

Abstract
Objectives
N-myc downstream-regulated gene-1 (NDRG1) is a hypoxia-inducible and differentiation-related protein and candidate biomarker in pancreatic cancer. As NDRG1 expression is lost in high-grade tumors, the effects of the differentiating histone deacetylase inhibitor trichostatin A (TSA) were examined in human pancreatic cancer cell lines representing different tumor grades.
Methods
PANC-1 (poorly differentiated) and Capan-1 (moderately to well-differentiated) cells were treated with TSA. Effects were assessed in vitro by microscopic analysis, colorimetric assays, cell counts, real-time polymerase chain reaction, and Western blotting.
Results
Treatment of PANC-1 cells over 4 days with 0.5 μM TSA restored cellular differentiation, inhibited proliferation, and enhanced p21Cip1 protein expression. Trichostatin A upregulated NDRG1 mRNA and protein levels under normoxia from day 1 and by 6-fold by day 4 (P < 0.01 at all time points). After 24 hours under hypoxia, NDRG1 expression was further increased in differentiated cells (P < 0.01). Favorable changes were identified in the expression of other hypoxia-regulated genes.
Conclusions
Histone deacetylase inhibitors offer a potential novel epidrug approach for pancreatic cancer by reversing the undifferentiated phenotype and allowing patients to overcome resistance and better respond to conventional cytotoxic treatments.
Key Words: N-myc downstream-regulated gene-1, pancreatic cancer, hypoxia, cellular differentiation, epigenetic regulation
Pancreatic ductal adenocarcinoma, subsequently referred to simply as “pancreatic cancer,” is the most common subtype of human pancreatic cancer. Pancreatic cancer affects both men and women and is highly aggressive, with a 5-year survival rate of only about 5%. Pancreatic cancer is not usually diagnosed until it reaches an advanced stage and becomes symptomatic, and only 15% to 20% of all patients are candidates for surgical resection.1,2 Even the most advanced chemotherapeutic regimens are ineffective,3,4 and survival from pancreatic cancer has not substantially improved over the past 40 years. In fact, death rates from pancreatic cancer have increased, and it is the only cancer for which deaths are predicted to continue to increase.5
Among patients eligible for surgical resection, important independent tumor-specific prognostic factors include tumor size and degree of differentiation.6 Pancreatic tumors are generally hypoxic due to their avascular morphology. Hypoxia can contribute significantly to their aggressive behavior through hypoxia-induced expression of proangiogenic factors, such as vascular endothelial growth factor (VEGF) and the inflammatory cytokine interleukin-8.7,8 Additionally, pancreatic cancers express high levels of the hypoxia-inducible transcription factor hypoxia-inducible factor 1 (HIF-1),2 whose target genes encourage an aggressive phenotype by promoting tumor growth, invasion, and metastasis and favor dedifferentiation.9
N-myc downstream-regulated gene 1 (NDRG1) is a protein induced by cellular stress, not least hypoxia, through HIF1-dependent and -independent mechanisms and by cellular differentiation. N-myc downstream-regulated gene 1 has been proposed as a tumor biomarker due to its high expression in malignant tissues but not normal tissues of the same origin. In pancreatic cancer, NDRG1 expression is related to the differentiation state of the tumor, and NDRG1 has been hypothesized a novel indicator of pancreatic malignancy as hypoxia is a general feature of these tumors.2 In particular, the mechanism by which undifferentiated cancer cells lose this response warrants further investigation.
Cancer is a genetic disease characterized by inherited or sporadic mutations in tissue homeostasis, cell cycle control, and apoptosis genes. It is also an epigenetic disease,10,11 as evidenced by the presence of genetic alterations in chromatin-remodeling enzymes or their aberrant activation or inactivation that deregulates the epigenetic landscape.12–15 Given that cellular differentiation is also regulated by epigenetic mechanisms, altered pancreatic cancer differentiation may result from epigenetic aberrations.
Epidemiological studies have shown that the incidence of all cancers is increasing, in part due to chronic exposure to environmental factors or exposure in utero. Animal studies have shown that tissue morphology may be altered due to epigenetic changes affecting gene expression in various organs, such as in the mammary glands of postnatal and adult rats. These gene expression patterns occur throughout life,16–19 and cancer may consequently develop at puberty or adulthood. In this context of “environmental epigenetics,” the correlation between diet, epigenetics, and pancreatic cancer has been reviewed.20 There is significant evidence that certain dietary factors are associated with pancreatic cancer, implying a role for epigenetic gene regulation. Several lifestyle factors have also been identified that might affect human health via epigenetic mechanisms.21
Recent studies have shown the potential of epigenetics-based therapies (epidrugs) in pancreatic cancer, and there are encouraging results with the combination of the histone deacetylase (HDAC) inhibitor suberanilohydroxamic acid (vorinostat) with chemotherapeutics and/or radiation.22–24 Trichostatin A (TSA), a hydroxamic acid, is now mainly used as a reference compound in HDAC inhibitor and cancer research.25 This study shows, for the first time, the restoration of cellular differentiation coupled to cell growth inhibition in vitro by histone or nonhistone protein acetylation, which increased NDRG1 expression and restored responsiveness to hypoxia. N-myc downstream-regulated gene 1 can be considered a marker of restored pancreatic cancer differentiation. Further, the poorly differentiated PANC-1 cell line treated with TSA represents a good model of cellular differentiation, in particular for the investigation of the relationship between the cell cycle, differentiation, and epigenetic mechanisms.
MATERIALS AND METHODS
Cell Lines and Culture Conditions
The human pancreatic cancer cell lines Capan-1 (moderately to well-differentiated adenocarcinoma) and PANC-1 (poorly differentiated adenocarcinoma) were purchased from the American Type Culture Collection (LGC Promochem, Molsheim, France). Cells were cultured in Dulbecco Modified Eagle’s Medium supplemented with 20% heat- inactivated fetal bovine serum for Capan-1 and 10% heat-inactivated fetal bovine serum for PANC-1. Both media were supplemented with 100 units/mL penicillin and 1% streptomycin (Life Technologies, Thermo Fisher Scientific, Paisley, Scotland) and incubated at 37°C in a humidified atmosphere with 5% CO2.
Chemical Treatment and Exposure to Hypoxia
PANC-1 or Capan-1 cells were treated with TSA, a class I and II HDAC inhibitor (Sigma-Aldrich, Buchs, Switzerland) and solubilized in dimethylsulfoxide (DMSO) as a 10-mM stock solution. Controls were either untreated or treated (vehicle) with equal DMSO concentrations. PANC-1 or Capan-1 cells were placed under hypoxia (1.5% O2, 5% CO2) for 24 hours before harvest (hypoxic chamber, Ruskinn Biotrace International, Bridgend, United Kingdom). All drugs were stored at −80°C. Drugs were prepared to obtain final dilutions for cellular assays immediately before use.
Acid Extraction of Histones
Cells were suspended in a buffer containing 10-mM hydroxyethyl piperazineethanesulfonic acid pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol, 1.5 mM phenylmethylsulfonyl fluoride, and 0.2 M HCl and lysed on ice for 30 minutes. Dialysis was performed using a Slide-A-Lyzer cassette (Pierce, Thermo Fisher Scientific AG, Reinach, Switzerland) as instructed by the manufacturer. Proteins were quantified using the Bio-Rad Protein Assay (Bio-Rad, Reinach, Switzerland), and equal amounts (30 μg) were separated by 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) for western blotting analysis. Primary antibodies were purchased from Cell Signaling (Allschwil, Switzerland): 1:1000 rabbit antihistone 2B (H2B; 8135), rabbit antihistone 4 (H4; 2592), rabbit antiacetylated H2B (lys5; 2574), and rabbit antiacetylated H4 (lys8; 2594). The antirabbit (1:3000) horseradish peroxidase-labeled secondary antibody was from Dako (Baar, Switzerland).
Alkaline Phosphatase Assay
Viable cells were adjusted to 5 × 104 in each well. Cell suspensions were lysed by adding 1% (vv−1) Triton X-100 in 1× lysis buffer. Total protein concentrations were determined by the Bio-Rad Protein Assay. The absorbance of the product (p-nitrophenylphosphate) was measured at 405 nm using a spectrophotometric plate reader. After incubating the plate at 37°C, enzyme activity was measured at an end-point of 30 minutes and expressed as alkaline phosphatase (ALP)/total protein (ng/mL). A standard curve was used as a reference. The ALP activity in treated cells was compared with that in the control.
Cell Proliferation Assay
The effect of TSA on pancreatic cancer cell line PANC-1 viability was quantified using the methylthiazolyldiphenyl-tetrazolium bromide assay. Absorption was determined at 570 nm. The DMSO solution was used as a blank reference. Sets of six wells were used for each drug concentration along with controls treated with the same final concentration of DMSO. The mean absorbance (A) from 6 wells was calculated. Cell proliferation was calculated as a percentage: (Aexperiment/Acontrol) × 100%.
HDAC Assay
Histone deacetylase activity was measured using the colorimetric HDAC activity assay from BioVision (Lucerne, Switzerland) according to the manufacturer’s instructions. One hundred twenty micrograms of total cell lysate were diluted in 85 μL of ddH2O. Subsequently, the reaction was stopped by adding 10 μL of lysine developer and left for additional 30 minutes at 37°C. Samples were then read using a spectrophotometric plate reader at 405 nm.
RNA Extraction and Real-Time PCR
Total RNA was isolated from PANC-1 or Capan-1 cells untreated or treated with TSA. RNA was extracted with TRIzol (Life Technologies) according to the manufacturer’s instructions. One microgram of total RNA was DNase treated (Promega, Madison, Wis.) and reverse transcribed into cDNA using a commercial kit (Qiagen, Hilden, Germany). FAM dye-labeled Taqman MGB probes and unlabeled PCR primers for human NDRG1, VEGF, LOX (lysyl oxidase), GLUT1 (glucose transporter 1), and CAIX (carbonic anhydrase 9) were purchased from Applied Biosystems (Warrington, UK). Internal ribosomal RNA 18S was used as an internal positive control with a VIC dye-labeled Taqman MGB probe (Applied Biosystems). Real-time PCR was performed using the ABI PRISM 7000 Sequence Detector System (Applied Biosystems). cDNA was amplified for 40 cycles with denaturation at 95°C for 15 seconds and a combined annealing-extension step at 60°C for 1 minute. Mean cycle threshold (Ct) values were calculated for 18S and the reporter gene. Ct values for NDRG1, VEGF, LOX, GLUT1, and CAIX were normalized against the 18S control probe to calculate ∆Ct values. ∆∆Ct values were calculated by subtracting the ∆Ct value of control cells under normoxia from the ∆Ct values of the treated (differentiated) cells under normoxia or control or treated cells under hypoxia. Fold increases were calculated using the formula 2-(∆∆Ct).
Protein Extraction and Western Blotting
Cells were lysed in a buffer (10 mM Tris pH 8.0, 1 mM EDTA pH 8.0, 150 mM NaCl, and 0.5% NP-40 with the inhibitors 1 mM phenylmethylsulfonyl fluoride, 1× protease inhibitor cocktail (P-8340; Sigma-Aldrich), 1 mM NaF, and 10 mM Na3VO4), and cell extracts were cleared by centrifugation. Total protein concentrations were determined using the Bio-Rad Protein Assay, and equal amounts (20 μg) were separated by 10% SDS-PAGE. Proteins were transferred to nitrocellulose membranes using a semidry transfer system (Bio-Rad), blocked with 5% nonfat dry milk in 50 mM Tris (pH 7.5), 150 mM NaCl, and 0.1% Tween-20 for 1 hour, and incubated overnight at 4°C with primary antibody: sheep anti-NDRG1 1:6000, AB-160, Kinasource (Dundee, Scotland); sheep anti-phospho-NDRG1 (Thr 346, Thr 356, Thr 366) 1:3000, Kinasource PB-025; mouse anti-p21Cip1 1:2000, Cell Signaling; rabbit anti-CAII (carbonic anhydrase 2) 1:5000, Santa Cruz Biotechnology (Heidelberg, Germany); rabbit anti-β-actin 1:1000, Sigma A5060; or mouse anti-α-tubulin 1:2000, Santa Cruz Biotechnology. Membranes were then incubated with secondary antibody: antisheep (1:3000, Dako); antimouse (1:5000; Pierce); or antirabbit (1:3000; Dako) horseradish peroxidase-labeled secondary antibodies for 1 hour at room temperature. Protein bands were detected using a chemiluminescent detection system.
Statistics
To compare real-time PCR data, ∆Ct values were analyzed by one-way analysis of variance (ANOVA) followed by Dunnett multiple comparisons test using SPSS (version 11.5) (SPSS (Switzerland) Limited, Zürich, Switzerland). A P value less than 0.05 was considered significant.
RESULTS
PANC-1 and Capan-1 Cell Lines Are Characterized by Distinct Hypoxia-Regulated Gene Expression Patterns
PANC-1 and Capan-1 cell lines were derived from human pancreatic adenocarcinomas of different tumor grades. Capan-1 cells were derived from well-differentiated tumors, whereas PANC-1 were derived from poorly-differentiated tumors. Capan-1 cells were previously shown to upregulate NDRG1 under hypoxia, while there was no NDRG1 induction in PANC-1 cells despite strong detection of the HIF-1α subunit in both tumor cell lines cultured under hypoxic conditions.2
To investigate whether the lack of NDRG1 expression in the poorly differentiated PANC-1 cells in response to hypoxia was gene- or cell line-specific, other hypoxia-inducible genes were examined. There was a significant increase in VEGF and LOX mRNA expression in untreated PANC-1 cells under hypoxic conditions (7- and 12-fold induction, respectively; P < 0.05 versus Capan-1 cell group), whereas there was a 30-fold upregulation of NDRG1 mRNA expression in the well-differentiated Capan-1 cells under the same culture conditions (P < 0.001 versus PANC-1 cell group) (Fig. 1). These findings suggested that some hypoxia-induced genes in PANC-1 cells are functional and the lack of NDRG1 upregulation was not due to a cell line dysfunction.
FIGURE 1.
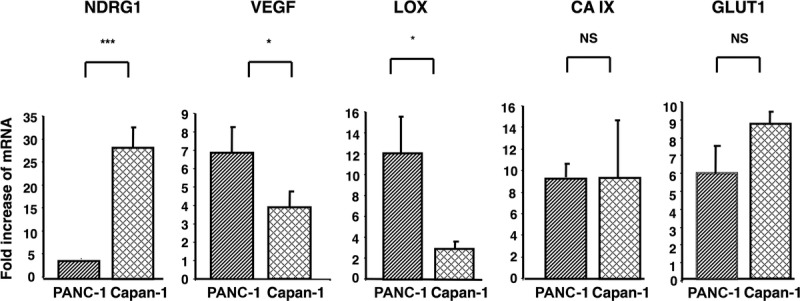
The differentiation status of the pancreatic cancer cell lines PANC-1 (poorly differentiated) and Capan-1 (well-differentiated) in response to hypoxia. The expression of several hypoxia-inducible genes was evaluated by real-time PCR. The 2 cell lines were cultured without treatment and exposed to hypoxia (1.5% O2) for 24 hours. Columns: means of values of three independent experiments; bars, standard deviation (SD). *P < 0.05, ***P < 0.001, NS, not significant.
The HDAC Inhibitor TSA Restores Pancreatic Cancer Cell Differentiation and Upregulates NDRG1
Histone deacetylase activity was measured in PANC-1 cells incubated with TSA using a colorimetric assay. Treated cells displayed 53% of the HDAC activity compared to control. In control cells incubated with vehicle alone (DMSO), HDAC activity was also reduced but only to 78.5% (Fig. 2A). Incubation of PANC-1 cells with TSA for 12 hours led to the strong upregulation of H2B (lysine 5) and H4 (lysine 8) acetylation compared to control cells treated with vehicle alone (Fig. 2B). Morphological changes were observed in PANC-1 cells as soon as 1 day after exposure to 0.5 μM TSA; after 2 days of treatment, morphological changes were even more evident and characterized by cell elongation and filamentous protrusions that were maintained up to 4 days (Fig. 3A). CA II protein was detectable by western blotting after 1 day of TSA treatment, and its level increased over the course of the experiment to reach a maximum at day 4 (Fig. 3A). Likewise, another marker of differentiation, ALP, showed increased activity over time (Fig. 3B). PANC-1 cell growth was inhibited by TSA after 1 day and increased up to 4 days, at which time it was 55% of control cells incubated with vehicle alone (Fig. 4A). p21Cip1 protein expression was induced after 1 day in PANC-1 cells treated with TSA, and its expression level remained stable up to 4 days (Fig. 4B). Taken together, these data show that TSA induces the differentiation of PANC-1 cells by altering the acetylation state of histones and is coupled with cell growth arrest.
FIGURE 2.
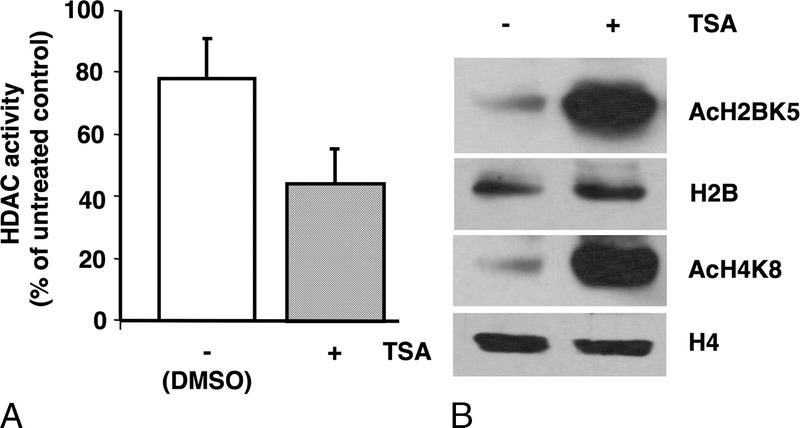
Impact of TSA on HDAC activity and histone acetylation. A, TSA diminishes HDAC activity after 8 hours. Columns, means of triplicate values of two independent experiments; bars, SD. B, TSA increases histone acetylation after 12 hours. Proteins were quantitated and fractionated by SDS-PAGE, transferred to nitrocellulose membranes, and probed with rabbit antibodies against total H4 (11 kDa) and H2B (14 kDa) and acetylated histones AcH4 (11 kDa) and AcH2B (14 kDa).
FIGURE 3.
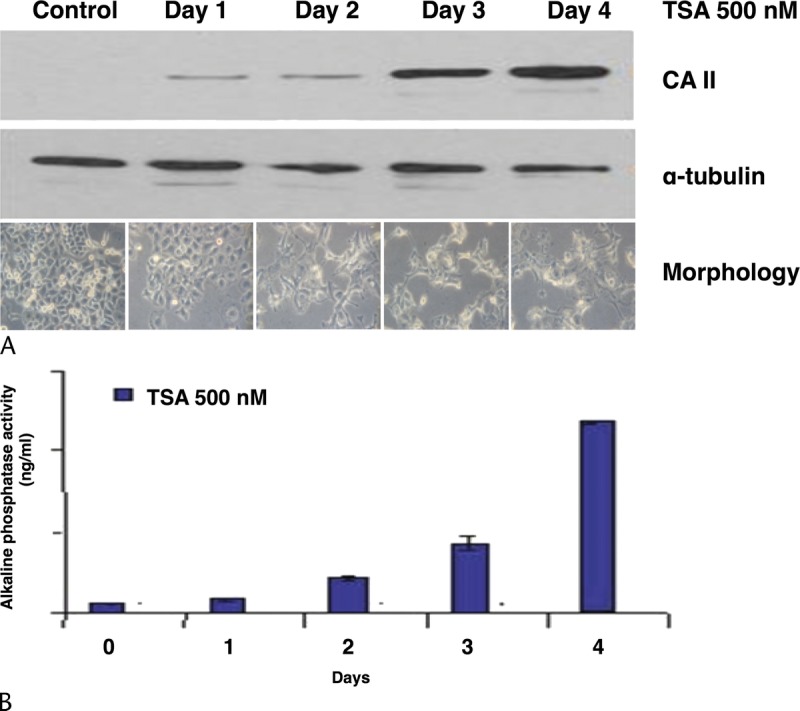
Impact of TSA on morphology and differentiation. A, Temporal expression of carbonic anhydrase 2 (CA II, 30 kDa). α-tubulin (50 kDa) was used as a loading control. Below blots are photomicrographs showing changes in morphology over time. Original magnification, ×200. B, Measurement of ALP activity over time. The results shown are the average of 3 measurements.
FIGURE 4.
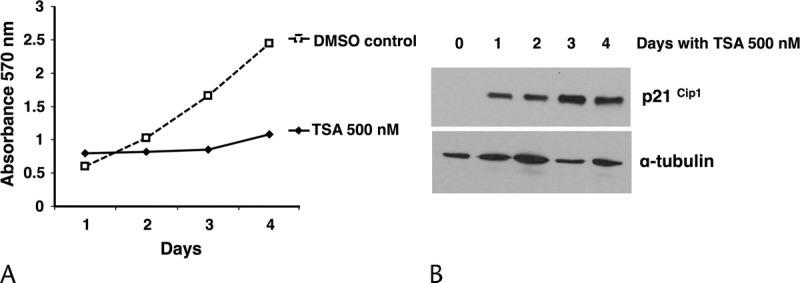
Impact of TSA on cell growth and the cell cycle regulator p21Cip1. A, Cell proliferation was evaluated by the MTT assay. Cell viability is expressed as the ratio of the number of viable cells with TSA treatment compared to that without treatment. Cells growing in DMSO were also analyzed as vehicle control. Means (SD) of six replicates are plotted. B, Temporal expression of p21Cip1 (21 kDa); α-tubulin was used as a loading control. The results shown are from 1 representative experiment out of 3 independently performed experiments. MTT, methylthiazolyldiphenyl-tetrazolium bromide.
The TSA-differentiated PANC-1 cells showed a 3- to 6-fold increase in NDRG1 gene expression relative to control over 4 days (P < 0.01) (Fig. 5A). Consistent with the mRNA analysis, NDRG1 protein expression increased after 1 day of TSA treatment and its level was maintained throughout the 4-day experiment. Interestingly, phosphorylated NDRG1 (Thr346,356,366) was induced at day 3 and increased further at day 4, by which time the expressed form of the NDRG1 protein was fully shifted towards phosphorylation (Fig. 5B). The NDRG1 gene appears to be regulated through epigenetic modifications.
FIGURE 5.
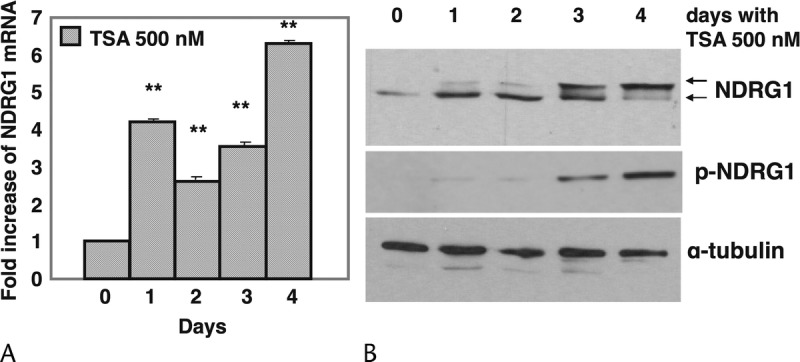
Regulation of NDRG1 expression by histone acetylation. PANC-1 cells were treated with TSA and monitored for 4 days in normal culture conditions (21% O2). A, RNA was isolated and real-time PCR performed. Ct values for NDRG1 were normalized against the internal ribosomal RNA (18S) control probe to calculate ΔCt values. Columns: means of three values; bars, SD. The results shown are from one representative experiment out of three independently performed experiments. **P < 0.01 compared to control. B, Expression of NDRG1 protein by western blotting. The same membrane used for CA II protein expression (Fig. 3A) was probed with a sheep anti-phospho-NDRG1 (>43 kDa) antibody. To determine total NDRG1, the membrane was stripped and reprobed with a sheep anti-NDRG1 (43 kDa) antibody.
PANC-1 Cells Differentiated by TSA Regain Hypoxic Responsiveness
To confirm that NDRG1 expression is dependent on the cellular differentiation state, poorly- differentiated PANC-1 cells were induced to differentiate by TSA and their ability to increase NDRG1 expression under hypoxia tested. NDRG1 gene expression was upregulated 4.8-fold in TSA-differentiated PANC-1 cells cultured at 21% O2 (normoxic conditions) compared with controls. After 24 hours under hypoxia, NDRG1 was increased 17-fold in differentiated PANC-1 cells (P < 0.01 versus undifferentiated controls) (Fig. 6A). There was a corresponding increase in NDRG1 protein expression in differentiated PANC-1 cells under normoxic conditions compared to control cells and the induced form of NDRG1 was mostly phosphorylated. Protein expression of NDRG1 and phospho-NDRG1 increased steadily in differentiated cells after 24 hours under hypoxia compared to differentiated cells in normoxia. CA II protein expression confirmed that the cells were differentiated (Fig. 6B). To provide additional evidence that NDRG1 upregulation by hypoxia was dependent on cellular differentiation, a further series of experiments were performed. PANC-1 cells were treated with TSA for 3 days and then divided into 2 groups: either continuous treatment with TSA or removal of the agent from the medium for a further 3 days. Cells cultured in medium without TSA for the final 3 days started to regrow, lost their differentiated phenotype, and returned to a control-like morphology (Fig. 6C). Consistent with these morphological changes, the expression of the differentiation marker CA II increased in the presence of TSA and substantially decreased when TSA was removed, confirming that, in the absence of TSA, cells reverted to their original undifferentiated phenotype. As expected, differentiated PANC-1 cells had higher NDRG1 protein levels, which were further increased by hypoxia along with induction of its phosphorylated form. In the absence of TSA, NDRG1 expression was substantially decreased, reverting back to control levels and losing its phosphorylated form (Fig. 6D). The upregulation of NDRG1 and its restored responsiveness to hypoxia appear to be related to the cellular differentiation state, whose restoration was mediated by TSA.
FIGURE 6.
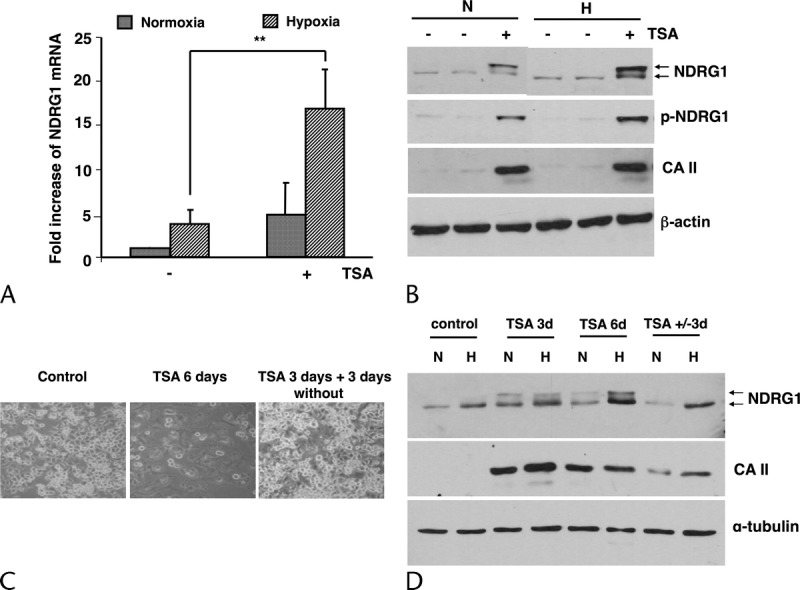
TSA-mediated differentiation of PANC-1 cells is required for the expression of NDRG1 under hypoxia. A, Expression of NDRG1 mRNA levels was determined by real-time PCR as described in the Materials and Methods. Cells were treated with TSA for 3 days under normoxia including 24 hours under hypoxia (1.5% O2). Columns: means of three values of three separate experiments; bars, SD. **P < 0.01. B, Expression of phospho-NDRG1, total NDRG1, and CA II proteins in cells monitored for 4 days. Total proteins were transferred to nitrocellulose membranes and probed with a sheep anti-phospho-NDRG1 antibody. The membrane was stripped and reprobed with a sheep anti-NDRG1 antibody and with a rabbit anti-CA II antibody. β-actin (42 kDa) was used to compare loading. The result shown is representative of three independently performed experiments. C, Photomicrograph showing the cell phenotype in each condition. D, Expression of CA II and NDRG1 proteins. α-tubulin was used as a loading control.
TSA-Differentiated PANC-1 Cells Display Changes in Hypoxia-Regulated Gene Expression Associated With a Lower Tumor Grade
The next question asked was whether the cellular differentiation state determines the induction of other established hypoxia-dependent genes. After 3 days of treatment with 0.5 μM TSA, including 24 hours under hypoxia, the master hypoxia regulator HIF-1α was stabilized in hypoxic PANC-1 cells (data not shown). However, while HIF-1α protein was expressed, VEGF mRNA expression was markedly reduced to 1.7-fold compared to 4.5-fold for controls (60% inhibition). This decrease in VEGF expression in differentiated PANC-1 cells prompted the investigation of whether this was a gene-specific or cell line-specific phenomenon.
The expression of other hypoxia-inducible genes was assessed. PANC-1 and Capan-1 cells were treated with TSA at 0.5 μM for 3 days and exposed to hypoxia for the last 24 hours of treatment. In well-differentiated Capan-1 cells, there was no change in cell morphology on expression of the evaluated genes (Fig. 7, upper row).
FIGURE 7.
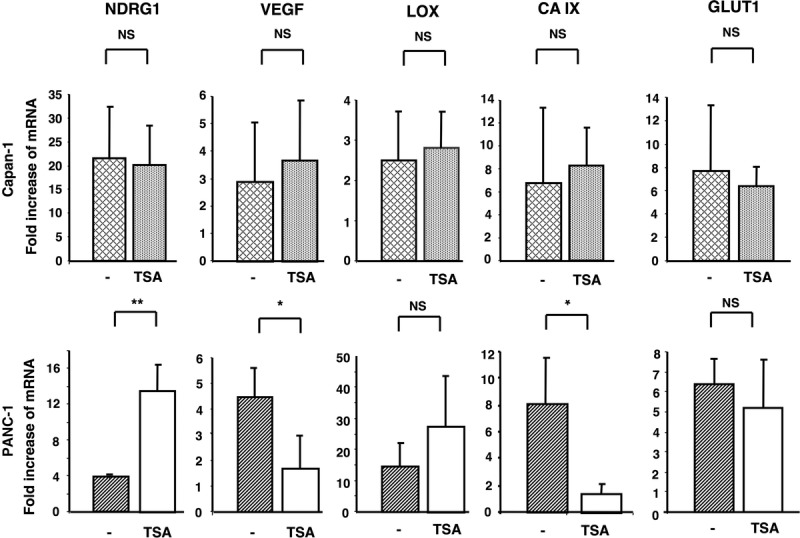
Impact of TSA-induced differentiation on Capan-1 (well-differentiated) cells (upper row) and PANC-1 (poorly-differentiated) cells (lower row) in response to hypoxia. The 2 cell lines were untreated or treated for 3 days with TSA and exposed to hypoxia (1.5% O2) for the last 24 hours of treatment. Ct values for each hypoxia-inducible gene were normalized against the internal ribosomal RNA (18S) control probe to calculate ΔCt values and compared with untreated cells cultured under normoxic conditions. Columns: means of three values of three separate experiments; bars, SD. *P < 0.05, **P < 0.01.
Genes that have been epigenetically silenced in cancer cells are preferentially activated by such epigenetic agents.26 After TSA treatment, many Capan-1 cells were noted to float in the medium (having lost adherence), which is an indication of cell death (data not shown). Therefore, TSA-treated Capan-1 cells, which are moderately to well-differentiated, may undergo cell death rather than differentiation in response to TSA treatment. In contrast, there was a significant decrease in CAIX and VEGF mRNA expression in TSA-differentiated PANC-1 cells (8 in control versus 1.5 in TSA-treated cells, P < 0.05; 4.5 in control versus 1.7 in TSA-treated cells, P < 0.05, respectively), whereas NDRG1 mRNA was significantly upregulated (4 in control versus 15 in TSA-treated cells) (Fig. 7, lower row).
DISCUSSION
Pancreatic cancer cell differentiation and tumor grade have previously been shown to determine the ability of cancer cells to express NDRG1 in response to hypoxia.2 Similar observations have been made in colorectal and prostatic cancers, suggesting that NDRG1 may be a potential biomarker of the differentiation status of pancreatic cancer cells and tumor aggression.
Here, it was confirmed that the differentiation state predicts the capability to upregulate NDRG1 and respond to hypoxia. TSA, a potent and reversible class I and II HDAC inhibitor,25 was applied to poorly differentiated PANC-1 cells and the morphological and gene expression changes assessed. Further, removal of TSA from the medium reverted PANC-1 cells to an undifferentiated phenotype and caused them to lose NDRG1 protein expression. Transfection of PANC-1 cells with NDRG1 did not result in cellular differentiation (data not shown). Trichostatin A–induced histone acetylation in PANC-1 cells and differentiation was confirmed by increased expression of pancreatic differentiation markers. Further, NDRG1 expression was restored in TSA-differentiated pancreatic cancer cells, implying epigenetic regulatory mechanisms. Concomitantly, there was cell growth arrest associated with induced p21Cip1 expression.
It has previously been shown that HDAC inhibitor-mediated cell growth arrest is correlated with transcriptional activation of the cell cycle inhibitor p21Cip1.27,28 Increased expression of p21Cip1 is associated with accumulation of acetylated histones around the p21Cip1 locus, suggesting that its reexpression here was probably due to the epigenetic regulation of the p21Cip1 promoter.29 This elevated expression was independent of p53, because TP53 is mutated in PANC-1 cells.30 Even though the exact regulation of NDRG1 expression is still unknown, various mechanisms have been suggested. Inhibition of DNA methylation and histone deacetylation enhances NDRG1 expression in other pancreatic cancer cell lines under normoxic conditions. We found that NDRG1 mRNA and protein expression were not affected by treatment with the methyltransferase inhibitor 5-aza-2′deoxycytidine alone or in combination with TSA (not shown). Angst and colleagues31 proposed an indirect mechanism for HDAC-mediated repression of the NDRG1 promoter via protein stabilization. In contrast, here TSA treatment enhanced both message levels and protein expression, suggesting that TSA may directly affect histone acetylation of NDRG1. However, the possibility that NDRG1 reactivation in PANC-1 cells by pharmacologic reversal of histone/nonhistone protein deacetylation via an indirect mechanism cannot be excluded. Besides histone proteins, HDAC also deacetylates nonhistone proteins, which affects NDRG1 transcription. Therefore, modification of acetylation may act on other genes controlled by the epigenetic machinery and involved in NDRG1 regulation. Reactivation of NDRG1 may also be a consequence of a differentiated phenotype in PANC-1 cells resulting from global gene expression changes. Further work is necessary to determine whether NDRG1 gene reactivation occurs as a consequence of a direct or indirect mechanism involving either promoter acetylation or a regulated transcription factor that influences NDRG1 expression.
Examination of the temporal expression of total and phosphorylated NDRG1 showed that its phosphorylated form was induced on day 3 and that phosphorylation increased thereafter. This suggests a potential relationship between cell differentiation status and NDRG1 phosphorylation. Although NDRG1 is known to be phosphorylated by several protein kinases such as serum and glucocorticoid-regulated kinase 1 (SGK1), glycogen synthase kinase 3 beta (GSK3β),32 protein kinase A (PKA), or calcium/calmodulin-dependent protein kinase II (CaMK-II), the functional relevance of phosphorylation has remained elusive. However, NDRG1 phosphorylation by protein kinases PKA and CaMK-II may be directly linked to the degranulation-enhancing capacity of NDRG1 in mast cells.33 NDRG1 protein expression is also temporally and spatially regulated during the cell cycle,34 suggesting a functional role for phosphorylated NDRG1 in the control of microtubule organization and cell abscission during mitosis.
Cellular phenotype is thought to influence the expression of genes regulated by hypoxia. Consistent with this, the present data showed that untreated poorly differentiated PANC-1 cells upregulated VEGF and LOX transcription under hypoxia to a greater extent than well-differentiated Capan-1 cells. LOX codes for the extracellular matrix protein lysyl oxidase and is involved in metastasis,35 as well as facilitating migration and cell-matrix adhesion formation in invasive breast cancer cells. The current results further support this observation, as PANC-1 cells differentiated by TSA demonstrated an inverse profile of genes dependent on hypoxia. CAIX expression was reduced, consistent with reports that the absence of CAIX protein expression detected by immunohistochemistry correlates with a low histological grade.36 Similarly, VEGF mRNA levels were downregulated by hypoxia compared to untreated controls. These findings support the idea that differentiated PANC-1 cells display an expression profile of hypoxia-regulated genes similar to that of a well-differentiated cell line.
The relationship between differentiation and cell division needs further investigation. These findings show that TSA-induced cell growth arrest was coupled with p21Cip1 expression and restoration of epithelial differentiation. Cancer cells are characterized by high proliferative activity and cellular de-differentiation; in fact, both events are linked. Cancer cells are often compared to induced pluripotent stem cells and embryonic stem cells because they share some similarities including a faster cell cycle with a short G1 phase and self-renewal capacity, while differentiated somatic cells have a long cell cycle with a longer G1 phase than induced pluripotent stem cells.37 The maintenance of the undifferentiated state is permitted by rapid proliferation, which serves to inhibit differentiation.38 A reduction in the overall amount of cyclin-dependent kinase (CDK) activity during G1 is associated with differentiation. Cyclin-dependent kinase inhibitors have consistently been shown to accumulate in many differentiated cell types in mice39 and can be induced by triggering differentiation and cell cycle exit. The CDKI p21Cip1 is 1 member of the Cip/Kip family, and it negatively regulates the kinases that phosphorylate the retinoblastoma protein (Rb) by binding to cyclin/CDK complexes, inhibiting CDK activity and arresting cells in G1.40 Histone deacetylases are directly implicated at the interface of the cell cycle and differentiation, and exact determination of their effects, in particular on master regulators or these processes, would be novel. Epigenetic alterations in one or several factors may occur and prevent normal regulation and interaction between the different molecular participants.
In conclusion, differentiation of pancreatic cancer cells by the HDAC inhibitor TSA reversed the poorly differentiated phenotype and cell behavior associated with changes in hypoxia-regulated gene expression. This epidrug strategy may allow cancer cells to overcome drug resistance and better respond to conventional treatments. Restoration of NDRG1 expression may represent a biomarker of malignant pancreatic tumors undergoing redifferentiation and redirecting toward a lower tumor grade. The use of the human ductal PANC-1 cell line treated with TSA represents a useful tool to study cellular differentiation through epigenetic mechanisms.
ACKNOWLEDGMENT
The author wishes to thank Professor G. Packham from the Cancer Sciences Division of the University of Southampton, United Kingdom, for his critical review of this manuscript, and gratefully acknowledges editorial assistance from Nextgenediting (www.nextgenediting.com).
Footnotes
The author declares no conflict of interest.
This work was supported by the Werner and Hedy Berger-Janser Foundation for Cancer Research, No. 1/2004.
REFERENCES
- 1.Warren KW, Christophi C, Armendariz R, et al. Current trends in the diagnosis and treatment of carcinoma of the pancreas. Am J Surg. 1983;145:813–818. [DOI] [PubMed] [Google Scholar]
- 2.Angst E, Sibold S, Tiffon C, et al. Cellular differentiation determines the expression of the hypoxia-inducible protein NDRG1 in pancreatic cancer. Br J Cancer. 2006;95:307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:1039–1049. [DOI] [PubMed] [Google Scholar]
- 5.Malvezzi M, Bertuccio P, Levi F, et al. European cancer mortality predictions for the year 2014. Ann Oncol. 2014;25:1650–1656. [DOI] [PubMed] [Google Scholar]
- 6.Sohn TA, Yeo CJ. The molecular genetics of pancreatic ductal carcinoma: a review. Surg Oncol. 2000;9:95–101. [DOI] [PubMed] [Google Scholar]
- 7.Shi Q, Abbruzzese JL, Huang S, et al. Constitutive and inducible interleukin 8 expression by hypoxia and acidosis renders human pancreatic cancer cells more tumorigenic and metastatic. Clin Cancer Res. 1999;5:3711–3721. [PubMed] [Google Scholar]
- 8.Büchler P, Reber HA, Büchler M, et al. Hypoxia-inducible factor 1 regulates vascular endothelial growth factor expression in human pancreatic cancer. Pancreas. 2003;26:56–64. [DOI] [PubMed] [Google Scholar]
- 9.Büchler P, Reber HA, Lavey RS, et al. Tumor hypoxia correlates with metastatic tumor growth of pancreatic cancer in an orthotopic murine model. J Surg Res. 2004;120:295–303. [DOI] [PubMed] [Google Scholar]
- 10.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. [DOI] [PubMed] [Google Scholar]
- 11.Iacobuzio-Donahue CA. Epigenetic changes in cancer. Annu Rev Pathol. 2009;4:229–249. [DOI] [PubMed] [Google Scholar]
- 12.Muraoka M, Konishi M, Kikuchi-Yanoshita R, et al. p300 gene alterations in colorectal and gastric carcinomas. Oncogene. 1996;12:1565–1569. [PubMed] [Google Scholar]
- 13.Grignani F, De Matteis S, Nervi C, et al. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature. 1998;391:815–818. [DOI] [PubMed] [Google Scholar]
- 14.Lin RJ, Nagy L, Inoue S, et al. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature. 1998;391:811–814. [DOI] [PubMed] [Google Scholar]
- 15.Maleszewska M, Kaminska B. Deregulation of histone-modifying enzymes and chromatin structure modifiers contributes to glioma development. Future Oncol. 2015;11:2587–2601. [DOI] [PubMed] [Google Scholar]
- 16.Moral R, Wang R, Russo IH, et al. Effect of prenatal exposure to the endocrine disruptor bisphenol A on mammary gland morphology and gene expression signature. J Endocrinol. 2008;196:101–112. [DOI] [PubMed] [Google Scholar]
- 17.El Sheikh H, Meduri G, Phrakonkham P, et al. Abnormal peripubertal development of the rat mammary gland following exposure in utero and during lactation to a mixture of genistein and the food contaminant vinclozolin. Reprod Toxicol. 2011;32:15–25. [DOI] [PubMed] [Google Scholar]
- 18.El Sheikh H, Toullec A, Vacher S, et al. In utero and lactational exposure to vinclozolin and genistein induces genomic changes in the rat mammary gland. J Endocrinol. 2013;216:245–263. [DOI] [PubMed] [Google Scholar]
- 19.Dhimolea E, Wadia PR, Murray TJ, et al. Prenatal exposure to BPA alters the epigenome of the rat mammary gland and increases the propensity to neoplastic development. PLoS One. 2014;9:e99800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weisbeck A, Jansen RJ. Nutrients and the pancreas: an epigenetic perspective. Nutrients. 2017;9 pii: E283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alegría-Torres JA, Baccarelli A, Bollati V. Epigenetics and lifestyle. Epigenomics. 2011;3:267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mazur PK, Herner A, Mello SS, et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015;21:1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park SJ, Kim SM, Moon JH, et al. SAHA, an HDAC inhibitor, overcomes erlotinib resistance in human pancreatic cancer cells by modulating E-cadherin. Tumour Biol. 2016;37:4323–4330. [DOI] [PubMed] [Google Scholar]
- 24.Chan E, Arlinghaus LR, Cardin DB, et al. Phase I trial of vorinostat added to chemoradiation with capecitabine in pancreatic cancer. Radiother Oncol. 2016;119:312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshida M, Horinouchi S, Beppu T. Trichostatin A and trapoxin: novel chemical probes for the role of histone acetylation in chromatin structure and function. Bioessays. 1995;17:423–430. [DOI] [PubMed] [Google Scholar]
- 26.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu T, Kuljaca S, Tee A, et al. Histone deacetylase inhibitors: multifunctional anticancer agents. Cancer Treat Rev. 2006;32:157–165. [DOI] [PubMed] [Google Scholar]
- 28.Johnstone RW, Licht JD. Histone deacetylase inhibitors in cancer therapy: is transcription the primary target? Cancer Cell. 2003;4:13–18. [DOI] [PubMed] [Google Scholar]
- 29.Chen YX, Fang JY, Zhu HY, et al. Histone acetylation regulates p21WAF1 expression in human colon cancer cell lines. World J Gastroenterol. 2004;10:2643–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butz J, Wickstrom E, Edwards J. Characterization of mutations and loss of heterozygosity of p53 and K-ras2 in pancreatic cancer cell lines by immobilized polymerase chain reaction. BMC Biotechnol. 2003;3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Angst E, Dawson DW, Nguyen A, et al. Epigenetic regulation affects N-myc downstream-regulated gene 1 expression indirectly in pancreatic cancer cells. Pancreas. 2010;39:675–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murray JT, Campbell DG, Morrice N, et al. Exploitation of KESTREL to identify NDRG family members as physiological substrates for SGK1 and GSK3. Biochem J. 2004;384:477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sugiki T, Murakami M, Taketomi Y, et al. N-myc downregulated gene 1 is a phosphorylated protein in mast cells. Biol Pharm Bull. 2004;27:624–627. [DOI] [PubMed] [Google Scholar]
- 34.Dong Z, Arnold RJ, Yang Y, et al. Modulation of differentiation-related gene 1 expression by cell cycle blocker mimosine, revealed by proteomic analysis. Mol Cell Proteomics. 2005;4:993–1001. [DOI] [PubMed] [Google Scholar]
- 35.Erler JT, Giaccia AJ. Lysyl oxidase mediates hypoxic control of metastasis. Cancer Res. 2006;66:10238–10241. [DOI] [PubMed] [Google Scholar]
- 36.Chia SK, Wykoff CC, Watson PH, et al. Prognostic significance of a novel hypoxia-regulated marker, carbonic anhydrase IX, in invasive breast carcinoma. J Clin Oncol. 2001;19:3660–3668. [DOI] [PubMed] [Google Scholar]
- 37.Kareta MS, Sage J, Wernig M. Crosstalk between stem cell and cell cycle machineries. Curr Opin Cell Biol. 2015;37:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li VC, Kirschner MW. Molecular ties between the cell cycle and differentiation in embryonic stem cells. Proc Natl Acad Sci U S A. 2014;111:9503–9508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galderisi U, Jori FP, Giordano A. Cell cycle regulation and neural differentiation. Oncogene. 2003;22:5208–5219. [DOI] [PubMed] [Google Scholar]
- 40.Harper JW, Elledge SJ. Cdk inhibitors in development and cancer. Curr Opin Genet Dev. 1996;6:56–64. [DOI] [PubMed] [Google Scholar]