dependent on D1 receptor activation of the cyclic adenosine monophosphate-protein kinase A (cAMP-PKA) cascade. Repeated exposure to cocaine occludes iLTP suggesting that cocaine selectively induces this synaptic potentiation increasing GABA release from NAc D1-MSN GABAergic terminals onto VTA GABAergic neurons. This in turn results in long-lasting disinhibiton of dopamine neurons by cocaine thereby increasing firing of dopamine neurons which then facilitates the induction of locomotor sensitization and occludes CPP by cocaine.90 As mentioned earlier, there are two distinct inhibitory NAc projections from D1MSNs to the VTA which can result in either disinhibiton of dopamine neurons via GABAA receptors (this is where iLTP has been studied) or direct inhibition of dopamine neurons via GABAB receptors.60 Whether chronic opiates act similarly or differently compared to cocaine in induction of iLTP at NAc D1-MSN GABAA synapses onto VTA GABAergic is worth pursuing. Interestingly, KOR activation appears to have a strong influence on the function of D1 and D2MSNs in the NAc. Bonci’s team recently demonstrated a pathway-specific modulation of glutamatergic afferents to the NAc by KORs.91 They showed that a KOR agonist is able to induce LTD at glutamatergic synapses originating from the basolateral amygdala in the NAc. This LTD occurs on D1MSNs. On the other hand, KOR activation modulates synaptic transmission between MSNs, preferentially inducing LTD of inhibitory synapses onto D2MSNs from D1MSN collaterals. Overall, this pathway-and cell-specific KOR activation results in a change in the excitatory/inhibitory balance toward inhibition of D1MSNs and disinhibiton (excitation) of D2MSNs.91 This could then affect the inhibitory drive from NAc to the VTA (i.e., GABAA or GABAB signaling on GABAergic interneurons or dopamine neurons of the VTA, respectively). Whether acute or chronic exposure to opiates differentially dysregulates KOR signaling within the NAc and disrupts LTD at these distinct glutamatergic inputs onto D1 and D2MSNs is an important question, given that each of these distinct NAc-VTA circuits encode opposite motivational aspect of behavior (i.e., in general the D1 pathway seems to mediate positive reward while the D2 pathway encode negative reinforcement and aversion92). Most of the synaptic plasticity described above (e.g., glutamatergic LTP, LTDGABA, LTPGABA, inhibitory STDP) have been studied at synapses onto presumably dopamine neurons that project mainly to the shell of NAc (based on their anatomical location and physiological characteristics).40,49,77 Future optogenetic studies will enable us to verify whether these distinct inputs onto different subpopulation of VTA neurons are able to exhibit different forms of activity-dependent plasticity and how opiates would modulate the plasticity in a cell- and pathway-specific manner. Finally, it is important to bear in mind that drug-induced plasticity seems to be transient when the route of administration is passive even in response to repeated drug exposures,93 urging the field of addiction research to also incorporate robust and translationally relevant animal models of addiction such as rodent opioid self-administration for future studies of drug-induced plasticity.
Opiates, BDNF Signaling, and Structural Plasticity in the VTA
Consistent with the decreased activity of dopamine neurons during morphine withdrawal, a reduction in the size of dopamine neurons mediated through downregulation of the insulin receptor substrate 2 (IRS2)-thymoma viral proto-oncogene (Akt) signaling pathway in the VTA has been observed.94 This opiate-induced structural plasticity can be reversed by intra-VTA infusion of brain-derived neurotrophic factor (BDNF)95 suggesting that the disruption of BDNF signaling by opiates may be responsible for the structural changes in the VTA. This is not surprising as BDNF can positively modulate neuronal excitability, and synaptic transmission and expression is rapidly enhanced by neuronal activity.96 Increased BDNF signaling within the VTA could reverse opiate withdrawal-induced structural plasticity (VTA dopamine neuron shrinkage) and restore VTA dopamine activity. On the other hand, opiate-induced neuroplasticity may be supported by local VTA BDNF signaling since upregulation of BDNF expression is a common finding after the administration of many drugs of abuse in the mesolimbic system particularly during prolonged drug withdrawal.9,97,98 Consistently, recent work from van der Kooy’s team suggests that opiate withdrawal increases BDNF signaling and that the BDNF-induced neuronal plasticity mediates the aversive state of withdrawal. The authors demonstrate that local knockdown of the expression of BDNF receptor tropomyosin-receptor-kinase type B (TrkB) in the VTA blocks the shift from inhibitory to excitatory GABAA receptor signaling on GABAergic VTA neurons induced by chronic opiate administration and withdrawal suggesting a critical role for BDNF signaling in the establishment of an opiate-dependent state and aversive withdrawal motivation.99 However, Nestler’s team discovered a counteractive and opposite role for BDNF where suppression of BDNF-TrkB signaling in the VTA promotes the rewarding effects of morphine through changes in K+ channel expression and VTA dopamine neuron excitability.100 Significant differences in experimental procedures and levels of localized knockdown of BDNF and its receptor (which are not specific to VTA dopamine or GABAergic neurons in both studies) in addition to different behavioral paradigms and the heterogeneity of dopamine neuron populations in the VTA may explain the discrepancy. In line with the idea of VTA BDNF as a negative modulator of drug-induced reward,100 a recent work identified a novel mechanism involving chronic opiate activation of microglial-BDNF-TrkB signaling in the VTA augmenting inhibition of VTA dopamine neurons and blunting the rewarding effects of cocaine in opioid-dependent animals through a reduction in the expression and function of the KCl cotransporter KCC2 within VTA GABAergic neurons.101 Taken together, these studies highlight the potential importance of local BDNF signaling in the VTA in chronic opiate-induced plasticity that underlie negative and positive motivational aspects of opiate addiction. In spite of the critical role of BDNF in induction and maintenance of synaptic plasticity and in opiate addiction, evidence for modulation of BDNF signaling in different forms of activity-dependent synaptic plasticity (LTP, LTD, and STDP) in the VTA in response to morphine and other drugs of abuse is lacking.
CONCLUSION
In this review, we have attempted to highlight the literature regarding synaptic modifications induced by opiates and how these neuroadaptations contribute to long lasting changes in the VTA circuitry. Various synaptic and epigenetic mechanisms are targeted by opiates in the VTA. Studying the interaction of different forms of plasticity and their link to the genesis of addictive behavior in response to opiates is going to be a critical goal of future studies. The development of circuit based approaches such as optogenetic manipulations and the study of the link between plasticity and epigenetic modifications will allow further understanding of the precise mechanisms underlying drug and opiate addiction.
Acknowledgments
We are grateful to Dr. Brian Cox for helpful and critical discussions for the present work.
Funding
This work was supported by the National Institutes of Health (NIH)-National Institute of Drugs of Abuse (NIDA) Grant R01 DA039533 to F.S.N.
ABBREVIATIONS
- OUD
Opioid use disorder
- VTA
ventral tegmental area
- NAc
nucleus accumbens
- PFC
prefrontal cortex
- LDTg
laterodorsal tegmentum
- RMTg
rostromedial tegmental area
- LHb
lateral habenula
- GIRKs
G protein inwardly rectifying K+ channels
- MSNs
medium spiny neurons
- BNST
bed nucleus of stria terminalis
- LTP
Long-term potentiation
- LTD
long-term depression
- AKAP79/150
A-kinase anchoring protein 79/150
- eCB
endocannabinoids
- PKA
protein kinase A
- STDP
spike-timing dependent plasticity
- MOR
Mu-opioid receptor
- DOR
delta-opioid receptor
- KOR
Kappa-opioid receptor
- HDAC
histone deacetylase
- BDNF
brain-derived neurotrophic factor
Footnotes
Author Contributions
L.D.L. and F.S.N. prepared the figures and wrote the paper.
The opinions and assertions contained herein are the private opinions of the authors and are not to be construed as official or reflecting the views of the Uniformed Services University of the Health Sciences or the Department of Defense or the Government of the United States. The funding agencies did not contribute to writing this article or deciding to submit it.
The authors declare no competing financial interest.
References
- 1.Wise RA, Koob GF. The development and maintenance of drug addiction. Neuropsychopharmacology. 2014;39:254–262. doi: 10.1038/npp.2013.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schuckit MA. Treatment of Opioid-Use Disorders. N. Engl. J. Med. 2016;375:357–368. doi: 10.1056/NEJMra1604339. [DOI] [PubMed] [Google Scholar]
- 3.Volkow ND, Collins FS. The Role of Science in Addressing the Opioid Crisis. N. Engl. J. Med. 2017;377:391. doi: 10.1056/NEJMsr1706626. [DOI] [PubMed] [Google Scholar]
- 4.Jones CM. Heroin use and heroin use risk behaviors among nonmedical users of prescription opioid pain relievers - United States, 2002–2004 and 2008–2010. Drug Alcohol Depend. 2013;132:95–100. doi: 10.1016/j.drugalcdep.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 5.Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu. Rev. Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- 6.Pignatelli M, Bonci A. Role of Dopamine Neurons in Reward and Aversion: A Synaptic Plasticity Perspective. Neuron. 2015;86:1145–1157. doi: 10.1016/j.neuron.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 7.Wolf ME. Addiction: making the connection between behavioral changes and neuronal plasticity in specific pathways. Mol. Interventions. 2002;2:146–157. doi: 10.1124/mi.2.3.146. [DOI] [PubMed] [Google Scholar]
- 8.Harnett MT, Bernier BE, Ahn KC, Morikawa H. Burst-timing-dependent plasticity of NMDA receptor-mediated transmission in midbrain dopamine neurons. Neuron. 2009;62:826–838. doi: 10.1016/j.neuron.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat. Rev. Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- 10.Gerdeman GL, Partridge JG, Lupica CR, Lovinger DM. It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci. 2003;26:184–192. doi: 10.1016/S0166-2236(03)00065-1. [DOI] [PubMed] [Google Scholar]
- 11.Kasanetz F, Deroche-Gamonet V, Berson N, Balado E, Lafourcade M, Manzoni O, Piazza PV. Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science (Washington, DC, U. S.) 2010;328:1709–1712. doi: 10.1126/science.1187801. [DOI] [PubMed] [Google Scholar]
- 12.Dacher M, Nugent FS. Opiates and plasticity. Neuropharmacology. 2011;61:1088–1096. doi: 10.1016/j.neuropharm.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 13.Nutt DJ, Lingford-Hughes A, Erritzoe D, Stokes PR. The dopamine theory of addiction: 40 years of highs and lows. Nat. Rev. Neurosci. 2015;16:305–312. doi: 10.1038/nrn3939. [DOI] [PubMed] [Google Scholar]
- 14.Kourrich S, Calu DJ, Bonci A. Intrinsic plasticity: an emerging player in addiction. Nat. Rev. Neurosci. 2015;16:173–184. doi: 10.1038/nrn3877. [DOI] [PubMed] [Google Scholar]
- 15.Gipson CD, Kupchik YM, Kalivas PW. Rapid, transient synaptic plasticity in addiction. Neuropharmacology. 2014;76:276–286. doi: 10.1016/j.neuropharm.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.George O, Le Moal M, Koob GF. Allostasis and addiction: role of the dopamine and corticotropin-releasing factor systems. Physiol. Behav. 2012;106:58–64. doi: 10.1016/j.physbeh.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koob GF, Volkow ND. Neuropsychopharmacology. 2010;35:217. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Volkow ND, Fowler JS, Wang GJ. The addicted human brain viewed in the light of imaging studies: brain circuits and treatment strategies. Neuropharmacology. 2004;47:3–13. doi: 10.1016/j.neuropharm.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 19.Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the ’dark side’ of drug addiction. Nat. Neurosci. 2005;8:1442–1444. doi: 10.1038/nn1105-1442. [DOI] [PubMed] [Google Scholar]
- 20.Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- 21.Koob GF, Stinus L, Le Moal M, Bloom FE. Opponent process theory of motivation: neurobiological evidence from studies of opiate dependence. Neurosci. Biobehav. Rev. 1989;13:135–140. doi: 10.1016/s0149-7634(89)80022-3. [DOI] [PubMed] [Google Scholar]
- 22.Fields HL, Margolis EB. Understanding opioid reward. Trends Neurosci. 2015;38:217–225. doi: 10.1016/j.tins.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pignatelli M, Bonci A. Role of Dopamine Neurons in Reward and Aversion: A Synaptic Plasticity Perspective. Neuron. 2015;86:1145–1157. doi: 10.1016/j.neuron.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 24.Grueter BA, Rothwell PE, Malenka RC. Integrating synaptic plasticity and striatal circuit function in addiction. Curr. Opin. Neurobiol. 2012;22:545–551. doi: 10.1016/j.conb.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zlebnik NE, Cheer JF. Drug-Induced Alterations of Endocannabinoid-Mediated Plasticity in Brain Reward Regions. J. Neurosci. 2016;36:10230–10238. doi: 10.1523/JNEUROSCI.1712-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen BT, Hopf FW, Bonci A. Synaptic plasticity in the mesolimbic system: therapeutic implications for substance abuse. Ann. N. Y. Acad. Sci. 2010;1187:129–139. doi: 10.1111/j.1749-6632.2009.05154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. U. S. A. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res. Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- 30.Lammel S, Lim BK, Malenka RC. Reward and aversion in a heterogeneous midbrain dopamine system. Neuropharmacology. 2014;76:351–359. doi: 10.1016/j.neuropharm.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daglish MR, Williams TM, Wilson SJ, Taylor LG, Eap CB, Augsburger M, Giroud C, Brooks DJ, Myles JS, Grasby P, Lingford-Hughes AR, Nutt DJ. Brain dopamine response in human opioid addiction. Br. J. Psychiatry. 2008;193:65–72. doi: 10.1192/bjp.bp.107.041228. [DOI] [PubMed] [Google Scholar]
- 32.Watson BJ, Taylor LG, Reid AG, Wilson SJ, Stokes PR, Brooks DJ, Myers JF, Turkheimer FE, Nutt DJ, Lingford-Hughes AR. Investigating expectation and reward in human opioid addiction with [(11) C]raclopride PET. Addict Biol. 2014;19:1032–1040. doi: 10.1111/adb.12073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zijlstra F, Veltman DJ, Booij J, van den Brink W, Franken IH. Neurobiological substrates of cue-elicited craving and anhedonia in recently abstinent opioid-dependent males. Drug Alcohol Depend. 2009;99:183–192. doi: 10.1016/j.drugalcdep.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 34.Dobi A, Margolis EB, Wang HL, Harvey BK, Morales M. Glutamatergic and nonglutamatergic neurons of the ventral tegmental area establish local synaptic contacts with dopaminergic and nondopaminergic neurons. J. Neurosci. 2010;30:218–229. doi: 10.1523/JNEUROSCI.3884-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hnasko TS, Hjelmstad GO, Fields HL, Edwards RH. Ventral tegmental area glutamate neurons: electro-physiological properties and projections. J. Neurosci. 2012;32:15076–15085. doi: 10.1523/JNEUROSCI.3128-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laviolette SR, Gallegos RA, Henriksen SJ, van der Kooy D. Opiate state controls bi-directional reward signaling via GABAA receptors in the ventral tegmental area. Nat. Neurosci. 2004;7:160–169. doi: 10.1038/nn1182. [DOI] [PubMed] [Google Scholar]
- 37.Bellone C, Luscher C. Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat. Neurosci. 2006;9:636–641. doi: 10.1038/nn1682. [DOI] [PubMed] [Google Scholar]
- 38.Borgland SL, Malenka RC, Bonci A. Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J. Neurosci. 2004;24:7482–7490. doi: 10.1523/JNEUROSCI.1312-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dong Y, Saal D, Thomas M, Faust R, Bonci A, Robinson T, Malenka RC. Cocaine-induced potentiation of synaptic strength in dopamine neurons: behavioral correlates in GluRA(−/−) mice. Proc. Natl. Acad. Sci. U. S. A. 2004;101:14282–14287. doi: 10.1073/pnas.0401553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nugent FS, Penick EC, Kauer JA. Opioids block long-term potentiation of inhibitory synapses. Nature. 2007;446:1086–1090. doi: 10.1038/nature05726. [DOI] [PubMed] [Google Scholar]
- 41.Faleiro LJ, Jones S, Kauer JA. Rapid synaptic plasticity of glutamatergic synapses on dopamine neurons in the ventral tegmental area in response to acute amphetamine injection. Neuropsychopharmacology. 2004;29:2115–2125. doi: 10.1038/sj.npp.1300495. [DOI] [PubMed] [Google Scholar]
- 42.Guan YZ, Ye JH. Ethanol Blocks Long-Term Potentiation of GABAergic Synapses in the Ventral Tegmental Area Involving mu-Opioid Receptors. Neuropsychopharmacology. 2010;35:1841. doi: 10.1038/npp.2010.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–357. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- 44.Melis M, Camarini R, Ungless MA, Bonci A. Long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J. Neurosci. 2002;22:2074–2082. doi: 10.1523/JNEUROSCI.22-06-02074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 46.Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- 47.Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–1031. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron. 2006;49:589–601. doi: 10.1016/j.neuron.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 49.Dacher M, Nugent FS. Morphine-induced modulation of LTD at GABAergic synapses in the ventral tegmental area. Neuropharmacology. 2011;61:1166–1171. doi: 10.1016/j.neuropharm.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 50.Authement ME, Langlois LD, Kassis H, Gouty S, Dacher M, Shepard RD, Cox BM, Nugent FS. Morphine-induced synaptic plasticity in the VTA is reversed by HDAC inhibition. J. Neurophysiol. 2016;116:1093–1103. doi: 10.1152/jn.00238.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J. Neurosci. 1992;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matsui A, Williams JT. Opioid-Sensitive GABA Inputs from Rostromedial Tegmental Nucleus Synapse onto Midbrain Dopamine Neurons. J. Neurosci. 2011;31:17729–17735. doi: 10.1523/JNEUROSCI.4570-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jalabert M, Bourdy R, Courtin J, Veinante P, Manzoni OJ, Barrot M, Georges F. Neuronal circuits underlying acute morphine action on dopamine neurons. Proc. Natl. Acad. Sci. U. S. A. 2011;108:16446–16450. doi: 10.1073/pnas.1105418108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jhou TC, Fields HL, Baxter MG, Saper CB, Holland PC. The rostromedial tegmental nucleus (RMTg), a GABAergic afferent to midbrain dopamine neurons, encodes aversive stimuli and inhibits motor responses. Neuron. 2009;61:786–800. doi: 10.1016/j.neuron.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stamatakis AM, Jennings JH, Ung RL, Blair GA, Weinberg RJ, Neve RL, Boyce F, Mattis J, Ramakrishnan C, Deisseroth K, Stuber GD. A unique population of ventral tegmental area neurons inhibits the lateral habenula to promote reward. Neuron. 2013;80:1039–1053. doi: 10.1016/j.neuron.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Margolis EB, Fields HL. Mu Opioid Receptor Actions in the Lateral Habenula. PLoS One. 2016;11:e0159097. doi: 10.1371/journal.pone.0159097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Root DH, Mejias-Aponte CA, Zhang S, Wang HL, Hoffman AF, Lupica CR, Morales M. Single rodent mesohabenular axons release glutamate and GABA. Nat. Neurosci. 2014;17:1543–1551. doi: 10.1038/nn.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shabel SJ, Proulx CD, Piriz J, Malinow R. Mood regulation. GABA/glutamate co-release controls habenula output and is modified by antidepressant treatment. Science (Washington, DC, U. S.) 2014;345:1494–1498. doi: 10.1126/science.1250469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J. Neurosci. 2006;26:2788–2797. doi: 10.1523/JNEUROSCI.4331-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Edwards NJ, Tejeda HA, Pignatelli M, Zhang S, McDevitt RA, Wu J, Bass CE, Bettler B, Morales M, Bonci A. Circuit specificity in the inhibitory architecture of the VTA regulates cocaine-induced behavior. Nat. Neurosci. 2017;20:438–448. doi: 10.1038/nn.4482. [DOI] [PubMed] [Google Scholar]
- 61.Margolis EB, Lock H, Chefer VI, Shippenberg TS, Hjelmstad GO, Fields HL. Kappa opioids selectively control dopaminergic neurons projecting to the prefrontal cortex. Proc. Natl. Acad. Sci. U. S. A. 2006;103:2938–2942. doi: 10.1073/pnas.0511159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Margolis EB, Hjelmstad GO, Bonci A, Fields HL. Kappa-opioid agonists directly inhibit midbrain dopaminergic neurons. J. Neurosci. 2003;23:9981–9986. doi: 10.1523/JNEUROSCI.23-31-09981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Margolis EB, Hjelmstad GO, Fujita W, Fields HL. Direct Bidirectional mu-Opioid Control of Midbrain Dopamine Neurons. J. Neurosci. 2014;34:14707–14716. doi: 10.1523/JNEUROSCI.2144-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tong ZY, Overton PG, Clark D. Stimulation of the prefrontal cortex in the rat induces patterns of activity in midbrain dopaminergic neurons which resemble natural burst events. Synapse (Hoboken, NJ, U. S.) 1996;22:195–208. doi: 10.1002/(SICI)1098-2396(199603)22:3<195::AID-SYN1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 65.Baimel C, Borgland SL. Orexin Signaling in the VTA Gates Morphine-Induced Synaptic Plasticity. J. Neurosci. 2015;35:7295–7303. doi: 10.1523/JNEUROSCI.4385-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bellone C, Luscher C. mGluRs induce a long-term depression in the ventral tegmental area that involves a switch of the subunit composition of AMPA receptors. European journal of neuroscience. 2005;21:1280–1288. doi: 10.1111/j.1460-9568.2005.03979.x. [DOI] [PubMed] [Google Scholar]
- 67.Jones S, Kornblum JL, Kauer JA. Amphetamine blocks long-term synaptic depression in the ventral tegmental area. J. Neurosci. 2000;20:5575–5580. doi: 10.1523/JNEUROSCI.20-15-05575.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thomas MJ, Malenka RC, Bonci A. Modulation of long-term depression by dopamine in the mesolimbic system. J. Neurosci. 2000;20:5581–5586. doi: 10.1523/JNEUROSCI.20-15-05581.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gutlerner JL, Penick EC, Snyder EM, Kauer JA. Novel protein kinase A-dependent long-term depression of excitatory synapses. Neuron. 2002;36:921–931. doi: 10.1016/s0896-6273(02)01051-6. [DOI] [PubMed] [Google Scholar]
- 70.Haj-Dahmane S, Shen RY. Regulation of plasticity of glutamate synapses by endocannabinoids and the cyclic-AMP/protein kinase A pathway in midbrain dopamine neurons. J. Physiol. 2010;588:2589–2604. doi: 10.1113/jphysiol.2010.190066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nugent FS, Niehaus JL, Kauer JA. PKG and PKA Signaling in LTP at GABAergic Synapses. Neuropsychopharmacology. 2009;34:1829–1842. doi: 10.1038/npp.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Graziane NM, Polter AM, Briand LA, Pierce RC, Kauer JA. Kappa opioid receptors regulate stress-induced cocaine seeking and synaptic plasticity. Neuron. 2013;77:942–954. doi: 10.1016/j.neuron.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Niehaus JL, Murali M, Kauer JA. Drugs of abuse and stress impair LTP at inhibitory synapses in the ventral tegmental area. European journal of neuroscience. 2010;32:108–117. doi: 10.1111/j.1460-9568.2010.07256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Polter AM, Barcomb K, Chen RW, Dingess PM, Graziane NM, Brown TE, Kauer JA. Constitutive activation of kappa opioid receptors at ventral tegmental area inhibitory synapses following acute stress. eLife. 2017;6:e23785. doi: 10.7554/eLife.23785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dacher M, Gouty S, Dash S, Cox BM, Nugent FS. A-kinase anchoring protein-calcineurin signaling in long-term depression of GABAergic synapses. J. Neurosci. 2013;33:2650–2660. doi: 10.1523/JNEUROSCI.2037-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kodangattil JN, Dacher M, Authement ME, Nugent FS. Spike timing-dependent plasticity at GABAergic synapses in the ventral tegmental area. J. Physiol. 2013;591:4699–4710. doi: 10.1113/jphysiol.2013.257873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Authement ME, Kodangattil JN, Gouty S, Rusnak M, Symes AJ, Cox BM, Nugent FS. Histone Deacetylase Inhibition Rescues Maternal Deprivation-Induced GABAergic Meta-plasticity through Restoration of AKAP Signaling. Neuron. 2015;86:1240–1252. doi: 10.1016/j.neuron.2015.05.024. [DOI] [PubMed] [Google Scholar]
- 78.Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr. Opin. Pharmacol. 2008;8:57–64. doi: 10.1016/j.coph.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haggarty SJ, Tsai LH. Probing the role of HDACs and mechanisms of chromatin-mediated neuroplasticity. Neurobiol. Learn. Mem. 2011;96:41–52. doi: 10.1016/j.nlm.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Christie MJ. Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. British journal of pharmacology. 2008;154:384–396. doi: 10.1038/bjp.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol. Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- 82.De Vries TJ, Shippenberg TS. Neural systems underlying opiate addiction. J. Neurosci. 2002;22:3321–3325. doi: 10.1523/JNEUROSCI.22-09-03321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brown RM, Lawrence AJ. Neurochemistry underlying relapse to opiate seeking behaviour. Neurochem. Res. 2009;34:1876–1887. doi: 10.1007/s11064-009-9967-y. [DOI] [PubMed] [Google Scholar]
- 84.Bossert JM, Ghitza UE, Lu L, Epstein DH, Shaham Y. Neurobiology of relapse to heroin and cocaine seeking: an update and clinical implications. Eur. J. Pharmacol. 2005;526:36–50. doi: 10.1016/j.ejphar.2005.09.030. [DOI] [PubMed] [Google Scholar]
- 85.Martini L, Whistler JL. The role of mu opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr. Opin. Neurobiol. 2007;17:556–564. doi: 10.1016/j.conb.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 86.von Zastrow M. Regulation of opioid receptors by endocytic membrane traffic: mechanisms and translational implications. Drug Alcohol Depend. 2010;108:166–171. doi: 10.1016/j.drugalcdep.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aston-Jones G, Harris GC. Brain substrates for increased drug seeking during protracted withdrawal. Neuropharmacology. 2004;47:167–179. doi: 10.1016/j.neuropharm.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 88.Fitzgerald LW, Ortiz J, Hamedani AG, Nestler EJ. Drugs of abuse and stress increase the expression of GluR1 and NMDAR1 glutamate receptor subunits in the rat ventral tegmental area: common adaptations among cross-sensitizing agents. J. Neurosci. 1996;16:274–282. doi: 10.1523/JNEUROSCI.16-01-00274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Manzoni OJ, Williams JT. Presynaptic regulation of glutamate release in the ventral tegmental area during morphine withdrawal. J. Neurosci. 1999;19:6629–6636. doi: 10.1523/JNEUROSCI.19-15-06629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bocklisch C, Pascoli V, Wong JC, House DR, Yvon C, de Roo M, Tan KR, Lüscher C. Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science (Washington, DC, U. S.) 2013;341:1521–1525. doi: 10.1126/science.1237059. [DOI] [PubMed] [Google Scholar]
- 91.Tejeda HA, Wu J, Kornspun AR, Pignatelli M, Kashtelyan V, Krashes MJ, Lowell BB, Carlezon WA, Jr, Bonci A. Pathway- and Cell-Specific Kappa-Opioid Receptor Modulation of Excitation-Inhibition Balance Differentially Gates D1 and D2 Accumbens Neuron Activity. Neuron. 2017;93:147–163. doi: 10.1016/j.neuron.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hikida T, Yawata S, Yamaguchi T, Danjo T, Sasaoka T, Wang Y, Nakanishi S. Pathway-specific modulation of nucleus accumbens in reward and aversive behavior via selective transmitter receptors. Proc. Natl. Acad. Sci. U. S. A. 2013;110:342–347. doi: 10.1073/pnas.1220358110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM, Chou JK, Bonci A. Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron. 2008;59:288–297. doi: 10.1016/j.neuron.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Russo SJ, Bolanos CA, Theobald DE, DeCarolis NA, Renthal W, Kumar A, Winstanley CA, Renthal NE, Wiley MD, Self DW, Russell DS, Neve RL, Eisch AJ, Nestler EJ. IRS2-Akt pathway in midbrain dopamine neurons regulates behavioral and cellular responses to opiates. Nat. Neurosci. 2007;10:93–99. doi: 10.1038/nn1812. [DOI] [PubMed] [Google Scholar]
- 95.Sklair-Tavron L, Shi WX, Lane SB, Harris HW, Bunney BS, Nestler EJ. Chronic morphine induces visible changes in the morphology of mesolimbic dopamine neurons. Proc. Natl. Acad. Sci. U. S. A. 1996;93:11202–11207. doi: 10.1073/pnas.93.20.11202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Russo SJ, Mazei-Robison MS, Ables JL, Nestler EJ. Neurotrophic factors and structural plasticity in addiction. Neuropharmacology. 2009;56:73–82. doi: 10.1016/j.neuropharm.2008.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vargas-Perez H, Ting-A-Kee R, Walton CH, Hansen DM, Razavi R, Clarke L, Bufalino MR, Allison DW, Steffensen SC, van der Kooy D. Ventral tegmental area BDNF induces an opiate-dependent-like reward state in naive rats. Science (Washington, DC, U. S.) 2009;324:1732–1734. doi: 10.1126/science.1168501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thomas MJ, Malenka RC. Synaptic plasticity in the mesolimbic dopamine system. Philos. Trans. R. Soc., B. 2003;358:815–819. doi: 10.1098/rstb.2002.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vargas-Perez H, Bahi A, Bufalino MR, Ting-A-Kee R, Maal-Bared G, Lam J, Fahmy A, Clarke L, Blanchard JK, Larsen BR, Steffensen S, Dreyer JL, van der Kooy D. BDNF Signaling in the VTA Links the Drug-Dependent State to Drug Withdrawal Aversions. J. Neurosci. 2014;34:7899–7909. doi: 10.1523/JNEUROSCI.3776-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koo JW, Mazei-Robison MS, Chaudhury D, Juarez B, LaPlant Q, Ferguson D, Feng J, Sun H, Scobie KN, Damez-Werno D, Crumiller M, Ohnishi YN, Ohnishi YH, Mouzon E, Dietz DM, Lobo MK, Neve RL, Russo SJ, Han MH, Nestler EJ. BDNF is a negative modulator of morphine action. Science (Washington, DC, U. S.) 2012;338:124–128. doi: 10.1126/science.1222265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Taylor AM, Castonguay A, Ghogha A, Vayssiere P, Pradhan AA, Xue L, Mehrabani S, Wu J, Levitt P, Olmstead MC, De Koninck Y, Evans CJ, Cahill CM. Neuroimmune Regulation of GABAergic Neurons Within the Ventral Tegmental Area During Withdrawal from Chronic Morphine. Neuropsychopharmacology. 2016;41:949–959. doi: 10.1038/npp.2015.221. [DOI] [PMC free article] [PubMed] [Google Scholar]