

Abstract
Exenatide, a glucagon‐like peptide‐1 receptor agonist, induces insulin secretion. Its role in insulin clearance has not been adequately examined. Carcinoembryonic antigen‐related cell adhesion molecule 1 (CEACAM1) promotes hepatic insulin clearance to maintain insulin sensitivity. Feeding C57BL/6J mice a high‐fat diet down‐regulates hepatic Ceacam1 transcription to cause hyperinsulinemia, insulin resistance, and hepatic steatosis, as in Ceacam1 null mice (Cc1 –/–). Thus, we tested whether exenatide regulates Ceacam1 expression in high‐fat diet‐fed mice and whether this contributes to its insulin sensitizing effect. Exenatide (100 nM) induced the transcriptional activity of wild‐type Ceacam1 promoter but not the constructs harboring block mutations of peroxisome proliferator‐activated receptor response element and retinoid X receptor alpha, individually or collectively, in HepG2 human hepatoma cells. Chromatin immunoprecipitation analysis demonstrated binding of peroxisome proliferator‐activated receptor gamma to Ceacam1 promoter in response to rosiglitazone and exenatide. Consistently, exenatide induced Ceacam1 messenger RNA expression within 12 hours in the absence but not in the presence of the glucagon‐like peptide‐1 receptor antagonist exendin 9‐39. Exenatide (20 ng/g body weight once daily intraperitoneal injection in the last 30 days of feeding) restored hepatic Ceacam1 expression and insulin clearance to curb diet‐induced metabolic abnormalities and steatohepatitis in wild‐type but not Cc1 –/– mice fed a high‐fat diet for 2 months. Conclusion: Exenatide promotes insulin clearance in parallel with insulin secretion to prevent chronic hyperinsulinemia and the resulting hepatic steatosis, and this contributes to its insulin sensitizing effect. Our data further highlight the relevance of physiologic insulin metabolism in maintaining insulin sensitivity and normal lipid metabolism. (Hepatology Communications 2018;2:35–47)
Abbreviations
- Cc1–/–
global Ceacam1 null mouse
- Cc1+/+
global Ceacam1 wild‐type mouse
- CEACAM1
carcinoembryonic antigen‐related cell adhesion molecule 1
- DMSO
dimethyl sulfoxide
- Fasn
fatty acid synthase
- GLP‐1
glucagon‐like peptide‐1
- hCEACAM1
human CEACAM1
- HF
high‐fat
- HF‐S
high‐fat saline‐treated
- IRE‐BP1
insulin response element‐binding protein 1
- mRNA
messenger RNA
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NEFA
nonesterified fatty acid
- PPAR
peroxisome proliferator‐activated receptor
- PPRE
peroxisome proliferator‐activated receptor response element
- qRT‐PCR
quantitative reverse‐transcription polymerase chain reaction
- RD
regular diet
- RD‐S
regular diet saline‐treated
- RXR
human retinoid X receptor
- SREBP‐1c
sterol regulatory element binding protein 1c
Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most common liver disease in the United States, with more than 30%‐40% of Americans being affected and nearly 12% manifesting the progressive form nonalcoholic steatohepatitis (NASH).1 It is a heterogeneous disease ranging from benign steatosis and steatohepatitis (NAFLD) to include chicken‐wire bridging fibrosis and apoptosis (NASH).
Currently, there is no definitive pharmacologic treatment of NASH. Weight loss constitutes a primary treatment, but patients usually regain weight.2 Despite persistent controversy,3 insulin resistance is believed to constitute a major risk factor for NAFLD/NASH,4 building a basis for focusing therapeutically on restoring insulin sensitivity. While insulin‐sensitizing drugs, such as pioglitazone, a peroxisome proliferator‐activated receptor gamma (PPARγ) agonist, reduces inflammation,5 it can also cause weight gain and increased water retention.6, 7 The long‐acting glucagon‐like peptide‐1 (GLP‐1) receptor agonist exenatide, a synthetic analog of exendin‐4 that induces glucose‐dependent insulin secretion and inhibits glucagon secretion from pancreatic α‐cells,8, 9 reverses steatohepatitis in some animal models of NAFLD/NASH.10, 11, 12 While combining it with pioglitazone is effective in reducing hemoglobin A1c levels in patients with uncontrolled type 2 diabetes,13 the data supporting its clinical relevance and safety in the treatment of NASH remain limited.14
Effective treatment of NASH requires a thorough understanding of its pathogenesis. However, this has been limited by the lack of an animal model that replicates all features of human NASH. Mice with the null mutation of the carcinoembryonic antigen‐related cell adhesion molecule 1 (Ceacam1) gene (Cc1 –/– mice)15 and with liver‐specific inactivation of CEACAM116 develop impaired insulin clearance, consistent with the role of CEACAM1 in increasing the rate of receptor‐mediated insulin uptake into the hepatocyte for degradation, following its phosphorylation by the insulin receptor tyrosine kinase.17 Impaired insulin clearance causes chronic hyperinsulinemia, insulin resistance, and de novo lipogenesis. In addition to hepatic steatosis, this leads to increased lipid repartitioning to white adipose tissue and visceral obesity. Ceacam1 mutants also develop inflammation in the liver and, remarkably, a NASH‐characteristic chicken‐wire pattern of fibrosis with a regular diet, which is a unique feature compared to other mouse models. When fed a high‐fat (HF) diet, Cc1 –/– and liver‐specific inactivation of CEACAM1 mice develop all key features of NASH, including macrosteatosis, steatohepatitis, apoptosis, and advanced chicken‐wire fibrosis.18, 19
HF feeding of C57BL/6 mice for 21‐30 days reduces hepatic CEACAM1 levels by > 50% to cause impairment of hepatic insulin clearance and insulin resistance in addition to hepatosteatosis.20 In contrast, adenoviral‐mediated redelivery of wild‐type but not phosphorylation‐defective mutant CEACAM1 reverses diet‐induced metabolic abnormalities.21 Similarly, liver‐specific transgenic overexpression of CEACAM1 protects against metabolic and histopathologic abnormalities caused by an HF diet, including hepatosteatosis, inflammation, and profibrosis.20 This promotes the induction of hepatic CEACAM1 levels as a potential therapeutic target for the prevention and/or treatment of NAFLD/NASH.
The mechanisms underlying the ameliorating effect of exenatide on hepatic steatosis remain elusive. Thus, we herein investigated whether exenatide increases hepatic insulin clearance by inducing Ceacam1 transcription to prevent hyperinsulinemia and reverse steatohepatitis.
Materials and Methods
MICE MAINTENANCE
Three‐month‐old C57BL/6.Cc1 −/− and Cc1 +/+ littermates were fed ad libitum a regular diet (RD) or HF diet deriving 45%:35%:20% calories from fat, carbohydrate, and protein, respectively (D12451; Research Diets) for 1 month20 prior to receiving an intraperitoneal injection/day of saline or exenatide (20 ng/g body weight per day) (507‐77; California Peptide Research) for 5‐30 days while maintained on the same diet.22 All procedures were approved by the University of Toledo Institutional Animal Care and Utilization Committee.
INSULIN INTERNALIZATION IN PRIMARY HEPATOCYTES
Hepatocytes were isolated by perfusing liver (1 mL/minute) with collagenase type II (1 mg/mL) (Worthington)20 and treated with exenatide (100 nM) or saline for 24 hours before internalization of human [125I]insulin (PerkinElmer Life Sciences) was assayed as described.20, 23
FATTY ACID SYNTHASE ACTIVITY
As described,24 livers were homogenized and the supernatant added to a reaction mix containing 0.1 µCi [14C]malonyl‐coenzyme A (Perkin‐Elmer). Fatty acid synthase (Fasn) activity was calculated as counts per minute of [14C]incorporated/µg cell lysate.
LUCIFERASE ASSAY
As described,25 human hepatocellular carcinoma cells (HepG2) were cultured overnight in 12‐well plates to reach ∼60%‐70% confluence. Transfection was performed with promoter constructs and the pRL‐thymidine kinase Renilla luciferase promoter (Promega) using Lipofectamine 2000 (Invitrogen). After 24 hours, cells were serum starved before being treated for 24 hours with dimethyl sulfoxide (DMSO), 1 μM rosiglitazone (Sigma), 1 μM pioglitazone (Sigma),26 100 nM insulin, and 100 nM exenatide. Luciferase activity was assessed using the Dual‐Luciferase Reporter Assay System (Promega).
CHROMATIN IMMUNOPRECIPITATION ASSAY
HepG2 cells were transfected with glutathione S‐transferase–PPARγ and human retinoid X receptor alpha (RXRα) (Addgene) constructs using Lipofectamine 2000 as above. Cells were serum starved and treated with DMSO (vehicle), 1 μM rosiglitazone (Sigma), or 100 nM exenatide. A chromatin immunoprecipitation assay was performed following the manufacturer's instructions (#9003; Cell Signaling Technology). PPARγ antibody (H‐100) (sc‐7196; Santa Cruz Biotechnology) was used to immunoprecipitate PPARγ, and normal rabbit immunoglobulin G and histone H3 (D2B12) XP rabbit monoclonal antibody were used as negative and positive controls, respectively. Immunoprecipitated and input samples were evaluated by quantitative reverse‐transcription polymerase chain reaction (qRT‐PCR) analysis (StepOne Plus; Applied Biosystems) using primers for RPL30 (kit) as control and human CEACAM1 (Supporting Fig. S1B,C). Human adipose differentiation‐related protein and human glyceraldehyde‐3‐phosphate dehydrogenase were also analyzed as positive and negative targets of PPARγ, respectively.27 Results are expressed as fold enrichment (immunoprecipitated/input).
STATISTICAL ANALYSIS
Data were analyzed by one‐way analysis of variance or the two‐tailed Student t test using GraphPad Prism 6 software. P < 0.05 was considered statistically significant.
Results
EXENATIDE INDUCES CEACAM1 EXPRESSION AND PROMOTES INSULIN INTERNALIZATION IN MOUSE PRIMARY HEPATOCYTES
As expected,20 qRT‐PCR analysis revealed ∼3‐fold lower Ceacam1 messenger RNA (mRNA) levels in saline‐treated primary hepatocytes derived from HF‐fed Cc1 +/+ mice (HF‐S) than from saline‐treated RD‐fed mice (RD‐S) (Fig. 1A, panel i). Exenatide treatment for 24 hours elevated Ceacam1 mRNA levels in hepatocytes derived from HF‐fed mice (Fig. 1A, panel i). Consistent with our previous study,23 the decline in Ceacam1 mRNA level in HF‐S hepatocytes is likely mediated by a Pparα‐dependent mechanism, as supported by the corresponding rise in Pparα mRNA levels (Fig. 1A, panel ii). Whereas exenatide restored the Pparα mRNA level (Fig. 1A, panel ii), it also induced that of Pparγ in HF‐fed mice (Fig. 1A, panel iii), as expected.10 This suggests that the remarkable rise in Ceacam1 mRNA in HF‐exenatide hepatocytes is mediated by the combined effect of removing the negative effect of Pparα in addition to the reciprocal increase in the Pparγ level (and presumably activity).
Figure 1.
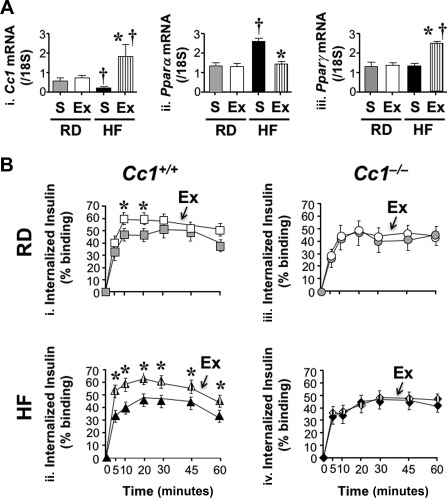
Effect of exenatide on insulin uptake in murine primary hepatocytes. (A) Primary hepatocytes were isolated from Cc1 +/+ mice fed an RD or HF diet for 1 month (n = 6 mice/treatment) and treated with exenatide or saline for 24 hours before performing qRT‐PCR analysis in triplicate to measure (i) Ceacam1, (ii) Pparα, and (iii) Pparγ mRNA levels normalized to 18S. Values are expressed as mean ± SEM; *P < 0.05 Ex versus S/feeding group; † P < 0.05 HF versus RD/treatment group. (B) We measured [125I]insulin internalization in triplicate as percent of specifically bound ligand in primary hepatocytes from RD‐fed or HF‐fed mice treated with Ex (white/hatched) or S (gray/black) for 24 hours; (i) squares, RD‐fed Cc1 +/+; (ii) triangles, HF‐fed Cc1 +/+; (iii) circles, RD‐fed Cc1 –/–; (iv) diamonds, HF‐fed Cc1 –/–; n = 6 mice/genotype per treatment. Values are expressed as mean ± SEM; *P < 0.05 Ex versus S/feeding group. Abbreviations: Ex, exenatide; S, saline.
Because CEACAM1 maintains insulin sensitivity by increasing the rate of insulin‐receptor endocytosis followed by its clearance,17 we then investigated whether exenatide promotes insulin uptake. We found exenatide significantly elevated [125I]‐insulin internalization in hepatocytes from Cc1 +/+ mice, in particular when HF‐fed (Fig. 1B, panel ii; HF‐exenatide [hatched triangles] versus HF‐S [black triangles]; P < 0.05). In contrast, exenatide did not affect insulin uptake in hepatocytes from RD‐ or HF‐fed Cc1 –/–‐ mice (Fig. 1B, panel iii and iv). Thus, exenatide up‐regulates insulin internalization in hepatocytes in a CEACAM1‐dependent fashion.
EXENATIDE INDUCES CEACAM1 PROMOTER ACTIVITY IN HEPG2 CELLS
We then tested whether exenatide regulates mouse Ceacam1 promoter activity in HepG2 cells. Mouse Ceacam1 promoter contains a functional PPAR response element/RXR recognition site (PPRE‐RXRα) between nucleotides –557 and –543.25 Alignment of orthologous Ceacam1 promoters from different species by CLUSTALW identified conserved PPRE‐RXRα and insulin response element‐binding protein 1 (IRE‐BP1)28 consensus sequences in many species, including mice and humans (Supporting Fig. S1A). Because exenatide induces PPARγ expression in hepatocytes from rats with NASH,10 we then assessed the effect of exenatide and the PPARγ agonists rosiglitazone and pioglitazone on mouse Ceacam1 promoter activity in HepG2 cells. In a luciferase reporter assay,25 rosiglitazone and exenatide induced the luciferase activity of the PPREx3‐thymidine kinase positive control individually or combined to the same extent (rosiglitazone/exenatide versus rosiglitazone and exenatide) without affecting that of the empty vector (Fig. 2A). They also activated mouse Ceacam1 promoter activity by ∼2.5‐fold compared to the vehicle‐treated mouse (Fig. 2A; –1,100). Mutating the functional PPRE‐RXRα sequence (–Δ2), PPRE (–ΔPPRE), and RXRα (–ΔRXRα)25 abolished their positive effect on Ceacam1 promoter activity (Fig. 2A). Pioglitazone exerted a comparable effect to rosiglitazone on the Ceacam1 promoter (Supporting Fig. S2). As expected from the up‐regulatory effect of insulin on rat Ceacam1 promoter activity,29 insulin activated the –1,100 mouse Ceacam1 promoter (Supporting Fig. S3). This effect was completely abolished in the –Δ2 mutant. Because the most proximal AGG sequence between nucleotides –557 and –555 in the IRE‐BP1 site is mutated in this construct, the data point to the important role of this site in insulin's regulation of Ceacam1 transcriptional activity. Combining insulin and exenatide exerted a synergistic effect (Fig. 2; Supporting Fig. S3; insulin/exenatide‐treated versus vehicle‐treated –1,100). In contrast, combining exenatide with pioglitazone did not alter the effect of PPARγ activation on the Ceacam1 promoter (Supporting Fig. S2; pioglitazone/exenatide‐treated versus vehicle‐treated –1,100). This demonstrated that exenatide did not compete off or synergize with pioglitazone in the regulation of Ceacam1 promoter activity.
Figure 2.
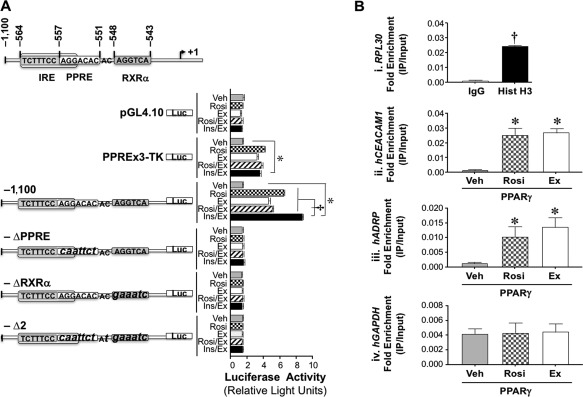
Regulation of Ceacam1 expression in HepG2 cells. (A) Wild‐type (−1,100) and block mutants (small letters) of the active PPRE‐RXR site (–Δ2), PPRE (–ΔPPRE), and RXRα (–ΔRXRα) in mouse Ceacam1 were subcloned into a pGL4.10 promoterless plasmid to assess luciferase activity in triplicate in response to DMSO (vehicle, light gray), rosiglitazone (checkered), exenatide (white), rosiglitazone plus exenatide plus (hatched), and insulin plus exenatide (black). PPREx3‐TK‐luc was used as positive and PGL4.10 as negative controls. Luciferase light units are expressed as mean ± SEM in relative light units; *P < 0.05 treatment versus vehicle, †P < 0.05 Ins/Ex versus other treatments. (B) HepG2 cells were transfected with pGST‐PPARγ and hRXRα and treated with DMSO (vehicle, light gray), rosiglitazone (checkered), or exenatide (white) before ChIP analysis was carried out in triplicate by immunoprecipitating with α‐PPARγ or with normal rabbit IgG (dark gray) and Hist H3 (black) as negative and positive controls, respectively. Immunorprecipitated and input samples were evaluated by qRT‐PCR analysis using primers for RPL30 (kit control) and hCEACAM1. We also analyzed hADRP and hGAPDH as positive and negative targets of PPARγ, respectively. Results are expressed as fold enrichment (IP/Input). Values are expressed as mean ± SEM; *P < 0.05 versus Veh; † P < 0.05 Hist H3 versus IgG. Abbreviations: ChIP, chromatin immunoprecipitation; Ex, exenatide; hADRP, human adipose differentiation‐related protein; hGAPDH, human glyceraldehyde‐3‐phosphate dehydrogenase; Hist H3, histone H3; IgG, immunoglobulin G; Ins/Ex, insulin plus exenatide; IP, immunprecipitated; luc, luciferase; pGST, glutathione S‐transferase plasmid; Rosi, rosiglitazone; Rosi/Ex, rosiglitazone plus exenatide; TK, thymidine kinase; Veh, vehicle.
A chromatin immunoprecipitation assay using a primer set from the human (h)CEACAM1 promoter showed that liganded PPARγ binds to the CEACAM1 gene in HepG2 cells treated with rosiglitazone or exenatide to a higher extent than DMSO (vehicle)‐treated cells (Fig. 2B, panel ii). A similar observation was made for the positive control, human adipose differentiation‐related protein, a known target of PPARγ (Fig. 2B, panel iii).27 in contrast, the primer set amplifying human glyceraldehyde‐3‐phosphate dehydrogenase, a nontarget of PPARγ, did not detect any binding (Fig. 2B, panel iv). Together, this revealed that activated PPARγ bound to the hCEACAM1 gene to up‐regulate its transcription.
Consistently, exenatide induced CEACAM1 mRNA levels in HepG2 cells after 12 hours of treatment (Fig. 3A, panel ii). This was preceded (6 hours) by an increase in GLP‐1R mRNA levels (Fig. 3A, panel i). It was also associated with reduction in PPARα and a reciprocal increase in PPARγ mRNA levels (Fig. 3B, panel iii and iv, respectively). The positive effect of exenatide on GLP‐1R, CEACAM1, and PPARγ expression, and its reciprocal effect on PPARα mRNA were mediated by its receptor, as these effects were abolished by pre‐incubating cells with exendin 9‐39 GLP‐1R antagonist (Fig. 3B).
Figure 3.
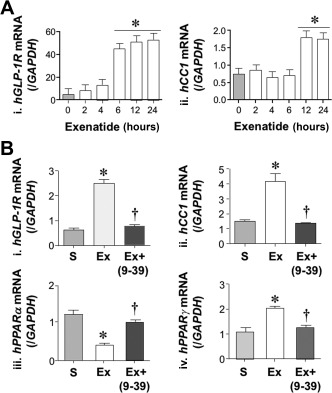
Effect of exenatide on CEACAM1 and GLP‐1R mRNA levels in HepG2 cells. (A) HepG2 cells were treated with saline (light gray) or exenatide (white) for 0‐24 hours before qRT‐PCR analysis to measure (i) hGLP‐1R and (ii) hCEACAM1 (CC1) mRNA levels normalized to iGAPDH (n = 5/group in triplicate). Values are expressed as mean ± SEM; *P < 0.05 treatment group versus basal level. (B) mRNA levels of (i) hGLP‐1R, (ii) hCEACAM1 (hCC1), (iii) hPPARα, and (iv) hPPARγ were analyzed in HepG2 as above except for pre‐incubation with exendin 9‐39 (dark gray). *P < 0.05 versus S, † P < 0.05 Ex + exendin 9‐39 (Ex+ 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39) versus Ex alone. Abbreviations: Ex, exenatide; Ex 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, exendin fragment 9‐39; h, human; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; /GAPDH, normalized to GAPDH.
EXENATIDE SUPPRESSES FOOD INTAKE AND INDUCES ACUTE‐PHASE INSULIN SECRETION IN MICE
One month of HF feeding increased body weight in Cc1 +/+ and Cc1 −/− mice (Supporting Table S2). As expected,8 exenatide treatment for 5 days reduced daily food intake in both mouse lines (Fig. 4A, panel i and ii), causing a decrease in body weight (Supporting Table S2). It also significantly induced glucose‐stimulated insulin secretion in RD‐fed Cc1 +/+ mice (Fig. 4B, panel i; RD‐exenatide [white] versus RD‐S [gray squares]; P < 0.05) and HF‐fed Cc1 +/+ mice (Fig. 4B, panel ii; HF‐exenatide [hatched] versus HF‐S [black triangles]; P < 0.05). Similarly, exenatide induced glucose‐stimulated insulin secretion in RD‐fed Cc1 −/− mice (Fig. 4B, panel iii; RD‐exenatide [white] versus RD‐S [gray circles]; P < 0.05) and HF‐fed Cc1 −/− mice (Fig. 4B, panel iv; HF‐exenatide [hatched] versus HF‐S [black diamonds]; P < 0.05). However, 5 days of exenatide injection did not restore insulin clearance, as shown by the steady‐state C‐peptide/insulin molar ratio (Supporting Table S2), nor did it affect blood glucose or plasma nonesterified fatty acids (NEFAs) and triacylglycerol levels in either mouse group (Supporting Table S2).
Figure 4.
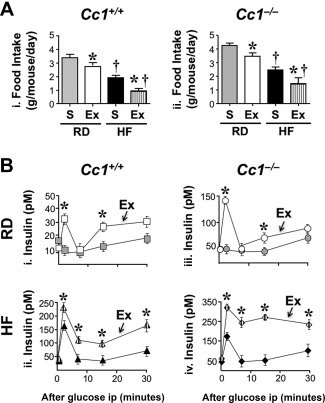
Effect of exenatide on food intake and insulin secretion. Mice were fed an RD or HF diet for 1 month and injected intraperitoneally once daily with saline or exenatide (20 ng/g body weight/day) for 5 days. (A) Daily food intake in RD‐S (gray), RD‐Ex (white), HF‐S (black), and HF‐Ex (vertically hatched bars) (n = 6/genotype per feeding per treatment). Values are expressed as mean ± SEM; *P < 0.05 Ex versus S/feeding group. †P < 0.05 HF versus RD/treatment group. (B) At the end of feeding, acute phase insulin release was assessed in duplicate 0‐30 minutes after glucose injection: (i) squares, RD‐fed Cc1+/+; (ii) triangles, HF‐fed Cc1 +/+; (iii) circles, RD‐fed Cc1 –/–; (iv) diamonds, HF‐fed Cc1 –/–. Values are expressed as mean ± SEM; *P < 0.05 Ex versus S/group. Abbreviations: Ex, exenatide; ip, intraperitoneally.
EXENATIDE TREATMENT FOR 4 WEEKS REVERSES DIET‐INDUCED IMPAIRMENT OF INSULIN CLEARANCE
Three weeks of exenatide treatment reduced but did not fully restore body weight, total fat mass, visceral adiposity, plasma NEFAs, hyperglycemia, and hepatic triacylglycerol levels in HF‐fed Cc1 +/+ mice (Supporting Table S3). It did not reverse hyperinsulinemia or restore the C‐peptide/insulin molar ratio (Supporting Table S3). Consistently, it failed to elevate Ceacam1 mRNA (Supporting Fig. S4A, panel i) and protein levels (Supporting Fig. S4A, panel ii) in HF‐fed mice, as shown by immunoblotting with α‐CEACAM1 antibody, normalized to α‐tubulin (Supporting Fig. S4A, panel ii). Consistent with its insulin releasing effect, exenatide treatment for 3 weeks induced CEACAM1 phosphorylation (p) in RD‐fed but not HF‐fed Cc1 +/+ mice, as shown by immunoblotting with α‐pCC1 (Supporting Fig. S4A, panel ii). Moreover, this treatment did not curb insulin intolerance in HF‐fed Cc1 +/+ (Supporting Fig. S4B, panel i) or Cc1 −/− mice (Supporting Fig. S4B, panel ii; and accompanying graphs representing the area under the curve, HF‐exenatide [hatched] versus HF‐S [black bars]).
In contrast, extending exenatide treatment to a fourth week while maintaining mice on the HF diet completely reversed diet‐induced visceral obesity and plasma NEFAs without fully restoring body weight or total fat mass (Table 1). Interestingly, this fully recovered Ceacam1 mRNA levels (Fig. 5A, panel i; HF‐exenatide [hatched] versus RD‐S [gray bars]). Western blot analysis confirmed full restoration of the protein content and phosphorylation of hepatic CEACAM1 Cc1 +/+ mice despite continuous HF feeding (Fig. 5A, panel ii). This translated into full recovery of insulin clearance by exenatide in HF‐fed Cc1 +/+ mice, as assessed by the higher C‐peptide/insulin molar ratio (Fig. 5B, panel i) and normal insulin levels (Table 1) in HF‐exenatide relative to HF‐S. In Cc1 –/– mice, however, 4 weeks of exenatide did not normalize insulin clearance (Fig. 5B, panel iv) or levels (Table 1). In parallel, this regimen also curbed the negative effect of HF on insulin (Fig. 5B, panel ii) and glucose tolerance (Fig. 5B, panel iii) in Cc1 +/+ (HF‐exenatide [hatched] versus HF‐S [black triangles]; P < 0.05) but not Cc1 –/– mice (Fig. 5B, panel v‐vii, respectively; HF‐exenatide [hatched] versus HF‐S [black diamonds]). The data demonstrate that the up‐regulatory effect of exenatide on insulin metabolism and action was mediated, at least in part, by inducing CEACAM1 expression.
Table 1.
Effect of 4 Weeks of Exenatide Treatment on Plasma Biochemistry And Fat Composition
| RD‐S | RD‐Ex | HF‐S | HF‐Ex | |
|---|---|---|---|---|
| a) Cc1 +/+ | ||||
| Body weight (g) | 29.6 ± 0.60 | 27.5 ± 0.24 | 36.5 ± 1.28† | 31.5 ± 0.39*† |
| % Fat mass (NMR) | 7.23 ± 1.01 | 5.53 ± 0.57 | 21.5 ± 0.97† | 13.9 ± 1.22*† |
| % Visceral fat (WAT/BW) | 0.71 ± 0.16 | 0.68 ± 0.22 | 2.38 ± 0.45† | 1.01 ± 0.25* |
| % BAT/BW | 0.42 ± 0.02 | 0.46 ± 0.05 | 0.21 ± 0.03† | 0.33 ± 0.04*† |
| % Lean mass (NMR) | 67.7 ± 0.86 | 68.5 ± 0.55 | 55.0 ± 0.60† | 60.5 ± 1.08*† |
| Plasma NEFA (mEq/L) | 0.78 ± 0.03 | 0.65 ± 0.03* | 0.92 ± 0.07† | 0.61 ± 0.05* |
| Plasma triacylglycerol (mg/dL) | 65.8 ± 3.70 | 43.6 ± 2.50* | 68.9 ± 7.40 | 59.2 ± 2.90* |
| Hepatic triacylglycerol (μg/mg) | 36.6 ± 7.06 | 40.3 ± 3.55 | 61.1 ± 6.20† | 30.3 ± 5.11* |
| Plasma insulin (pM) | 51.1 ± 2.99 | 54.6 ± 2.68 | 96.7 ± 15.5† | 57.0 ± 4.49* |
| Plasma C‐peptide (pM) | 201 ± 29.0 | 190 ± 16.6 | 228 ± 14.3 | 295 ± 60.8*† |
| C‐peptide/Insulin molar ratio | 4.93 ± 0.31 | 4.26 ± 0.29 | 3.02 ± 0.38† | 6.54 ± 0.40*† |
| Fasting blood glucose (mg/dL) | 79 ± 2 | 56 ± 6* | 121 ± 7† | 90 ± 10*† |
| Fed blood glucose (mg/dL) | 101 ± 2 | 84 ± 3* | 152 ± 8† | 122 ± 7*† |
| b) Cc1 –/– | ||||
| Body weight (g) | 30.9 ± 0.59 | 28.0 ± 0.59 | 38.1 ± 0.57† | 35.2 ± 0.46*† |
| % Fat mass (NMR) | 10.7 ± 1.76 | 8.72 ± 1.16 | 25.3 ± 1.04† | 20.6 ± 1.25*† |
| % Visceral fat (WAT/BW) | 0.89 ± 0.25 | 0.92 ± 0.18 | 3.75 ± 0.52† | 3.02 ± 0.41*† |
| % BAT/BW | 0.31 ± 0.04 | 0.36 ± 0.05 | 0.16 ± 0.02† | 0.24 ± 0.05*† |
| % Lean mass (NMR) | 63.7 ± 1.41 | 64.2 ± 0.77 | 50.9 ± 0.43† | 55.5 ± 0.94*† |
| Plasma NEFA (mEq/L) | 0.92 ± 0.05 | 0.87 ± 0.09 | 1.31 ± 0.07† | 1.26 ± 0.09† |
| Plasma triacylglycerol (mg/dL) | 63.9 ± 4.35 | 60.6 ± 3.63 | 66.0 ± 3.53 | 63.6 ± 5.05 |
| Hepatic triacylglycerol (μg/mg) | 55.9 ± 3.86 | 48.9 ± 4.81 | 99.3 ± 9.89† | 90.5 ± 7.56† |
| Plasma insulin (pM) | 136 ± 4.28 | 135 ± 5.05 | 195 ± 1.24† | 183 ± 9.49† |
| Plasma C‐peptide (pM) | 436 ± 32.3 | 498 ± 72.5 | 538 ± 75.7† | 524 ± 107† |
| C‐peptide/Insulin molar ratio | 3.21 ± 0.61 | 3.67 ± 0.66 | 2.76 ± 1.86† | 2.86 ± 0.35† |
| Fasting blood glucose (mg/dL) | 84 ± 5 | 98 ± 8 | 112 ± 26† | 121 ± 12† |
| Fed blood glucose (mg/dL) | 113 ± 4 | 121 ± 7 | 185 ± 8† | 171 ± 7† |
Mice (3 months of age) were fed an RD or HF diet for 2 months. During the last month of feeding, mice were injected intraperitoneally once daily with either saline or exenatide (20 ng/g BW) (n = 8‐10/genotype per feeding per treatment). Whole body composition was assessed by NMR technology (Bruker Minispec). Values are expressed as mean ± SEM; *P < 0.05 Ex versus S/feeding group; † P < 0.05 HF versus RD/treatment group.
Abbreviations: BAT, brown adipose tissue; BW, body weight; EX, exenatide; NMR, nuclear magnetic resonance; WAT, white adipose tissue.
Figure 5.
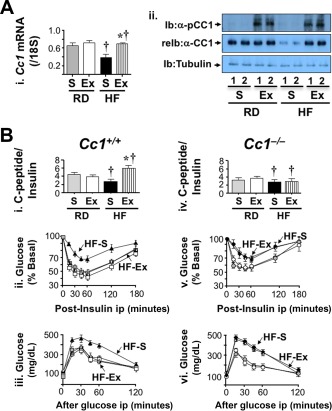
Effect of 4 weeks of exenatide treatment on insulin clearance. Mice were fed an RD or HF diet for 2 months and injected daily with exenatide in the last month. (A) (i) Cc1 +/+ liver lysates were analyzed by qRT‐PCR to measure Ceacam1 mRNA levels normalized to 18S (n = 5/each group in duplicate). Values are expressed as mean ± SE; *P < 0.05 Ex versus S/feeding group; †P < 0.05 HF versus RD/treatment group. (ii) Western blot analysis by immunoblotting with phospho‐CEACAM1 followed by re‐immunoblotting with CEACAM1 antibody. The lower part of the gel was immunoblotted with α‐tubulin for protein normalization. Gels represent more than two experiments performed on different mice per feeding per treatment group. (B) Retro‐orbital venous blood was drawn from overnight‐fasted mice to measure C‐peptide/insulin molar ratio (i and iv) in Cc1 +/+ and Cc1 –/– mice, respectively. Following a 7‐hour fasting period, mice were injected intraperitoneally with (ii and v) insulin or (iii and vi) glucose to assess insulin and glucose tolerance, respectively. Values are expressed as mean ± SEM (n = 7‐8/time point for each group); *P < 0.05 Ex versus S. Abbreviations: α‐CC1, CEACAM1 antibody; Ex, exenatide; Ib, immunoblotting; ip, intraperitoneally; pCC1, phospho‐CEACAM1; reIb, re‐immunoblotting.
EFFECT OF EXENATIDE ON LIPID METABOLISM AND INFLAMMATORY MARKERS
HF caused an increase in hepatic triacylglycerol content in both mouse lines (Table 1; HF‐S versus RD‐S). Exenatide decreased hepatic triacylglycerol levels (Table 1) and reversed hepatic steatosis in HF‐fed Cc1 +/+ but not HF‐fed Cc1 –/– mice (Table 1). This is consistent with the histologic analysis by hematoxylin and eosin stain of the liver (Fig. 6A) revealing a marked reduction of the diffuse fat infiltration detected in HF‐fed Cc1 +/+ by exenatide treatment for 4 weeks (Fig. 6A; HF‐exenatide versus HF‐S [iv versus ii]). In contrast, exenatide failed to reverse fat deposition in Cc1 –/– livers whether mice were fed RD or HF (Fig. 6A, panel vii versus v or viii versus vi, respectively). Additionally, HF induced mRNA levels of genes involved in de novo lipogenesis (sterol regulatory element binding protein 1c [SREBP‐1c] and Fasn) and fatty acid transport (clusters of differentiation [Cd]36, fatty acid transport protein [Fatp]‐1, and Fatp‐4) in both mouse groups (Table 2; HF‐S versus RD‐S). Exenatide treatment normalized the level of these genes in Cc1 +/+ but not Cc1 –/– mice (Table 2).
Figure 6.
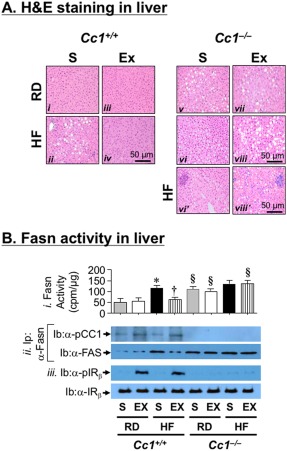
Effect of 4 weeks of exenatide on hepatic lipid metabolism. (A) Liver histology was assessed in H&E‐stained sections (n = 7‐8/genotype per feeding per treatment). Representatives from (i and v) S‐treated RD‐fed, (ii, vi, and vi') S‐treated HF‐fed, (iii and vii) Ex‐treated RD‐fed, and (iv, viii, and viii') Ex‐treated HF‐fed mice are shown. Of note, vi' and viii', from the same groups as vi and viii mice, highlight the infiltration of inflammatory foci. (B) (i) Fasn activity measured in triplicate by [14C]‐malonyl‐coenzyme A incorporation (n = 5/genotype per treatment group). Values are expressed as mean ± SEM; *P < 0.05 HF‐S versus RD‐S/genotype; † P < 0.05 Ex versus S in HF‐fed Cc1 +/+, §P < 0.05 Cc1 –/– versus Cc1+/+/feeding group per treatment group. (ii) Liver lysates were immunoprecipitated with α‐Fasn followed by immunoblotting with α‐phospho‐CEACAM1 antibody. (iii) Western blot of liver lysates to assess insulin receptor phosphorylation (Ib: α‐pIRβ) and level (Ib: α‐IRβ). Abbreviations: α‐pCC1, α‐phospho‐CEACAM1 antibody; cpm, counts per minute; H&E, hematoxylin and eosin; Ib, immunoblotting; Ip, immunoprecipitation; IR, insulin receptor; pIR, insulin receptor phosphorylation.
Table 2.
Effect Of 4 Weeks Of Exenatide Treatment On The Mrna Levels Of Genes Involved In Lipid Metabolism, and Inflammation In The Liver Of Male Mice
| RD‐S | RD‐Ex | HF‐S | HF‐Ex | |
|---|---|---|---|---|
| Cc1 +/+ | ||||
| Lipid metabolism | ||||
| Srebp‐1c | 0.74 ± 0.07 | 0.79 ± 0.06 | 3.23 ± 0.21† | 0.88 ± 0.05* |
| Fasn | 0.35 ± 0.03 | 0.45 ± 0.03 | 2.61 ± 0.15† | 0.54 ± 0.07* |
| Cd36 | 0.43 ± 0.04 | 0.48 ± 0.03 | 3.25 ± 0.22† | 0.52 ± 0.05* |
| Fatp‐1 | 0.77 ± 0.05 | 0.65 ± 0.03 | 2.04 ± 0.13† | 0.69 ± 0.05* |
| Fatp‐4 | 0.89 ± 0.04 | 0.76 ± 0.06 | 2.62 ± 0.19† | 0.74 ± 0.06* |
| Inflammation | ||||
| F4/80 | 0.79 ± 0.04 | 0.75 ± 0.04 | 2.55 ± 0.17† | 0.80 ± 0.06* |
| Cd68 | 0.87 ± 0.09 | 0.81 ± 0.07 | 3.25 ± 0.20† | 0.99 ± 0.07* |
| Il‐1β | 3.51 ± 0.32 | 3.57 ± 0.30 | 9.07 ± 0.37† | 4.35 ± 0.26* |
| Il‐6 | 4.15 ± 0.24 | 4.57 ± 0.18 | 8.95 ± 0.27† | 4.88 ± 0.28* |
| Ifn‐γ | 6.47 ± 0.20 | 6.59 ± 0.26 | 13.0 ± 0.43† | 6.49 ± 0.25* |
| Tnf‐α | 2.65 ± 0.26 | 2.79 ± 0.25 | 6.70 ± 0.23† | 2.39 ± 0.17* |
| Cc1 –/– | ||||
| Lipid metabolism | ||||
| Srebp‐1c | 1.42 ± 0.16 | 1.65 ± 0.11 | 4.36 ± 0.16† | 3.93 ± 0.15† |
| Fasn | 0.82 ± 0.05 | 0.80 ± 0.05 | 1.53 ± 0.06† | 1.57 ± 0.09† |
| Cd36 | 0.74 ± 0.05 | 0.76 ± 0.03 | 3.25 ± 0.22† | 3.28 ± 0.20† |
| Fatp‐1 | 0.93 ± 0.04 | 0.90 ± 0.04 | 2.75 ± 0.20† | 2.84 ± 0.20† |
| Fatp‐4 | 0.97 ± 0.04 | 0.96 ± 0.04 | 3.42 ± 0.15† | 3.68 ± 0.19† |
| Inflammation | ||||
| F4/80 | 1.93 ± 0.16 | 1.83 ± 0.17 | 4.79 ± 0.21† | 5.08 ± 0.20† |
| Cd68 | 1.34 ± 0.11 | 1.83 ± 0.13 | 5.96 ± 0.27† | 5.86 ± 0.35† |
| Il‐1β | 6.16 ± 0.26 | 6.11 ± 0.26 | 12.4 ± 0.36† | 12.2 ± 0.51† |
| Il‐6 | 6.22 ± 0.24 | 6.57 ± 0.18 | 12.5 ± 0.40† | 12.0 ± 0.38† |
| Ifn‐γ | 8.18 ± 0.25 | 8.39 ± 0.17 | 15.9 ± 0.26† | 16.3 ± 0.30† |
| Tnf‐α | 4.53 ± 0.21 | 4.34 ± 0.18 | 10.0 ± 0.32† | 10.2 ± 0.24† |
Mice (3 months of age) were fed an RD or HF diet for 2 months. During the last month of feeding, mice were given saline or exenatide (20 ng/g BW) once daily by intraperitoneal injections (n = 5/genotype per feeding per treatment). Liver extracts were analyzed by qRT‐PCR normalized to 18S in triplicate. Values are expressed as mean ± SEM; *P < 0.05 Ex versus S/feeding group; † P < 0.05 HF versus RD/treatment group.
Abbreviations: BW, body weight; Cd, clusters of differentiation; Ex, exenatide; Fatp, fatty acid transport protein; Il, interleukin; Ifn, interferon; Tnf, tumor necrosis factor.
Consistent with increased hyperinsulinemia‐driven Fasn transcription by SREBP‐1c activation,30 HF induced Fasn levels, as shown by immunoblotting proteins in the Fasn immunopellet with α‐Fasn antibody (Fig. 6B, panel ii; HF‐S versus RD‐S). This led to increased Fasn activity (Fig. 6B, panel i; HF‐S versus RD‐S). Exenatide treatment for 4 weeks activated the insulin receptor (Fig. 6B, panel iii; pIRβ) to cause CEACAM1 phosphorylation (Fig. 6B, panel ii; pCC1) and binding to Fasn, as evidenced by detecting pCC1 in the α‐Fasn immunopellet of lysates derived from HF‐exenatide versus HF‐S Cc1 +/+ livers (Fig. 6B, panel ii). As expected,24 this lowered Fasn activity in HF‐exenatide‐treated Cc1 +/+ livers (Fig. 6B, panel i). This effect of exenatide on Fasn activity was not detected in Cc1 –/– mice.
Consistent with an enhanced proinflammatory state with an increased energy supply,31 qRT‐PCR analysis showed elevation of inflammatory markers (interleukin‐1β, interleukin‐6, interferon‐γ) in addition to increasing the macrophage pool (F4/80 and Cd68) and activation (tumor necrosis factor α) in the liver of untreated HF‐fed Cc1 +/+ and Cc1 –/– mice (Table 2; HF‐S versus RD‐S). Exenatide treatment for 4 weeks lowered the mRNA levels of these inflammatory markers in HF‐fed Cc1 +/+ but not Cc1 –/– mice (Table 2; HF‐exenatide versus HF‐S). This is consistent with persistence of the foci of inflammatory cell infiltrates in the hepatic lobules of Cc1 –/– mice despite 4 weeks of exenatide treatment, as illustrated by hematoxylin and eosin stain analysis (Fig. 6A, panel viii' versus vi').
Discussion
The role of exenatide in suppressing appetite and inducing glucose‐stimulated insulin secretion is well documented.8, 9 The current study identifies a novel positive effect of exenatide on insulin clearance in parallel with insulin secretion to prevent chronic hyperinsulinemia and the subsequent increase in hepatic de novo lipogenesis.30
Exenatide promotes insulin clearance by inducing the expression of CEACAM1, which increases the rate of receptor‐mediated insulin uptake in a phosphorylation‐dependent manner.17 Null mutation of Ceacam1 and its liver‐specific inactivation impair insulin clearance to develop chronic hyperinsulinemia followed by insulin resistance and enhanced hepatic de novo lipogenesis, an event mainly caused by activating SREBP‐1c transcriptional activation of lipogenic genes, such as Fasn.30 Moreover, under conditions of hyperinsulinemia, pulsatility of insulin32 and its negative effect on Fasn activity mediated by CEACAM1 phosphorylation are abolished.24 Combined, this leads to increased hepatic lipid production and accumulation in addition to its redistribution to white adipose tissue to cause visceral obesity followed by lipolysis. In contrast, liver‐specific transgenic reconstitution of CEACAM1 reverses the abnormal metabolic phenotype of Cc1 –/– mice.33 Reduction of hepatic CEACAM1 plays a key role in the pathogenesis of diet‐induced insulin resistance, hepatic steatosis, and visceral adiposity,20 and adenoviral‐mediated redelivery of wild‐type but not the phosphorylation‐defective CEACAM1 to the liver reverses these metabolic derangements.21 Thus, it is conceivable that restoring hepatic CEACAM1 content (and phosphorylation) by exenatide contributes substantially to its preventive effect on diet‐induced insulin resistance and hepatic steatosis.
Compromised hepatic insulin extraction in human subjects is associated with obesity,34, 35 diet‐induced insulin resistance,36 type 2 diabetes,37 metabolic syndrome,38, 39 and fatty liver disease.40 At the molecular level, hepatic CEACAM1 levels are markedly reduced in patients with high‐grade NAFLD, insulin resistance, and obesity,41, 42 with hepatic steatosis positively correlating with the loss of CEACAM1.41 Hepatic CEACAM1 is also low in rat models of obesity and insulin resistance with impaired insulin clearance,42 including rats selectively bred for low aerobic capacity, which manifest features of metabolic syndrome, insulin resistance, and NAFLD/NASH. Consistent with the beneficial effect of body weight loss and exercise in NAFLD/NASH treatment,43 caloric restriction reversed the metabolic abnormalities in selectively bred rats together with restoring hepatic CEACAM1 levels.44 Thus, it is reasonable to predict that up‐regulating CEACAM1‐dependent pathways constitutes a promising therapeutic approach to prevent insulin resistance and hepatic steatosis.
The most common NASH pharmacological therapy involves the use of PPARγ agonists. While the clinical benefit of exenatide solo or in combination with pioglitazone awaits further investigations,14 exenatide increases PPARγ expression10 to promote lipid repartition to adipose tissue, thus alleviating hepatic steatosis and suppressing associated inflammation.45 The current study also showed that exenatide reduces lipolysis and fatty acids supply to the hepatocytes, leading to normalization of Pparα‐mediated fatty acid β‐oxidation in HF‐fed mice.
Mechanistically, exenatide, like rosiglitazone, bound to the conserved active PPRE‐RXRα site in the Ceacam1 promoter25 to induce its transcription to facilitate hepatic insulin clearance and suppress inflammation.31, 45 The protective effect of exenatide on insulin clearance against an HF diet in mice agrees with studies by Li et al.46 In contrast, exenatide did not modulate insulin clearance in RD‐fed wild‐type mice in support of its reported lack of effect on insulin extraction in healthy subjects and in nondiabetic first‐degree relatives of patients with type 2 diabetes.47
The conserved PPRE‐RXRα sequence also contains nucleotides from the juxtaposed IRE‐BP1 fragment that is critical in insulin‐induced activation of the Ceacam1 promoter. Consistent with the conservation of these sequences, insulin induces rat Ceacam1 promoter activity.29 Thus, it is conceivable that in vivo exenatide activates Ceacam1 transcription by inducing PPARγ and secretion of insulin that can bind to the Ceacam1 promoter. This requires ligand‐induced expression of GLP‐1R in hepatocytes, as emphasized by the blunted effect of exenatide on CEACAM1 mRNA levels in the presence of exendin 9‐39 GLP‐1R antagonist in HepG2 cells. While GLP‐1R expression in human hepatocytes remains debatable, our data agree with the reported direct effect of GLP‐1R activation on steatosis in human hepatocytes48 and by decreased GLP‐1R expression in the liver of NASH patients.10
Several mechanisms can underlie the effect of exenatide on CEACAM1 expression and action (phosphorylation) in vivo. In HF‐fed Cc1 +/+ mice, exenatide may reverse insulin resistance by restoring insulin pulsatility.49, 50 This could lead to increased CEACAM1 phosphorylation, followed by its enhanced internalization as part of the insulin‐receptor complex and its binding to Fasn to reduce its activity. This limits the effect of an HF diet on hepatic de novo lipogenesis and restricts lipid repartitioning to the white adipose tissue, reducing visceral adiposity and NEFA output and sustaining insulin sensitivity. On the other hand, it is also possible that exenatide initially induced CEACAM1 expression in HF‐fed Cc1 +/+ mice not only by activating Ceacam1 promoter by PPARγ and insulin, but also by preventing lipolysis and NEFA release from adipose tissue. This would relieve the Ceacam1 promotor from the inhibitory effect of PPARα activation by fatty acids.23 By inducing CEACAM1 expression, exenatide promoted hepatic insulin clearance to limit chronic hyperinsulinemia, thus restoring insulin pulsatility and sensitivity and lowering Fasn activity and hepatic steatosis. Regardless of the underlying mechanisms, the current study proposes that inducing hepatic CEACAM1 expression constitutes a novel mechanism contributing to the amelioration of hepatic steatosis by GLP‐1 analogs.
Our study demonstrated that exenatide induced CEACAM1 expression to protect insulin clearance and reduce hepatic de novo lipogenesis in response to an HF diet. The physiologic implication of increased insulin extraction in the face of elevated insulin secretion by exenatide is to maintain homeostatic plasma insulin levels, an essential determinant of insulin sensitivity and lipid homeostasis.23 Sustained insulin resistance and hepatic steatosis in Cc1 –/– mice despite reduction of food intake and a more pronounced release of insulin by exenatide further supports the relevance of CEACAM1‐mediated hepatic insulin clearance in regulating systemic insulin action.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1117/full
Supporting Information Slide 1
Supporting Information Slide 2
Supporting Information Slide 3
Supporting Information Slide 4
Supporting Information 1
Supporting Information 2
Supporting Information 3
Acknowledgment
We thank Melissa W. Kopfman and Zachary N. Smiley at the Najjar Laboratory at the University of Toledo College of Medicine for their technical assistance in exenatide injections and in the generation and maintenance of mice.
Potential conflict of interest: Nothing to report.
Supported by grants from the National Institutes of Health (R01‐DK054254, R01‐DK083850, and R01‐HL112248 to S.M.N. and R01‐DK101711 to V.P.) and by the American Diabetes Association Junior Faculty Development (Award 1‐17‐JDF‐055 to K.Y.L.); also supported by fellowships from the American Heart Association (14POST20480294 to L.R.), the Endocrine Society Summer Fellowship (to C.C.M.), and the Middle‐East Diabetes Research Center (to H.E.G. and S.S.G.); partially supported by the John J. Kopchick Ph.D. Ohio Heritage Foundation Eminent Research Chair Fund (to S.M.N.).
REFERENCES
- 1. Spengler EK, Loomba R. Recommendations for diagnosis, referral for liver biopsy, and treatment of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Mayo Clin Proc 2015;90:1233‐1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Promrat K, Kleiner DE, Niemeier HM, Jackvony E, Kearns M, Wands JR, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010;51:121‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Green RM. NASH‐‐hepatic metabolism and not simply the metabolic syndrome. Hepatology 2003;38:14‐17. [DOI] [PubMed] [Google Scholar]
- 4. Chitturi S, Abeygunasekera S, Farrell GC, Holmes‐Walker J, Hui JM, Fung C, et al. NASH and insulin resistance: insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology 2002;35:373‐379. [DOI] [PubMed] [Google Scholar]
- 5. Bell LN, Wang J, Muralidharan S, Chalasani S, Fullenkamp AM, Wilson LA, et al.; Nonalcoholic Steatohepatitis Clinical Research Network . Relationship between adipose tissue insulin resistance and liver histology in nonalcoholic steatohepatitis: a pioglitazone versus vitamin E versus placebo for the treatment of nondiabetic patients with nonalcoholic steatohepatitis trial follow‐up study. Hepatology 2012;56:1311‐1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, et al. A placebo‐controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 2006;355:2297‐2307. [DOI] [PubMed] [Google Scholar]
- 7. Aithal GP, Thomas JA, Kaye PV, Lawson A, Ryder SD, Spendlove I, et al. Randomized, placebo‐controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology 2008;135:1176‐1184. [DOI] [PubMed] [Google Scholar]
- 8. Drucker DJ. Deciphering metabolic messages from the gut drives therapeutic innovation: the 2014 Banting Lecture. Diabetes 2015;64:317‐326. [DOI] [PubMed] [Google Scholar]
- 9. D'Alessio D. Is GLP‐1 a hormone: whether and when? J Diabetes Investig 2016;7(Suppl. 1):50‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Svegliati‐Baroni G, Saccomanno S, Rychlicki C, Agostinelli L, De Minicis S, Candelaresi C, et al. Glucagon‐like peptide‐1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high‐fat diet in nonalcoholic steatohepatitis. Liver Int 2011;31:1285‐1297. [DOI] [PubMed] [Google Scholar]
- 11. Trevaskis JL, Griffin PS, Wittmer C, Neuschwander‐Tetri BA, Brunt EM, Dolman CS, et al. Glucagon‐like peptide‐1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am J Physiol Gastrointest Liver Physiol 2012;302:G762‐G772. [DOI] [PubMed] [Google Scholar]
- 12. Ding X, Saxena NK, Lin S, Gupta NA, Anania FA. Exendin‐4, a glucagon‐like protein‐1 (GLP‐1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006;43:173‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abdul‐Ghani M, Migahid O, Megahed A, DeFronzo RA, Zirie M, Jayyousi A. Efficacy of exenatide plus pioglitazone vs basal/bolus insulin in T2DM patients with very high HbA1c. J Clin Endocrinol Metab 2017;102:2162‐2170. [DOI] [PubMed] [Google Scholar]
- 14. Le TA, Loomba R. Management of non‐alcoholic fatty liver disease and steatohepatitis. J Clin Exp Hepatol 2012;2:156‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. DeAngelis AM, Heinrich G, Dai T, Bowman TA, Patel PR, Lee SJ, et al. Carcinoembryonic antigen‐related cell adhesion molecule 1: a link between insulin and lipid metabolism. Diabetes 2008;57:2296‐2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Poy MN, Yang Y, Rezaei K, Fernstrom MA, Lee AD, Kido Y, et al. CEACAM1 regulates insulin clearance in liver. Nat Genet 2002;30:270‐276. [DOI] [PubMed] [Google Scholar]
- 17. Choice CV, Howard MJ, Poy MN, Hankin MH, Najjar SM. Insulin stimulates pp120 endocytosis in cells co‐expressing insulin receptors. J Biol Chem 1998;273:22194‐22200. [DOI] [PubMed] [Google Scholar]
- 18. Lee SJ, Heinrich G, Fedorova L, Al‐Share QY, Ledford KJ, Fernstrom MA, et al. Development of nonalcoholic steatohepatitis in insulin‐resistant liver‐specific S503A carcinoembryonic antigen‐related cell adhesion molecule 1 mutant mice. Gastroenterology 2008;135:2084‐2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ghosh S, Kaw M, Patel PR, Ledford KJ, Bowman TA, McInerney MF, et al. Mice with null mutation of Ceacam I develop nonalcoholic steatohepatitis. Hepat Med 2010;2010:69‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Al‐Share QY, DeAngelis AM, Lester SG, Bowman TA, Ramakrishnan SK, Abdallah SL, et al. Forced hepatic overexpression of CEACAM1 curtails diet‐induced insulin resistance. Diabetes 2015;64:2780‐2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Russo L, Ghadieh HE, Ghanem SS, Al‐Share QY, Smiley ZN, Gatto‐Weis C, et al. Role for hepatic CEACAM1 in regulating fatty acid metabolism along the adipocyte‐hepatocyte axis. J Lipid Res 2016;57:2163‐2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liang CP, Han S, Li G, Tabas I, Tall AR. Impaired MEK signaling and SERCA expression promote ER stress and apoptosis in insulin‐resistant macrophages and are reversed by exenatide treatment. Diabetes 2012;61:2609‐2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ramakrishnan SK, Russo L, Ghanem SS, Patel PR, Oyarce AM, Heinrich G, et al. Fenofibrate decreases insulin clearance and insulin secretion to maintain insulin sensitivity. J Biol Chem 2016;291:23915‐23924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Najjar SM, Yang Y, Fernstrom MA, Lee SJ, Deangelis AM, Rjaily GA, et al. Insulin acutely decreases hepatic fatty acid synthase activity. Cell Metab 2005;2:43‐53. [DOI] [PubMed] [Google Scholar]
- 25. Ramakrishnan SK, Khuder SS, Al‐Share QY, Russo L, Abdallah SL, Patel PR, et al. PPARalpha (peroxisome proliferator‐activated receptor alpha) activation reduces hepatic CEACAM1 protein expression to regulate fatty acid oxidation during fasting‐refeeding transition. J Biol Chem 2016;291:8121‐8129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang LH, Kamanna VS, Ganji SH, Xiong XM, Kashyap ML. Pioglitazone increases apolipoprotein A‐I production by directly enhancing PPRE‐dependent transcription in HepG2 cells. J Lipid Res 2010;51:2211‐2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tachibana K, Kobayashi Y, Tanaka T, Tagami M, Sugiyama A, Katayama T, et al. Gene expression profiling of potential peroxisome proliferator‐activated receptor (PPAR) target genes in human hepatoblastoma cell lines inducibly expressing different PPAR isoforms. Nucl Recept 2005;3:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chahal J, Chen CC, Rane MJ, Moore JP, Barati MT, Song Y, et al. Regulation of insulin‐response element binding protein‐1 in obesity and diabetes: potential role in impaired insulin‐induced gene transcription. Endocrinology 2008;149:4829‐4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Najjar S, Boisclair Y, Nabih Z, Philippe N, Imai Y, Suzuki Y, et al. Cloning and characterization of a functional promoter of the rat pp120 gene, encoding a substrate of the insulin receptor tyrosine kinase. J Biol Chem 1996;271:8809‐8817. [DOI] [PubMed] [Google Scholar]
- 30. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002;109:1125‐1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Najjar SM, Russo L. CEACAM1 loss links inflammation to insulin resistance in obesity and non‐alcoholic steatohepatitis (NASH). Semin Immunopathol 2014;36:55‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Matveyenko AV, Liuwantara D, Gurlo T, Kirakossian D, Dalla Man C, Cobelli C, et al. Pulsatile portal vein insulin delivery enhances hepatic insulin action and signaling. Diabetes 2012;61:2269‐2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Russo L, Muturi HT, Ghadieh HE, Ghanem SS, Bowman TA, Noh HL, et al. Liver‐specific reconstitution of CEACAM1 reverses the metabolic abnormalities caused by its global deletion in male mice. Diabetologia 2017; doi: 10.1007/s00125-017-4432-y. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Meistas MT, Margolis S, Kowarski AA. Hyperinsulinemia of obesity is due to decreased clearance of insulin. Am J Physiol 1983;245:E155‐E159. [DOI] [PubMed] [Google Scholar]
- 35. Jones CN, Abbasi F, Carantoni M, Polonsky KS, Reaven GM. Roles of insulin resistance and obesity in regulation of plasma insulin concentrations. Am J Physiol Endocrinol Metab 2000;278:E501‐E508. [DOI] [PubMed] [Google Scholar]
- 36. Bakker LEH, van Schinkel LD, Guigas B, Streefland TCM, Jonker JT, van Klinken JB, et al. A 5‐day high‐fat, high‐calorie diet impairs insulin sensitivity in healthy, young South Asian men but not in Caucasian men. Diabetes 2014; 63:248‐258. [DOI] [PubMed] [Google Scholar]
- 37. Lee CC, Haffner SM, Wagenknecht LE, Lorenzo C, Norris JM, Bergman RN, et al. Insulin clearance and the incidence of type 2 diabetes in Hispanics and African Americans: the IRAS Family Study. Diabetes Care 2013;36:901‐907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Marini MA, Frontoni S, Succurro E, Arturi F, Fiorentino TV, Sciacqua A, et al. Differences in insulin clearance between metabolically healthy and unhealthy obese subjects. Acta Diabetol 2014;51:257‐261. [DOI] [PubMed] [Google Scholar]
- 39. Pivovarova O, Bernigau W, Bobbert T, Isken F, Mohlig M, Spranger J, et al. Hepatic insulin clearance is closely related to metabolic syndrome components. Diabetes Care 2013;36:3779‐3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Finucane FM, Sharp SJ, Hatunic M, Sleigh A, De Lucia Rolfe E, Aihie Sayer A, et al. Liver fat accumulation is associated with reduced hepatic insulin extraction and beta cell dysfunction in healthy older individuals. Diabetol Metab Syndr 2014;6:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee W. The CEACAM1 expression is decreased in the liver of severely obese patients with or without diabetes. Diagn Pathol 2011;6:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Heinrich G, Muturi HT, Rezaei K, Al‐Share QY, DeAngelis AM, Bowman TA, et al. Reduced hepatic carcinoembryonic antigen‐related cell adhesion molecule 1 level in obesity. Front Endocrinol (Lausanne) 2017;8:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McCarthy EM, Rinella ME. The role of diet and nutrient composition in nonalcoholic fatty liver disease. J Acad Nutr Diet 2012;112:401‐409. [DOI] [PubMed] [Google Scholar]
- 44. Bowman TA, Ramakrishnan SK, Kaw M, Lee SJ, Patel PR, Golla VK, et al. Caloric restriction reverses hepatic insulin resistance and steatosis in rats with low aerobic capacity. Endocrinology 2010;151:5157‐5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sathyanarayana P, Jogi M, Muthupillai R, Krishnamurthy R, Samson SL, Bajaj M. Effects of combined exenatide and pioglitazone therapy on hepatic fat content in type 2 diabetes. Obesity (Silver Spring) 2011;19:2310‐2315. [DOI] [PubMed] [Google Scholar]
- 46. Li L, Yang G, Li Q, Tan X, Liu H, Tang Y, et al. Exenatide prevents fat‐induced insulin resistance and raises adiponectin expression and plasma levels. Diabetes Obes Metab 2008;10:921‐930. [DOI] [PubMed] [Google Scholar]
- 47. Meier JJ, Holst JJ, Schmidt WE, Nauck MA. Reduction of hepatic insulin clearance after oral glucose ingestion is not mediated by glucagon‐like peptide 1 or gastric inhibitory polypeptide in humans. Am J Physiol Endocrinol Metab 2007;293:E849‐E856. [DOI] [PubMed] [Google Scholar]
- 48. Gupta NA, Mells J, Dunham RM, Grakoui A, Handy J, Saxena NK, et al. Glucagon‐like peptide‐1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010;51:1584‐1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ritzel R, Schulte M, Porksen N, Nauck MS, Holst JJ, Juhl C, et al. Glucagon‐like peptide 1 increases secretory burst mass of pulsatile insulin secretion in patients with type 2 diabetes and impaired glucose tolerance. Diabetes 2001;50:776‐784. [DOI] [PubMed] [Google Scholar]
- 50. Gastaldelli A, Brodows RG, D'Alessio D. The effect of chronic twice daily exenatide treatment on beta‐cell function in new onset type 2 diabetes. Clin Endocrinol (Oxf) 2014;80:545‐553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1117/full
Supporting Information Slide 1
Supporting Information Slide 2
Supporting Information Slide 3
Supporting Information Slide 4
Supporting Information 1
Supporting Information 2
Supporting Information 3