

Abstract
Purpose of review
To highlight and emphasize how new knowledge of mechanisms linked to the IL-23/IL-17 pathway are relevant to the pathophysiology of axial spondyloarthritis (axSpA) and to demonstrate how molecules in IL-23/IL-17 pathway provide novel therapeutic targets for axSpA patients.
Recent findings
Similar to AS, the increased frequency of Th17 cells in nr-axSpA patients underscores the concept that these disorders can be viewed on a spectrum. Recent findings suggest that the contribution of IL-23/IL-17 signaling pathways possibly differ in male and female AS patients. The finding that IL-17 and IL-22 secreting type 3 innate lymphoid cells (ILC3), are increased in AS patients point to their potential role in the pathogenesis of axSpA. Reports of dysbiosis in the gut microbiome of AS patients supports previous work indicating a possible causal relationship between altered gut flora, ileocolonic inflammation and axSpA. Of important clinical relevance, are results from clinical trials supporting the efficacy and safety of agents that block IL-12/23 (ustekinumab) and IL-17 (secukinumab and ixekizumab) in AS patients.
Summary
Recent studies further establish the central position of the IL-23/IL-17 pathway in the pathogenesis of axSpA. Targeting IL-23/IL-17 pathway appears to be a safe and effective strategy for treatment of axSpA patients.
Keywords: ankylosing spondylitis, antagonists, axial spondyloarthritis, clinical trial, gut, IL-17, IL-22, IL-23, innate lymphoid cells, microbiome, non-radiographic axial spondyloarthritis, secukinumab, Th17, Ustekinumab
Introduction
The term axial spondyloarthritis (axSpA) emerged in the revised Assessment in SpondyloArthritis international Society (ASAS) diagnostic criteria published in 2009 [1]. Looking beyond the NY criteria, which are limited to patients with radiographic changes, the ASAS criteria included a wider spectrum of patients with inflammatory back disease. A subsequent population based study in the United States found the prevalence of SpA to be 1%, significantly higher than previous estimates and somewhat puzzling to practicing rheumatologists who evaluate far more rheumatoid arthritis (RA) than inflammatory back pain patients [2]. It has now became apparent from several studies that focusing on ankylosing spondylitis (AS), which requires defined radiographic features in the sacroiliac joints, excludes a large population of patients with non-radiographic axSpA (nr-axSpA). These patients are more likely to be female and demonstrate normal C-reactive protein (CPR) levels. Moreover, patients with nr-axSpA demonstrate similar disease activity and quality of life indices to patients with AS and in one observational study, similar response and adherence to anti-TNF agents [3]. Based on these observations, some experts in the field eschew creating distinct disease subsets (nr-axSpA, AS) in favor of viewing these as different presentations of a single disease or as a continuum. The finding that about 5% of patients with nr-axSpA go on to develop AS at 2 years provides support that the diseases may be distinct with unique phenotypes that would be best appreciated over the course of time.
Layered on this background of controversy regarding natural history, diagnosis and disease classification are the considerable advances in our understanding of disease pathogenesis of axial SpA. In particular, the discovery that IL-23/IL-17 pathway is a central axis in the development and persistence of the disease unleashed a search for new targets that now form the basis for drug development in this area. Convincing evidence from animal models, translational studies and recent clinical trials underscore the importance and complexity of this pathway. In this review, we will discuss major recent advancements in the field about our understanding of IL-17/IL-23 molecular mechanisms and provide examples of how these discoveries catalyzed development of effective therapies.
Brief overview of IL-23/IL-17 pathway and major players
IL-17 is an immunologically important cytokine that was first identified as a transcript from murine lymphoid cells [4]. Subsequent studies established IL-17 not only as a key mediator of immune response against pathogens but also as a potent mediator of inflammation and tissue damage in several autoimmune and inflammatory diseases [5]. Over the years, many other similar proteins have been discovered. Currently, the IL-17 family consists of six ligand members (IL-17A to IL-17F) that bind to five different receptors; IL-17RA, IL-17RB, IL-17RC, IL-17RD and IL-17RE that have been identified. Initially, a subset of CD4+ T cells, now popularly known as Th17, was thought to be the major source of IL-17, but recent studies revealed that many other cell subsets including CD8+ cytotoxic T cells [6], γδ T cells [7], natural killer (NK) cells and innate lymphoid cells (ILCs) can also secrete high levels of IL-17 and other related cytokines, such as IL-22 and TNF-α. A model that incorporates elements of the IL-23/IL-17 pathway in axSpA pathogenesis is depicted in Figure 1.
Figure 1. The IL-17/IL-23 pathway in axSpA pathogenesis.
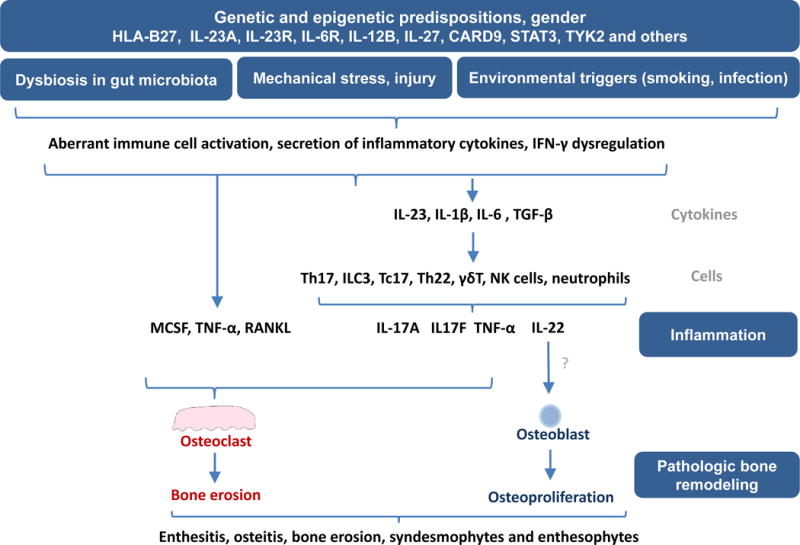
The convergence of genetic and epigenetic factors, environmental triggers, mechanical stress, and gut dysbiosis promote an inflammatory response in the gut and joints through activation of dendritic cells, macrophages, synovial fibroblasts and other resident cells. In the resulting inflammatory response, these cells secrete an array of inflammatory cytokines such as IL-1β, IL-6, IL-23 and TGF-β. Elevated levels of IL-1β, IL-6, IL-23 and TGF-β generate Th17 cell differentiation resulting in an increased frequency of IL-17 and IL-22 secreting CD4+ cells along with Th22, CD8+IL17+ (Tc17), type 3 innate lymphoid cells (ILC3), NK cells and neutrophils. IL-17 secreted by these cells, together with increased levels of RANKL and TNF-α, leads to increased osteoclastogenesis and bone erosion whereas IL-22 may drive pathologic new bone formation, resulting in altered bone remodeling and radiographic damage in axSpA.
Specific subsets of ILCs i.e. type 3 ILCs (ILC3) were also recognized as another major source of IL-17A, IL-17F, IL-22 and other related cytokines [8]. Importantly, differentiation of IL-17 secreting cells is critically dependent on the presence of key inflammatory cytokines, IL-1β, IL-6, IL-23 and TGF-β. Among these, IL-23 appeared to be of great significance as studies revealed that IL-23 not only stabilizes the phenotype of Th17 cells [9, 10], but also determines their function and pathogenic capacity [11]. IL-23 is predominantly secreted by activated macrophages and dendritic cells and plays deterministic functions in the differentiation of various subsets of IL-17 secreting cells [7, 11].
The importance of IL-23/IL-17 pathway in the pathogenesis axSpA
For well over a decade, the dominant paradigm of AS pathogenesis has been centered on the dominance of the Th1 pathway and TNF-α. The efficacy of anti-TNF agents for axial disease at a time when no other therapies were active for this manifestation provided strong support for this model. The discovery of IL-23 and other members of the IL-17 pathway, however, unveiled alternative mechanisms and targets. While preclinical studies suggested this pathway to be operative in both, RA and spondyloarthritis, human translational reports and clinical trials demonstrated that IL-23 and IL-17 are key cytokines in only axSpA and PsA. These exciting discoveries provide unparalleled opportunities to decipher disease pathogenesis and to uncover new therapeutic targets.
Findings from genetic, epigenetic, in vitro and recent animal studies
The earliest evidence pointing to the involvement of IL-23/IL-17 pathway in axSpA came from genetic association studies which showed that various polymorphisms in the IL-23 receptor (IL23R) gene were associated with AS [12]. Genome wide association revealed that IL23R variant rs11209026 (Arg381Gln) offers protection against AS [12] by showing decreased STAT3 phosphorylation [13] which ultimately leads to a selective impairment of IL-17 production [14]. Many other IL-23/IL-17 pathway related genes such as IL-6R, IL-12B, IL-27, CARD9, STAT3 and TYK2, also have been implicated in AS [15] indicating the potential importance of the IL-I7/IL-23 pathway in the axSpA. Uddin et al. applied an integrative genomics approach to show that AS risk alleles overlap with global immune related pathways and Th17 related genes in AS pathogenesis [16]**. Integration of genetic and epigenetic fine mapping of causal variants related to autoimmune diseases further revealed the relatedness of AS, psoriasis and inflammatory bowel disease; all three disorders demonstrated a Th17 cell specific epigenetic signature for the known variants [17]**.
Previous animal studies established the essential role of IL-23/IL-17 pathway in ankylosing enthesitis and bone remodeling [18, 19]. Animal studies also indicated that IL-23-driven inflammation can lead to new bone formation in AS [20]. The finding from the proteoglycan-induce spondylitis (PGISp) mouse model of AS further suggests that inflammation and new bone formation unfold sequentially and not in a parallel sequence. Inflammation precedes intervertebral disc destruction and serves as a prerequisite for axial disease progression [21]*. Findings from this study further suggest that inflammation drives the increased osteoproliferation which subsequently leads to altered bone phenotypes in AS [21]*. Additional support for the importance of IL-17 in spondylitis is supported by prophylactic administration of anti-IL-17 antibodies which block development of ankylosis in male SKG1 mice [22]. Taken together, these studies indicate that IL-23/IL-17 pathway is centrally involved in the pathogenesis of axSpA and that targeting this pathway may be an effective strategy in this group of disorders.
Preclinical and translational studies supporting the importance of IL-17, IL-22 and IL-23 secreting cells in axSpA
Some of the earliest evidence indicating the pivotal role of a dysregulated IL-23/IL-17 pathway in axSpA is derived from studies of human blood cells and joint tissues. Analysis of samples from AS patients revealed increased numbers of Th17 [23, 24], Th22 [23] and γδ T cells [25] in the peripheral blood and an increased level of IL-17 [26-28] and IL-23 [27, 29, 30] in the serum and synovium. It has been also reported that the KIR3DL2+ Th17 cells, responsive to HLA-B27 homodimers, are increased in the peripheral blood and synovium of AS patients [31] and the interaction of KIR3DL2 with HLA-B27 dimers promote survival and the IL-17 production capacity of the KIR3DL2 expressing Th17 cells [31]. Most of these previous reports of Th17 cells in AS focused on patients with established disease whereas pathogenic events in the early phase of SpA were not emphasized. Now, a recent report suggests, that in nr-ax-SpA patients, a higher number of Th17 cells expressing TCRαβ+CD161+, a memory phenotype marker, are present in the peripheral blood of the nr-axSpA patients compared to healthy controls [32]*. It is important to mention that this finding contradicts earlier data that reported either no difference [33] or decreased frequencies of Th17 cells in peripheral blood of nr-axSpA patients [34] compared to the healthy controls. These divergent findings may be due to differences in gender composition, genetic background or immunologic methods. Additional studies are necessary to resolve these conflicting results.
It is well established that compared to females with AS, men are more likely to have radiographic changes in the SI joints and increased CRP but the two sexes experience similar levels of pain and functional impairment [35]. The underlying biology responsible for these differences is unknown. Investigators in Toronto examined sexual dimorphism in the Th17 signature of males and females with AS [36]**. They analyzed cells present in the whole blood collected from disease matched cohorts of male and female patients with age matched healthy controls. The identified distinctive upregulation of Th17 cells and related gene signatures in male AS patients while female AS patients were comparable to healthy controls. Interestingly, in this case, the sex bias in terms of IL-17 pathway related gene signatures were shown to be independent of sex hormone levels. Thus, this new findings may reveal the key pathways responsible for the divergent phenotypes in the sexes. Additional studies are required, however, to better understand the underlying biology responsible for the sexual dimorphism in the Th17 signature in AS.
Past evidence from human studies also suggested that in AS, apart from Th17, innate immune cells can also serve as major source of IL-17 especially in target tissues [33, 37]. The recognition that innate lymphoid cells (ILC) promote inflammation in a range of disorders is now apparent. In relation to SpA, emerging evidence indicates that ILC3 cells that respond to IL-23 also serve as major sources of IL-17, IL-22 and other related cytokines and this population is expanded in the joints of PsA but not RA patients [38]. In AS patients, IL-17 and IL-22 secreting NKp44+ILC3 cells were expanded in the peripheral blood, gut, synovial fluid and bone marrow [39]**. Interestingly, ILC3 cells present in peripheral blood, synovial fluid and bone marrow of AS patients expressed the α3β7 integrin indicating these cells likely originated in the gut. The ILC3 cells produced distinct cytokines in individual tissue compartments, although IL-22 secretion was elevated in most specimens while only a small percentage of cells in peripheral blood expressed IL-17 alone or in combination with IL-22. These data highlight the potential importance of innate immune cells in disease pathogenesis and raise the possibility that they may serve as therapeutic targets.
The role of the gut microbiome and intestinal inflammation in axSpA
A growing body of literature supports strong links between gut inflammation and spondylitis (reviewed in [40, 41]) and implicate both HLA-B27 alleles and the IL-23/IL-17 axis as key pathogenic factors. Studies in transgenic rats carrying the human HLA-B27 and β2 microglobulin genes revealed that intestinal inflammation in these rats concurrently occur with elevated expression of IL-23 and IL-17 in the colonic tissues [42]. Recent studies further revealed altered gut microbiota in transgenic HLA-B27 rats compared to wild type littermates [43]. Of particular relevance was the finding of dysbiosis in the gut microbiome of AS patients characterized by higher abundance of five families of bacteria, Bacteroidaceae, Lachnospiraceae, Porphyromonadaceae, Rikenellaceae, and Ruminococcaceae, and decrease abundance of two families of bacteria, Prevotellaceae and Veillonellaceae, in the terminal ileum of AS patients compared to healthy controls [44]** Altered bacterial flora have also been reported in PsA [45] and IBD [46-48]. Interactions of cell surface ligands with specific microbes can induce differentiation of IL-17 and IL-22 secreting cells [49, 50] programming their pathogenic capacity [51]. Collectively, these findings implicate microbial dysbiosis as triggers of altered homeostasis through the IL-23/IL-17 pathway resulting into gut and joint inflammation.
In support of a gut-spondylitis connection are reports of a high prevalence of gut inflammation in axSpA identified in the Ghent Inflammatory Arthritis and spoNdylitis cohorT (GIANT) patient cohort [52]. Ileocolonscopy combined with MRI imaging studies showed that the degree of spinal inflammation in axSpA patents correlated well with chronic but not acute gut inflammation in patients without gastrointestinal symptoms [53]. Increased expression of IL-23 had been already noted in the gut of AS patients when compared to healthy controls [54]. A recent study further revealed that AS patients display altered DCs and T cell populations that further implicate role of IL-23 in gut inflammation [55]*. In another report, IL-17 and IL-22 producing ILC3 cells were present in greater frequency in the gut of AS patients compared to healthy controls [39]. These ILC3 cells appeared to be predominantly expanded in the more inflamed gut of the AS patients and TNF-blocking agents were shown to be effective in reducing their abundance in the gut [39]. These above mentioned results indicate the existence of a gut-joint/spine axis in AS whereby ILC3 may be critical effectors secreting IL-17, IL-22 and possibly other inflammatory cytokines.
Targeting IL-23/IL-17 pathway for the treatment of axial SpA
As previously mentioned, anti-TNF-α blocking agents were the only effective way to treat axSpA patients who failed first line of therapy with NSAIDs and physical therapy for over a decade. Moreover, up to 50% of patients with active axSpA do not achieve primary endpoints despite treatment with NSAIDs and/or anti-TNF-α blocking agents [56]**. Moreover, recent clinical trials also demonstrated that targeting other cytokines or molecules such as IL-1 [57], IL-6 [58], or enzyme phosphodiesterase 4 (PDE4) [59] are not effective in axSpA. Fortunately, results from clinical trials show that targeting the IL-23/IL-17 pathway holds great promise for treatment of axSpA (Figure 2). Approved therapeutic drugs along with agents currently in clinical trials and in the pipeline that target molecules in the IL-23/IL-17 pathways are discussed below and outlined in Table 1.
Figure 2. Targeting IL-17/IL-23 pathway with neutralizing agents.

IL-23 is a heterodimer of p40 (IL-12p40) and p19 (IL-23p19). Ustekinumab binds to IL-12p40 and blocks the binding of both IL-12 and IL-23 to their receptors. BI655066 binds sIL-23p19 and block the binding of IL-23 to IL-23R. Secukinumab and ixekizumab bind IL-17A and block the binding of both IL-17A and IL-17A/F to their receptor. Bimekizumab and MSB0010841 bind and neutralize both IL-17A and IL-17F isoforms. Brodalumab binds IL-17RA and blocks the binding of IL-17A, IL-17A/F, IL-17F, IL-17C, and IL-17E to their receptors. Fezakinumab binds and neutralizes IL-22.
Table 1.
Therapeutic agents targeting IL-23/IL-17 pathway
| Agents: FDA approved or in clinical trials | |||
|---|---|---|---|
| Agents | Type | Target/mechanism | Current status |
| Secukinumab | mAb | IL-17A | FDA approved |
| Ixekizumab | mAb | IL-17A | Phase III |
| Ustekinumab | mAb | p40 subunit of IL-12 and IL-23 | Phase II, completed |
| BI655066 | mAb | p19 subunit of IL-23 | Phase II |
| Agents with potential efficacy in axSpA | |||
| Agents | Type | Target/mechanism | Current status |
| Brodalumab | mAb | IL-17RA | No trial data |
| Bimekizumab | mAb | IL-17A and IL-17F | No trial data |
| MSB0010841 | Nanobody | IL-17A and IL-17F | No trial data |
| Fezakinumab | mAb | IL-22 | No trial data |
| Tildrakizumab | mAb | p19 subunit of IL-23 | No trial data |
| Guselkumab | mAb | p19 subunit of IL-23 | No trial data |
| ABT-122 | Dual-variable-domain immunoglobulin | Binds to TNF-α and IL-17A | No trial data |
| COVA322 | Bispecific antibody fusion protein | Binds to TNF-α and IL-17A | No trial data |
| Apilimod | Small molecule | Suppresses synthesis of IL-23 and IL-12 | No trial data |
| CB300 | Small molecule | Suppresses production of IL-17A and other proinflammatory cytokines | Preclinical studies |
mAb, monoclonal antibody; FDA, Food and Drug Administration
IL-23 antagonists: Ustekinumab and BI655066
IL-23 is composed of two protein subunits, p19 and p40. While the p19 subunit is unique to IL-23, the p40 subunit also associates with the protein subunit, p35 to form IL-12. Therapeutic agents have been developed neutralize IL-23 by binding to either of these two subunits. Ustekinumab is a monoclonal antibody that binds to the p40 subunit and serves as a dual antagonist of IL-12 and IL-23. Ustekinumab is effective and well tolerated in patients with plaque psoriasis [60] and in PsA patients [61. Indeed, the drug is approved in the United States for both diseases. Importantly, ustekinumab inhibits radiographic progression in PsA patients {Kavanaugh, 2014 #86]. In a phase II proof of concept study (TOPAS), the efficacy and safety of ustekinumab in AS was analyzed in 20 patients. In this study, 65% of the patients achieved the ASAS40 response and 55% reached BASDAI50 response at week 24 [62]. Ustekinumab administration was also shown to be substantially effective in reducing active inflammation of the sacroiliac joints and the spine (41% and 31% reductions respectively) in treatment responders based on MRI assessment [62]*. In an ongoing clinical trials (ClinicalTrials.gov ID - NCT02407223) the efficacy and safety of ustekinumab to treat patients with active nr-axSpA as well as patients with active radiographic axSpA, who are naive or refractory to anti-TNF-α agents (ClinicalTrials.gov ID - NCT02437162 and NCT02438787 respectively) are underway. Similarly, a fully human IgG1 monoclonal antibody against p19 subunit of IL-23, BI655066 is under investigation in 48 week phase III randomized, double-blind, placebo-controlled clinical trial in patients with AS (ClinicalTrials.gov ID - NCT02047110).
IL-17 antagonists: secukinumab, ixekizumab and brodalumab
At present two antibodies that inhibit IL-17A have been developed: secukinumab is approved for AS and ixekizumab is in phase III trials. Secukinumab is a fully human monoclonal antibody (mAb) that selectively binds neutralizes IL-17A. In an earlier phase 2 randomized double blinded placebo-controlled trial, secukinumab was effective in controlling symptoms of AS [63]. Subsequent phase 3 trials provided further proof of its efficacy in AS patients [64]**. In the MEASURE 1 trial, in 371 patients with active AS, patients given intravenous loading followed by monthly subcutaneous dosing. The primary outcome measure, ASAS20 response rate at week 16, was 61%, 60%, and 29% respectively for patients treated with 150 mg or 75 mg of secukinumab or placebo, showing significant difference between active drug and placebo (p<0.001) [64]**. In MEASURE 2, the study design used subcutaneous loading and monthly dosing, and the ASAS 20 response rates at 16 weeks were 61% and 41% with secukinumab at a doses of 150 mg and 75 mg and 28% for placebo [64]**. A recent study using MRI endpoints further revealed that secukinumab treatment reduces inflammation in AS patients detected even after two years [65]. Taken together, these 2 studies demonstrated that secukinumab is efficacious for treatment of AS patients and additional studies are planned for patients with nr-axSpA. Further studies will be also needed to compare efficacy of secukinumab with established treatment regiments with approved anti-TNF agents. Nonetheless, secukinumab will likely benefit patients with axSpA and is also effective in anti-TNF inadequate responders although the response is lower than in biologic naive patients. Similar to secukinumab, ixekizumab is another humanized monoclonal antibody that binds and neutralizes IL-17A. A phase III trial of ixekizumab in AS (COAST-W, ClinicalTrials.gov ID - NCT02696798) is underway. A phase III clinical trial to test the efficacy and safety of an anti-IL-17R monoclonal antibody, brodalumab, in PsA were cancelled (ClinicalTrials.gov ID - NCT02429882) due to increased suicidal ideation and completed suicides noted in psoriasis clinical trials [66]. Similarly, clinical trials to test the efficacy of an anti-IL-17R monoclonal antibody, brodalumab, with proven efficacy in psoriasis and PsA were planned (ClinicalTrials.gov ID - NCT02429882) but the study was cancelled due to safety concerns noted in psoriasis and PsA trials.
Novel therapeutic strategies to inhibit the IL-23/IL-17 pathway
To date, major efforts have been completed or are underway to inhibit IL-17 and IL-23. Therapies targeting IL-22 have not been tested in axSpA patients. An anti-IL-22 antibody, ILV-094, is available and trials are currently in progress in RA (ClinicalTrials.gov ID - NCT00883896), psoriasis (ClinicalTrials.gov ID - NCT00563524) and atopic dermatitis (ClinicalTrials.gov ID - NCT01941537). Similarly, agents that target both IL-17 A and IL-17F will likely be assessed in patients with axSpA. Another promising approach is dual agents that simultaneously target TNF-α as well as IL-17A. IL-17 and TNF-α play crucial role in the pathogenesis of axial SpA and thus targeting both molecules with a single agent presents a promising albeit challenging treatment strategy. Recently, one such dual-specific immunoglobulin (DVD-Ig) molecule targeting IL-17 and TNF-α, ABT-122, was developed and is currently under investigation in phase I and II clinical trials. COVA322 is another such antibody fusion protein that can bind to TNF-α and IL-17A [67]. These dual agents will be tested for efficacy and safety axSpA in future clinical trials. Lastly, CBP30, a selective small molecule inhibitor of CBP/p300 bromodomains, was shown to be effective in suppressing Th17 cytokine production from T cells of AS patients as well as of healthy controls [68]. Future clinical trials are anticipated.
Future directions
Despite the many advances associated with elucidation of IL-23/IL-17 pathway in axSpA pathogenesis, many challenges remain which hold great promise for additional advances and improved therapeutic opportunities (Table 2). A major challenge is how specific differences in IL-23/IL-17 pathway gene signatures in male and female axSpA patients may translate into more personalized treatment strategies. Also, our understanding of the steps leading to new bone formation in axSpA are still very limited although animal studies point to a possible contribution from IL-22. Further studies will be needed to clarify the role of not only IL-22 but also IL-17A, IL-17F, -23 and other key cytokines in osteoproliferation- a process that limits function in axSpA. Current studies provide a tantalizing glimpse of the relationship between gut dysbiosis and axial inflammation but we require confirmatory data from both preclinical studies and human tissues to confirm the specifics of the proposed gut-axial disease axis. Moreover, it is imperative from both a pathogenesis and therapeutic perspective to determine if alteration in gut microbial composition drives immunological and pathological alterations or is a result of these changes. It will also be of central importance to define the complex relationship between TNF-α and IL-23/IL-17 pathways in order to develop more effective treatment strategies, possibly combining these agents, in the near future. Similarly, the efficacy and safety of IL-17F, IL-22 and IL-23 inhibition require investigation in patients with axSpA. One of the biggest challenges in this group of disorders which targets young adults most commonly is successful strategies to ensure early diagnosis and several approaches are underway around the globe to address it. Future studies will undoubtedly uncover additional cellular and serum biomarkers and imaging strategies to correctly diagnose axSpA in the early stages. Similarly, such biomarkers will facilitate patient stratification to optimize treatment and to monitor therapeutic response.
Table 2.
Topics for future investigation in axSpA
|
Conclusion
Over the last five years, we have witnessed how discoveries at the bench which redefined immunology can also improve human health. New knowledge regarding the importance of the IL-23/IL-17 pathway in axSpA paved the way for new therapeutic options which are either in the clinic or in clinical trials. Despite these advances, many unresolved issues remain and require further investigation. Recent efforts are directed towards development of agents that block additional molecules in this pathway and we expect that some of these will prove successful therapies in axSpA. Additional studies are required to further delineate the role of innate cells (ILC3, NK, CD4−) cells and gut inflammation in axSpA pathogenesis and how cytokines such as IL-17, IL-22 and IL-23 regulate inflammation and bone formation in axSpA. Finally, identification of new biomarkers will not only permit earlier diagnosis but also help direct treatment selection of biologic therapies including those that target the IL-23/IL-17 pathway in axSpA.
Key points.
Converging data support the concept that nr-axSpA and AS can be viewed in a disease continuum in which the IL-23/IL-17 pathway is pivotal in disease pathogenesis
An increased frequency of circulating Th17 cells are a feature of both AS and nr-axSpA.
Studies in axSpA patients support the involvement of ILC3, IL-17A, IL-17F, IL-22, IL-23 dysbiosis of gut microbiota and subclinical gut inflammation in SpA pathogenesis
Recent findings suggests that the contribution of Th17 cells and related signaling pathways to inflammation differ in male and female AS patients
Data from clinical trials indicate that ustekinumab and the IL-17 antagonists secukinumab and ixekizumab are effective and safe therapies for AS.
Identification of patients at an earlier stage of disease and biomarkers in the IL-23/IL-17 pathway may permit stratification of patients to improve outcomes in axSpA.
Acknowledgments
None
Financial support and sponsorship
Christopher Ritchlin’s research works are supported by NIH CTSI Incubator Grant (5UL1TR000042-09) and RO1 AR069000.
Footnotes
Conflicts of interest
Christopher Ritchlin has research grants from Amgen, Abbvie, and UCB. He is also a consultant for Amgen, Abbvie, Boehringer Ingelheim, Celgene, Janssen, and Novartis. Ananta Paine has no conflicts of interest.
Contributor Information
Ananta Paine, Allergy, Immunology & Rheumatology Division, University of Rochester Medical Center, Rochester, New York 14623.
Christopher Ritchlin, Allergy, Immunology & Rheumatology Division, University of Rochester Medical Center, Rochester, New York 14623.
References
- 1.Rudwaleit M, van der Heijde D, Landewe R, et al. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part II): validation and final selection. Ann Rheum Dis. 2009;68:777–783. doi: 10.1136/ard.2009.108233. [DOI] [PubMed] [Google Scholar]
- 2.Reveille JD, Witter JP, Weisman MH. Prevalence of axial spondylarthritis in the United States: estimates from a cross-sectional survey. Arthritis Care Res (Hoboken) 2012;64:905–910. doi: 10.1002/acr.21621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallman JK, Kapetanovic MC, Petersson IF, et al. Comparison of non-radiographic axial spondyloarthritis and ankylosing spondylitis patients - baseline characteristics, treatment adherence, and development of clinical variables during three years of anti-TNF therapy in clinical practice. Arthritis Res Ther. 2015;17:378. doi: 10.1186/s13075-015-0897-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rouvier E, Luciani MF, Mattei MG, et al. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J Immunol. 1993;150:5445–5456. [PubMed] [Google Scholar]
- 5.Patel DD, Kuchroo VK. Th17 Cell Pathway in Human Immunity: Lessons from Genetics and Therapeutic Interventions. Immunity. 2015;43:1040–1051. doi: 10.1016/j.immuni.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 6.Huber M, Heink S, Pagenstecher A, et al. IL-17A secretion by CD8+ T cells supports Th17-mediated autoimmune encephalomyelitis. J Clin Invest. 2013;123:247–260. doi: 10.1172/JCI63681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sutton CE, Lalor SJ, Sweeney CM, et al. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Gasteiger G, Rudensky AY. Interactions between innate and adaptive lymphocytes. Nat Rev Immunol. 2014;14:631–639. doi: 10.1038/nri3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu C, Yosef N, Thalhamer T, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496:513–517. doi: 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Awasthi A, Riol-Blanco L, Jager A, et al. Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J Immunol. 2009;182:5904–5908. doi: 10.4049/jimmunol.0900732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghoreschi K, Laurence A, Yang XP, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2010;467:967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wellcome Trust Case Control C, Australo-Anglo-American Spondylitis C. Burton PR, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007;39:1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarin R, Wu X, Abraham C. Inflammatory disease protective R381Q IL23 receptor polymorphism results in decreased primary CD4+ and CD8+ human T-cell functional responses. Proc Natl Acad Sci U S A. 2011;108:9560–9565. doi: 10.1073/pnas.1017854108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Meglio P, Di Cesare A, Laggner U, et al. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PloS one. 2011;6:e17160. doi: 10.1371/journal.pone.0017160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown MA, Kenna T, Wordsworth BP. Genetics of ankylosing spondylitis-insights into pathogenesis. Nat Rev Rheumatol. 2016;12:81–91. doi: 10.1038/nrrheum.2015.133. [DOI] [PubMed] [Google Scholar]
- **16.Uddin M, Codner D, Hasan SM, et al. Integrated genomics identifies convergence of ankylosing spondylitis with global immune mediated disease pathways. Sci Rep. 2015;5:10314. doi: 10.1038/srep10314. Employing an integrative genomics approach, this study revealed that AS risk genes overlap with global immune related pathways and reported that STAT2/3 and other Th17 related genes likely are central to AS pathogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **17.Farh KK, Marson A, Zhu J, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337–343. doi: 10.1038/nature13835. Integrating genetic and epigenetic fine mapping of causal variants related to autoimmune diseases, this recent study revealed a correlation between AS, psoriasis and inflammatory bowel disease. These disorders share enriched Th17 cell specific epigenetic signatures for the known causal variants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glatigny S, Fert I, Blaton MA, et al. Proinflammatory Th17 cells are expanded and induced by dendritic cells in spondylarthritis-prone HLA-B27-transgenic rats. Arthritis and rheumatism. 2012;64:110–120. doi: 10.1002/art.33321. [DOI] [PubMed] [Google Scholar]
- 19.Abe Y, Ohtsuji M, Ohtsuji N, et al. Ankylosing enthesitis associated with up-regulated IFN-gamma and IL-17 production in (BXSB × NZB) F(1) male mice: a new mouse model. Mod Rheumatol. 2009;19:316–322. doi: 10.1007/s10165-009-0166-0. [DOI] [PubMed] [Google Scholar]
- 20.Sherlock JP, Joyce-Shaikh B, Turner SP, et al. IL-23 induces spondyloarthropathy by acting on ROR-gammat+ CD3+CD4-CD8- entheseal resident T cells. Nat Med. 2012;18:1069–1076. doi: 10.1038/nm.2817. [DOI] [PubMed] [Google Scholar]
- *21.Tseng HW, Pitt ME, Glant TT, et al. Inflammation-driven bone formation in a mouse model of ankylosing spondylitis: sequential not parallel processes. Arthritis Res Ther. 2016;18:35. doi: 10.1186/s13075-015-0805-0. Utilizing the peptidoglycan-induced spondylitis (PGISp) mouse model of AS, this study demonstrated that inflammation precedes the tissue damage and osteoproliferation responsible for the altered bone phenotypes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ebihara S, Date F, Dong Y, Ono M. Interleukin-17 is a critical target for the treatment of ankylosing enthesitis and psoriasis-like dermatitis in mice. Autoimmunity. 2015;48:259–266. doi: 10.3109/08916934.2014.976630. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L, Li YG, Li YH, et al. Increased frequencies of Th22 cells as well as Th17 cells in the peripheral blood of patients with ankylosing spondylitis and rheumatoid arthritis. PloS one. 2012;7:e31000. doi: 10.1371/journal.pone.0031000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen H, Goodall JC, Hill Gaston JS. Frequency and phenotype of peripheral blood Th17 cells in ankylosing spondylitis and rheumatoid arthritis. Arthritis and rheumatism. 2009;60:1647–1656. doi: 10.1002/art.24568. [DOI] [PubMed] [Google Scholar]
- 25.Kenna TJ, Davidson SI, Duan R, et al. Enrichment of circulating interleukin-17-secreting interleukin-23 receptor-positive gamma/delta T cells in patients with active ankylosing spondylitis. Arthritis and rheumatism. 2012;64:1420–1429. doi: 10.1002/art.33507. [DOI] [PubMed] [Google Scholar]
- 26.Wendling D, Cedoz JP, Racadot E, Dumoulin G. Serum IL-17, BMP-7, and bone turnover markers in patients with ankylosing spondylitis. Joint Bone Spine. 2007;74:304–305. doi: 10.1016/j.jbspin.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 27.Mei Y, Pan F, Gao J, et al. Increased serum IL-17 and IL-23 in the patient with ankylosing spondylitis. Clin Rheumatol. 2011;30:269–273. doi: 10.1007/s10067-010-1647-4. [DOI] [PubMed] [Google Scholar]
- 28.Liu W, Wu YH, Zhang L, et al. Elevated serum levels of IL-6 and IL-17 may associate with the development of ankylosing spondylitis. Int J Clin Exp Med. 2015;8:17362–17376. [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Lin Z, Wei Q, et al. Expression of IL-23 and IL-17 and effect of IL-23 on IL-17 production in ankylosing spondylitis. Rheumatol Int. 2009;29:1343–1347. doi: 10.1007/s00296-009-0883-x. [DOI] [PubMed] [Google Scholar]
- 30.Ugur M, Baygutalp NK, Melikoglu MA, et al. Elevated serum interleukin-23 levels in ankylosing spondylitis patients and the relationship with disease activity. Nagoya J Med Sci. 2015;77:621–627. [PMC free article] [PubMed] [Google Scholar]
- 31.Bowness P, Ridley A, Shaw J, et al. Th17 cells expressing KIR3DL2+ and responsive to HLA-B27 homodimers are increased in ankylosing spondylitis. J Immunol. 2011;186:2672–2680. doi: 10.4049/jimmunol.1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *32.Jansen DT, Hameetman M, van Bergen J, et al. IL-17-producing CD4+ T cells are increased in early, active axial spondyloarthritis including patients without imaging abnormalities. Rheumatology (Oxford) 2015;54:728–735. doi: 10.1093/rheumatology/keu382. This study showed elevated frequencies of Th17 cells in the peripheral blood of nr-axSpA patients compared to healthy individuals. [DOI] [PubMed] [Google Scholar]
- 33.Appel H, Maier R, Wu P, et al. Analysis of IL-17(+) cells in facet joints of patients with spondyloarthritis suggests that the innate immune pathway might be of greater relevance than the Th17-mediated adaptive immune response. Arthritis Res Ther. 2011;13:R95. doi: 10.1186/ar3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bautista-Caro MB, Arroyo-Villa I, Castillo-Gallego C, et al. Decreased Th17 and Th1 cells in the peripheral blood of patients with early non-radiographic axial spondyloarthritis: a marker of disease activity in HLA-B27(+) patients. Rheumatology (Oxford) 2013;52:352–362. doi: 10.1093/rheumatology/kes267. [DOI] [PubMed] [Google Scholar]
- 35.van der Horst-Bruinsma IE, Zack DJ, Szumski A, Koenig AS. Female patients with ankylosing spondylitis: analysis of the impact of gender across treatment studies. Ann Rheum Dis. 2013;72:1221–1224. doi: 10.1136/annrheumdis-2012-202431. [DOI] [PubMed] [Google Scholar]
- **36.Gracey E, Yao Y, Green B, et al. Sexual Dimorphism in the Th17 Signature of Ankylosing Spondylitis. Arthritis Rheumatol. 2015 doi: 10.1002/art.39464. Findings from this study suggest that the contribution of Th17 cells and related signaling pathways to inflammation and disease pathogenesis differ in male and female AS patients. [DOI] [PubMed] [Google Scholar]
- 37.Noordenbos T, Yeremenko N, Gofita I, et al. Interleukin-17-positive mast cells contribute to synovial inflammation in spondylarthritis. Arthritis and rheumatism. 2012;64:99–109. doi: 10.1002/art.33396. [DOI] [PubMed] [Google Scholar]
- 38.Leijten EF, van Kempen TS, Boes M, et al. Brief report: enrichment of activated group 3 innate lymphoid cells in psoriatic arthritis synovial fluid. Arthritis Rheumatol. 2015;67:2673–2678. doi: 10.1002/art.39261. [DOI] [PubMed] [Google Scholar]
- **39.Ciccia F, Guggino G, Rizzo A, et al. Type 3 innate lymphoid cells producing IL-17 and IL-22 are expanded in the gut, in the peripheral blood, synovial fluid and bone marrow of patients with ankylosing spondylitis. Ann Rheum Dis. 2015;74:1739–1747. doi: 10.1136/annrheumdis-2014-206323. This study demonstrates those higher frequencies of IL-17 and IL-22 producing NKp44+ILC3 cells are present in the peripheral blood, gut, synovial fluid and bone marrow of AS patients when compared healthy controls. ILC3 cells appeared to be predominantly expanded in the inflamed gut of the AS patients indicating the existence of a gut-joint/spine axis in axSpA whereby ILC3 may be critical effectors. [DOI] [PubMed] [Google Scholar]
- 40.Stoll ML. Gut microbes, immunity, and spondyloarthritis. Clin Immunol. 2015;159:134–142. doi: 10.1016/j.clim.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 41.Costello ME, Elewaut D, Kenna TJ, Brown MA. Microbes, the gut and ankylosing spondylitis. Arthritis Res Ther. 2013;15:214. doi: 10.1186/ar4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DeLay ML, Turner MJ, Klenk EI, et al. HLA-B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis and rheumatism. 2009;60:2633–2643. doi: 10.1002/art.24763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin P, Bach M, Asquith M, et al. HLA-B27 and human beta2-microglobulin affect the gut microbiota of transgenic rats. PloS one. 2014;9:e105684. doi: 10.1371/journal.pone.0105684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **44.Costello ME, Ciccia F, Willner D, et al. Intestinal dysbiosis in ankylosing spondylitis. Arthritis Rheumatol. 2014 doi: 10.1002/art.38967. Using 16s ribosomal RNA gene sequencing, this study showed that microbial communities in the terminal ileum of AS patients differ significantly from healthy individuals. Intestinal dysbiosis in the gut microbiome of AS patients further supports a possible causal relationship between altered gut microbiota, ileocolonic inflammation and axSpA. [DOI] [PubMed] [Google Scholar]
- 45.Scher JU, Ubeda C, Artacho A, et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol. 2015;67:128–139. doi: 10.1002/art.38892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frank DN, St Amand AL, Feldman RA, et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sokol H, Leducq V, Aschard H, et al. Fungal microbiota dysbiosis in IBD. Gut. 2016 doi: 10.1136/gutjnl-2015-310746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang Y, Torchinsky MB, Gobert M, et al. Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature. 2014;510:152–156. doi: 10.1038/nature13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zielinski CE, Mele F, Aschenbrenner D, et al. Pathogen-induced human TH17 cells produce IFN-gamma or IL-10 and are regulated by IL-1beta. Nature. 2012;484:514–518. doi: 10.1038/nature10957. [DOI] [PubMed] [Google Scholar]
- 52.Van Praet L, Van den Bosch FE, Jacques P, et al. Microscopic gut inflammation in axial spondyloarthritis: a multiparametric predictive model. Ann Rheum Dis. 2013;72:414–417. doi: 10.1136/annrheumdis-2012-202135. [DOI] [PubMed] [Google Scholar]
- 53.Van Praet L, Jans L, Carron P, et al. Degree of bone marrow oedema in sacroiliac joints of patients with axial spondyloarthritis is linked to gut inflammation and male sex: results from the GIANT cohort. Ann Rheum Dis. 2014;73:1186–1189. doi: 10.1136/annrheumdis-2013-203854. [DOI] [PubMed] [Google Scholar]
- 54.Ciccia F, Bombardieri M, Principato A, et al. Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis and rheumatism. 2009;60:955–965. doi: 10.1002/art.24389. [DOI] [PubMed] [Google Scholar]
- *55.Wright PB, McEntegart A, McCarey D, et al. Ankylosing spondylitis patients display altered dendritic cell and T cell populations that implicate pathogenic roles for the IL-23 cytokine axis and intestinal inflammation. Rheumatology (Oxford) 2016;55:120–132. doi: 10.1093/rheumatology/kev245. Findings from this study suggest that CD14−CD16+ mononuclear cells may contribute to AS pathogenesis by promoting Th17 response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **56.Ward MM, Deodhar A, Akl EA, et al. American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network 2015 Recommendations for the Treatment of Ankylosing Spondylitis and Nonradiographic Axial Spondyloarthritis. Arthritis & rheumatology. 2016;68:282–298. doi: 10.1002/art.39298. Updated treatment recommendations for axSpA based on the GRADE system performed by experts in the field and methodologists. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haibel H, Rudwaleit M, Listing J, Sieper J. Open label trial of anakinra in active ankylosing spondylitis over 24 weeks. Ann Rheum Dis. 2005;64:296–298. doi: 10.1136/ard.2004.023176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sieper J, Braun J, Kay J, et al. Sarilumab for the treatment of ankylosing spondylitis: results of a Phase II, randomised, double-blind, placebo-controlled study (ALIGN) Ann Rheum Dis. 2015;74:1051–1057. doi: 10.1136/annrheumdis-2013-204963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pathan E, Abraham S, Van Rossen E, et al. Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in ankylosing spondylitis. Ann Rheum Dis. 2013;72:1475–1480. doi: 10.1136/annrheumdis-2012-201915. [DOI] [PubMed] [Google Scholar]
- 60.Langley RG, Lebwohl M, Krueger GG, et al. Long-term efficacy and safety of ustekinumab, with and without dosing adjustment, in patients with moderate-to-severe psoriasis: results from the PHOENIX 2 study through 5 years of follow-up. Br J Dermatol. 2015;172:1371–1383. doi: 10.1111/bjd.13469. [DOI] [PubMed] [Google Scholar]
- 61.Weitz JE, Ritchlin CT. Ustekinumab : targeting the IL-17 pathway to improve outcomes in psoriatic arthritis. Expert Opin Biol Ther. 2014;14:515–526. doi: 10.1517/14712598.2014.890587. [DOI] [PubMed] [Google Scholar]
- *62.Poddubnyy D, Hermann KG, Callhoff J, et al. Ustekinumab for the treatment of patients with active ankylosing spondylitis: results of a 28-week, prospective, open-label, proof-of-concept study (TOPAS) Ann Rheum Dis. 2014;73:817–823. doi: 10.1136/annrheumdis-2013-204248. Presented results from the proof-of-concept trial demonstrated the efficacy and safety of ustekinumab in patients with active AS. [DOI] [PubMed] [Google Scholar]
- 63.Baeten D, Baraliakos X, Braun J, et al. Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double-blind, placebo-controlled trial. Lancet. 2013;382:1705–1713. doi: 10.1016/S0140-6736(13)61134-4. [DOI] [PubMed] [Google Scholar]
- **64.Baeten D, Sieper J, Braun J, et al. Secukinumab, an Interleukin-17A Inhibitor, in Ankylosing Spondylitis. The New England journal of medicine. 2015;373:2534–2548. doi: 10.1056/NEJMoa1505066. Presented results from two phase III clinical trials demonstrating that secukinumab, an anti-IL-17 antibody is safe and effective for treatment of AS. [DOI] [PubMed] [Google Scholar]
- 65.Baraliakos X, Borah B, Braun J, et al. Long-term effects of secukinumab on MRI findings in relation to clinical efficacy in subjects with active ankylosing spondylitis: an observational study. Ann Rheum Dis. 2015 doi: 10.1136/annrheumdis-2015-207544. [DOI] [PubMed] [Google Scholar]
- 66.Lebwohl M, Strober B, Menter A, et al. Phase 3 Studies Comparing Brodalumab with Ustekinumab in Psoriasis. The New England journal of medicine. 2015;373:1318–1328. doi: 10.1056/NEJMoa1503824. [DOI] [PubMed] [Google Scholar]
- 67.Silacci M, Lembke W, Woods R, et al. Discovery and characterization of COVA322, a clinical-stage bispecific TNF/IL-17A inhibitor for the treatment of inflammatory diseases. MAbs. 2016;8:141–149. doi: 10.1080/19420862.2015.1093266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hammitzsch A, Tallant C, Fedorov O, et al. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc Natl Acad Sci U S A. 2015;112:10768–10773. doi: 10.1073/pnas.1501956112. [DOI] [PMC free article] [PubMed] [Google Scholar]