

Abstract
Breast cancer is the most prevalent cancer diagnosis in women. We aimed to identify biomarkers for breast cancer prognosis. mRNA expression profiling was performed using Gene Chip Human Transcriptome Array 2.0. Microarray analysis and series test of cluster (STC) analysis were used to screen the differential expressed mRNAs and the expression trend of genes. Immumohistochemical staining with 100 clinical specimens was used to validate two differentially expressed genes, ITGA11 and Jab1. In the present study, significantly enriched Gene Ontology (GO) terms and pathways were identified. 26 model profiles were used to summarize the expression pattern of differentially expressed genes. Results of immunohistochemistry were consistent with those of the microarray, in that ITGA11 and Jab1 were differentially expressed with the same trend. Survival analyses using the Kaplan–Meier method demonstrated that breast cancer patients with high levels of either ITGA11 or Jab1 had a significant association with worse prognosis. Our study identified ITGA11 and Jab1 as novel biomarkers for breast cancer.
Keywords: breast cancer, microarray, ITGA11, Jab1/COPS5, biomarker
INTRODUCTION
Breast cancer is the most common cancer [1] and also the second leading cause of cancer deaths for women [2]. The lack of better adjuvant therapy remains to be a major challenge in reducing the burden of breast cancer patients. Nowadays, the tumor size, lymph node involvement, and distant metastasis (TNM) staging system of the American Joint Committee on Cancer (AJCC) has been widely recognized, but there is still a lack of worldwide-recognized system or reliable markers predicting the prognosis of breast cancer patients. While applying for neoadjuvant chemotherapy or endocrine therapy, clinicopathological parameters are usually unstable, which complicates the judgment of real prognosis. Therefore, there is a pressing need to find biomarkers for breast cancer which can help to develop better treatment solutions for breast cancer.
Intensive research has been focused on understanding the molecular mechanisms of breast cancer [3]. Many genetic changes that lead to abnormal cellular functions have been identified in breast cancer cells [4, 5]. Multiple factors in the tumor microenvironment further influence the cancer progression via a wide variety of receptors and the corresponding signal pathways [6], which also involve various oncogenes and anti-oncogenes [7, 8]. Microarray data analysis, which features high throughput and high sensitivity, has made it possible to test the expression changes of the whole genome [9]. There have been many reports on gene expression profiling in breast cancer [10, 11]. Therefore, the development of microarray analysis provides new insight in diagnosis and treatment of breast cancer.
Understanding new developments in transcriptome and pathways may identify novel biomarkers for cancer. Integrin α11 (ITGA11), a integrin family members, involve in various processes that influences the cell’s biological behavior, such as metastasis, embryogenesis, hemostasis, immune response, tissue repair, cancer growth, tumor angiogenesis, and resistance to therapy [12, 13]. Alterations in integrin disturb cancer cell adhesion and extracellular matrix assembly, which may further lead to tumor metastasis [14]. Integrins also interact with tyrosine kinase receptors which promote cancer cell proliferation, and differentiation [15].
c-Jun activation domain-binding protein 1 (Jab1), primarily identified as a c-Jun coactivator [16], is the fifth member of the constitutive photomorphogenesis 9 signalosome (COPS5) complex. Jab1/COPS5 regulates cell signal transduction, genetic transcription and protein stability [16]. Importantly, Jab1/COPS5 plays an important role in proliferation and invasiveness [17].
In the current study, we identified potential genes associated with breast cancer tumorigenesis by transcriptional network analysis and further validated ITGA11 from the STC analysis and a differentially expressed gene Jab1/COPS5 in clinical patients.
RESULTS
Patient characteristics
In the present study, patients’ median age was 50 years old (range 25–80). 35 cases (43.8%) were menopause. The main histological type was ductal in 74 (92.5%), lobular in 5 (6.25%), medullary in 1 (1.25%). 6 (7.5%) had a grade 1 tumor; 54 (67.5%) had a grade 2 tumor; 20 (25.8%) had a grade 3 tumor. Based on tumor staging system, most patients were defined as stage II (48, 60.0%) and stage I (10, 12.5%). 66 patients (82.5%) had lymph node involvement. 1 patients (1.3%) developed distant metastasis. The patients’ clinicopathologic characteristics were summarized in Table 1.
Table 1. Characteristics of breast cancer patients (n = 80).
| Variable | Number (%) |
|---|---|
| Age | |
| <60 | 66 (82.5%) |
| ≥60 | 14 (17.5%) |
| Histological type | |
| Ductal | 74 (92.5%) |
| Lobular & Medullary | 6 (7.5%) |
| Stage | |
| I | 10 (12.5%) |
| II | 48 (60.0%) |
| III & IV | 22 (27.5%) |
| Grade | |
| G1 | 6 (7.5%) |
| G2 | 54 (67.5%) |
| G3 | 20 (25.0%) |
| Tumor size | |
| T1 | 9 (11.3%) |
| T2 | 48 (60.0%) |
| T3 & T4 | 23 (28.7%) |
| Lymph node metastasis | |
| No | 14 (17.5%) |
| Yes | 66 (82.5%) |
| Distant metastasis | |
| No | 79 (99.8%) |
| Yes | 1 (1.3%) |
| HER2 | |
| negative | 38 (47.5%) |
| positive | 42 (52.5%) |
| ER | |
| negative | 40 (50%) |
| positive | 40 (50%) |
| PR | |
| negative | 46 (57.5%) |
| positive | 34 (42.5%) |
| Ki-67 | |
| <14% | 39 (48.8%) |
| ≥14% | 41 (51.2%) |
| Family history | |
| No | 56 (70.0%) |
| Yes | 24 (30.0%) |
| Menopause | |
| No | 45 (56.3%) |
| Yes | 35 (43.8%) |
Transcriptome array analysis of mRNA expression in breast cancer
HE Staining confirmed the breast cancer tissue and paired adjacent noncancerous breast tissue (Figure 1A). We used Affymetrix GeneChip Human Transcriptome Array 2.0 to analyze mRNA expression in the breast tissue, and applied the RVM t-test to filter the differentially expressed mRNAs, 509 mRNAs were found to be significantly down-regulated (fold change > 1.2, p < 0.05) while 1277 mRNA were markedly up-regulated (fold change > 1.2, p < 0.05) in breast cancer tissue compared with the adjacent noncancerous breast tissue (Figure 1B).
Figure 1. The mRNA profile differentiates breast cancer tissue from adjacent noncancerous breast tissue.
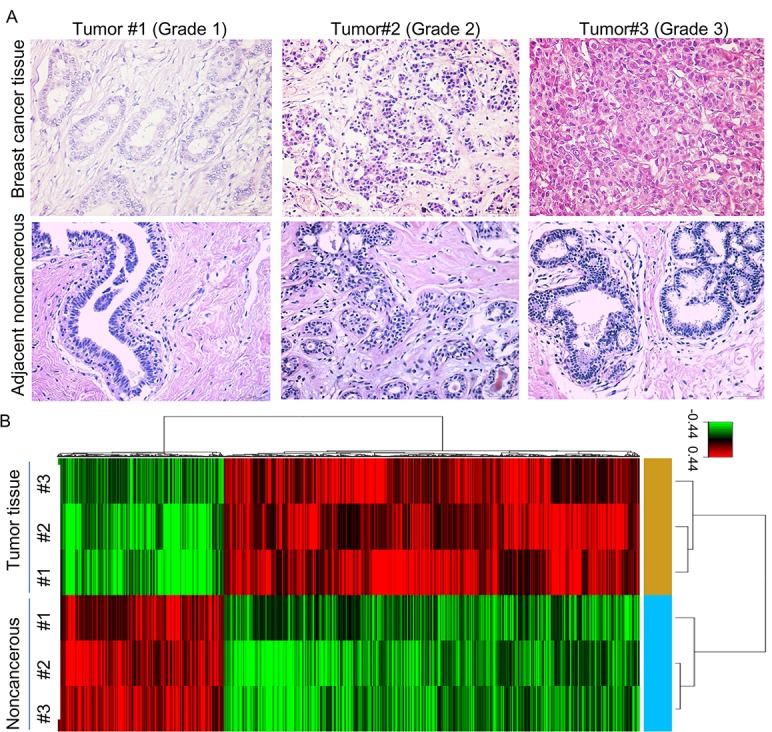
(A) HE staining of breast cancer tissue and adjacent noncancerous breast tissue. (B) Hierarchical clustering for the differentially expressed mRNAs (p < 0.05).
In order to identify mRNAs that are overrepresented in any functional class, the dysregulated mRNAs were subjected to functional enrichment analysis using FunRich software (Figures 2 and 3). According to the cell component, 19.3% of genes were categorized as cytoplasm, 11.8% of genes were identified in plasma membrane (Figure 2A). Among molecular functions, genes were enriched in transporter activity (3.9%), ubiquitin-specific protease activity (2.5%), protein serine/threonine kinase activity (2.2%) and extracellular matrix structural constituent (2.1%) (Figure 2B).
Figure 2. Functional enrichment analysis of genes using FunRich.

Enrichment of cell component (A) and molecular functions (B) in dysregulated mRNAs.
Figure 3. Functional enrichment analysis of genes using FunRich.

Pie graph of biological processes (A) and Bar graph of biological pathways (B) in genes are shown.
Further to this, genes involved in metabolism, energy pathways, cell growth and/or maintenance and protein metabolism were enriched in (Figure 3A). In the context of biological pathway, cell cycle, mitotic, polo-like kinase signaling events in the cell cycle, PLK1 signaling events membrane trafficking were significantly overrepresented in mRNAs (Figure 3B).
STC analysis
To further narrow the range of target genes with high significance, the tumor grade-serial expression pattern of significantly gene was investigated. Each profile consists of a cluster of multiple genes that have similar expression patterns with increasing tumor grade. As shown in Figure 4A, 26 model profiles were used to summarize the expression pattern of these genes. Each box represents a model profile. Among the 26 patterns, totally 10 expression patterns including profiles 23, 4, 22, 7, 14, 25, 26, 20, 17 and 9 showed significant P values (P < 0.001). Six of these clusters contained genes which were stable (profile 22 and 14) or gradually elevated (profiles 23, 25, 26 and 17), while genes in profile 4 had opposite effects to profile 23. Profile 9 contained genes which were suppressed at grade 1 points and then gradually increased expression levels at higher tumor grades points (Figure 4B).
Figure 4. STC analysis for differential genes related to tumor grade.

(A) The expression patterns of genes were analyzed and 26 model profiles were used to summarize. Each box represents a model expression profile. Each image corresponds to a different model grade expression profile. The upper number in the profile box is the model profile number and the P value. The horizontal axis represents group points, and the vertical axis shows the gene expression levels for the gene after Log normalized transformation. (B) Two expression patterns of genes showed significant P values (P < 0.001). The number of genes assigned to each model profile is used as the estimate of the number of co-expressed genes.
ITGA11 and Jab1 overexpressed in breast cancer patients
To validate the findings from our transcriptome array analysis, immunohistochemical staining of ITGA11 and Jab1/Cops5 was conducted in 80 breast cancer tissue and 20 noncancerous breast tissue. As expected, both ITGA11 and Jab1/Cops5 were overexpressed in breast cancer (Figure 5A and 5B). In agreement with the immunohistochemistry findings, the data from online database ONCOMINE indicated that ITGA11 and Jab1/Cops5 mRNA expression levels in breast cancer are much higher than those in normal breast tissue (Figure 5C).
Figure 5. IGTA11 and Jab1 expressed in breast cancer.

(A) Immunohistochemical analysis of IGTA11 and Jab1 in breast hyperplasia and breast cancer tissue. Original magnification, ×200. (B) The percentages of postive IGTA11 or positive Jab1 cell in noncancerous and cancerous tissues. (C) IGTA11 and Jab1 gene expression in normal breast and breast cancer using the Oncomine gene expression tool (https://www.oncomine.com). The clinical data were downloaded from Oncomine Data Portal.
Association between ITGA11 and Jab1 and clinicopathological parameters
Correlations between ITGA11 and Jab1/Cops5 and clinicopathological parameters were summarized in Table 2. Elevated ITGA11 levels were associated with higher tumor grade (G1 vs. G2 and G3, 20.0% vs. 34.9% and 42.1%, respectively, p = 0.044). Jab1 levels were not only associated with tumor grade (G1 vs. G2 and G3, 26.7% vs. 31.4% and 45.7%, respectively, p = 0.040), but also associated with ER status (negative vs. positive, 27.5% vs. 41.7%, p = 0.005) and PR status (negative vs. positive, 30.0% vs. 40.8%, p = 0.039). However, we did not observe any association between ITGA11/ Jab1 and histological type, tumor stage, family history nor menopause.
Table 2. Correlation between ITGA11/Jab1 and clinicopathological parameters.
| Variable | ITGA11 postive cells % | P value | Jab1 postive cells % | P value |
|---|---|---|---|---|
| Age | ||||
| <60 | 35.4 ± 20.2 | 0.854 | 34.5 ± 22.2 | 0.972 |
| ≥60 | 36.5 ± 15.4 | 34.8 ± 28.4 | ||
| Histological type | ||||
| Ductal | 36.3 ± 19.4 | 0.310 | 34.5 ± 23.6 | 0.921 |
| Lobular & Medullary | 27.8 ± 19.4 | 35.5 ± 19.4 | ||
| Stage | ||||
| I | 25.9 ± 13.2 | 0.210 | 26.1 ± 17.7 | 0.104 |
| II | 37.8 ± 17.8 | 39.0 ± 25.6 | ||
| III & IV | 35.2 ± 24.9 | 28.7 ± 17.7 | ||
| Grade | ||||
| G1 | 20.0 ± 10.5 | 0.044 | 26.7 ± 22.4 | 0.040 |
| G2 | 34.9 ± 16.0 | 31.4 ± 15.0 | ||
| G3 | 42.1 ± 26.4 | 45.7 ± 35.9 | ||
| Tumor size | ||||
| T1 | 26.7 ± 10.8 | 0.314 | 26.1 ± 15.5 | 0.100 |
| T2 | 37.4 ± 18.4 | 39.1 ± 25.9 | ||
| T3 & T4 | 35.3 ± 23.4 | 28.5 ± 17.3 | ||
| LN metastasis | ||||
| No | 30.0 ± 15.7 | 0.235 | 35.1 ± 24.4 | 0.922 |
| Yes | 36.8 ± 20.0 | 34.5 ± 23.1 | ||
| HER2 | ||||
| negative | 36.8 ± 22.7 | 0.597 | 36.9 ± 22.6 | 0.395 |
| positive | 34.5 ± 16.1 | 32.5 ± 23.8 | ||
| ER | ||||
| negative | 33.2 ± 18.3 | 0.262 | 27.5 ± 20.0 | 0.005 |
| positive | 38.1 ± 20.3 | 41.7 ± 24.2 | ||
| PR | ||||
| negative | 34.8 ± 16.5 | 0.646 | 30.0 ± 22.1 | 0.039 |
| positive | 36.8 ± 22.9 | 40.8 ± 23.5 | ||
| Ki-67 | ||||
| <14% | 36.3 ± 17.3 | 0.752 | 34.5 ± 25.8 | 0.985 |
| ≥14% | 34.9 ± 21.4 | 34.6 ± 20.7 | ||
| Family history | ||||
| No | 37.8 ± 21.3 | 0.123 | 37.4 ± 23.2 | 0.092 |
| Yes | 30.5 ± 13.1 | 27.9 ± 22.2 | ||
| Menopause | ||||
| No | 35.1 ± 20.5 | 0.773 | 36.6 ± 23.7 | 0.387 |
| Yes | 36.3 ± 18.2 | 32.0 ± 22.6 |
Abbreviations: LN: lymph node.
In agreement with the immunohistochemistry results, data from ONCOMINE indicated that ITGA11 and Jab1/Cops5 expression levels tend to be higher in breast cancer with higher grade (Figure 6A). Furthermore, RNA sequencing analysis of mRNA expression from the GEPIA online database revealed that ITGA11 was associated with Jab1/Cops5 in breast cancer patients (Figure 6B). In addition, our immunohistochemistry data in breast cancer tissue indicated that Jab1 level was associated with ITGA11 levels in breast cancer (Figure 6C) and both ITGA11 and Jab1 levels were correlated with tumor grade (Figure 6D).
Figure 6. IGTA11 and Jab1 associates with tumor grade in breast cancer.

(A) IGTA11 and Jab1 gene expression in breast cancer with different tumor grade using the Oncomine gene expression tool. (B) Overview of the two genes in breast cancer across online data GEPIA. (C) IGTA11 was associated with Jab1 expression in breast cancer from the immunohistochemistry data. (D) IGTA11 and Jab1 associated with tumor grade in breast cancer from the immunohistochemistry data. The R and P values were from Pearson Correlation.
Survival analysis
The median follow-up time was 64 months (range 10–120). In order to evaluate the prognostic influence of ITGA11 and Jab1/COPS5 expression, we carried out Kaplan–Meier analyses to compare grouped patients. The survival curves demonstrated that patients with high levels of ITGA11 or Jab1/COPS5 had a significant association with worse OS (p = 0.034, p = 0.007; Figure 7A and 7B).
Figure 7. IGTA11 and Jab1/COPS5 predict survival in breast cancer.

(A, B) Kaplan–Meier analyses of the association between IGTA11 and Jab1 protein expression and survival. (C, D) In vivo RNA sequencing data and Kaplan–Meier plots from GEPIA database were used to assess correlations between IGTA11 or Jab1/COPS5 gene expression and breast cancer patients survival. (E) Kaplan–Meier analyses of the association between endocrine therapy and survival.
We also investigated the relationship between ITGA11 and Jab1/COPS5 and survival of breast cancer patients in GEPIA database. The online data was consistent with the IHC data, suggesting high levels of either ITGA11 or Jab1/COPS5 was associated with worse survival in breast cancer (Figure 7C and 7D). However, we didn't find significant influence of endocrine therapy on the survival (Figure 7E). We also analysed the survival in different subtypes of breast cancer and found similar results (Supplementary Figures 1 and 2).
DISCUSSION
Breast cancer is the most frequent cancer among women worldwide. Screening and diagnosis of breast cancer at earlier stages are of great importance to improve patient survival and reduce treatment costs. However, the underlying mechanism regulating breast cancer aggressiveness remain poorly understood, and biomarkers for the detection of early-stage breast cancer are still lacking.
The advent of high throughput mRNA microarray analysis method makes it possible to detect the expression of thousands of mRNAs, which allows us to have a clearer picture of the global transcriptome in both cancer tissue and normal tissue [18]. In this study, we assessed the mRNA expression profiles in both breast cancer tissue and paired noncancerous breast tissue using microarray technique and explored their possible functions using GO analysis, KEGG pathway analysis and STC analysis. A number of mRNAs were significantly differentially expressed in breast cancer tissue compared with noncancerous breast tissue. In order to validate the microarray results, we further carried out independent measurement of ITGA11 and Jab1 protein levels in breast cancer and noncancerous tissue samples using immunohistochemistry. The immunohistochemistry results showed good consistency with microarray.
We also performed GO analysis, KEGG pathway analysis, and STC analysis to identify the enriched biological functions among the differentially expressed genes. It was found that the genes were participated in a variety of molecular functions, cellular components, and biological processes. Many pathways related to cancer have been identified by the pathway analysis, among which “cell cycle” and “PLK1 signaling events” are two of the most enriched pathways. Cell cycle is a highly organized and regulated process that ensures duplication of genetic material and cell division. Proliferation depends on progression through four distinct phases of the cell cycle, which is regulated by several cyclin-dependent kinases (CDKs) and their cyclin partners [19]. Cancer growth is caused by abrogation of appropriate cell-cycle control, and many cell-cycle kinases are amplified or overexpressed in cancer [19]. Polo-like kinase 1 (PLK1) plays a vital role in cell cycle progression through mitosis via its effects on chromosome segregation, spindle assembly and cytokinesis [20]. Inhibition of PLK1 delay acentriolar spindle formation during mitosis and promote apoptosis [21]. Further, PLK1 is an important regulator of the DNA damage checkpoint [22]. PLK1 is overexpressed in a variety of malignancy including breast cancer [23, 24]. Additionally, PLK1 overexpression is associated with poor prognosis in cancer patients [25]. These results demonstrated the reliability of our microarray study.
Integrins are heterodimeric cell surface adhesion receptors contains α and β subunits. Twenty-four distinct integrin heterodimers are expressed in mammals as a result of combinatorial association of 18 α and 8 β subunits [26]. Extracellular matrix (ECM) ligands can bind to the α subunit and activate intracellular signaling events via the β subunit to integrate extracellular and intracellular events necessary for cell motility and invasion [14]. Many integrins are expressed at low or undetectable levels in adult epithelia, but are up-regulated in tumors [26]. Integrin α11 (ITGA11) is expressed in many tissues in the embryo but disappears with maturation in adult tissues [27]. However, it has been proved that its expression is up-regulated in malignant conditions such as non-small-cell lung carcinoma, where it has been suggested to be connected to cancer cell growth [28, 29].
We found high expression of ITGA11 was correlated with poor prognosis in breast cancer patients, which is in agreement with previous studies that Integrin expression levels are correlated with prognosis in glioblastoma, melanoma, gastric cancer, cervical cancer, and ovarian cancer [30–33].
Aberrant overexpression of Jab1/COPS5 is demonstrated to play a role in the pathogenesis of several types of human cancers and correlate with poor cancer prognosis [34, 35]. Jab1/COPS5 isopeptidase activity is essential for human and murine mammary epithelial transformation and progression [36]. Jab1/COPS5 expression was low in or absent from normal breast tissue, while it was abnormally expressed in breast tumors [37]. Importantly, breast cancer patients with Jab1/COPS5-negative tumors had neither relapse nor disease progression at a median follow-up time of 70 months [38]. In line with these results, our study found that higher levels of Jab1 expression in breast cancer patients compared with that in non-cancerous tissue and Jab1 expression was associated with tumor grade, suggesting an role of Jab1 in tumor progress.
Taken together, we have identified ITGA11 and Jab1 as biomarkers in breast cancer. High throughput microarray data analysis may act as an efficient tool to discover more prognostic markers and therapeutic targets in breast cancer.
MATERIALS AND METHODS
Patients and tissue samples
Three breast cancer tissue and three paired adjacent noncancerous breast tissue specimens were from Anyang Tumor Hospital (Anyang, Henan, China). Other 80 cases of breast cancer patients and 20 cases of breast hyperplasia patients treated at Anyang Tumor Hospital from July 2007 to July 2012 were randomly included in this study for immunohistochemical analysis. Breast hyperplasia is a diagnostic category of proliferative disease that includes inflammatory hyperplasia, atypical ductal hyperplasia and atypical lobular hyperplasia. The inclusion criteria for the participants were: aged 18 years above; diagnosis of breast cancer. Exclusion criteria were: preoperative chemotherapy or radiotherapy; deficiency of clinical data or lack of follow up. The diagnosis of breast cancer and hyperplasia was confirmed pathologically. Patients who had preoperative diagnosis and had not received preoperative chemoradiotherapy were selected in our study based on the availability of archived paraffin-embedded tissue blocks for immunohistochemistry. Ethical approval from Anyang Tumor Hospital and informed consent from patients have been obtained. The clinical and pathological characteristics of 80 breast cancer patients were summarized in Table 1.
RNA isolation and transcriptome array
Total RNA in the samples was extracted using Trizol reagent. The integrity and concentration of all RNA samples were measured using the NanoDrop 1000 spectrophotometer. The total RNA extracted from three breast cancer tissue and three paired adjacent noncancerous breast tissue specimens were hybridized to an Affymetrix GeneChip Human Transcriptome Array 2.0. The arrays were scanned by GeneChip® Command Console® Software and the acquired array images were analyzed by Affymetrix GeneChip Operating Software.
Cluster analysis and series test of cluster (STC) analysis
Differential expressed genes from microarray data were screened by applying random variance model (RVM) t-test and considered to be down or up regulated with p < 0.05. The cluster analysis of genes was accordingly conducted through GCBI online system (https://www.gcbi.com.cn/gcuser/html/member/home). STC algorithm of gene expression was performed to profile the gene expression with grade malignancy series and to identify the most probable set of clusters generating grade malignancy series. Dynamic nature of gene expression profiles was taken into account in STC and therefore it can identify the number of distinct clusters. Fisher’s exact test was used to examine significant profiles, and p < 0.05 was considered as the threshold of significance.
Functional enrichment analysis
mRNAs identified in breast cancer were subjected to Gene Ontology (GO) and biological pathway enrichment analysis using FunRich tool (http://www.funrich.org) against human FunRich background database.
Analysis of clinical mRNA microarrays for the detection of correlations between ITGA11 and patients survival
Transcriptome data from patient samples of breast cancer were analyzed using the online database ONCOMINE (https://www.oncomine.org/resource/login.html) to investigate whether the expression of the markers are associated with tumor grade. RNA sequencing analysis and visualization platform GEPIA (http://gepia.cancer-pku.cn/) was used to determine whether the expression levels of ITGA11 and Jab1 were correlated in breast cancer. GEPIAwas used to determine whether the expression of ITGA11 and Jab1 was correlated with the breast cancer patients’ overall survival.
Immunohistochemical analysis
The tissues were processed routinely and stained with hematoxylin and eosin (HE) [39]. ITGA11 and Jab1 levels in the formalin-fixed, paraffin-embedded tissue were evaluated using immunohistochemical staining, as described in our previous work [40]. Briefly, the samples were sectioned and mounted on slides, following drying at 60°C for 1 hour. Slides were then deparaffinized in 2-xylene. To retrieval antigen, the slides were boiled for 3 minutes in 0.01 mol/L sodium citrate (pH 6.0) and then cooled at room temperature for 30 minutes. To block the Endogenous peroxidase activity, the slides were further immersed in 0.3% H2O2. Then the slides were incubated with the primary antibodies ITGA11 (Santa Cruz, sc-98740) and Jab1 (Santa Cruz, sc-13157) diluted at 1:200 overnight at 4°C and were detected by a secondary antibody kit (Dako Corp). ITGA11 and Jab1 expression were measured by counting no less than 400 tumor cells. Tumor cells were considered positive for markers when nuclear or cytoplasmic staining was present. The positivity represented the estimated fraction of positively stained cells (−, ≤5%; +, 5% to 25%; ++, 26% to 50%; +++, >50%). All experiments were performed in accordance with approved guidelines and regulations of Anyang Tumor Hospital.
Follow-up and statistical analysis
Overall survival (OS) was calculated as the period from initial diagnosis to death regardless of breast cancer related or not. Before closing the research database, the authors updated the follow-up data of patients who had not visited our outpatient department for more than three months. Patient follow-up was censored at the time of death or finalization of the study. Percent of ITGA11 positive cells and Jab1 positive cells were presented as means ± standard deviation (SD). Categorical variables were presented as numbers and percentages. Comparisons between groups were carried out with the T test or the one-way ANOVA and LSD tests for continuous variables. Multivariate survival analyses were performed to identify independent factors for overall survival. Kaplan–Meier method was applied for performing stratified overall survival analysis, followed by the log-rank test. It was regarded as statistically significant when P < 0.05. Calculations were performed using IBM SPSS statistics software 22.0.
SUPPLEMENTARY FIGURES
Abbreviations
- STC
series test of cluster
- GO
Gene Ontology
- HE
hematoxylin and eosin
- OS
Overall survival
- ECM
Extracellular matrix
- CDK
cyclin-dependent kinase
- PLK1
Polo-like kinase 1
Footnotes
Author contributions
YP conceived the study. YP and GL performed the experiments, analyzed the data and wrote the manuscript. YY, JZ and YY reviewed the medical records and collected data. YL analyzed the data and directed the project.
CONFLICTS OF INTEREST
None.
FUNDING
This work was supported by the funds from the National Natural Science Foundation of China (81372816).
REFERENCES
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. https://doi.org/10.3322/caac.21262 [DOI] [PubMed] [Google Scholar]
- 2.DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin. 2014;64:52–62. doi: 10.3322/caac.21203. https://doi.org/10.3322/caac.21203 [DOI] [PubMed] [Google Scholar]
- 3.Li Z, Dong M, Fan D, Hou P, Li H, Liu L, Lin C, Liu J, Su L, Wu L, Li X, Huang B, Lu J, et al. LncRNA ANCR down-regulation promotes TGF-beta-induced EMT and metastasis in breast cancer. Oncotarget. 2017;8:67329–67343. doi: 10.18632/oncotarget.18622. https://doi.org/10.18632/oncotarget.18622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, Tomiguchi M, Sueta A, Murakami K, Omoto Y, Iwase H. Analysis of ESR1 and PIK3CA mutations in plasma cell-free DNA from ER-positive breast cancer patients. Oncotarget. 2017;8:52142–52155. doi: 10.18632/oncotarget.18479. https://doi.org/10.18632/oncotarget.18479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma F, Li W, Liu C, Li W, Yu H, Lei B, Ren Y, Li Z, Pang D, Qian C. MiR-23a promotes TGF-beta1-induced EMT and tumor metastasis in breast cancer cells by directly targeting CDH1 and activating Wnt/beta-catenin signaling. Oncotarget. 2017;8:69538–69550. doi: 10.18632/oncotarget.18422. https://doi.org/10.18632/oncotarget.18422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desmedt C, Yates L, Kulka J. Catalog of genetic progression of human cancers: breast cancer. Cancer Metastasis Rev. 2016;35:49–62. doi: 10.1007/s10555-016-9609-1. https://doi.org/10.1007/s10555-016-9609-1 [DOI] [PubMed] [Google Scholar]
- 7.Guerrero-Zotano A, Mayer IA, Arteaga CL. PI3K/AKT/mTOR: role in breast cancer progression, drug resistance, and treatment. Cancer Metastasis Rev. 2016;35:515–24. doi: 10.1007/s10555-016-9637-x. https://doi.org/10.1007/s10555-016-9637-x [DOI] [PubMed] [Google Scholar]
- 8.Bayraktar S, Arun B. BRCA mutation genetic testing implications in the United States. Breast. 2017;31:224–32. doi: 10.1016/j.breast.2016.11.021. https://doi.org/10.1016/j.breast.2016.11.021 [DOI] [PubMed] [Google Scholar]
- 9.Zhu T, Gao YF, Chen YX, Wang ZB, Yin JY, Mao XY, Li X, Zhang W, Zhou HH, Liu ZQ. Genome-scale analysis identifies GJB2 and ERO1LB as prognosis markers in patients with pancreatic cancer. Oncotarget. 2017;8:21281–89. doi: 10.18632/oncotarget.15068. https://doi.org/10.18632/oncotarget.15068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makoukji J, Makhoul NJ, Khalil M, El-Sitt S, Aldin ES, Jabbour M, Boulos F, Gadaleta E, Sangaralingam A, Chelala C, Boustany RM, Tfayli A. Gene expression profiling of breast cancer in Lebanese women. Sci Rep. 2016;6:36639. doi: 10.1038/srep36639. https://doi.org/10.1038/srep36639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Yang X, Chen H, Wang X, Wang X, Fang Y, Jia Z, Gao J. A high-throughput method to detect RNA profiling by integration of RT-MLPA with next generation sequencing technology. Oncotarget. 2017;8:46071–80. doi: 10.18632/oncotarget.17551. https://doi.org/10.18632/oncotarget.17551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwartz MA, Ginsberg MH. Networks and crosstalk: integrin signalling spreads. Nat Cell Biol. 2002;4:E65–8. doi: 10.1038/ncb0402-e65. https://doi.org/10.1038/ncb0402-e65 [DOI] [PubMed] [Google Scholar]
- 13.Nam K, Son SH, Oh S, Jeon D, Kim H, Noh DY, Kim S, Shin I. Binding of galectin-1 to integrin beta1 potentiates drug resistance by promoting survivin expression in breast cancer cells. Oncotarget. 2017;8:35804–23. doi: 10.18632/oncotarget.16208. https://doi.org/10.18632/oncotarget.16208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–87. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 15.Hehlgans S, Haase M, Cordes N. Signalling via integrins: implications for cell survival and anticancer strategies. Biochim Biophys Acta. 2007;1775:163–80. doi: 10.1016/j.bbcan.2006.09.001. https://doi.org/10.1016/j.bbcan.2006.09.001 [DOI] [PubMed] [Google Scholar]
- 16.Claret FX, Hibi M, Dhut S, Toda T, Karin M. A new group of conserved coactivators that increase the specificity of AP-1 transcription factors. Nature. 1996;383:453–7. doi: 10.1038/383453a0. https://doi.org/10.1038/383453a0 [DOI] [PubMed] [Google Scholar]
- 17.Adler AS, Lin M, Horlings H, Nuyten DS, van de Vijver MJ, Chang HY. Genetic regulators of large-scale transcriptional signatures in cancer. Nat Genet. 2006;38:421–30. doi: 10.1038/ng1752. https://doi.org/10.1038/ng1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kadara H, Fujimoto J, Yoo SY, Maki Y, Gower AC, Kabbout M, Garcia MM, Chow CW, Chu Z, Mendoza G, Shen L, Kalhor N, Hong WK, et al. Transcriptomic architecture of the adjacent airway field cancerization in non-small cell lung cancer. J Natl Cancer Inst. 2014;106:dju004. doi: 10.1093/jnci/dju004. https://doi.org/10.1093/jnci/dju004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. 2017;17:93–115. doi: 10.1038/nrc.2016.138. https://doi.org/10.1038/nrc.2016.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Carcer G, Manning G, Malumbres M. From Plk1 to Plk5: functional evolution of polo-like kinases. Cell Cycle. 2011;10:2255–62. doi: 10.4161/cc.10.14.16494. https://doi.org/10.4161/cc.10.14.16494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X, Erikson RL. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc Natl Acad Sci USA. 2003;100:5789–94. doi: 10.1073/pnas.1031523100. https://doi.org/10.1073/pnas.1031523100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takaki T, Trenz K, Costanzo V, Petronczki M. Polo-like kinase 1 reaches beyond mitosis--cytokinesis, DNA damage response, and development. Curr Opin Cell Biol. 2008;20:650–60. doi: 10.1016/j.ceb.2008.10.005. https://doi.org/10.1016/j.ceb.2008.10.005 [DOI] [PubMed] [Google Scholar]
- 23.Takai N, Hamanaka R, Yoshimatsu J, Miyakawa I. Polo-like kinases (Plks) and cancer. Oncogene. 2005;24:287–91. doi: 10.1038/sj.onc.1208272. https://doi.org/10.1038/sj.onc.1208272 [DOI] [PubMed] [Google Scholar]
- 24.Iyer RS, Nicol SM, Quinlan PR, Thompson AM, Meek DW, Fuller-Pace FV. The RNA helicase/transcriptional co-regulator, p68 (DDX5), stimulates expression of oncogenic protein kinase, Polo-like kinase-1 (PLK1), and is associated with elevated PLK1 levels in human breast cancers. Cell Cycle. 2014;13:1413–23. doi: 10.4161/cc.28415. https://doi.org/10.4161/cc.28415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tokumitsu Y, Mori M, Tanaka S, Akazawa K, Nakano S, Niho Y. Prognostic significance of polo-like kinase expression in esophageal carcinoma. Int J Oncol. 1999;15:687–92. doi: 10.3892/ijo.15.4.687. [DOI] [PubMed] [Google Scholar]
- 26.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. https://doi.org/10.1038/nrc2748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Popova SN, Lundgren-Akerlund E, Wiig H, Gullberg D. Physiology and pathology of collagen receptors. Acta Physiol (Oxf) 2007;190:179–87. doi: 10.1111/j.1748-1716.2007.01718.x. https://doi.org/10.1111/j.1748-1716.2007.01718.x [DOI] [PubMed] [Google Scholar]
- 28.Zhu CQ, Popova SN, Brown ER, Barsyte-Lovejoy D, Navab R, Shih W, Li M, Lu M, Jurisica I, Penn LZ, Gullberg D, Tsao MS. Integrin alpha 11 regulates IGF2 expression in fibroblasts to enhance tumorigenicity of human non-small-cell lung cancer cells. Proc Natl Acad Sci USA. 2007;104:11754–9. doi: 10.1073/pnas.0703040104. https://doi.org/10.1073/pnas.0703040104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Navab R, Strumpf D, To C, Pasko E, Kim KS, Park CJ, Hai J, Liu J, Jonkman J, Barczyk M, Bandarchi B, Wang YH, Venkat K, et al. Integrin α11β1 regulates cancer stromal stiffness and promotes tumorigenicity and metastasis in non-small cell lung cancer. Oncogene. 2016;35:1899–908. doi: 10.1038/onc.2015.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang ZY, Xu KS, Wang JS, Yang GY, Wang W, Wang JY, Niu WB, Liu EY, Mi YT, Niu J. Integrin alphanvbeta6 acts as a prognostic indicator in gastric carcinoma. Clin Oncol (R Coll Radiol) 2008;20:61–6. doi: 10.1016/j.clon.2007.09.008. https://doi.org/10.1016/j.clon.2007.09.008 [DOI] [PubMed] [Google Scholar]
- 31.Elayadi AN, Samli KN, Prudkin L, Liu YH, Bian A, Xie XJ, Wistuba II, Roth JA, McGuire MJ, Brown KC. A peptide selected by biopanning identifies the integrin alphavbeta6 as a prognostic biomarker for nonsmall cell lung cancer. Cancer Res. 2007;67:5889–95. doi: 10.1158/0008-5472.CAN-07-0245. https://doi.org/10.1158/0008-5472.CAN-07-0245 [DOI] [PubMed] [Google Scholar]
- 32.Goldberg I, Davidson B, Reich R, Gotlieb WH, Ben-Baruch G, Bryne M, Berner A, Nesland JM, Kopolovic J. Alphav integrin expression is a novel marker of poor prognosis in advanced-stage ovarian carcinoma. Clin Cancer Res. 2001;7:4073–9. [PubMed] [Google Scholar]
- 33.Schittenhelm J, Schwab EI, Sperveslage J, Tatagiba M, Meyermann R, Fend F, Goodman SL, Sipos B. Longitudinal expression analysis of alphav integrins in human gliomas reveals upregulation of integrin alphavbeta3 as a negative prognostic factor. J Neuropathol Exp Neurol. 2013;72:194–210. doi: 10.1097/NEN.0b013e3182851019. https://doi.org/10.1097/NEN.0b013e3182851019 [DOI] [PubMed] [Google Scholar]
- 34.Pan Y, Yang H, Claret FX. Emerging roles of Jab1/CSN5 in DNA damage response, DNA repair, and cancer. Cancer Biol Ther. 2014;15:256–62. doi: 10.4161/cbt.27823. https://doi.org/10.4161/cbt.27823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pan Y, Claret FX. Targeting Jab1/CSN5 in nasopharyngeal carcinoma. Cancer Lett. 2012;326:155–60. doi: 10.1016/j.canlet.2012.07.033. https://doi.org/10.1016/j.canlet.2012.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adler AS, Littlepage LE, Lin M, Kawahara TL, Wong DJ, Werb Z, Chang HY. CSN5 isopeptidase activity links COP9 signalosome activation to breast cancer progression. Cancer Res. 2008;68:506–15. doi: 10.1158/0008-5472.CAN-07-3060. https://doi.org/10.1158/0008-5472.CAN-07-3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Esteva FJ, Sahin AA, Rassidakis GZ, Yuan LX, Smith TL, Yang Y, Gilcrease MZ, Cristofanilli M, Nahta R, Pusztai L, Claret FX. Jun activation domain binding protein 1 expression is associated with low p27(Kip1) levels in node-negative breast cancer. Clin Cancer Res. 2003;9:5652–9. [PubMed] [Google Scholar]
- 38.Kouvaraki MA, Rassidakis GZ, Tian L, Kumar R, Kittas C, Claret FX. Jun activation domain-binding protein 1 expression in breast cancer inversely correlates with the cell cycle inhibitor p27(Kip1) Cancer Res. 2003;63:2977–81. [PubMed] [Google Scholar]
- 39.Pan Y, Yuan Y, Liu G, Wei Y. P53 and Ki-67 as prognostic markers in triple-negative breast cancer patients. PLoS One. 2017;12:e0172324. doi: 10.1371/journal.pone.0172324. https://doi.org/10.1371/journal.pone.0172324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pan Y, Wang S, Su B, Zhou F, Zhang R, Xu T, Zhang R, Leventaki V, Drakos E, Liu W, Claret FX. Stat3 contributes to cancer progression by regulating Jab1/Csn5 expression. Oncogene. 2017;36:1069–79. doi: 10.1038/onc.2016.271. https://doi.org/10.1038/onc.2016.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.