

Abstract
Colorectal cancer (CRC) is the third most common solid internal malignancy among cancers. Early detection of cancer is key to increasing the survival rate of colorectal cancer patients. Overexpression of the EGFR protein is associated with CRC. We have designed a series of peptides that are highly specific for the extracellular domain of EGFR, based on our earlier studies on linear peptides. The previously reported linear peptide LARLLT, known to bind to EGFR, was modified with the goals of increasing its stability and its specificity towards EGFR. Peptide modifications, including D-amino acid substitution, cyclization, and chain reversal, were investigated. In addition, to facilitate labeling of the peptide with a fluorescent dye, an additional lysine residue was introduced onto the linear (KLARLLT) and cyclic peptides cyclo(KLARLLT) (Cyclo.L1). The lysine residue was also converted into an azide group in both a linear and reversed cyclic peptide sequences cyclo(K(N3)larllt)(Cyclo.L1.1) to allow for subsequent “click” conjugation. The cyclic peptides showed enhanced binding to EGFR by SPR. NMR and molecular modeling studies suggest that the peptides acquire a β-turn structure in solution. In vitro stability studies in human serum show that the cyclic peptide is more stable than the linear peptide.
Keywords: EGFR extracellular domain, cyclic peptide, linear peptide, surface plasmon resonance, colorectal cancer
Graphical Abstract
Based on the linear peptides that have an affinity for EGFR extracellular domain, cyclic peptides with enhanced affinity and stability were designed. Cyclic peptides exhibited serum stability and binding to EGFR protein. Such peptides can be conjugated with fluorescent labels for imaging EGFR overexpressed colon cancer.
Epidermal growth factor receptor (EGFR) has been implicated in tumor-specific targeted therapy and diagnosis. EGFR overexpression is frequently found in breast, lung, colon and ovarian cancers (1). Among the different types of cancers, colorectal cancer (CRC) is the third most common solid internal malignancy. Over 50,000 deaths in the U.S. are attributed to this disease, making it second only to lung cancer in U.S. cancer mortality (2). Although recent studies suggest that the overall CRC incidence in the U.S. is decreasing, driven by screening, the incidence in young adults under the age of 50 years in still increasing (3). Disease progression of colon cancer is typically slow and happens in a linear fashion from adenomatous polyps to carcinoma. Early stage detection by screening and removal of polyp adenomas can reduce the incidence of CRC by about 80% (2). Indeed, the advent of broader population screening has largely led to the improvement in mortality observed during the past 20 years (2). Since EGFR has a strong association with several cancers including CRC, with approximately 97% detected in all of the colon cancers, it is a convenient target for ligands, such as peptides and antibodies, for targeted drug or gene delivery. EGFR is over-expressed even on small cancers (< 5 mm) and on the flat, dysplastic, aberrant crypt foci that are believed to precede cancer development (4). These are the types of lesions most often missed by standard colonoscopy (5).
We have recently prepared and investigated EGFR-targeted porphyrin-peptide and phthalocyanine-peptide conjugates (6, 7). Two small peptides with sequences LARLLT (L1) and YHWYGYTPQNVI (L2) were used in these studies, due to their reported ability for EGFR targeting, both in vitro and in vivo (8, 9). The conjugates with L1 linked via a low molecular weight PEG spacer showed enhanced water solubility compared with the conjugates to the longer hydrophobic peptide. Furthermore, the LARLLT-bearing conjugates showed higher EGFR targeting ability, accumulating in EGFR over-expressing cells up to 17-fold compared with unconjugated fluorophore. These results suggest that fluorophore-LARLLT conjugates have substantially increased EGFR-targeting ability, and could be very useful for the early detection and diagnosis of CRC. Our previous studies also showed that the peptide L1 conjugates bind to EGFR with higher affinity compared with the L2-based conjugates (7). However, L1 is a linear peptide with limited in vivo stability (10, 11). Several strategies exist to improve the stability of peptides in vivo, including cyclization, N- and C-termini modification, and D-amino acid substitution (12, 13). In the present work, we investigated several strategies for modification of the L1 peptide. In addition to enhanced in vivo stability, we also introduced functionalization for easy conjugation to fluorophores, via the addition of a lysine residue or an azide-transformed lysine that can be used in “click” conjugations (14, 15). We investigated the EGFR-binding ability of the resulting peptides by surface plasmon resonance (SPR) and molecular docking studies. Our results reveal that the cyclic D-amino acid version of the linear L1 peptide binds to EGFR with higher affinity, and it is more stable in human serum than the linear peptide.
Materials and Methods
Materials
All the chemicals, reagents, solvents and cell lines were from commercial sources. Fmoc-protected amino acids, 2-(6-chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HCTU) and trifluoroacetic acid (TFA) were purchased from Advanced ChemTech, Louisville, KY. Chlorotrityl chloride resin (CTC) was purchased from ChemImpex, Wood Dale, IL. Diisopropylethyl amine (DIEA), methanol (MeOH), chloroform, acetic acid and azidoacetic acid were purchased from Sigma-Aldrich, St. Louis, MO. Dimethylformamide (DMF), dichloromethane (DCM) and triisopropylsilane (TIPS) were purchased from Protein Technologies, Tucson, AZ., N-[(Dimethylamino)-1H-1,2,3-triazolo-[4,5-b]pyridin-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate N-oxide (HATU) was purchased from ChemPep (Wellington, FL).
General Synthesis Procedure
All linear peptides were synthesized using solid phase peptide synthesis (SPPS) and Fmoc chemistry. Peptides were synthesized using Fmoc-PAL-PEG-PS on a 0.2 mmol scale, and a 4-fold excess of the Fmoc protected amino acids in the presence of HOBt and TBTU as the activating agents were used for SPPS. Final deprotection of Fmoc from the last amino acid was achieved by washing the peptidic resin beads five times with DMF and six times with DCM, followed by drying under vacuum for 6 hours. Peptide was cleaved from the resin using a cocktail consisting of 94% TFA/2.5% liquid phenol/2.5% water/1% TIPS. After washing, cold diethyl ether was added to the mixture to precipitate the peptide, and the mixture centrifuged and lyophilized. Peptides were purified using reversed-phase HPLC (Waters, MA USA). Analytical HPLC was carried out using a XBridge, 4.6 mm × 250 mm (Waters, MA, USA) column with a stepwise gradient. The purity of the peptides was determined by HPLC. A general procedure for the synthesis of peptides is provided in Scheme 1.
Scheme 1.

General synthesis scheme for linear and cyclic peptides.
Synthesis of peptides L1, and L1.1 – L1.7
Title compounds were synthesized on a Tribute peptide synthesizer (Protein Technologies, Tucson, AZ) utilizing a standard Fmoc peptide chemistry protocol on a 125 μmol scale using the previously loaded H-(d)Leu-CTC resin. Side-chain functionalities were protected with tert-butyl (Thr) and NG-2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl (Arg). Five fold excess of Fmoc-amino acids and HCTU, in the presence of 10 equivalents of DIEA were used for each of the coupling steps (10 min) with DMF as the solvent. After the synthesis of each sequence was complete, the final Fmoc groups were removed using 20% piperidine in DMF (1 × 5 min; 1 × 10 min). The resin from each synthesis was washed with DMF (5 × 30 sec) and DCM (5 × 30 sec). In the case of L1.4, azidoacetic acid was coupled using the same procedure as other amino acids. The peptides were simultaneously side chain deprotected and cleaved from the resin using TFA:TIPS:water (95:2.5:2.5) for 2 hours, then filtered to remove the resin. Cold diethyl ether was then added to the peptide solutions to precipitate the crude peptides. The peptides were centrifuged for 10 minutes (10,000 rpm) and the ether layers decanted. Fresh cold diethyl ether was added, and the pelleted peptides were re-suspended. The peptides were centrifuged again, and the procedure was repeated 5 times for each peptide. Each peptide pellet was dissolved in 5 mL of water containing 0.1% TFA, frozen and lyophilized to yield the crude peptide powders. Peptides were purified as described in section 2.2.
L1: Peptide Sequence: LARLLT-CONH2
Confirmation by MS(MALDI-TOF): m/z 685.390; calc’d. for C31H61N10O7+ 685.891. 1H NMR (500 MHz, DMSO-d6, 308K): δ 8.65 (d, J= 7.4 Hz, 2H), 8.10 (d, J = 7.9 Hz, 4H), 7.91 (d, J = 8.1 Hz, 2H), 7.52 (t, J = 5.8 Hz, 1H), 7.44 (d, J = 8.5 Hz, 2H), 7.09 (d, J = 9.0 Hz, 3H), 4.41 (t, J = 7.2 Hz, 2H), 4.33 (t, J = 7.8 Hz, 3H), 4.27 (d, J = 7.1 Hz, 1H), 4.07 (dd, J = 8.4 Hz, J= 3.3 Hz, 2H), 4.02 (s, 1H), 3.80 (s, 1H), 2.53-1.55 (m, 9H), 1.57-1.38 (m, 8H), 1.24 (d, J = 6.7 Hz, 5H), 1.00 (d, J = 6.3 Hz, 3H), 0.98-.52 (m, 18H).
L1.1: Peptide Sequence: larllt-COOH
Confirmation by MS (MALDI-TOF): m/z 686.451; calc’d for C31H60N9O8+ 686.875. 1H NMR (500 MHz, DMSO-d6) δ 8.71 (d, J = 7.5 Hz, 1H), 8.15 (d, J = 8.4 Hz, 1H), 7.97 (d, J = 8.6 Hz, 1H), 7.55 (s, 1H), 7.25 (s, 1H), 4.51-3.94 (m, 6H), 3.85-3.77 (m, 1H), 3.09 (t, J = 6.6 Hz, 2H), 1.72 (d, J = 13.4 Hz, 1H), 1.72-1.57 (m, 4H), 1.56-1.37 (m, 9H), 1.26 (d, J = 7.1 Hz, 3H), 1.03 (d, J = 6.1 Hz, 3H), 0.97-0.74 (m, 18H).
L1.2: Peptide Sequence: tllral-COOH
Confirmation by MS (MALDI-TOF): m/z 686.451; calc’d for C31H60N9O8+ 686.875. 1H NMR (500 MHz, DMSO-d6) δ 8.60 (d, J = 8.3 Hz, 1H), 8.27 (d, J = 8.2 Hz, 1H), 8.09 (s, 1H), 7.93 (d, J = 7.8 Hz, 1H), 7.25 (s, 1H), 4.41 (dd, J = 9.4 Hz, J = 5.7 Hz, 1H), 4.37-3.94 (m, 4H), 3.92-3.64 (m, 1H), 3.57 (d, J = 7.6 Hz, 1H), 3.20-2.91 (m, 2H), 1.73 (s, 1H), 1.71-1.57 (m, 4H), 1.59-1.32 (m, 9H), 1.24 (d, J = 7.1 Hz, 3H), 1.15 (d, J = 6.3 Hz, 3H), 1.00-0.58 (m, 18H).
L1.3 Peptide Sequence: tllarl-CONH2
Confirmation by MS (MALDI-TOF): m/z 685.361; calc’d for C31H61N10O7+ 685.891. 1H NMR (500 MHz, DMSO-d6, 308K) δ 8.53 (d, J = 8.0 Hz, 1H), 8.19 (d, J = 7.9 Hz, 1H), 8.09 (s, 3H), 7.93 (dd, J = 10.8 Hz, J = 7.3 Hz, 2H), 7.78 (d, J = 8.3 Hz, 1H), 7.54 (t, J = 5.7 Hz, 1H), 7.26 (s, 2H), 6.96 (s, 2H), 4.44-4.36 (m, 1H), 4.35-4.14 (m, 4H), 3.78 (t, J = 6.8 Hz, 1H), 3.58 (s, 1H), 3.09 (q, J = 6.5 Hz, 3H), 1.83-1.54 (m, 5H), 1.58-1.32 (m, 9H), 1.21 (d, J = 7.1 Hz, 4H), 1.14 (d, J = 6.3 Hz, 3H), 1.01-0.64 (m, 18H).
L1.4 Peptide Sequence: AAc-tllral-COOH
Confirmation by MS (MALDI-TOF): m/z 769.475; calc’d for C33H61N12O19+ 769.467. 1H NMR (500 MHz, DMSO-d6) δ 8.16 (d, J = 7.9 Hz, 1H), 8.11 (d, J = 8.2 Hz, 1H), 8.04 (d, J = 8.1 Hz, 1H), 7.98-7.83 (m, 1H), 7.44 (s, 1H), 4.46-4.06 (m, 6H), 3.96 (d, J = 2.6 Hz, 3H), 3.10 (t, J = 6.8 Hz, 2H), 1.76-1.56 (m, 4H), 1.59-1.38 (m, 9H), 1.23 (d, J = 7.1 Hz, 3H), 1.06 (d, J = 6.3 Hz, 3H), 0.90-0.74 (m, 18H).
L1.5 Peptide Sequence: K(N3)LARLLT-CONH2
Confirmation by MS (MALDI-TOF): m/z 839.421; calc’d. for C37H71N14O8+ 839.558. 1H NMR (500 MHz, DMSO-d6 308K) δ 8.45 (d, J = 8.2 Hz, 1H), 8.17 (d, J = 7.2 Hz, 1H), 8.06 (s, 4H), 7.91 (dd, J = 14.7 Hz, J = 8.2 Hz, 3H), 7.43 (d, J = 11.0 Hz, 2H), 7.08 (d, J = 12.7 Hz, 2H), 4.88 (t, J = 5.3 Hz, 1H), 4.46-4.24 (m, 6H), 4.13-3.97 (m, 3H), 3.79 (s, 1H), 3.09 (q, J = 6.5 Hz, 2H), 1.75-1.57 (m, 6H), 1.57-1.43 (m, 11H), 1.37 (t, J = 7.5 Hz, 2H), 1.20 (d, J = 7.1 Hz, 4H), 1.00 (d, J = 6.3 Hz, 4H), 0.93-0.80 (m, 18H).
L1.6 Peptide Sequence: GLARLLT-CONH2
Confirmation by MS (MALDI-TOF): m/z 742.991; calc’d. for C33H64N11O8+ 742.493. 1H NMR (500 MHz, DMSO-d6, 308K): δ 8.44 (d, J = 8.3 Hz, 1H), 8.30 (d, J = 7.2 Hz, 1H), 8.13 (d, J = 8.0 Hz, 2H), 8.01-7.86 (m, 4H), 7.43 (d, J = 8.5 Hz, 2H), 7.08 (d, J = 13.4 Hz, 3H), 4.57-4.37 (m, 1H), 4.38-4.17 (m, 4H), 4.11-3.96 (m, 3H), 3.84 (d, J = 5.6 Hz, 1H), 3.08 (q, J = 6.5 Hz, 2H), 1.72-1.54 (m, 5H), 1.54-1.33 (m, 10H), 1.20 (d, J = 7.1 Hz, 3H), 0.99 (d, J = 6.3 Hz, 4H), 0.97-0.72 (m, 18H).
L1.7 Peptide Sequence: KLARLLT-CONH2
Confirmation by MS (MALDI-TOF): m/z [M+H] 814.055; calc’d C37H73N12O82+ 814.547. 1H NMR (500 MHz, DMSO-d6, 308K) δ 8.49 (s, 1H), 8.25 (d, J = 7.3 Hz, 1H), 8.14 (d, J = 7.8 Hz, 4H), 7.97-7.87 (m, 3H), 7.52 (d, J = 5.5 Hz, 1H), 7.44 (d, J = 8.6 Hz, 2H), 7.08 (d, J = 9.8 Hz, 2H), 4.39 (q, J = 7.1 Hz, 2H), 4.37-4.24 (m, 5H), 4.10-4.03 (m, 2H), 3.78 (s, 1H), 3.08 (d, J = 7.2 Hz, 4H), 2.75 (t, J = 7.8 Hz, 2H), 1.70-1.55 (m, 6H), 1.56-1.42 (m, 10H), 1.26-1.17 (m, 6H), 0.99 (d, J = 6.3 Hz, 4H), 0.93-0.80 (m, 18H).
Synthesis of cyclic peptides Cyclo.L1 and Cyclo.L1.1
One gram of CTC resin (16) (1.2 mmol/g) was placed in a polypropylene reaction vessel fitted with a polypropylene frit. The resin was swollen with dry DCM (10 mL/gram of resin) for 30 minutes after which the solvent was removed. Fmoc-Leu-OH (297 mg, 0.84 mmol, 0.7 equivalents) was dissolved in dry DCM (10 mL) and DIEA (730 uL, 3.5 equivalents) was added. The amino acid solution was added to the resin and shaken for 1 hour. After this time, the resin was drained and washed with DMF (6 × 3 min). Unreacted sites on the resin were capped with DCM/MeOH/DIEA (80:15:5) twice for 15 minutes. The resin was washed again with DMF (5 × 30 sec) followed by DCM (5 × 30 sec). After drying the resin under vacuum overnight, the substitution level of the resin was determined by a quantitative Fmoc test (17). After the substitution level was determined (usually about 0.5 mmol/g), the Fmoc-Leu-CTC resin was deprotected using 20% piperidine in DMF (2 × 5 min) and washed with DMF (5 × 30 sec) followed by DCM (5 × 30 sec). The H-Leu-CTC resin was dried under vacuum and then stored at 4°C. The above procedure was repeated using Fmoc-(d)Leu-OH for peptide Cyclo.L1.1.
The Cyclo.L1 and Cyclo.L1.1 were prepared on a Tribute peptide synthesizer (Protein Technologies, Tucson, AZ) utilizing a standard Fmoc peptide chemistry protocol on a 100 μmol scale using the previously loaded H-Leu-CTC and H-(d)Leu-CTC resins, respectively. Side-chain functionalities were protected with tert-butyl (Thr), NG-2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl (Arg) and tert-butyloxycarbonyl (Lys). In the case of peptide Cyclo.L1.1, Fmoc-Lys(N3)-OH was used. Five fold excess of Fmoc-amino acids and HCTU, in the presence of 10 equivalents of DIEA, were used for each of the coupling steps (10 min) with DMF as the solvent. After the synthesis of each sequence was complete, the final Fmoc groups were removed using 20% piperidine in DMF (1 × 5 min; 1 × 10 min). The resin from each synthesis was washed with DMF (5 × 30 sec) and DCM (5 × 30 sec). The side chain protected peptides were cleaved from the resin with 5 mL of 1% TFA in DCM for 5 min. The cleavage reactions were repeated 10 times. The cleavage solutions for each respective peptide were combined and concentrated under vacuum. The residues were dissolved in water:acetonitrile containing 0.05% TFA (1:1, 20 mL), frozen and lyophilized to yield white solids. The peptides were dissolved in DMF to yield 2 mM solutions after which HATU (2.5 equivalents) and DIEA (5 equivalents) were added. The cyclization reactions were stirred for 3 hours, and then the peptides were placed under high vacuum to remove the DMF (usually overnight) to yield an oil. Protecting groups were removed from the peptides using TFA/water/TIPS (3 mL, 96:2:2) for 2.5 hours. Cold diethyl ether was then added to the peptide solutions to precipitate the crude cyclized peptides. The peptides were centrifuged for 10 minutes (10,000 rpm) and the ether layers decanted. Fresh cold diethyl ether was added, and the pelleted peptides were re-suspended. The peptides were centrifuged again, and the procedure was repeated 5 times for each peptide. After the final ether wash, the peptide pellets were dissolved in a minimal amount of water containing 0.1% TFA, frozen and lyophilized.
HPLC analysis was performed with a Waters 616 pump, Waters 2707 Autosampler, and 996 Photodiode Assay Detector which are controlled by Waters Empower 2 software. The separation was performed on an Agilent Zorbax 300 SB-C18 (5 um, 4.6 × 250 mm) with an Agilent guard column Zorbax 300 SB-C18 (5 um, 4.6 × 12.5 mm). Elution was done with a linear 5% to 55% gradient of solvent B (0.1% TFA in acetonitrile) into A (0.1% TFA in water) over 50 min at a 1 mL/min flow rate with UV detection at 215 nm. Preparative HPLC runs were performed with a Waters prep LC Controller, Waters Sample Injector, and 2489 UV/Visible Detector that are controlled by Waters Empower 2 software. The separation was performed on a Agilent Zorbax 300SB-C18 PrepHT column (7 um, 21.2× 250 mm) with Zorbax 300SB-C18 PrepHT guard column (7 um 21.2 × 10 mm) using a linear 5% to 55% gradient of solvent B (0.1% TFA in acetonitrile) into A (0.1% TFA in water) over 50 min at a 20 mL/min flow rate with UV detection at 215 nm. Fractions of high (>95%) HPLC purity with the expected mass were combined and lyophilized. High resolution mass spectra were obtained for Cyclo.L1 and Cyclo.L1.1. All analytical data are provided in the supporting information and Table S1.
Cyclo.L1. Peptide sequence: Cyclo(KLARLLT)
Confirmation by MS (MALDI-TOF): m/z 796.609; calc’d. for C37H70N11O8+ 796.540. 1H NMR (500 MHz, DMSO-d6) δ 8.51 (d, J = 8.9 Hz, 1H), 8.43 (d, J = 5.6 Hz, 1H), 8.30 (d, J = 7.4 Hz, 1H), 8.10 (s, 1H), 7.78 (t, J = 9.6 Hz, 2H), 4.32-4.27 (m, 2H), 4.24-4.20 (m, 6H), 4.03 (q, J = 7.3 Hz, 1H), 3.91-3.89 (m, 1H), 2.93 (d, J = 50.6 Hz, 6H), 2.77 (t, J = 8.4 Hz, 1H), 1.81-1.76 (m, 5H), 1.64-1.49 (m, 12H), 1.29 (d, J = 7.2 Hz, 4H), 1.09 (d, J = 6.3 Hz, 1H), 1.03 (d, J = 6.4 Hz, 4H), 0.97 (d, J = 6.6 Hz, 4H), 0.90-0.85 (m, 18H). Detailed 1H 2D NMR data with chemical shifts of protons with assignments are provided in the Supporting Information.
Cyclo.L1.1. Peptide sequence: Cyclo (K(N3)larllt)
Confirmation by MS (MALDI-TOF): m/z 822.616; calc’d. for C37H68N13O8+ 822.530. 1H NMR (500 MHz, DMSO-d6) δ 8.23 (dd, J = 21.2, 7.5 Hz, 1H), 8.07 (d, J = 37.3 Hz, 1H), 7.91 (d, J = 9.3 Hz, 2H), 7.53 (t, J = 5.4 Hz, 1H), 4.37- 4.32 (m, 4H), 4.16 (q, J = 7.3 Hz, 2H), 4.09 (dd, J = 9.8, 5.4 Hz, 3H), 4.16-4.01 (m, 4H), 3.94 (d, J = 7.9 Hz, 1H), 1.77 (m, 7H), 1.54-1.50 (m, 6H), 1.56 – 1.50 (m, 11H), 1.35 (d, J = 7.3 Hz, 4H), 1.07 (d, J = 5.9 Hz, 4H), 0.91-0.85 (m, 18H). Detailed 1H 2D NMR data with chemical shifts of protons with assignments are provided in the Supporting Information.
Circular dichroism spectroscopy
Circular dichroism (CD) data was collected (Jasco J-815 spectrometer) using 1 mm path length quartz cell. The linear and cyclic peptides were dissolved in water or methanol at a concentration of 1 mg/mL. Spectra were acquired at room temperature with an average of four scans in the wavelength range of 190 to 260 nm. The spectra were baseline corrected.
2D NMR of cyclic peptides
NMR studies of cyclic peptides Cyclo.L1 and Cyclo.L1.1 were carried out in 90% H2O/10 %D2O/DSS as well as in 100% DMSO-d6. NMR data were collected using a Bruker AV-III 500 MHz spectrometer with a liquid nitrogen cooled Prodigy TCI probe. The strongest solvent peak was suppressed by using excitation sculpting with gradients. 1D NMR experiments were performed at temperatures in the range 25 to 37 °C and 2D NMR experiments were performed at 25 °C. A DQF-COSY, TOCSY (80 msec mixing time), NOESY (150 and 300 msec mixing time) and ROESY (300 msec spin-lock time) were performed. Data were processed in SPARKY format, and analysis was done using SPARKY software (18).
Molecular modeling of peptides
Linear structures of the peptides were built using InsightII (BIOVIA Sandiego, CA). Linear structures were cyclized using NMR distance restraints obtained from NOESY/ROESY connectivities as described in our previous publications and literature (19–21). Peptide bond was formed once the distance of the N and C-termini were within 2 Å. Cyclized peptides were further subjected to simulated annealing procedure with NMR restraints. From high-temperature dynamics (900 K dynamics) 6 random structures were selected, and these structures were subjected to dynamics with NMR restraints at temperatures 800 K to 400 K in steps of 100 K with each step 10 ps duration. Structures from 400 K dynamics were used to soak 8 Å layer of water molecules and subjected to MD simulations for 20 ps. From this 300 K dynamics, 5–7 structures were chosen and subjected to energy minimization, first with steepest descent methods and then conjugate gradient method for 4000 steps with rms derivative of 0.5 kcal/mol-Å2. A total of 70 structures were generated. These structures were verified for NMR distance restraints, and structures that satisfied all the NMR distance restraints were considered as a probable structure of the peptide in solution. These structures were overlapped and represented using PyMol software.
Docking
Autodock Tools and Autodock (22) version 4.2 software was used for docking studies. Three-dimensional structures of EGFR in the open (3njp) (23) and closed (1nql) (24) conformations were obtained from the protein data bank. A three-dimensional structure of peptides was generated using InsightII (BIOVIA, San Diego, CA) as described above. Solvent molecules were removed from the protein file. Using the previously published procedure, the binding site of peptides on EGFR was assumed to be around Glu71, Asn134, and Gly177. A grid was created around the binding site with 126×126×126 Å3 box. Three-dimensional structures of peptides were used for docking studies with the Lamarckian genetic algorithm as described in our earlier studies (6). Briefly, 150 starting conformations with 10 million energy evaluations were done, and final 50 low-energy docked structures were analyzed. Final lowest energy docked structure was used for presentation using PyMol software (Schrodinger LLC, Portland, OR). Docking studies were carried out on a high-performance computer (HPC) at LSU, Baton Rouge via the Louisiana Optical Network Initiative (LONI).
Antiproliferative activity
Colon cancer cell line SW480 (ATCC® Number: CCL-228™) were purchased from ATCC. Cells were grown to confluency using the Leibovitz’s L-15 Medium. Antiproliferative activity was measured by CellTiter-Glo®, (25) cell viability assay. Nearly 1 × 104cells/well were seeded and incubated overnight at 37 °C and 5% CO2. Peptide solutions were prepared by dissolving 1.5 mg/mL of the peptide in deionized water and diluted using serum-free medium to prepare solutions of different concentrations of the peptide. For each concentration triplicate experiments were performed by incubating the peptide with cells for 72 h. Cells treated with 1% sodium dodecyl sulfate, and medium without the compound were used as controls. At the end of the experiment, CellTiter-Glo® reagent was added, and luminescence was measured using a plate reader. IC50 values were obtained from dose-response curves (GraphPad software, La Jolla, CA). Experiments were repeated at least three times to obtain standard deviation values.
SPR
Surface Plasmon Resonance was performed at 25°C using Biacore ×100 (GE Healthcare Biosciences, Piscataway, NJ), as described in detail in our previous publication (26). Briefly, the pure recombinant extracellular domain of EGFR protein (Leinco Technologies, St. Louis, MO) was immobilized on the CM5 SPR sensor chip (GE Healthcare Biosciences) at a rate of 5 μL/min in sodium acetate buffer (pH 4.5) using a standard amine coupling procedure. Before binding studies were performed, different concentrations of EGFR protein (0.1 to 1 μM) at different pH values including pH 4.0, 4.5, 5.0 and 5.0 (acetate buffers) were used for immobilization of the protein. We have selected the optimum concentration and pH value to obtain about 4000 to 5000 relative response units for protein immobilization and this was used as baseline with protein. Different peptides with concentrations ranging from 0 to 200 μM were used as analytes with HBS-EP (GE Healthcare Lifesciences, PA) as running buffer at a flow rate of 30 μL/min. The KD value was determined by fitting the obtained sensorgrams in Langmuir’s equation for 1:1 binding by using Biacore evaluation software. Curve fitting was analyzed by evaluating the chi squared value.
Stability of peptides in serum
Human serum (Innovative Research Novi, MI) was used according to approved guidelines, in a biosafety II cabinet with IBC certification. The detailed method for stability of peptides in serum was described in our previous publication (26). Peptide solutions were prepared in PBS at a concentration of 2 mg/mL, added to human serum in the ratio of 1: 9 and incubated at 37 °C. At different time points (0 min to 48 h), an aliquot of 100 μL was taken out and treated with 500 μL of cold acetonitrile for peptide extraction. Samples were analyzed by RP-HPLC as described previously (26). Relative intensity (AUC) for each time point, considering zero time point as 100% peptide concentration was plotted with respect to time, from triplicate experiments. The freeze-dried samples obtained from HPLC analysis were further analyzed by MALDI-TOF-MS using α-cyano-4hydroxy-cinnamic acid as the matrix gel.
Results
Peptide Design
Based on our previous studies using L1, we designed and synthesized the peptide derivatives shown in Table 1, with the aim to enhance the stability of L1. In the cyclic peptides with backbone cyclization, a lysine residue was introduced at the N–terminus for subsequent conjugation with a fluorophore. Similarly, for the linear peptide derivatives, a glycine or lysine residue was introduced to facilitate fluorophore conjugation. In addition, to improve peptide stability, several derivatives were synthesized with D-amino acids, with or without reversal of sequence, and N-termini stabilizing acetyl groups (Table 1).
Table 1.
Peptides designed and synthesized in this studya
| Code | Peptide sequence | Comment | bKD value for EGFR binding from SPR (mM) |
|---|---|---|---|
| L1 | LARLLT-CONH2 | Original sequence from Phage display* | 8.7 |
| Cyclo.L1 | Cyclo(KLARLLT) | Cyclic version of L1 with lysine | 1.16 |
| Cyclo.L1.1 | Cyclo(K(N3)larllt) | L1 cyclic D-amino acids and lysine with azide for conjugation | 5.09 |
| L1.1 | larllt-COOH | Linear all D-amino acids | N.D |
| L1.2 | tllral-COOH | Linear all D-amino acids reverse sequence | N.D |
| L1.3 | tllral-CONH2 | Linear all D-amino acids reverse sequence | N.D |
| L1.4 | AAc-tllral-COOH | Linear reverse sequence with D-amino acids, N-terminal azidoacetate. | N.D |
| L1.5 | K(N3)LARLLT-CONH2 | L1- peptide with azide | 4.21 |
| L1.6 | GLARLLT-CONH2 | L1-peptide with glycine | N.D |
| L1.7 | KLARLLT-CONH2 | L1-peptide with lysine | 1.25 |
Capital letters are used to represent L amino acids and small letters are used to represent D amino acids.
Song et al. FASEB J. (2009).
KD values were not determined for all the peptides accurately as Chi squared value was high for curve fitting.
The linear peptides likely exhibit unordered or flexible structure in solution. However, in the presence of EGFR, L1 might acquire a folded stable structure that maximizes interactions with the protein. To verify this, we performed docking studies of peptide L1 with EGFR. Several low energy docked structures of L1 were analyzed for the possible folded structures of the peptide. Figure 1 indicates that the L1 peptide acquires a folded structure in the presence of the protein. Careful observation of these docked structures suggests that there is a possibility of a turn structure in the peptide in the bound state. Based on this observation, and to increase the peptide stability, we investigated cyclization of the L1 peptide. However, the direct cyclization of L1 would result in a cyclic structure without the N or C-termini, and without functionality for subsequent conjugation to a fluorophore, such as a free amino group. Hence a lysine residue was introduced in the peptide sequence at the N-terminus. The side chain of Lys can be used for subsequent conjugation to a fluorophore via amide bond formation. In addition, we introduced an azide group at the lysine side chain to allow conjugation via “click” chemistry. To further increase the peptide stability and to facilitate the cyclization reaction, we also used D-amino acids with the exception of Lys-azide, in the preparation of peptide Cyclo.L1.1. (Figure 2 A&B)
Fig. 1.

Possible conformations acquired by the linear peptide L1-LARLLT in its binding pocket on EGFR. Structures shown in B) and C) are folded in such a way that N and C-termini are closer to form cyclic structure. Structure A) and D) show folding of backbone around A2 and R3 but the N-and C-are not in close contact. Structures D, E and F have extended backbone structures for the amino acids R3 to T6.
Fig. 2.

Design concept for peptides. A) Linear peptides with modification in chirality of amino acids, N and C-termini protection and reversal of sequence. B) Design of cyclic peptide for stability and conjugation.
Circular Dichroism (CD) Studies
To evaluate the changes in the overall conformation of the linear, cyclic and D-amino acids peptides, CD studies were conducted. The CD spectra of L1.7 and Cyclo.L1 peptides exhibited a negative band around 198 nm in water suggesting flexible backbone conformations for these peptides in water (27). Peptide Cyclo.L1.1 exhibited a positive band around 198 nm indicating a change in chirality of L to D amino acids in the peptide (Figure 3A). In methanol, the negative and positive bands of the peptides around 198 in water shifted to 200 nm. Furthermore, an additional CD band around 220 nm appeared, suggesting the possibility of a well-defined β-turn conformation of the peptides in an organic solvent (27) (Figure 3B). CD spectra of linear and cyclic peptides studied are provided in the supporting information.
Fig. 3.

CD spectra of linear and cyclic peptides in A) water and B) methanol. CD bands around 200 and 215 nm indicate possible secondary structure of the peptides in methanol solution.
SPR Studies
Among the peptides studied, L1, L1.3, L1.5, L1.6, L1.7, Cyclo.L1 and Cyclo.L1.1 (Table 1) exhibited binding to the EGFR protein in a concentration-dependent manner (Figures 4 and 5 and SI). Peptides L1.1, L1.2, and L1.4 did not show any binding to EGFR as seen by SPR. As expected and as previously reported (8, 9), the L1 peptide showed an affinity for binding to EGFR. Cyclic peptide Cyclo.L1 and the Cyclic peptide with azide side chain and D-amino acids Cyclo.L1.1 exhibited binding to EGFR (Figures 4 and 5).
Fig. 4.


SPR analysis of linear peptides. EGFR was immobilized on a CM5 sensor chip and different concentrations of the following linear peptides were injected to obtain the SPR response: A) L1), B) D amino acid sequence, L1.1), C) sequence with lysine residue, L1.7, and D) sequence with lysine azide, L1.5.
Fig. 5.

SPR analysis of cyclic peptides. EGFR was immobilized on CM5 sensor chip and different concentrations of the cyclic peptides were injected to obtain the SPR response. A) Cyclo.L1), B) Cyclo.L1.1.
The kinetics of binding of cyclic peptides to EGFR were analyzed assuming Langmuir 1:1 binding and the sensorgrams were fit using the Langmuir equation to provide the KD values. The Cyclo.L1 and Cyclo.L1.1 peptides were found to have KD values of 1 and 5 μM, respectively, while the linear peptide L1 exhibited a KD value of 8 μM, indicating relatively higher binding affinity of the cyclic peptides to EGFR. It should be noted that the immobilized EGFR did not have EGF bound to it and hence we assume that the EGFR was mainly in a closed conformation. Peptide L1 is known to bind to EGFR in both the open and closed EGFR conformations. In order to evaluate the specificity of Cyclo.L1.1 binding to EGFR, SPR analysis was also carried out using the homologous proteins HER2 and HER3 (28). The analysis indicated that Cyclo.L1.1 binds to HER2 with a KD value of 40 μM, and to HER3 with a KD value of 50 μM.
NMR Studies
To investigate the 3D structures of Cyclo.L1 and Cyclo.L1.1, the 1D and 2D NMR spectra of these cyclic peptides were obtained, both in water and in DMSO-d6. The amide resonances of both the peptides were dispersed around 1 ppm, suggesting well-defined conformations for the peptides in solution. All the amino acid resonances could be identified using DQF-COSY and TOCSY spectra. Sequence-specific assignments were performed using NOESY and ROESY spectra (29) (Figure 6). Both peptides have three leucine amino acids, and the amide resonances of two leucine amino acids were merged, indicating similar chemical environment for those amides in solution. The NMR spectra of the peptides in water exhibited limited spatial connectivities in ROESY and NOESY spectra, and hence detailed 2D NMR studies were carried out in DMSO-d6. Both cyclic peptides have similar amino acids in the sequence, however, Cyclo.L1.1 has all D-amino acids except for lysine, and the L-Lys has an azide group at the side chain. Most of the amino acids exhibited similar chemical shifts for the amide resonances in both peptides, except for K1 and A3. The chemical shift, temperature dependence of amide resonances, and coupling constants of amide resonances are provided in the Supporting Information. For Cyclo.L1, the temperature dependence of the chemical shift for the amide resonances of L2, R4, L5, and L6 was ≤ 4.3 ppb/K, indicating that the amide protons of these amino acid residues were solvent shielded or involved in intramolecular hydrogen bonding (30). In the case of Cyclo.L1.1, the l2, l6 and t7 amide protons appear to be intramolecularly hydrogen bonded or solvent shielded. For Cyclo.L1, the 3JHNα coupling constants of L2 to L6 were ≤ 5Hz, whereas T7 has a coupling constant > 8 Hz suggesting that the peptide has a well-defined turn structure. In the case of Cyclo.L1.1, the 3JHNα coupling constants of l2, a3 and t7 were ≤ 5Hz and for K1, r4, l5 and l6 the coupling constant values were > 8 Hz, suggesting an extended structure of the peptide. In NOESY spectra, the peptide Cyclo.L1 exhibited spatial connectivity between the NH of R4-L5, the NH of T7-K1. Other spatial connectivities were sequential and within the same residue (Figure 6A&B). In the case of Cyclo.L1.1, spatial connectivity was observed between the NH of t7-l6,5, the NH of l2-K1 and other spatial connectivities were sequential or within the same amino acid residues (Figure 6C&D).
Fig. 6.
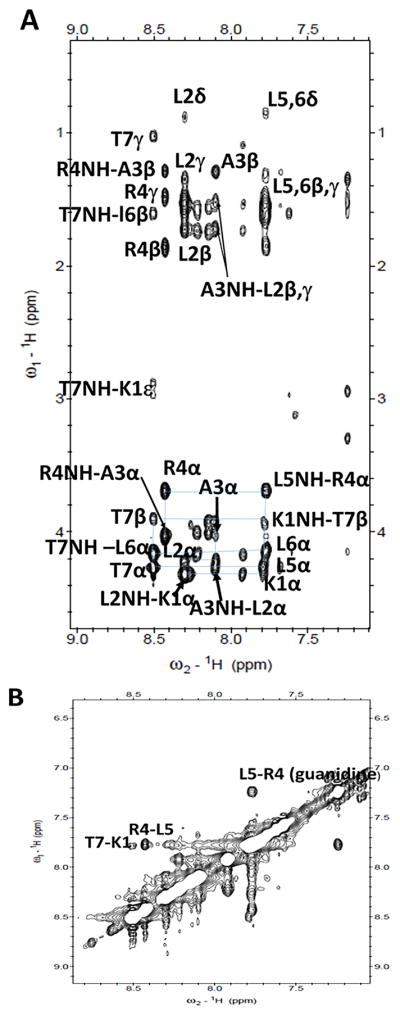

2D 1H NMR data for cyclo.L1 and Cyclo.L1.1 showing folded structure of the peptide. A) Finger print region (NH-CαH) of NOESY of Cyclo.L1. B) NH-NH region of Cyclo.L1. C) Finger print region (NH-CαH) of NOESY of Cyclo.L1.1. D) NH-NH region of Cyclo.L1.1.
Molecular Modeling
Based on the NMR-derived NOESY/ROESY distance restraints, possible 3D structures of peptides Cyclo.L1 and Cyclo.L1.1 were proposed and investigated. For Cyclo.L1, 43 NMR-based distance restraints were obtained, and for Cyclo.L1.1 40 distance restraints were obtained. Based on these restraints, the structures of these peptides were elucidated using distance-restrained MD simulations and energy minimization calculations. The structure of the Cyclo.L1 peptide exhibits two β–turns stabilized by intramolecular hydrogen bonding (Figure 7A). The first β-turn (31) occurs at T7-K1-L2-A3 with Φ, Ψ values of −40, −50 and −73, −12 at K1 and L2, respectively. This turn is stabilized by hydrogen bonding between the C=O of T7 and the NH of A3. The second β-turn is at R4-L5-L6 and T7 with Φ, Ψ values of −69, −94, −87, −4 at L5 and L6, respectively. The second turn is also stabilized by intramolecular hydrogen bonding between C=O of R4 and the NH of T7. There is one additional hydrogen bond between C=O of T7 and the NH of R4. The participation of amide protons in intramolecular hydrogen bonding between the C=O of R4 with the NH of T7, and the C=O of T7 with the NH of R4, is supported by the low-temperature coefficient chemical shift of R4 and T7 amides (Supporting Information). Overall, the structure of the peptide features a stable backbone conformation, and the 25 overlapped structures of Cyclo.L1 show that the average rms deviation of the backbone structure is 1.25 ± 0.36 Å (Figure 7C). The side chains of L2, A3, L5, L6 and T7 are oriented on one face of the peptide backbone plane, and R4 is on the opposite side of this peptide backbone plane. The K1 side chain is folded in the plane of the peptide backbone.
Fig. 7.

Proposed structure of peptides A) Cyclo.L1 and B) Cyclo.L1.1 in solution based on NMR data. Backbone structures of 25 superimposed C) Cyclo.L1 and D) Cyclo.L1.1. The L amino acids are shown in capital letters and D amino acids are shown in small letters.
The structure of peptide Cyclo.L1.1 is slightly different from that of Cyclo.L1. The backbone structure of Cyclo.L1.1 (Figure 7B) does not exhibit well-defined β-turns, as in the case of Cyclo.L1. The first β-turn is at l2-a3-r4-l5 with Φ, Ψ values of −46, 126, 70, 23 at a3 and r4, respectively. The hydrogen bond is between C=O of l2 and the NH of l5. The second β-turn is at l5-l6-t7-K1 with Φ, Ψ values of 140, −58, −44, 111. This turn is not stabilized by hydrogen bonding as the NH of K1 moved away from the C=O of l5. There is a γ-turn type of hydrogen bond between the C=O of l5 and the NH of t7. The hydrogen bonding between the C=O of l5 and the NH of t7 is supported by the low-temperature coefficient of the amide proton of t7. Unlike the Cyclo.L1 peptide conformation, the turn structure in Cyclo.L1.1 is not stabilized by hydrogen bonds. This is also supported by the high-temperature coefficient of the chemical shifts of K1, a3, r4, and l5. Most of the side chains face one side of the peptide backbone plane, except for the K1 side chain. This may be because K1 is an L-amino acid residue while the remaining amino acids in the peptide have D-chirality. The average rms deviation of the backbone of 25 overlapped structures is 1.60 ± 0.54 Å (Figure 7D).
To model the binding of these peptides to EGFR, docking studies were carried out using Autodock (22). The linear peptide L1 is known to bind to the domain I of the EGFR protein, away from the EGR binding pocket (32, 33). Cyclo.L1 was docked near the presumed L1 binding site, as reported in our previous studies (6). The low energy docked structure of Cyclo.L1 is shown in Figure 8A. The lowest energy docked structure (−2 kcal/mol) forms six hydrogen bond interactions with the EGFR protein, stabilizing the ligand-receptor interactions. The hydrogen bonds are between the NH of L6 and the C=O of Gln184, the C=O of L6 and the NH of Leu186, the C=O of R4 and the Lys185 side chain amine group, the NH of L2 and the C=O of Arg200, the side chain of T7 and the C=O of Leu186, and between the side chain of R4 and the C=O of Glu180. The peptide-EGFR interaction is also stabilized by hydrophobic interactions between the L6 side chain and Pro171 and Leu186, as well as between the L5 side chain and Arg4, Cys170, and Cys183. The lowest energy docked structure of Cyclo.L1.1 was on EGFR domain I (Figure 8B). In terms of docking energy, both peptides were within the error of docking energy (2 kcal/mol) (22). The docking site on domain I of EGFR is the same as the site occupied by the Cyclo.L1 peptide. However, Cyclo.L1.1 is stabilized by two hydrogen bond interactions, at t7 side chain with the C=O of Leu186, and the K1 side chain with Gln184. The hydrophobic interactions were between the l2 side chain and Leu186, Ile189, Ile190 and the K1 side chain with Glu181 side chain, l5 side chains with Arg200 side chain and the Thr217 methyl groups (Figure 8B).
Fig. 8.

Proposed model of A) Cyclo.L1 and B) Cyclo.L1.1 binding to EGFR based on docking studies.
Antiproliferative activity
To evaluate whether the designed peptides cause any antiproliferative activity, peptide Cyclo.L1.1 was selected to be evaluated for its ability to inhibit growth in EGFR overexpressing SW480 cells. From the dose response curve, the IC50 value was found to be >100 μM suggesting that this peptide does not cause any growth inhibition and hence it is not toxic to EGFR overexpressing cells.
Stability of Peptides in Serum
The stability in human serum of the linear and cyclic peptides KLARLLT (L1.7) and cyclo(KLARLLT) (Cyclo.L1) were compared, and the results obtained are shown in Figure 9. In these studies, the peptides were incubated in human serum at 37 °C, and samples were analyzed using HPLC at different time points up to 48 h. The area under the curve (AUC) at each time point of the peptide peak was plotted vs time to assess the stability, assuming the AUC at 0 h as 100%. The linear peptide L1.7 exhibited a sharp decay curve (Figure 9A) indicating fast degradation of this peptide, with only 10% intact after 48 h and a half-life of about 5 h. On the other hand, the cyclic peptide Cyclo.L1 exhibited slower degradation, with nearly 60% of the peptide intact after 48 h (Figure 9B). The degradation profile for Cyclo.L1 was constant after 6 h, suggesting that there is a possibility of peptide binding to serum proteins (34), however, further studies are needed to differentiate the amount of degradation of cyclic peptide and serum bound peptide. The samples were also analyzed using mass spectrometry to confirm the presence of intact peptide at the various time points. The molecular ion (m/z) corresponding to the peptide molecular weight was observed for all the samples collected at the different time points, indicating the presence of intact peptide at all time points, in agreement with the HPLC results.
Fig. 9.

Serum stability of A) linear peptide L1 and B) cyclic peptide Cyclo.L1 by HPLC. Data is plotted from three experiments and error bars indicate ±SD.
Discussion
The main goal of our studies was to design peptide derivatives of L1 with enhanced stability and binding affinity to EGFR domain I. Peptides that are specific for extracellular receptors can be used for targeting drugs to those receptors and/or for conjugation with fluorescent probes for molecular imaging. In the present work we designed and investigated derivatives of the linear peptide L1, known to bind to the EGFR extracellular domain I. The designed peptides (Table 1) were modified at the N-terminus, or with an azide-containing Lys side chain, to facilitate their subsequent conjugation to fluorescent probes, such as 4,4-difluoro-4-bora-3a,4a-diaza-s-indacenes (BODIPYs), using either amide bond formation or click chemistry. Our previous studies showed that peptide L1 conjugated to a phthalocyanine or porphyrin increases the uptake of the fluorophore by up to 17-fold in cells over-expressing EGFR (6). However, one main disadvantage of peptides is their susceptibility to degradation in vivo (11, 12). Peptides can be designed with improved stability by modification of their structure, for example by cyclization via a disulfide bond or main chain cyclization, and by substitution with D-amino acids at crucial positions (13). In addition, conformational constraints, such as a β-turn (35), can be used for optimization of the binding activity of peptide ligands. Changing the chirality of the amino acids in L1 and/or reversing the sequence as in L1.1 and L1.2 (Table 1), do not favor the binding of the peptide to EGFR. Furthermore, changing the chirality, reversing the sequence, and capping the N-terminus of the peptide (L1.4) also do not favor EGFR-binding. However, changing the chirality of amino acids, reversing the sequence, and introducing C-terminal amidation, resulted in retaining of the binding activity, as seen in L1.3. The addition of amino acids such as Gly or Lys at the N-terminal of the L1 peptide, and modification of the terminal Lys residue with azide, as in L1.5–L1.7, did not change the binding affinity of the peptide to EGFR. Therefore, Gly or Lys amino acid spacers can be introduced to facilitate fluorescence labeling of peptide L1 without affecting its binding affinity to EGFR. A cyclic peptide Cyclo.L1 with Lys in the sequence showed binding to the EGFR extracellular domain. An analog of cyclic peptide Cyclo.L1.1 with D-amino acids and an azide group in the Lys side chain, was also shown to bind to EGFR. The peptide L1 is known to target EGFR in vitro and in vivo in a mouse model (9) and it does not cause any antiproliferative activity or toxicity. To evaluate whether the cyclization of this peptide induces cytotoxicity, the antiproliferative activity of Cyclo.L1.1 was evaluated in the colon cancer cell line SW480 known to overexpress EGFR (36). The IC50 value determined for Cyclo.L1.1 was >100 μM indicating that this cyclic peptide does not modulate EGFR signaling processes, and does not cause cytotoxicity.
As mentioned above, the main goal of our studies was to design peptides that exhibit high EGFR binding activity, and good stability in serum for in vivo studies. Cyclo.L1 was evaluated for its serum stability using human serum in vitro, and its stability was compared to its linear analog L1 using HPLC and MS techniques. These stability studies clearly suggested that Cyclo.L1 is more stable compared with linear peptide L1 in serum. From the stability profile of Cyclo.L1 it is clear that within the first 2 h nearly 40% of the intact peptide was lost. Possible reasons for the loss of 40% of peptide could be the existence of a racemic mixture and loss of one form of the peptide, and the binding of the peptide to serum proteins. Considering the synthesis procedure and the CD spectra that clearly represent the relative chirality of the peptide (Figure 3 and Supporting Information), we hypothesize that binding of the peptide to serum proteins is the most likely cause of nearly 40% loss of the peptide observed in the stability studies. We rule out the proteolysis process as the intact peptide amount was constant after 4 h and up to 48 h. To differentiate between serum protein binding and proteolysis, future studies will investigate serum protein binding by both SPR and dialysis.
The design of cyclic peptides was based on conformational studies of the parent linear peptide in bound form with EGFR using docking models. Linear peptides with flexible structure may change their conformation upon binding to the receptor and therefore may exhibit different conformation in the free state compared with the bound state. On the other hand, cyclic peptides with constrained structure may not change their conformation significantly upon binding to the receptor as their conformation is restricted (37). Hence, the design of cyclic peptides is important in terms of binding to the receptor. Upon cyclization, if the conformation is not suitable for binding, the binding affinity of the cyclic peptide to the receptor decreases significantly compared with the linear peptide. The structure of the peptides derived from NMR and modeling studies indicated that peptide Cyclo.L1 exhibited two β-turns and these β-turns were stabilized by hydrogen bonds. Cyclo.L1.1 exhibited β-turns, however, NMR and molecular modeling studies suggested that the β-turn was not stabilized by hydrogen bonds. Thus, in solution, the backbone conformation of Cyclo.L1.1 was different from that of Cyclo.L1. We believe that introduction of D-amino acids in the peptide, with exception of Lys, lead to this change in conformation. The conformations of cyclic hexapeptides have been studied extensively. Usually, cyclic hexapeptides acquire well-defined backbone structures and cyclic hexapeptides containing trans-amide bonds exhibit two β-turns stabilized by intramolecular hydrogen bonds (38). The occurrence of intramolecular hydrogen bonds in cyclic peptides also depends on the overall backbone conformation of the peptide. Nielsen et al. (39) studied cyclic hexapeptides with different number of proline residues in the peptide sequence. Their study suggested that cyclic hexapeptides without a proline have relatively flexible backbone conformations in solution compared with cyclic peptide with prolines. Cyclic peptides with anticancer properties and orally available have been reported. The stability and increase in anticancer activity of a nine amino acid residue peptide compared to the linear peptide were attributed to its conformational constraints (40–42). Compared with the linear peptide L1, Cyclo.L1 and Cyclo.L1.1 acquire restricted conformations that are suitable for binding to the EGFR receptor. This conformational locking is achieved by cyclization of the linear peptide. To compare the conformation of linear peptide in bound form to the structure of cyclic peptides derived from NMR data, we overlapped the NMR-derived structure of Cyclo.L1 and its docked structure, with those obtained for the linear peptide L1. Figure 10A compares the structures of the L1 linear peptide in the EGFR-docked conformation with the NMR-derived structure of Cyclo.L1. One can see that the backbone of RLLT overlaps with the cyclic peptides. Similarly, the final docked structure of Cyclo.L1 overlaps with the linear LARLLT docked structure, showing a good overlap of the peptide backbones (Figure 10B). The overlapped structure suggests that the linear and cyclic peptides fold in a similar conformation in the presence of EGFR in the bound form. Therefore, the cyclization of peptide L1 results in a conformation that is ready to bind to the EGFR protein. Docking results suggest that Cyclo.L1 and Cyclo.L1.1 bind to the domain I of EGFR and this site is away from the EGF binding site. Furthermore, cyclic peptides bind to both the open and close conformations of EGFR (28), since the docking site on domain I is not affected by these conformational changes in EGFR. The model proposed based on docking studies is also supported by SPR studies that indicate that Cyclo.L1 and Cyclo.L1.1 bind to EGFR in the absence of EGF ligand, indicating that the conjugates also bind to EGFR in a closed conformation. Previous studies on affibody molecules designed to bind to EGFR have suggested that molecules that are designed to bind to domain I may bind to the open or closed conformations of EGFR (43). Overall, these results suggest that both cyclic peptides based on linear L1, Cyclo.L1, and Cyclo.L1.1, specifically bind to EGFR, and therefore could be used in the design of fluorescent EGFR-targeting conjugates.
Fig. 10.

Comparison of the EGFR-docked structures of linear peptide L1 and the NMR derived and docked cyclic peptide structures. A) Overlapped of two L1 structures docked to EGFR (red and blue sticks) with the NMR-based structure of Cyclo.L1 (cyan sticks); B) Overlapped of two L1 structures docked to EGFR (green and red sticks) with the low energy docked structure of Cyclo.L1 (cyan sticks).
Conclusions
Structure-activity studies of linear and cyclic versions of the L1 peptide, known to specifically bind to EGFR, were conducted using CD, SPR, NMR, molecular dynamics, and Autodock. Additional amino acid residues (Gly, Lys) were added to the linear peptide sequence, and the effects of change of amino acid chirality and reversal of sequence were also investigated. The L1 peptide was cyclized to improve its stability and affinity for the EGFR protein, and a Lys residue with or without azide modification, and reversed sequence were introduced for subsequent conjugation to a fluorophore. The cyclized peptides exhibited higher affinity for the EGFR protein compared with the linear peptides, as indicated by SPR analysis. Detailed CD, NMR, and molecular modeling studies suggested that the peptides acquire a β-turn structure in solution. A model was proposed for the cyclic peptide-EGFR interaction using a docking method. The cyclic peptides appear to bind to the domain I of EGFR, away from the EGF binding pocket. Stability studies in human serum using HPLC and mass spectrometry indicated that the cyclic peptides are more stable than the linear peptides. Our studies suggest that the cyclic peptides are efficient ligands for EGFR, and their conjugation to a fluorescent tag could lead to an efficient strategy for the detection of CRC and other cancers that have high EGFR expression.
Supplementary Material
Acknowledgments
The work was supported by funding from the National Institutes of Health grant number R01 CA179902 and R25GM069743. The authors acknowledge the LSU facilities for peptide synthesis, NMR and mass spectrometry. The computational studies were carried out using the LSU HPC facility, via the Louisiana Optical Network Initiative (LONI).
Abbreviations Used
- ACN
Acetonitrile
- BODIPY
4,4-difluoro-4-bora-3a,4a-diaza-s-indacenes
- CD
circular dichroism
- CTC
chlorotrityl chloride resin
- DCM
dichloromethane
- DIEA
diisopropylethylamine
- DMF
dimethylformamide
- EDTA
ethylenediaminetetraacetic acid
- EDC
N-ethyl N-(dimethylaminopropyl)
- EGFR
epidermal growth factor receptor
- Fmoc
fluoroenylmethyloxycarbonyl
- HATU
N-[(Dimethylamino)-1H-1,2,3-triazolo-[4,5-b]pyridine-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate N-oxide
- HCTU
2-(6- Chloro- 1H–benzotriazole-1-yl)-1,1,3,3- tetramethylaminium hexafluorophosphate
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HOBt
Hydroxybenzotriazole
- HPC
high performace computer
- HPLC
high performance liquid chromatography
- MALDI-TOF
matrix-assisted laser desorption/ionization-time of flight
- MD
molecular dynamics
- MeOH
methanol
- MS
mass spectrometry
- NHS
N-hydroxysuccinimide
- NMR
nuclear magnetic resonance
- PBS
phosphate buffered saline
- SPPS
solid phase peptide synthesis
- SPR
Surface Plasmon Resonance
- TBTU
O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate
- TFA
trifluoroacetic acid
- TIPS
triisopropylsilane
- UV
ultra violet
Footnotes
Supplementary data related to this article can be found in the online version of the article. HPLC, Mass Spectrometry, MS-MS, 1D and 2D 1H NMR data, Table of chemical shift values for amino acids in the cyclic peptides are included in the supporting information.
Conflict of Interest
No conflict of interest to declare
References
- 1.Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- 2.van den Broek FJ, Fockens P, Dekker E. Review article: New developments in colonic imaging. Aliment Pharmacol Ther. 2007;26(Suppl 2):91–9. doi: 10.1111/j.1365-2036.2007.03494.x. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Fedewa SA, Anderson WF, Miller KD, Ma J, Rosenberg PS, et al. Colorectal Cancer Incidence Patterns in the United States, 1974–2013. JNCI: Journal of the National Cancer Institute. 2017;109 doi: 10.1093/jnci/djw322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dougherty U, Sehdev A, Cerda S, Mustafi R, Little N, Yuan W, et al. Epidermal growth factor receptor controls flat dysplastic aberrant crypt foci development and colon cancer progression in the rat azoxymethane model. Clin Cancer Res. 2008;14:2253–62. doi: 10.1158/1078-0432.CCR-07-4926. [DOI] [PubMed] [Google Scholar]
- 5.Baxter NN, Goldwasser MA, Paszat LF, Saskin R, Urbach DR, Rabeneck L. Association of colonoscopy and death from colorectal cancer. Ann Intern Med. 2009;150:1–8. doi: 10.7326/0003-4819-150-1-200901060-00306. [DOI] [PubMed] [Google Scholar]
- 6.Ongarora BG, Fontenot KR, Hu X, Sehgal I, Satyanarayana-Jois SD, Vicente MG. Phthalocyanine-peptide conjugates for epidermal growth factor receptor targeting. J Med Chem. 2012;55:3725–38. doi: 10.1021/jm201544y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fontenot KR, Ongarora BG, LeBlanc LE, Zhou Z, Jois SD, Vicente MG. Targeting of the epidermal growth factor receptor with mesoporphyrin IX-peptide conjugates. J Porphyr Phthalocyanines. 2016;20:352–66. doi: 10.1142/S1088424616500115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Z, Zhao R, Wu X, Sun Y, Yao M, Li J, et al. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J. 2005;19:1978–85. doi: 10.1096/fj.05-4058com. [DOI] [PubMed] [Google Scholar]
- 9.Song S, Liu D, Peng J, Deng H, Guo Y, Xu LX, et al. Novel peptide ligand directs liposomes toward EGF-R high-expressing cancer cells in vitro and in vivo. FASEB J. 2009;23:1396–404. doi: 10.1096/fj.08-117002. [DOI] [PubMed] [Google Scholar]
- 10.Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug Discov Today. 2010;15:40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 11.Jenssen H, Aspmo SI. Serum stability of peptides. Methods Mol Biol. 2008;494:177–86. doi: 10.1007/978-1-59745-419-3_10. [DOI] [PubMed] [Google Scholar]
- 12.Hess S, Ovadia O, Shalev DE, Senderovich H, Qadri B, Yehezkel T, et al. Effect of structural and conformation modifications, including backbone cyclization, of hydrophilic hexapeptides on their intestinal permeability and enzymatic stability. J Med Chem. 2007;50:6201–11. doi: 10.1021/jm070836d. [DOI] [PubMed] [Google Scholar]
- 13.Tugyi R, Uray K, Ivan D, Fellinger E, Perkins A, Hudecz F. Partial D-amino acid substitution: Improved enzymatic stability and preserved Ab recognition of a MUC2 epitope peptide. Proc Natl Acad Sci U S A. 2005;102:413–8. doi: 10.1073/pnas.0407677102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi JY, Lee BC. Click Reaction: An Applicable Radiolabeling Method for Molecular Imaging. Nucl Med Mol Imaging. 2015;49:258–67. doi: 10.1007/s13139-015-0377-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew Chem Int Ed Engl. 2001;40:2004–21. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 16.Ieronymaki M, Androutsou ME, Pantelia A, Friligou I, Crisp M, High K, et al. Use of the 2-chlorotrityl chloride resin for microwave-assisted solid phase peptide synthesis. Biopolymers. 2015;104:506–14. doi: 10.1002/bip.22710. [DOI] [PubMed] [Google Scholar]
- 17.Chan WC, White PD. Fmoc solid phase peptide synthesis. Oxford University Press; 2000. [Google Scholar]
- 18.Goddard T, Kneller D. Sparky 3. University of California; San Francisco, USA: 2008. [Google Scholar]
- 19.Kang TS, Radic Z, Talley TT, Jois SD, Taylor P, Kini RM. Protein folding determinants: structural features determining alternative disulfide pairing in alpha- and chi/lambda-conotoxins. Biochemistry. 2007;46:3338–55. doi: 10.1021/bi061969o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gokhale A, Weldeghiorghis TK, Taneja V, Satyanarayanajois SD. Conformationally constrained peptides from CD2 to modulate protein-protein interactions between CD2 and CD58. J Med Chem. 2011;54:5307–19. doi: 10.1021/jm200004e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sutcliffe MJ. Structure determination from NMR data II. Computational approaches. Oxford University Press; New York: 1993. [Google Scholar]
- 22.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–91. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu C, Mi LZ, Grey MJ, Zhu J, Graef E, Yokoyama S, et al. Structural evidence for loose linkage between ligand binding and kinase activation in the epidermal growth factor receptor. Mol Cell Biol. 2010;30:5432–43. doi: 10.1128/MCB.00742-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferguson KM, Berger MB, Mendrola JM, Cho HS, Leahy DJ, Lemmon MA. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol Cell. 2003;11:507–17. doi: 10.1016/s1097-2765(03)00047-9. [DOI] [PubMed] [Google Scholar]
- 25.Tolliday N. High-throughput assessment of Mammalian cell viability by determination of adenosine triphosphate levels. Curr Protoc Chem Biol. 2010;2:153–61. doi: 10.1002/9780470559277.ch100045. [DOI] [PubMed] [Google Scholar]
- 26.Sable R, Durek T, Taneja V, Craik DJ, Pallerla S, Gauthier T, et al. Constrained Cyclic Peptides as Immunomodulatory Inhibitors of the CD2:CD58 Protein-Protein Interaction. ACS Chem Biol. 2016;11:2366–74. doi: 10.1021/acschembio.6b00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perczel A, Park K, Fasman GD. Analysis of the circular dichroism spectrum of proteins using the convex constraint algorithm: a practical guide. Anal Biochem. 1992;203:83–93. doi: 10.1016/0003-2697(92)90046-a. [DOI] [PubMed] [Google Scholar]
- 28.Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TP, Leahy DJ, et al. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12:541–52. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 29.Wuthrich K. NMR of proteins and nucleic acids. Wiley; 1986. [Google Scholar]
- 30.Andersen NH, Neidigh JW, Harris SM, Lee GM, Liu Z, Tong H. Extracting Information from the Temperature Gradients of Polypeptide NH Chemical Shifts. 1. The Importance of Conformational Averaging. J Am Chem Soc. 1997;119:8547–61. [Google Scholar]
- 31.Rose GD, Gierasch LM, Smith JA. Turns in peptides and proteins. Adv Protein Chem. 1985;37:1–109. doi: 10.1016/s0065-3233(08)60063-7. [DOI] [PubMed] [Google Scholar]
- 32.Ongarora BG, Fontenot KR, Hu X, Sehgal I, Satyanarayana-Jois SD, Vicente MGH. Phthalocyanine–Peptide Conjugates for Epidermal Growth Factor Receptor Targeting. J Med Chem. 2012;55:3725–38. doi: 10.1021/jm201544y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song S, Liu D, Peng J, Deng H, Guo Y, Xu LX, et al. Novel peptide ligand directs liposomes toward EGF-R high-expressing cancer cells in vitro and in vivo. FASEB J. 2009;23:1396–404. doi: 10.1096/fj.08-117002. [DOI] [PubMed] [Google Scholar]
- 34.Bohnert T, Gan LS. Plasma protein binding: from discovery to development. J Pharm Sci. 2013;102:2953–94. doi: 10.1002/jps.23614. [DOI] [PubMed] [Google Scholar]
- 35.Andersen NH, Olsen KA, Fesinmeyer RM, Tan X, Hudson FM, Eidenschink LA, et al. Minimization and optimization of designed beta-hairpin folds. J Am Chem Soc. 2006;128:6101–10. doi: 10.1021/ja054971w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang JL, Qu XJ, Russell PJ, Goldstein D. Regulation of epidermal growth factor receptor in human colon cancer cell lines by interferon alpha. Gut. 2004;53:123–9. doi: 10.1136/gut.53.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duffy FJ, Devocelle M, Shields DC. Computational approaches to developing short cyclic peptide modulators of protein-protein interactions. Methods Mol Biol. 2015;1268:241–71. doi: 10.1007/978-1-4939-2285-7_11. [DOI] [PubMed] [Google Scholar]
- 38.McHugh SM, Rogers JR, Yu H, Lin YS. Insights into How Cyclic Peptides Switch Conformations. J Chem Theory Comput. 2016;12:2480–8. doi: 10.1021/acs.jctc.6b00193. [DOI] [PubMed] [Google Scholar]
- 39.Nielsen DS, Lohman RJ, Hoang HN, Hill TA, Jones A, Lucke AJ, et al. Flexibility versus Rigidity for Orally Bioavailable Cyclic Hexapeptides. Chembiochem. 2015;16:2289–93. doi: 10.1002/cbic.201500441. [DOI] [PubMed] [Google Scholar]
- 40.Bennett JA, DeFreest L, Anaka I, Saadati H, Balulad S, Jacobson HI, et al. AFPep: an anti-breast cancer peptide that is orally active. Breast Cancer Res Treat. 2006;98:133–41. doi: 10.1007/s10549-005-9140-5. [DOI] [PubMed] [Google Scholar]
- 41.Jacobson HI, Andersen TT, Bennett JA. Development of an active site peptide analog of alpha-fetoprotein that prevents breast cancer. Cancer Prev Res. 2014;7:565–73. doi: 10.1158/1940-6207.CAPR-13-0405. [DOI] [PubMed] [Google Scholar]
- 42.Bryan A, Joseph L, Bennett JA, Jacobson HI, Andersen TT. Design and synthesis of biologically active peptides: a ‘tail’ of amino acids can modulate activity of synthetic cyclic peptides. Peptides. 2011;32:2504–10. doi: 10.1016/j.peptides.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jost C, Schilling J, Tamaskovic R, Schwill M, Honegger A, Pluckthun A. Structural basis for eliciting a cytotoxic effect in HER2-overexpressing cancer cells via binding to the extracellular domain of HER2. Structure. 2013;21:1979–91. doi: 10.1016/j.str.2013.08.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.