

Abstract
Vaccination represents a cost-effective weapon for disease prevention and has proven to dramatically reduce the incidences of several diseases that once were responsible for significant mortality and morbidity worldwide. The nasal cavity constitutes the initial stage of the respiratory system and the first contact with inhaled pathogens. The intranasal (IN) route for vaccine administration is an attractive alternative to injection, due to the ease of administration as well as better patient compliance. Many published studies have demonstrated the safety and effectiveness of IN immunization with liquid vaccines. Currently, two liquid IN vaccines are available and both contain live attenuated influenza viruses. FluMist® was approved in 2003 in the United States, and Nasovac® H1N1 vaccine was approved in India in 2010. Preclinical studies showed that IN immunization with dry powder vaccines (DPVs) is feasible. Although there is not a commercially available DPV yet, DPVs have the inherent advantage of being relatively more stable than liquid vaccines. This review focuses on recent developments of DPVs as next-generation IN vaccines.
Keywords: Mucosal immune responses, NALT, Dry powder, Devices, Mucoadhesives
Graphical abstract
1. Nasal immune systems and its role in intranasal immunization
The nose is responsible for conditioning the inspired air and protecting the airways from large and potentially harmful particles, as well as functions as a sensory organ responsible for olfaction [1]. The air inhaled through the nose moves from the nasopharynx to the oropharynx, and then larynx and trachea until it reaches the deep lung [2].
The nasal cavity is divided by the nasal septum into two paired compartments, further separated by the folds of the superior, middle, and inferior concha that form three ducts called superior, middle, and inferior meatus [3] (Fig. 1). Based on the increased turbulence and air flow resistance, two major deposition areas of inhaled particles can be identified in the nasal cavity, the middle concha and the internal ostium [4].
Figure 1.
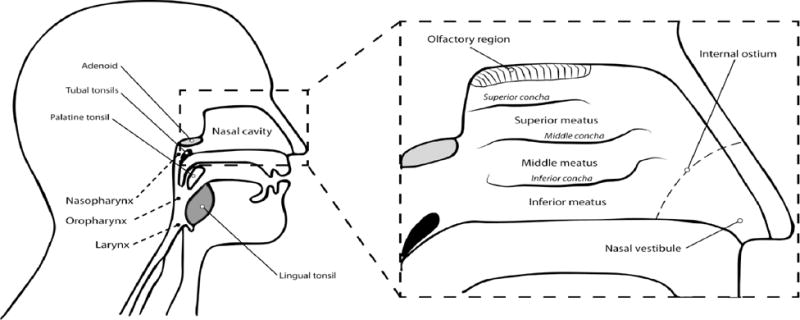
Representation of the upper airways. Enlarged is a cross-section of the upper airway with details of the nasal cavity [2,3]. The adenoid, tubals, palatine tonsil and lingual tonsil are collectively called as the pharyngeal lymphoid ring or the Waldeyer’s ring. Modified from Perry and Whyte with copyright permission [109].
The mucous membrane lining the nasal cavity secretes mucus that works as an innate immunity mechanism by washing away or entrapping potential pathogens [5]. Nasal secretions also contain protective elements such as immunoglobulins [6], lactoferrin [7, 8], and immune cells [9, 10]. The mucosal surfaces present inductive sites that can initiate an immune response to specific antigens independent from the systemic immune system. These inductive sites are collectively described as mucosa-associated lymphoid tissue (MALT) [11]. The nose-associated lymphoid tissue (NALT) is a subdivision of the MALT [12]. The term NALT in rodents refers to a pair of aggregated lymphoid tissue localized in the nose (i.e., the bottom of the nasal ducts) [13]. The Waldeyer’s ring, a well-known group of tonsils that includes the adenoid, tubal, palatine, and lingual tonsils, is the key lymphoid tissue in human nose [14]. A post-mortem study by Debertin et al. provided the first evidence of the existence of a NALT, in addition to the Walderyer’s ring, in young children [15]. This study in young children found disseminated aggregates of lymphoid tissue in the nasal cavity in 38% of the cases, mainly located in the superior meatus (30.1%), the middle concha (26.4%), the inferior concha (13.5%), and the superior concha (10.4%). In a recent review, Pabst stated that there is not reported data on the frequency of NALT in adolescents and adults [16].
Upon nasal vaccination, antigens can follow different routes that were reviewed by Kuper et al. [17]. While soluble antigens can potentially penetrate the nasal mucosa and contact antigen-presenting cells (APCs) such as dendritic cells and macrophages in the mucosa, particulate antigens can be either cleared by the mucociliary system or taken up by microfold (M) cells in the NALT. The route followed by the antigens in DPVs should mainly resemble the fate of particulate antigens. The uptake of antigens by APCs leads to the activation of T and B cells. The activated B cells differentiate into plasma cells, which leave the follicles and secrete the immunoglobulin A (IgA) class of antibodies into the lumen, where they can interact and neutralize the specific antigens. This interaction leads to the formation of IgA-antigen complexes, which are easily entrapped in the mucus and then eliminated by the ciliated epithelial cells [18]. Also, lymphocytes activated in a mucosal surface can reach remote mucosal sites through the lymphatic system and transfer the immunity in a response called “the common mucosal immune system” [19]. The studies by Johansson et al. demonstrated that in humans an IN vaccine could provide immunity to the cervicovaginal mucosa [20], but not to the gut mucosa [21].
M cells are specialized cells that have been extensively studied in the gut-associated lymphoid tissue (GALT) for its role in the transport of antigens across the mucous membrane to underlying lymphoid cells [22]. Fujimura found M cells in excised human adenoid tissue [23]. These cells could take up influenza A virus, suggesting that M cells are sites of antigen uptake for induction of mucosal immunity after IN immunization [24]. Finally, antigen particles taken up by NALT are drained into lymph nodes. These antigens may induce the production of serum IgA and IgG in systemic lymphoid organs [17, 25]. However, the systemic immune response elicited by mucosal vaccination is usually weaker than that induced by parenteral immunization [26]. Therefore, the immune response provoked by DPV formulations is generally characterized by measuring specific sIgA and serum IgG titers as markers of local and systemic immune responses, respectively [27].
2. Advantages and disadvantages of intranasal immunization
2.1. Intranasal immunization versus parenteral injection
Most of the pathogens enter the body by penetrating mucosal surfaces, making the nasal mucosa one of the most promising routes for vaccinations due to the accessibility of the mucosal tissue, the improved patient compliance, and the potential for self-administration [28]. Moreover, in light of the continuous search for novel approaches for heat-stable and needle-free vaccines, the IN route of immunization denotes a great versatile strategy. Specifically, needle-free vaccines can potentially decrease the costs of vaccination, as they do not require trained professionals for administration and waste disposal, and represent a viable approach for mass vaccination campaigns. In addition, IN route allows for the administration of both liquid and dry vaccine formulations.
A hurdle in the development of nasal vaccine formulations is the limited time available for antigen absorption, due to the rapid mucociliary clearance of foreign particles [29]. This obstacle has been addressed by using mucoadhesives, which can increase the immunogenicity of vaccines by prolonging the residence of the antigens at the immune effector sites [30]. Chitosan is one of the most extensively studied mucoadhesives in IN vaccine formulations [31–33]. Its mechanism of action is based on its capacity to increase vaccine residence time as well as open tight junctions transiently in mucosal membranes, which improve antigen penetration [34]. Chitosan also enhanced the response to a parenteral immunization [33, 35]. It has been proposed that the observed increase in immune response induced by vaccine formulations that contain chitosan could be due to the presence of impurities in the chitosan [27]. Glycol chitosan has also been tested as a mucoadhesive in liquid IN vaccines. In preclinical studies, vaccine formulations containing glycol chitosan have shown better mucosal uptake and reduced nasal clearance [36], as well as stronger immunogenicity [37], as compared to those containing chitosan.
2.2. Intranasal versus pulmonary immunization
Currently, there are seven non-parenteral vaccines approved by the US Food and Drug Administration (FDA) [38]. Five of these products are for oral administration (i.e., adenovirus, cholera, rotavirus and typhoid vaccines) and two are liquid vaccines for intranasal administration (influenza vaccines). As compared to the nasal route, the pulmonary route offers a much larger mucosal surface area that has been extensively explored for local as well as systemic delivery of both liquid and powder drug formulations [39]. For vaccines, the high vascularization of the alveolar tissue may facilitate the systemic delivery of antigens with a greater potential of achieving both respiratory and systemic immunization [40]. The delivery of DPVs by inhalation has been explored in vivo for tuberculosis, hepatitis, influenza, and measles with promising results [41–46]. The safety and tolerability of a dry powder measles vaccine administered by inhalation were also confirmed in a clinical trial [47].
The main challenge of this route of administration is the low efficiency of the drug delivery achieved by current products. Both the formulation and the device used to administer the formulation determine the critical particle size for lung deposition [48]. To reach the deep lung, the aerosolized particles should have an ideal aerodynamic size between 1 and 5 μm. Particles above this size tend to impact on the surface of the upper airways [49]. For vaccine delivery, the later may not be completely disadvantageous because these particles still can reach the lymphoid tissue in the oropharyngeal region. However, if aiming for pulmonary and systemic immunization, advanced processing is required to obtain a high fine particle fraction (FPF) as well as delivery devices that can prevent or reduce the loss of product in the mouth and throat [50]. Saluja et al. developed two influenza DPVs with FPF values of 37% and 23% that were able to trigger significantly higher serum IgG titers than IM immunization in a mouse model [42]. The animals were immunized through an oropharyngeal tube.
3. Advantages and disadvantages of dry powder vaccines
FluMist® (MedImmune, LLC) is the first live attenuated influenza virus IN vaccine that was successfully approved and commercialized in the US and Europe (as Fluenz®) [51]. Nasovac® (Serum Institute of India, Ltd.) is an IN flu vaccine approved in India [52]. The inactivated IN influenza virus vaccine Nasalflu® (Berna Biotech AG, Switzerland) was available for a short period of time before it was discontinued in 2001 due to a potential association with partial facial paralysis [53]. Other vaccines for IN administration have been extensively studied against infectious diseases (e.g., measles, meningitis, tuberculosis, and pneumonia) [33, 54–60], or autoimmune diseases (e.g., arthritis) [61]. However, most of these formulations are liquids such as drops, sprays, and even emulgels.
Liquid vaccine, similar to other biologic products, are susceptible to physical, chemical, and thermal instability that can affect their potency and efficacy by undergoing degradation, aggregation, and/or hydrolysis [62]. Therefore, to stabilize them, liquid vaccines need preservatives, buffers, and cold-chain for storage and transportation [54]. Dry powder vaccine formulations can potentially eliminate these requirements, leading to more stable, more efficacious, and less expensive vaccines, which would be highly advantageous in particular for remote areas in tropical developing countries because these vaccines would become available for the population in the areas [63]. Data from many studies evidenced the superior storage stability of DPVs at different temperatures and humidities, as compared to liquid vaccines [27, 64–67]. Most of these studies evaluated the stability of the stored vaccines after reconstitution using in vitro assays, but Wang et al. instead demonstrated that an IN anthrax DPV stored for 2 years at room temperature was as immunogenic as the fresh DPV [68].
DPVs, as other dry powder formulations for inhalation, need to be protected from humidity [69]. The composition and quality of the packaging system are key elements for the chemical and physical stability of dry powder formulations [70]. Suitable containers for DPVs may well follow the packaging used in the recently available drug powders for nasal delivery [71]. In these intranasal products, the drugs are protected from humidity either in sealed pouches with desiccant (e.g., Onzetra Xsail®, sumatriptan nasal powder) or within the device with a built-in desiccant chamber (e.g., Rhinocort Turbuhaler®, budesonide nasal powder).
4. Dry powder vaccines tested in vivo
DPV formulations usually contain the same components of traditional vaccines (i.e., antigen and adjuvant), in addition to bulking agents, stabilizers, and mucoadhesives. Spray drying (SD) and freeze-drying (FD) are the most commonly used pharmaceutical processes to produce dry powders of biologic compounds. Spray freeze-drying (SFD) uses the same principle of liquid atomization of SD, but the atomized liquid is instantly frozen in liquid nitrogen and then lyophilized. SD was compared to SFD in the preparation of DPV formulations for pulmonary delivery, and it was noted that both methods produce stable vaccines with good inhalation properties [42]. Thin-film freeze-drying was also reported as a method to successfully convert liquid vaccines containing aluminum salts to DPVs [72]. A more recent approach instead is based on the encapsulation of the vaccine components in mucoadhesive particles, and then the particles are converted into a dry powder [73–76].
Finally, carrier-based formulations have been proposed as an alternative method to improve the nasal deposition of DPVs and consequently reduce the lung deposition of vaccine particles. Again, the carrier-based vaccines are converted into a dry powder for IN immunization. Sugar alcohols [76], polylactic acid-polyethylene glycol nanoparticles [77], and poly (lactic-co-glycolic) acid microspheres [78] are some examples of carriers tested for IN vaccines. Table 1 summarizes the different DPVs investigated for IN administration in preclinical and clinical studies. Below is described in detail each type of vaccine.
Table 1.
Intranasal dry powder vaccines tested in vivo.
| Disease | Antigen | Method of production | Adjuvant | Other excipients | Device | Model | Findings | References |
|---|---|---|---|---|---|---|---|---|
| Diphtheria | CRM197 formaldehyde treated (50 μg/dose) |
N/S | Chitosan and mannitol | Monopowder (Valois) | Human |
|
(Mills et al., 2003) | |
|
| ||||||||
| CRM197 formaldehyde treated (50 μg/dose) |
N/S | Chitosan, phosphate salts, and mannitol | Monopowder (Valois) | Human |
|
(McNeela et al., 2004) | ||
|
| ||||||||
| Influenza | Whole inactivated virus strain H1N1 (100 μg/dose) |
FD | Chitosan and trehalose | Plastic housing unit attached to a syringe (WO 2002055133 A3) | Rat |
|
(Huang et al., 2004) | |
|
| ||||||||
| Whole inactivated virus strain H1N1 (5 μg/dose) |
SFD | Chitosan, trehalose, HPMC-HMW, CMC-HMW and SA. | Lab-made device | Rat |
|
(Garmise et al., 2007) | ||
|
| ||||||||
| Whole heat-inactivated virus strain H3N2 (50 μg/dose) |
FD | LTR192G | Starch and polyacrylic acid | Lab-made device | Rabbit |
|
(Coucke et al., 2009) | |
|
| ||||||||
| Whole UV-inactivated virus strain H1N1 (45 μg/dose) |
Nanoencapsulation | CpG or Quillaja saponin | Chitosan | Lab-made device | Rabbit |
|
(Dehghan et al., 2014) | |
|
| ||||||||
| Meningitis | Meningococcal C oligosaccharide-CRM197 conjugated (10 μg/dose) |
N/S | Chitosan | Combitips plus® (Eppendorf) | Human |
|
(Huo et al., 2005) | |
|
| ||||||||
| Anthrax | rPA (50 μg/dose) |
FD or SFD | CpG | Chitosan and trehalose | Plastic housing unit attached to a syringe (WO 2002055133 A3) | Rabbit |
|
(Mikszta et al., 2005) |
|
| ||||||||
| rPA (10 μg/dose) |
SFD | CpG | Chitosan and trehalose | Plastic housing unit attached to a syringe (WO 2002055133 A3) | Rabbit |
|
(Huang et al., 2007) | |
|
| ||||||||
| rPA (90 μg/dose) combined with free capsule peptide (18 μg/dose) or with rPA- conjugate (90 μg/dose) | FD | MPL | Chitosan and mannitol | Monopowder (Valois) | Rabbit |
|
(Wimer-Mackin et al., 2006) | |
|
| ||||||||
| rPA (50–150 μg/dose) alone or combined with BSA-conjugate (150 μg/dose) | FD | MPL | Chitosan and mannitol | Monopowder (Valois) | Rabbit |
|
(Klas et al., 2008) | |
|
| ||||||||
| rPA (30 μg/dose) | SFD | C48/80 | Trehalose | Unit-dose system (Aptar) | Rabbit |
|
(Wang et al., 2012) | |
|
| ||||||||
| Tetanus | Tetanus toxoid (40 Lf/dose) | Microencapsulation | Quillaja saponin | Alginate and cross-linked dextran | Lab-made | Rabbit |
|
(Tafaghodi and Rastegar, 2010) |
|
| ||||||||
| Gastroenteritis | Norovirus virus-like particles (GI.1) (10 μg/dose) |
SD | Gardiquimod | GelSite® | Lab-made | Guinea pig |
|
(Velasquez et al., 2011) |
|
| ||||||||
| Norovirus virus-like particles GI and GII.4 (0.1–100 μg/dose) |
FD | GelSite® | Unit-dose system (Aptar) | Guinea pig |
|
(Springer et al., 2016) | ||
|
| ||||||||
| Norovirus virus-like particles GI and GII.4 (5100 μg/dose) | FD | GelSite® | Unit-dose system (Aptar) | Guinea pig |
|
(Ball et al, 2017) | ||
|
| ||||||||
| Norovirus virus-like particles (GI.1) (5–100 μg/dose) | FD | MPL | Chitosan, sucrose, and mannitol | UniDose DP (Bespak) | Human |
|
(El-Kamary et al., 2010) | |
|
| ||||||||
| Norovirus virus-like particles (GI.1) (50–100 μg/dose) | FD | MPL | Chitosan, sucrose, and mannitol | UniDose DP (Bespak) | Human |
|
(El-Kamary et al., 2010, Ramirez et al., 2012) | |
|
| ||||||||
| Norovirus virus-like particles (GI.1) (100 μg/dose) |
FD | MPL | Chitosan, sucrose, and mannitol | UniDose DP (Bespak) | Human |
|
(Atmar et al., 2011) | |
CRM197: cross-reacting material of the diphtheria toxin; FD: Freeze-drying; IN: intranasal; IM: intramuscular; SFD: Spray-freeze drying; HPMC-HMW: Hydroxypropyl methylcellulose, high molecular weight; CMC-HMW: Carboxymethylcellulose sodium, high molecular weight; SA: Sodium alginate; LTR192G: Heat-labile enterotoxin R192G mutant; rPA: recombinant Protective Antigen of B. anthracis; ID: intradermal; rPA conjugate; Capsule peptide conjugated to rPA; MPL: Monophosphoryl lipid A; BSA-conjugate: Capsule peptide conjugated to bovine serum albumin; GelSite®: Aloe vera derived polysaccharide polymer; Lf: Flocculation units; N/S: not specified
4.1. Influenza
Influenza represents the leading cause of respiratory diseases in humans [79]. The first reported influenza DPV formulation was developed using whole, inactivated influenza viruses [64]. This IN vaccine generated a potent nasal mucosal immune response in rats, in addition to a systemic immune response that was comparable to that induced through intramuscular (IM) injection. Data from that study also demonstrated that chitosan increased the immune response induced by the DPV [64]. Garmise et al. prepared and characterized influenza DPVs by testing the polydispersity of particles and the flow properties of DPV formulations containing whole, inactivated influenza viruses, lactose or trehalose, and chitosan [80]. In another study, the research group focused on identifying the effect of different mucoadhesive compounds on the DPV formulations and found a consistent trend between the nasal residence time of the formulations and the strength of the resultant immune response. Maximal mucosal and systemic responses were observed when using sodium alginate and carboxymethylcellulose-high molecular weight [27].
Different ratios of the bioadhesives, starch, and polyacrylic acid, were evaluated as carriers of the viral influenza antigen by Coucke et al. [81]. These powder formulations were able to induce systemic immune responses. It was observed that the levels of IgG titers induced by the IN vaccine had a dose-dependent correlation with the polyacrylic acid content.
Dehghan et al. observed the highest increase in IgG titers after the administration of chitosan nanospheres loaded with whole UV-inactivated influenza virus A together with synthetic CpG oligodeoxynucleotides (CpG ODNs) or Quillaja saponin as adjuvants in a rabbit model, demonstrating the feasibility of including vaccine adjuvants in IN DPV formulations [73, 82].
4.2. Diphtheria and Meningitis
Diphtheria is a disease caused by a toxin produced by Corynebacterium diphtheria. The cross-reacting material (CRM197) of the diphtheria toxin, not toxic but still antigenic, is used extensively as a licensed polysaccharide antigen carrier in human vaccines [83]. CRM197 can be further treated with formaldehyde to increase resistance to proteolytic degradation [84]. McNeela et al. found that the addition of chitosan to a liquid formulation of CRM197 treated with 0.18% formaldehyde resulted in significantly enhanced immunogenicity measured by specific IgG titers in guinea pigs [33]. In another study, the group demonstrated that the vaccine formulated as a DPV was well tolerated in humans [85]. This IN vaccine induced significantly higher secretory IgA levels and similar protective levels of serum IgA and IgG, compared to those induced by IM immunization, and the same DPV caused a strong Th2-biased response in humans after IN immunization [86].
Menjugate-C® is a meningitis C vaccine developed and commercialized in Europe by Chiron Corp. (now Novartis Vaccines and Diagnostics, Inc.). It contains meningococcal C oligosaccharide conjugated to CRM197 in a powder form to be resuspended before IM administration [87]. Huo et al. used this vaccine powder mixed with chitosan for nasal immunization in a clinical study [88]. IN administration of the powder was well tolerated by the subjects, and the meningococcal-specific serum IgG and IgA levels observed 28 days after the administration were comparable to those seen after IM administration.
4.3. Viral gastroenteritis
Noroviruses are recognized as the main cause of epidemics of gastroenteritis in children and adults. The genetically variable Norovirus genus present three genogroups (GI, GII, and GIV) that can infect humans [89]. The expression of Norwalk virus (GI.1) capsid proteins without the viral RNA allows the development of immunogenic but noninfectious virus-like particles for vaccine formulations [90]. The immunogenicity of the norovirus virus-like particles (NV VLPs) was later confirmed by intranasal administration using a liquid formulation in mice[91].
El-Kamary et al. conducted a phase 1 clinical study to evaluate the safety and immunogenicity of a DPV containing NV VLPs, monophosphoryl lipid A (MPL) as adjuvant and chitosan as a mucoadhesive [92]. This DPV was highly immunogenic and well tolerated with only mild side effects. Peripheral blood mononuclear cells (PBMCs) from subjects in these clinical studies were collected to analyze B memory cell responses [93]. The B memory cell frequencies determined by flow cytometry were correlated with the serum levels of NV VLP-specific serum antibodies [93]. A significant NV-specific IgA and IgG responses were obtained in all subjects immunized with 100 μg/dose. In a different clinical study, the same DPV containing NV VLP, MPL, and chitosan provided significant protection against gastroenteritis after exposure to a homologous virus [94].
Velasquez et al. formulated a vaccine powder containing these NV VLPs in combination with a gelling agent derived from Aloe vera (GelSite®) and gardiquimod as an adjuvant. In situ gelation as well as mucosal and systemic immunity were shown in guinea pigs when this formulation was given IN [29]. Two novel adjuvant-free norovirus vaccines have been developed containing NV VLPs from genogroups I and II.4 with GelSite® as a mucoadhesive. The preclinical study conducted by Springer and colleagues reported that these DPVs showed maximal immunogenicity with smaller doses (greater than 15 μg) in guinea pigs [95]. Recently, the same group reported a bivalent DPV containing the antigens GI and GII.4 [96].
4.4. Anthrax
This lethal disease is caused by an exotoxin produced by Bacillus anthracis. Investigated IN vaccine formulations include those containing the recombinant protective antigen (rPA) protein and synthetic CpG oligonucleotides (as an adjuvant) [97, 98]. Mikszta et al. evaluated the protective effect of this vaccine in rabbits [99]. IN administration of the DPV showed better protection against anthrax (100% of survival) than IN liquid vaccine (67% survival). In a following study, the same complete protection was observed when a lower dose of this DPV was used to immunize rabbits [97]. Finally, this DPV formulation allowed the maintenance of protein integrity for one month at ambient temperature and in accelerated stability studies [65].
Another antigen used for anthrax vaccines is a peptide from the capsule of B. anthracis. DPVs were developed by lyophilization with mannitol and MPL using rPA in combination with either the free peptide or the peptide conjugated to rPA (rPA-conjugated). The powder was then resuspended to obtain liquid vaccines or mixed with chitosan to prepare DPVs for IN administration [100]. These vaccines were used to immunize rabbits exposed later to a lethal aerosol spore challenge. All DPVs demonstrated 100% protection, but only the animals immunized with rPA plus rPA-conjugated capsular peptide did not show signs of illness after 14 days. Also, the DPVs induced serum IgG titers that were higher than those induced by IN liquid vaccination and similar to that induced by IM vaccination. Klas et al. tested a different formulation containing rPA and the capsule peptide mentioned above in rabbits [101]. The peptide was conjugated to bovine serum albumin (BSA-conjugated), lyophilized with mannitol and MPL, and then mixed with chitosan. Results after an aerosol spore challenge showed that a single IN immunization could protect the rabbits against the lethal spore challenge, up to 9 weeks post-treatment. Wang et al. used a different adjuvant, the mast cell activator C48/80, to prepare an rPA DPV without mucoadhesives [68]. IN administration of a freshly prepared DPV or a DPV stored for 2 years at room temperature induced immune responses comparable to those induced by IM administration of a liquid vaccine.
4.5. Tetanus
Tetanus toxoid (TT) has been used as a model antigen for IN vaccine development [74, 102]. Tafaghodi and Rastegar used TT and Quillaja saponin encapsulated in alginate microspheres with and without cross-linked dextran mucoadhesive microspheres for nasal immunization in rabbits [103]. These microspheres were tested for morphology, particle size, and in vitro release of TT and Quillaja saponin. They observed the highest mucosal and systemic immune responses when dextran microspheres were added to the formulation, which was converted into a dry powder and filled into a polyethylene tube (2 mm in diameter) connected to a syringe for IN administration.
5. Devices used for intranasal delivery of dry powder vaccines
The IN FluMist® Quadrivalent vaccine is a liquid formulation in a prefilled intranasal sprayer. However, dry powders for IN immunization require a different type of device to aerosolize the product. Dry powder inhalers (DPIs) are examples of commercially available inhalers developed for pulmonary drug delivery and have been on the market for decades. Depending on the aerosolization mechanism, DPIs are broadly classified as active or passive devices. Active devices have an aerosolization mechanism, while in passive devices the aerosolization is driven by patient’s inhalation [104]. Following this classification criterion, most of the devices tested for IN delivery of DPVs can be classified as active devices. In fact, during the administration of IN vaccines inhalation should be prevented to keep the formulation in the nasal cavity to target the lymphoid tissues in the nasal cavity and prevent lung deposition.
In general, these devices for DPV delivery use a pneumatic force to expel the powder into the nasal cavity, which resembles the mechanism of action of a syringe. Indeed, most of the lab-made devices used for preclinical studies are based on syringes [64]. Some examples of commercially available devices for IN delivery of powders are the Monopowder single-dose disposable device (Valois Pharmaceutical Division), the Unit-dose system (AptarGroup, Inc.), and the Powder-Jet® device (RPC Bramlage GmbH). The Unit-dose system has been used to intranasally dose DPVs in preclinical studies in rabbits and guinea pigs [68, 95]. The Monopowder single-dose disposable device has been used for intranasal DPV administration in preclinical and clinical studies [85, 86, 100, 101]. The Powder-Jet®, different from the other devices, is a multidose system and was used by Trows and Scherließ to characterize their carrier-based DPV formulations [76]. A detailed review of devices for nasal drug delivery is available [105]. Since vaccines are aimed for long-term immunization, there is no clear need for multi-dose chambers nor reusability, which are important aspects of devices used for chronic diseases. An ideal IN DPV product should be pre-loaded in a ready-to-use, disposable device to prevent manipulation and loss of the product. The packaging of the device should be moisture-sealed to ensure protection from humidity until use. Onzetra Xsail® (Avanir Pharmaceuticals, Inc.), approved by the FDA in 2016, is a sumatriptan powder for nasal delivery with the breath-powered device OptiNose® (OptiNose US, Inc.) [106]. This passive device is actuated when the patient blows air in the mouthpiece that releases the drug into the nasal cavity through the nosepiece. Even though OptiNose® has not been tested for DPVs delivery, it sets the precedent of an approved device that can be used for the study of future DPVs. Also, blowing through the mouth causes the closure of the soft palate between the nasal and the oral cavity, which prevents powder from flowing into the oropharynx and lungs [107, 108]. Therefore, this characteristic could be helpful to evaluate in clinical studies the immune responses triggered at the nasal mucosa without the contribution of additional responses from the airway mucosa outside the nasal cavity.
A list of the main findings, key challenges, and potential future directions of IN DPVs research can be found in Table 2.
Table 2.
Summary of key findings, main challenges, and future directions in intranasal DPV research.
| Key findings in the literature about IN DPVs | Main challenges of IN DPVs | Prediction/future directions of IN DPVs |
|---|---|---|
|
|
|
6. Conclusion
Vaccines are a primary weapon to safeguard public health, and now even more important, with the continuous development of antimicrobial-resistant microorganisms. Injectable vaccines present some disadvantages such as the requirement of professional assisted administration, cold chain during storage and transport, as well as the costs of waste disposal. Needle-free IN vaccination represents an attractive approach to address these issues, safely and efficiently. The success with the IN liquid FluMist® and Nasovac® vaccines demonstrated the viability of IN immunization from both marketing and vaccinology perspectives. It is true that there is not an FDA-approved DPV yet, but the increasing amount of preclinical and clinical studies, as well as the approval of IN dry powder products, have shown the interest of the pharmaceutical industry in this new type of vaccines and route of administration. Preclinical studies showed that IN immunization with DPVs is feasible. Finally, intranasal immunization with DPVs has many advantages over with liquid vaccines, but different devices that can actively deliver the powder into the nasal cavity are needed to efficiently administer DPVs intranasally.
Acknowledgments
Tania Bahamondez-Canas would like to thank the CONICYT (Becas Chile) for the scholarship to support her Ph.D. studies. Z.C.’s research is supported by grants from the U.S. National Institutes of Health (CA135274, CA179362), the National Natural Science Foundation of China (81460454), and the Inner Mongolia Natural Science Fund (2014ZD05). The authors would also like to thank Daniel Moraga-Espinoza for his help in drawing Figure 1 and the figure in the graphic abstract.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
The authors report no conflict of interest.
References
- 1.Elad D, Wolf M, Keck T. Air-conditioning in the human nasal cavity. Respir Physiol Neurobiol. 2008;163:121–127. doi: 10.1016/j.resp.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Boiselle PM, Lynch DA. CT of the airways. Humana Press; Totowa, NJ, USA: 2008. [Google Scholar]
- 3.Ogle OE, Weinstock RJ, Friedman E. Surgical anatomy of the nasal cavity and paranasal sinuses. Oral Maxillofac Surg Clin North Am. 2012;24:155–166. vii. doi: 10.1016/j.coms.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 4.Kublik H, Vidgren M. Nasal delivery systems and their effect on deposition and absorption. Advanced drug delivery reviews. 1998;29:157–177. doi: 10.1016/s0169-409x(97)00067-7. [DOI] [PubMed] [Google Scholar]
- 5.Jones N. The nose and paranasal sinuses physiology and anatomy. Adv Drug Deliv Rev. 2001;51:5–19. doi: 10.1016/s0169-409x(01)00172-7. [DOI] [PubMed] [Google Scholar]
- 6.Kirkeby L, Rasmussen TT, Reinholdt J, Kilian M. Immunoglobulins in nasal secretions of healthy humans: structural integrity of secretory immunoglobulin A1 (IgA1) and occurrence of neutralizing antibodies to IgA1 proteases of nasal bacteria. Clin Diagn Lab Immunol. 2000;7:31–39. doi: 10.1128/cdli.7.1.31-39.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riechelmann H, Deutschle T, Friemel E, Gross HJ, Bachem M. Biological markers in nasal secretions. Eur Respir J. 2003;21:600–605. doi: 10.1183/09031936.03.00072003. [DOI] [PubMed] [Google Scholar]
- 8.Cole AM, Dewan P, Ganz T. Innate antimicrobial activity of nasal secretions. Infect Immun. 1999;67:3267–3275. doi: 10.1128/iai.67.7.3267-3275.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morinaka S, Nakamura H. Inflammatory cells in nasal mucosa and nasal polyps. Auris Nasus Larynx. 2000;27:59–64. doi: 10.1016/s0385-8146(99)00038-3. [DOI] [PubMed] [Google Scholar]
- 10.Linder A, Karlsson-Parra A, Hirvela C, Jonsson L, Koling A, Sjoberg O. Immunocompetent cells in human nasal polyps and normal mucosa. Rhinology. 1993;31:125–129. [PubMed] [Google Scholar]
- 11.Cesta MF. Normal structure, function, and histology of mucosa-associated lymphoid tissue. Toxicol Pathol. 2006;34:599–608. doi: 10.1080/01926230600865531. [DOI] [PubMed] [Google Scholar]
- 12.Brandtzaeg P, Kiyono H, Pabst R, Russell MW. Terminology: nomenclature of mucosa-associated lymphoid tissue. Mucosal Immunol. 2008;1:31–37. doi: 10.1038/mi.2007.9. [DOI] [PubMed] [Google Scholar]
- 13.Pabst R. Lymphatic tissue of the nose (NALT) and larynx (LALT) in species comparison: human, rat, mouse. Pneumologie. 2010;64:445–446. doi: 10.1055/s-0030-1255509. [DOI] [PubMed] [Google Scholar]
- 14.Hellings P, Jorissen M, Ceuppens JL. The Waldeyer’s ring. Acta Otorhinolaryngol Belg. 2000;54:237–241. [PubMed] [Google Scholar]
- 15.Debertin A, Tschernig T, Tönjes H, Kleemann W, Tröger H, Pabst R. Nasal-associated lymphoid tissue (NALT): frequency and localization in young children. Clinical & Experimental Immunology. 2003;134:503–507. doi: 10.1111/j.1365-2249.2003.02311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pabst R. Mucosal vaccination by the intranasal route. Nose-associated lymphoid tissue (NALT)-Structure, function and species differences. Vaccine. 2015;33:4406–4413. doi: 10.1016/j.vaccine.2015.07.022. [DOI] [PubMed] [Google Scholar]
- 17.Kuper CF, Koornstra PJ, Hameleers DM, Biewenga J, Spit BJ, Duijvestijn AM, van Breda Vriesman PJ, Sminia T. The role of nasopharyngeal lymphoid tissue. Immunol Today. 1992;13:219–224. doi: 10.1016/0167-5699(92)90158-4. [DOI] [PubMed] [Google Scholar]
- 18.Goldsby RA, Kindt TJ, Osborne BA, Kuby J. Kuby immunology. 4th. W.H. Freeman; New York: 2000. [Google Scholar]
- 19.Holmgren J, Czerkinsky C. Mucosal immunity and vaccines. Nat Med. 2005 doi: 10.1038/nm1213. [DOI] [PubMed] [Google Scholar]
- 20.Johansson EL, Wassen L, Holmgren J, Jertborn M, Rudin A. Nasal and vaginal vaccinations have differential effects on antibody responses in vaginal and cervical secretions in humans. Infect Immun. 2001;69:7481–7486. doi: 10.1128/IAI.69.12.7481-7486.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johansson EL, Bergquist C, Edebo A, Johansson C, Svennerholm AM. Comparison of different routes of vaccination for eliciting antibody responses in the human stomach. Vaccine. 2004;22:984–990. doi: 10.1016/j.vaccine.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 22.Clark MA, Jepson MA, Hirst BH. Exploiting M cells for drug and vaccine delivery. Advanced Drug Delivery Reviews. 2001;50:81–106. doi: 10.1016/s0169-409x(01)00149-1. [DOI] [PubMed] [Google Scholar]
- 23.Fujimura Y. Evidence of M cells as portals of entry for antigens in the nasopharyngeal lymphoid tissue of humans. Virchows Archiv. 2000;436:560–566. doi: 10.1007/s004289900177. [DOI] [PubMed] [Google Scholar]
- 24.Fujimura Y, Takeda M, Ikai H, Haruma K, Akisada T, Harada T, Sakai T, Ohuchi M. The role of M cells of human nasopharyngeal lymphoid tissue in influenza virus sampling. Virchows Arch. 2004;444:36–42. doi: 10.1007/s00428-003-0898-8. [DOI] [PubMed] [Google Scholar]
- 25.Neutra MR, Kozlowski PA. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol. 2006;6:148–158. doi: 10.1038/nri1777. [DOI] [PubMed] [Google Scholar]
- 26.Czerkinsky C, Anjuere F, McGhee JR, George-Chandy A, Holmgren J, Kieny MP, Fujiyashi K, Mestecky JF, Pierrefite-Carle V, Rask C, Sun JB. Mucosal immunity and tolerance: relevance to vaccine development. Immunol Rev. 1999;170:197–222. doi: 10.1111/j.1600-065X.1999.tb01339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garmise RJ, Staats HF, Hickey AJ. Novel dry powder preparations of whole inactivated influenza virus for nasal vaccination. AAPS PharmSciTech. 2007;8:2–10. doi: 10.1208/pt0804081. [DOI] [PubMed] [Google Scholar]
- 28.Garmise RJ, Hickey AJ. Dry powder nasal vaccines as an alternative to needle-based delivery. Critical Reviews™ in Therapeutic Drug Carrier Systems. 2009;26 doi: 10.1615/critrevtherdrugcarriersyst.v26.i1.10. [DOI] [PubMed] [Google Scholar]
- 29.Velasquez LS, Shira S, Berta AN, Kilbourne J, Medi BM, Tizard I, Ni Y, Arntzen CJ, Herbst-Kralovetz MM. Intranasal delivery of Norwalk virus-like particles formulated in an in situ gelling, dry powder vaccine. Vaccine. 2011;29:5221–5231. doi: 10.1016/j.vaccine.2011.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garg NK, Mangal S, Khambete H, Tyagi RK. Mucosal delivery of vaccines: role of mucoadhesive/biodegradable polymers. Recent Pat Drug Deliv Formul. 2010;4:114–128. doi: 10.2174/187221110791185015. [DOI] [PubMed] [Google Scholar]
- 31.Illum L, Jabbal-Gill I, Hinchcliffe M, Fisher A, Davis S. Chitosan as a novel nasal delivery system for vaccines. Advanced drug delivery reviews. 2001;51:81–96. doi: 10.1016/s0169-409x(01)00171-5. [DOI] [PubMed] [Google Scholar]
- 32.Roy K, Mao HQ, Huang SK, Leong KW. Oral gene delivery with chitosan–DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nature medicine. 1999;5:387–391. doi: 10.1038/7385. [DOI] [PubMed] [Google Scholar]
- 33.McNeela EA, O’Connor D, Jabbal-Gill I, Illum L, Davis SS, Pizza M, Peppoloni S, Rappuoli R, Mills KH. A mucosal vaccine against diphtheria: formulation of cross reacting material (CRM 197) of diphtheria toxin with chitosan enhances local and systemic antibody and Th2 responses following nasal delivery. Vaccine. 2000;19:1188–1198. doi: 10.1016/s0264-410x(00)00309-1. [DOI] [PubMed] [Google Scholar]
- 34.Dodane V, Khan M Amin, Merwin JR. Effect of chitosan on epithelial permeability and structure. Int J Pharm. 1999;182:21–32. doi: 10.1016/s0378-5173(99)00030-7. [DOI] [PubMed] [Google Scholar]
- 35.Ghendon Y, Markushin S, Vasiliev Y, Alkopova I, Koptiaeva I, Krivtsov G, Borisova O, Ahmatova N, Kurbatova E, Mazurina S, Gervazieva V. Evaluation of Properties of Chitosan as an Adjuvant for Inactivated Influenza Vaccines Administered Parenterally. J Med Virol. 2009;81:494–506. doi: 10.1002/jmv.21415. [DOI] [PubMed] [Google Scholar]
- 36.Pawar D, Jaganathan K. Mucoadhesive glycol chitosan nanoparticles for intranasal delivery of hepatitis B vaccine: enhancement of mucosal and systemic immune response. Drug delivery. 2016;23:185–194. doi: 10.3109/10717544.2014.908427. [DOI] [PubMed] [Google Scholar]
- 37.Gogev S, de Fays K, Versali MF, Gautier S, Thiry E. Glycol chitosan improves the efficacy of intranasally administrated replication defective human adenovirus type 5 expressing glycoprotein D of bovine herpesvirus 1. Vaccine. 2004;22:1946–1953. doi: 10.1016/j.vaccine.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 38.Malcolmson RJ, Embleton JK. Dry powder formulations for pulmonary delivery. Pharmaceutical Science & Technology Today. 1998;1:394–398. [Google Scholar]
- 39.Ruge CA, Kirch J, Lehr CM. Pulmonary drug delivery: from generating aerosols to overcoming biological barriers—therapeutic possibilities and technological challenges. The Lancet Respiratory Medicine. 2013;1:402–413. doi: 10.1016/S2213-2600(13)70072-9. [DOI] [PubMed] [Google Scholar]
- 40.Patton JS, Fishburn CS, Weers JG. The lungs as a portal of entry for systemic drug delivery. Proceedings of the American Thoracic Society. 2004;1:338–344. doi: 10.1513/pats.200409-049TA. [DOI] [PubMed] [Google Scholar]
- 41.Amorij JP, Saluja V, Petersen AH, Hinrichs WL, Huckriede A, Frijlink HW. Pulmonary delivery of an inulin-stabilized influenza subunit vaccine prepared by spray-freeze drying induces systemic, mucosal humoral as well as cell-mediated immune responses in BALB/c mice. Vaccine. 2007;25:8707–8717. doi: 10.1016/j.vaccine.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 42.Saluja V, Amorij J, Kapteyn J, de Boer A, Frijlink H, Hinrichs W. A comparison between spray drying and spray freeze drying to produce an influenza subunit vaccine powder for inhalation. Journal of Controlled Release. 2010;144:127–133. doi: 10.1016/j.jconrel.2010.02.025. [DOI] [PubMed] [Google Scholar]
- 43.Garcia-Contreras L, Wong YL, Muttil P, Padilla D, Sadoff J, DeRousse J, Germishuizen WA, Goonesekera S, Elbert K, Bloom BR. Immunization by a bacterial aerosol. Proceedings of the National Academy of Sciences. 2008;105:4656–4660. doi: 10.1073/pnas.0800043105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swart RL de, LiCalsi C, Quirk AV, Amerongen G van, Nodelman V, Alcock R, Yüksel S, Ward GH, Hardy JG, Vos H. Measles vaccination of macaques by dry powder inhalation. Vaccine. 2007;25:1183–1190. doi: 10.1016/j.vaccine.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 45.Muttil P, Prego C, Garcia-Contreras L, Pulliam B, Fallon JK, Wang C, Hickey AJ, Edwards D. Immunization of guinea pigs with novel hepatitis B antigen as nanoparticle aggregate powders administered by the pulmonary route. The AAPS journal. 2010;12:330–337. doi: 10.1208/s12248-010-9192-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu D, Garcia-Contreras L, Muttil P, Padilla D, Xu D, Liu J, Braunstein M, McMurray DN, Hickey AJ. Pulmonary immunization using antigen 85-B polymeric microparticles to boost tuberculosis immunity. The AAPS journal. 2010;12:338–347. doi: 10.1208/s12248-010-9193-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Agarkhedkar S, Kulkarni PS, Winston S, Sievers R, Dhere RM, Gunale B, Powell K, Rota PA, Papania M. Safety and immunogenicity of dry powder measles vaccine administered by inhalation: a randomized controlled Phase I clinical trial. Vaccine. 2014;32:6791–6797. doi: 10.1016/j.vaccine.2014.09.071. [DOI] [PubMed] [Google Scholar]
- 48.Labiris N, Dolovich M. Pulmonary drug delivery. Part II: the role of inhalant delivery devices and drug formulations in therapeutic effectiveness of aerosolized medications. British journal of clinical pharmacology. 2003;56:600–612. doi: 10.1046/j.1365-2125.2003.01893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carvalho TC, Peters JI, Williams RO. Influence of particle size on regional lung deposition–what evidence is there? International Journal of Pharmaceutics. 2011;406:1–10. doi: 10.1016/j.ijpharm.2010.12.040. [DOI] [PubMed] [Google Scholar]
- 50.Shoyele SA, Cawthorne S. Particle engineering techniques for inhaled biopharmaceuticals. Advanced drug delivery reviews. 2006;58:1009–1029. doi: 10.1016/j.addr.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 51.Billich A. Technology evaluation: FluMist, University of Michigan. Curr Opin Mol Ther. 2000;2:340–344. [PubMed] [Google Scholar]
- 52.Kulkarni PS, Raut SK, Dhere RM. A post-marketing surveillance study of a human live-virus pandemic influenza A (H1N1) vaccine (Nasovac ((R))) in India. Hum Vaccin Immunother. 2013;9:122–124. doi: 10.4161/hv.22317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mutsch M, Zhou W, Rhodes P, Bopp M, Chen RT, Linder T, Spyr C, Steffen R. Use of the inactivated intranasal influenza vaccine and the risk of Bell’s palsy in Switzerland. New England journal of medicine. 2004;350:896–903. doi: 10.1056/NEJMoa030595. [DOI] [PubMed] [Google Scholar]
- 54.Tlaxca JL, Ellis S, Remmele RL., Jr Live attenuated and inactivated viral vaccine formulation and nasal delivery: Potential and challenges. Adv Drug Deliv Rev. 2014 doi: 10.1016/j.addr.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 55.Zhang J, Jex E, Feng T, Sivko GS, Baillie LW, Goldman S, Van Kampen KR, Tang DC. An adenovirus-vectored nasal vaccine confers rapid and sustained protection against anthrax in a single-dose regimen. Clin Vaccine Immunol. 2013;20:1–8. doi: 10.1128/CVI.00280-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamanaka H, Hoyt T, Yang X, Golden S, Bosio CM, Crist K, Becker T, Maddaloni M, Pascual DW. A nasal interleukin-12 DNA vaccine coexpressing Yersinia pestis F1-V fusion protein confers protection against pneumonic plague. Infect Immun. 2008;76:4564–4573. doi: 10.1128/IAI.00581-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liashenko VA, Krasnova VP, Youminova NV. Measles IgA in the nasal washings of adult volunteers and children immunized intranasally with measles vaccine L-16. Hum Antibodies. 1999;9:143–148. [PubMed] [Google Scholar]
- 58.Haneberg B, Dalseg R, Wedege E, Hoiby EA, Haugen IL, Oftung F, Andersen SR, Naess LM, Aase A, Michaelsen TE, Holst J. Intranasal administration of a meningococcal outer membrane vesicle vaccine induces persistent local mucosal antibodies and serum antibodies with strong bactericidal activity in humans. Infect Immun. 1998;66:1334–1341. doi: 10.1128/iai.66.4.1334-1341.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Larbig M, Mansouri E, Freihorst J, Tummler B, Kohler G, Domdey H, Knapp B, Hungerer KD, Hundt E, Gabelsberger J, von Specht BU. Safety and immunogenicity of an intranasal Pseudomonas aeruginosa hybrid outer membrane protein F-I vaccine in human volunteers. Vaccine. 2001;19:2291–2297. doi: 10.1016/s0264-410x(00)00550-8. [DOI] [PubMed] [Google Scholar]
- 60.Derrick SC, Kolibab K, Yang A, Morris SL. Intranasal administration of BCG induces superior protection against an aerosol infection with Mycobacterium tuberculosis in mice. Clinical and Vaccine Immunology. 2014 doi: 10.1128/CVI.00394-14. CVI.00394-00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tarkowski A, Sun JB, Holmdahl R, Holmgren J, Czerkinsky C. Treatment of experimental autoimmune arthritis by nasal administration of a type II collagen-cholera toxoid conjugate vaccine. Arthritis Rheum. 1999;42:1628–1634. doi: 10.1002/1529-0131(199908)42:8<1628::AID-ANR10>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 62.Pfleiderer M. Stability of vaccines – Bridging from stability data to continuous safety and efficacy throughout shelf life – An always reliable approach? Biologicals. 2009;37:364–368. doi: 10.1016/j.biologicals.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 63.Lydon P, Zipursky S, Tevi-Benissan C, Djingarey MH, Gbedonou P, Youssouf BO, Zaffran M. Economic benefits of keeping vaccines at ambient temperature during mass vaccination: the case of meningitis A vaccine in Chad. Bulletin of the World Health Organization. 2014;92:86–92. doi: 10.2471/BLT.13.123471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang J, Garmise RJ, Crowder TM, Mar K, Hwang CR, Hickey AJ, Mikszta JA, Sullivan VJ. A novel dry powder influenza vaccine and intranasal delivery technology: induction of systemic and mucosal immune responses in rats. Vaccine. 2004;23:794–801. doi: 10.1016/j.vaccine.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 65.Jiang G, Joshi SB, Peek LJ, Brandau DT, Huang J, Ferriter MS, Woodley WD, Ford BM, Mar KD, Mikszta JA, Hwang CR, Ulrich R, Harvey NG, Middaugh CR, Sullivan VJ. Anthrax vaccine powder formulations for nasal mucosal delivery. J Pharm Sci. 2006;95:80–96. doi: 10.1002/jps.20484. [DOI] [PubMed] [Google Scholar]
- 66.Jones RM, Burke M, Dubose D, Chichester JA, Manceva S, Horsey A, Streatfield SJ, Breit J, Yusibov V. Stability and pre-formulation development of a plant-produced anthrax vaccine candidate. Vaccine. 2017;35:5463–5470. doi: 10.1016/j.vaccine.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 67.Saboo S, Tumban E, Peabody J, Wafula D, Peabody DS, Chackerian B, Muttil P. Optimized Formulation of a Thermostable Spray-Dried Virus-Like Particle Vaccine against Human Papillomavirus. Molecular pharmaceutics. 2016;13:1646–1655. doi: 10.1021/acs.molpharmaceut.6b00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang SH, Kirwan SM, Abraham SN, Staats HF, Hickey AJ. Stable dry powder formulation for nasal delivery of anthrax vaccine. J Pharm Sci. 2012;101:31–47. doi: 10.1002/jps.22742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Price R, Young PM, Edge S, Staniforth JN. The influence of relative humidity on particulate interactions in carrier-based dry powder inhaler formulations. International Journal of Pharmaceutics. 2002;246:47–59. doi: 10.1016/s0378-5173(02)00359-9. [DOI] [PubMed] [Google Scholar]
- 70.FDA. Guidance for the industry: Metered Dose inhalers (MDI) and Dry Powder Inhalers (DPI) drug products. 1998 [Google Scholar]
- 71.Tiozzo Fasiolo L, Manniello MD, Tratta E, Buttini F, Rossi A, Sonvico F, Bortolotti F, Russo P, Colombo G. Opportunity and challenges of nasal powders: Drug formulation and delivery. European Journal of Pharmaceutical Sciences. doi: 10.1016/j.ejps.2017.09.027. [DOI] [PubMed] [Google Scholar]
- 72.Li X, Thakkar SG, Ruwona TB, Williams RO, Cui Z. A method of lyophilizing vaccines containing aluminum salts into a dry powder without causing particle aggregation or decreasing the immunogenicity following reconstitution. Journal of Controlled Release. 2015;204:38–50. doi: 10.1016/j.jconrel.2015.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dehghan S, Tafaghodi M, Bolourieh T, Mazaheri V, Torabi A, Abnous K, Kheiri M Tavassoti. Rabbit nasal immunization against influenza by dry-powder form of chitosan nanospheres encapsulated with influenza whole virus and adjuvants. Int J Pharm. 2014;475:1–8. doi: 10.1016/j.ijpharm.2014.08.032. [DOI] [PubMed] [Google Scholar]
- 74.Vila A, Sanchez A, Evora C, Soriano I, Vila Jato JL, Alonso MJ. PEG-PLA nanoparticles as carriers for nasal vaccine delivery. J Aerosol Med. 2004;17:174–185. doi: 10.1089/0894268041457183. [DOI] [PubMed] [Google Scholar]
- 75.Yao W, Peng Y, Du M, Luo J, Zong L. Preventative vaccine-loaded mannosylated chitosan nanoparticles intended for nasal mucosal delivery enhance immune responses and potent tumor immunity. Mol Pharm. 2013;10:2904–2914. doi: 10.1021/mp4000053. [DOI] [PubMed] [Google Scholar]
- 76.Trows S, Scherließ R. Carrier-based dry powder formulation for nasal delivery of vaccines utilizing BSA as model drug. Powder Technology. 2016;292:223–231. [Google Scholar]
- 77.Vila A, Sanchez A, Evora C, Soriano I, McCallion O, Alonso M. PLA-PEG particles as nasal protein carriers: the influence of the particle size. International journal of pharmaceutics. 2005;292:43–52. doi: 10.1016/j.ijpharm.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 78.Zhao H, Wu B, Wu H, Su L, Pang J, Yang T, Liu Y. Protective immunity in rats by intranasal immunization with Streptococcus mutans glucan-binding protein D encapsulated into chitosan-coated poly (lactic-co-glycolic acid) microspheres. Biotechnology letters. 2006;28:1299–1304. doi: 10.1007/s10529-006-9086-7. [DOI] [PubMed] [Google Scholar]
- 79.Molinari NA, Ortega-Sanchez IR, Messonnier ML, Thompson WW, Wortley PM, Weintraub E, Bridges CB. The annual impact of seasonal influenza in the US: measuring disease burden and costs. Vaccine. 2007;25:5086–5096. doi: 10.1016/j.vaccine.2007.03.046. [DOI] [PubMed] [Google Scholar]
- 80.Garmise RJ, Mar K, Crowder TM, Hwang CR, Ferriter M, Huang J, Mikszta JA, Sullivan VJ, Hickey AJ. Formulation of a dry powder influenza vaccine for nasal delivery. AAPS PharmSciTech. 2006;7:E19. doi: 10.1208/pt070103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Coucke D, Schotsaert M, Libert C, Pringels E, Vervaet C, Foreman P, Saelens X, Remon JP. Spray-dried powders of starch and crosslinked poly (acrylic acid) as carriers for nasal delivery of inactivated influenza vaccine. Vaccine. 2009;27:1279–1286. doi: 10.1016/j.vaccine.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 82.Dehghan S, Kheiri M Tavassoti, Tabatabaiean M, Darzi S, Tafaghodi M. Dry-powder form of chitosan nanospheres containing influenza virus and adjuvants for nasal immunization. Arch Pharm Res. 2013;36:981–992. doi: 10.1007/s12272-013-0043-4. [DOI] [PubMed] [Google Scholar]
- 83.Rappuoli R. New and improved vaccines against diphtheria and tetanus. New generation vaccines. 1997:417–435. [Google Scholar]
- 84.Porro M, Saletti M, Nencioni L, Tagliaferri L, Marsili I. Immunogenic correlation between cross-reacting material (CRM197) produced by a mutant of Corynebacterium diphtheriae and diphtheria toxoid. Journal of Infectious Diseases. 1980;142:716–724. doi: 10.1093/infdis/142.5.716. [DOI] [PubMed] [Google Scholar]
- 85.Mills KH, Cosgrove C, McNeela EA, Sexton A, Giemza R, Jabbal-Gill I, Church A, Lin W, Illum L, Podda A, Rappuoli R, Pizza M, Griffin GE, Lewis DJ. Protective levels of diphtheria-neutralizing antibody induced in healthy volunteers by unilateral priming-boosting intranasal immunization associated with restricted ipsilateral mucosal secretory immunoglobulin a. Infect Immun. 2003;71:726–732. doi: 10.1128/IAI.71.2.726-732.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McNeela EA, Jabbal-Gill I, Illum L, Pizza M, Rappuoli R, Podda A, Lewis DJ, Mills KH. Intranasal immunization with genetically detoxified diphtheria toxin induces T cell responses in humans: enhancement of Th2 responses and toxin-neutralizing antibodies by formulation with chitosan. Vaccine. 2004;22:909–914. doi: 10.1016/j.vaccine.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 87.Jones D. Menjugate (Chiron), Current opinion in investigational drugs (London, England: 2000) 2001;2:47–49. [PubMed] [Google Scholar]
- 88.Huo Z, Sinha R, McNeela EA, Borrow R, Giemza R, Cosgrove C, Heath PT, Mills KH, Rappuoli R, Griffin GE, Lewis DJ. Induction of protective serum meningococcal bactericidal and diphtheria-neutralizing antibodies and mucosal immunoglobulin A in volunteers by nasal insufflations of the Neisseria meningitidis serogroup C polysaccharide-CRM197 conjugate vaccine mixed with chitosan. Infect Immun. 2005;73:8256–8265. doi: 10.1128/IAI.73.12.8256-8265.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Glass RI, Parashar UD, Estes MK. Norovirus gastroenteritis. New England Journal of Medicine. 2009;361:1776–1785. doi: 10.1056/NEJMra0804575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jiang X, Wang M, Graham DY, Estes MK. Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J Virol. 1992;66:6527–6532. doi: 10.1128/jvi.66.11.6527-6532.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guerrero RA, Ball JM, Krater SS, Pacheco SE, Clements JD, Estes MK. Recombinant Norwalk virus-like particles administered intranasally to mice induce systemic and mucosal (fecal and vaginal) immune responses. J Virol. 2001;75:9713–9722. doi: 10.1128/JVI.75.20.9713-9722.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.El-Kamary SS, Pasetti MF, Mendelman PM, Frey SE, Bernstein DI, Treanor JJ, Ferreira J, Chen WH, Sublett R, Richardson C, Bargatze RF, Sztein MB, Tacket CO. Adjuvanted intranasal Norwalk virus-like particle vaccine elicits antibodies and antibody-secreting cells that express homing receptors for mucosal and peripheral lymphoid tissues. J Infect Dis. 2010;202:1649–1658. doi: 10.1086/657087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ramirez K, Wahid R, Richardson C, Bargatze RF, El-Kamary SS, Sztein MB, Pasetti MF. Intranasal vaccination with an adjuvanted Norwalk virus-like particle vaccine elicits antigen-specific B memory responses in human adult volunteers. Clin Immunol. 2012;144:98–108. doi: 10.1016/j.clim.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 94.Atmar RL, Bernstein DI, Harro CD, Al-Ibrahim MS, Chen WH, Ferreira J, Estes MK, Graham DY, Opekun AR, Richardson C, Mendelman PM. Norovirus vaccine against experimental human Norwalk Virus illness. N Engl J Med. 2011;365:2178–2187. doi: 10.1056/NEJMoa1101245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Springer MJ, Ni Y, Finger-Baker I, Ball JP, Hahn J, DiMarco AV, Kobs D, Horne B, Talton JD, Cobb RR. Preclinical dose-ranging studies of a novel dry powder norovirus vaccine formulation. Vaccine. 2016;34:1452–1458. doi: 10.1016/j.vaccine.2016.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ball JP, Springer MJ, Ni Y, Finger-Baker I, Martinez J, Hahn J, Suber JF, DiMarco AV, Talton JD, Cobb RR. Intranasal delivery of a bivalent norovirus vaccine formulated in an in situ gelling dry powder. PLoS One. 2017;12:e0177310. doi: 10.1371/journal.pone.0177310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huang J, Mikszta JA, Ferriter MS, Jiang G, Harvey NG, Dyas B, Roy CJ, Ulrich RG, Sullivan VJ. Intranasal administration of dry powder anthrax vaccine provides protection against lethal aerosol spore challenge. Hum Vaccin. 2007;3:90–93. doi: 10.4161/hv.3.3.4011. [DOI] [PubMed] [Google Scholar]
- 98.Bode C, Zhao G, Steinhagen F, Kinjo T, Klinman DM. CpG DNA as a vaccine adjuvant. 2011 doi: 10.1586/erv.10.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mikszta JA, Sullivan VJ, Dean C, Waterston AM, Alarcon JB, Dekker JP, 3rd, Brittingham JM, Huang J, Hwang CR, Ferriter M, Jiang G, Mar K, Saikh KU, Stiles BG, Roy CJ, Ulrich RG, Harvey NG. Protective immunization against inhalational anthrax: a comparison of minimally invasive delivery platforms. J Infect Dis. 2005;191:278–288. doi: 10.1086/426865. [DOI] [PubMed] [Google Scholar]
- 100.Wimer-Mackin S, Hinchcliffe M, Petrie C, Warwood S, Tino W, Williams M, Stenz J, Cheff A, Richardson C. An intranasal vaccine targeting both the Bacillus anthracis toxin and bacterium provides protection against aerosol spore challenge in rabbits. Vaccine. 2006;24:3953–3963. doi: 10.1016/j.vaccine.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 101.Klas SD, Petrie CR, Warwood SJ, Williams MS, Olds CL, Stenz JP, Cheff AM, Hinchcliffe M, Richardson C, Wimer S. A single immunization with a dry powder anthrax vaccine protects rabbits against lethal aerosol challenge. Vaccine. 2008;26:5494–5502. doi: 10.1016/j.vaccine.2008.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vila A, Sanchez A, Janes K, Behrens I, Kissel T, Vila Jato JL, Alonso MJ. Low molecular weight chitosan nanoparticles as new carriers for nasal vaccine delivery in mice. Eur J Pharm Biopharm. 2004;57:123–131. doi: 10.1016/j.ejpb.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 103.Tafaghodi M, Rastegar S. Preparation and in vivo study of dry powder microspheres for nasal immunization. Journal of drug targeting. 2010;18:235–242. doi: 10.3109/10611860903434035. [DOI] [PubMed] [Google Scholar]
- 104.Crowder TM, Donovan MJ. Science and technology of Dry Powder Inhalers. In: Smyth HDC, Hickey AJ, editors. Controlled Pulmonary Drug Delivery. Springer; 2011. pp. 203–222. [Google Scholar]
- 105.Djupesland PG. Nasal drug delivery devices: characteristics and performance in a clinical perspective—a review. Drug delivery and translational research. 2013;3:42–62. doi: 10.1007/s13346-012-0108-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cady RK, McAllister PJ, Spierings ELH, Messina J, Carothers J, Djupesland PG, Mahmoud RA. A Randomized, Double-Blind, Placebo-Controlled Study of Breath Powered Nasal Delivery of Sumatriptan Powder (AVP-825) in the Treatment of Acute Migraine (The TARGET Study) Headache. 2015;55:88–100. doi: 10.1111/head.12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Djupesland PG, Skretting A, Winderen M, Holand T. Bi-directional nasal delivery of aerosols can prevent lung deposition. Journal of Aerosol Medicine-Deposition Clearance and Effects in the Lung. 2004;17:249–259. doi: 10.1089/jam.2004.17.249. [DOI] [PubMed] [Google Scholar]
- 108.Djupesland PG, Skretting A, Winderen M, Holand T. Breath actuated device improves delivery to target sites beyond the nasal valve. Laryngoscope. 2006;116:466–472. doi: 10.1097/01.MLG.0000199741.08517.99. [DOI] [PubMed] [Google Scholar]
- 109.Perry M, Whyte A. Immunology of the tonsils. Immunology today. 1998;19:414–421. doi: 10.1016/s0167-5699(98)01307-3. [DOI] [PubMed] [Google Scholar]