

Summary
Trypanosoma brucei employs multiple mechanisms to evade detection by its insect and mammalian hosts. The flagellar pocket (FP) is the exclusive site of uptake from the environment in trypanosomes and shields receptors from exposure to the host. The FP neck is tightly associated with the flagellum via a series of cytoskeletal structures that include the hook complex (HC) and the centrin arm. These structures are implicated in facilitating macromolecule entry into the FP and nucleating the flagellum attachment zone (FAZ), which adheres the flagellum to the cell surface. TbSmee1 (Tb927.10.8820) is a component of the HC and a putative substrate of polo-like kinase (TbPLK), which is essential for centrin arm and FAZ duplication. We show that depletion of TbSmee1 in the insect-resident (procyclic) form of the parasite causes a 40% growth decrease and the appearance of multinucleated cells that result from defective cytokinesis. Cells lacking TbSmee1 contain HCs with aberrant morphology and show delayed uptake of both fluid-phase and membrane markers. TbPLK localization to the tip of the new FAZ is also blocked. These results argue that TbSmee1 is necessary for maintaining HC morphology, which is important for the parasite’s ability to take up molecules from its environment.
Keywords: Trypanosoma brucei, cytoskeleton, cell cycle, flagellum, polo-like kinase, Flagellar pocket
Introduction
Trypanosoma brucei is an obligate extracellular pathogen that has one of the highest rates of endocytosis known, which reflects a high degree of adaptation to survival under the constant threat posed by the host immune system (Overath and Engstler, 2004; Engstler et al., 2007). The parasite causes human African trypanosomiasis (HAT) and nagana in cattle (Yaro et al., 2016; Büscher et al., 2017). The mammalian-infective bloodstream form of the parasite (BSF) employs antigenic variation and rapid clearance of surface-bound antibodies to evade detection by immune cells (Overath and Engstler, 2004; Engstler et al., 2007; Rudenko, 2011; Mugnier et al., 2015). To complete its life cycle, the parasite must be transmitted via bloodmeal to the tsetse fly, where it differentiates into an insect-infective procyclic form (PCF) that utilizes different strategies to colonize and disseminate within this host (Freeman, 1973; Gibson and Bailey, 2003; Dean et al., 2009; Rotureau et al., 2012). PCFs change their cell surface coat as they progress from the fly midgut to the salivary glands, where the parasite is transmitted via bite, restarting the cycle (Tetley et al., 1987; Roditi et al., 1989; Acosta-Serrano et al., 2001; Urwyler et al., 2007).
Both the BSF and PCF forms of the parasite have highly polarized cell bodies defined by an array of subpellicular microtubules that underlie the cell surface (Vickerman, 1969; Sherwin and Gull, 1989; Robinson et al., 1995). The parasite’s single flagellum is nucleated from a basal body that docks to a flask-like invagination of the plasma membrane near the posterior end of the cell, known as the flagellar pocket (FP), which also serves as the exclusive site for endo- and exo-cytosis (Grünfelder et al., 2003; Engstler et al., 2004; Lacomble et al., 2009; Field and Carrington, 2009; Lacomble et al., 2010). The FP also sequesters certain proteins, such as the transferrin receptor, which would otherwise be antigenic if presented on the cell surface (Salmon et al., 1994; Gadelha et al., 2009; Tiengwe et al., 2017). The neck of the FP is encircled by an electron-dense structure known as the flagellar pocket collar (FPC) (Bonhivers et al., 2008; Lacomble et al., 2009). Electron tomography has shown that the FPC is continuous except for a small disruption caused by a set of four microtubules known as the microtubule quartet (MtQ) (Figure 1) (Lacomble et al., 2009). The MtQ is nucleated between the pro and mature basal bodies and is thought to have the opposing polarity to microtubules present in the subpellicular microtubule array (Taylor and Godfrey, 1969; Robinson et al., 1995; Lacomble et al., 2009). The MtQ wraps around one side of the flagellar pocket and extends towards the anterior end of the cell, interpolating into the corset array. The disruption in the FPC caused by the MtQ forms a small channel that may serve as the primary means of entry into and exit from the FP (Gadelha et al., 2009). This channel may also function to exclude the passive uptake of material above the size of a low-density lipoprotein particle (approximately 22 nm).
Figure 1.
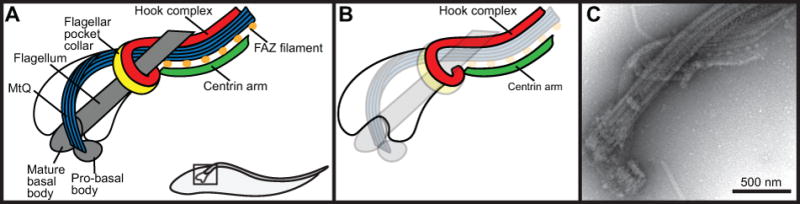
Representation of key cytoskeletal structures surrounding the T. brucei flagellar pocket. (A) Schematic of flagellar pocket associated cytoskeletal structures. (B) Schematic from left panel highlighting the hook complex and centrin arm. (C) Negative stain electron microscopy of an isolated flagellum
On the anterior side of the pocket neck is a cytoskeletal element that has been previously described as the “bilobe structure”. This structure closely associates with the MtQ and marks the origin of the flagellar attachment zone (FAZ), which contains a series of junctional complexes that attach the flagellum to the cell surface (Figure 1A) (Vickerman, 1969; Vaughan et al., 2008; Lacomble et al., 2009; Sunter and Gull, 2016). The bilobe structure was originally defined by the pan-centrin antibody 20H5 and subsequently by antibodies against two specific centrin proteins, TbCentrin2 and TbCentrin4, which localize to the bilobe structure, the basal body, and flagellum (He et al., 2005; Selvapandiyan et al., 2007; Shi et al., 2008). A homolog of the Toxoplasma gondii MORN1 (TgMORN1) protein was shown to partially colocalize with 20H5, although more detailed work employing immunoelectron microscopy showed that TbMORN1 localizes to a structure that has a hook-like extension that wraps around the flagellum and a shank that is adjacent to the centrin-containing structure (Morriswood et al., 2009; Esson et al., 2012). Further analysis showed that the hook portion of the structure is situated above the FPC (Figure 1A–C) (Esson et al., 2012). Other proteins such as LRRP1 and the putative tubulin cofactor TBCCD1 are also bilobe components, with LRRP1 localized to the HC (Gheiratmand and He, 2010; Esson et al., 2012). TBCCD1 did not localize specifically with markers of either structure (André et al., 2013).
Interestingly, TbMORN1 depletion causes a slow growth phenotype in PCFs but is essential in BSFs, producing “big eye” cells that are a hallmark of endocytic defects (Allen et al., 2003; Morriswood et al., 2009; Morriswood and Schmidt, 2015). This difference in phenotype mirrors the relative importance of endocytosis in the two life stages; BSFs constantly internalize and degrade antibodies bound to the cell surface coat while PCFs endocytose at a much slower rate (Natesan et al., 2007; Engstler et al., 2007). Depletion of TbCentrin2 or TbCentrin4 causes defects in FAZ assembly and cytokinesis, but since these proteins are components of several different structures it is difficult to dissect their specific function at the pocket neck (He et al., 2005; Shi et al., 2008; Min Wang et al., 2012). Because the TbMORN1-containing structure and the centrin-containing structure appear to be spatially and functionally distinct, we will adopt a recently-proposed nomenclature that provides individual names for each structure: the hook complex (HC), as defined by TbMORN1, and the centrin arm, as defined by TbCentrin4 (Morriswood and Schmidt, 2015; Morriswood, 2015).
Duplication of the FP is coordinated with the assembly of the new flagellum. While the cell is still at a single FP stage, the probasal body matures and nucleates a new flagellum that is attached to the old flagellum via a mobile transmembrane junction called the flagella connector (FC) (Moreira-Leite, 2001; Lacomble et al., 2010; Höög et al., 2016). Two new probasal bodies are then assembled to provide flagellum nucleation sites for the next round of cell division. The new basal body and flagellum then reposition via a rotation that places them closer to the posterior of the cell; this movement is tightly coupled to the production of a new flagellar pocket (Lacomble et al., 2010).
Assembly of a new centrin arm and HC occurs during FP duplication, although little is known about this process. TbMORN1- and LRRP1-positive “tendrils’” linking the HC to the basal body have been seen during cell division, suggesting that the duplication of the two structures may be linked (Esson et al., 2012). A new FAZ filament becomes visible towards the end of HC and centrin arm duplication. The polo-like kinase homolog in T. brucei (TbPLK) localizes to the FP region during the early stages of cell division and is then loaded onto the anterior tip of the new FAZ once the flagellum has exited the flagellar pocket (de Graffenried et al., 2008; Umeyama and Ching C Wang, 2008; Ikeda and de Graffenried, 2012). TbPLK has been implicated in the duplication of the centrin arm by phosphorylating the centrin-arm resident pool TbCentrin2 during cell division (de Graffenried et al., 2013; Hu et al., 2017). A TbCentrin2 mutant that mimics constitutive phosphorylation of serine 54 blocks duplication of the centrin arm and causes defects in FAZ duplication and cytokinesis (de Graffenried et al., 2013).
A better understanding of the functions of the HC and centrin arm will require the identification and characterization of additional components of these structures. We and others have employed methods such as proximity-dependent biotin identification (BioID), phosphoproteomics, and whole-genome GFP tagging to identify novel proteins that localize to organelles and structures proximal to the FP (Morriswood et al., 2013; McAllaster et al., 2015; Dean et al., 2017). Two BioID screens employing either TbMORN1 or TbPLK as the bait protein identified several novel components of both the HC and centrin arm, including proteins that localize to subdomains of both structures (Morriswood et al., 2013; McAllaster et al., 2015). One component (Tb927.10.8820) localized to the shank of the HC and was also identified as a putative TbPLK substrate via phosphoproteomics (Aslett et al., 2010; McAllaster et al., 2015). We have characterized the function of this protein in procyclic cells and show that it is essential for maintaining the morphology of the HC. We termed this protein TbSmee1 in order to highlight its localization and role on the hook complex (as an homage to Peter Pan). Cells lacking TbSmee1 show a decreased growth rate due to delayed cytokinesis and a diminished rate of uptake into the FP. Our results show that the cytoskeletal elements present at the top of the FP are involved in controlling entry into the compartment, which could serve as a mechanism for ensuring the import of necessary cargo and evading the host immune response.
Results
TbSmee1 localizes to the HC and new FAZ tip
TbSmee1 was previously shown to localize to the shank of the TbMORN1 hook in PCFs (Morriswood et al., 2013; McAllaster et al., 2015). To confirm and extend these results, we appended a triple-HA epitope tag to the 5’ end of one of the endogenous TbSmee1 alleles. We then performed 3-color immunofluorescence microscopy using antibodies against endogenous TbMORN1 and TbCentrin4 to mark the HC and centrin arm respectively, along with anti-HA antibody to label TbSmee1 (Figure 2A). TbSmee1 colocalized primarily with the shank region of the HC, showing minimal overlap with TbCentrin4. The HC and centrin arm are in close proximity to one another, which made it difficult to further refine the localization (Esson et al., 2012). To improve our resolution, we employed electron microscopy to resolve the HC and centrin arm. It has been previously shown that the HC and centrin arm remain tightly associated with the flagellum when cells are extracted with detergent followed by depolymerization of the subpellicular microtubules using 1 M KCl (Figure 1C) (Esson et al., 2012). We used these conditions along with a polyclonal antibody against TbSmee1 to confirm the localization of the protein using immuno-electron microscopy (iEM). Flagella were isolated from wild-type cells and labeled with antibodies against TbSmee1, followed by secondary antibodies conjugated to 10 nm gold particles (Figure 2B). The labeled samples were then negatively stained with aurothioglucose to distinguish the HC and centrin arms. Gold particles were visible along the shank of the HC, which confirmed our immunofluorescence results. TbSmee1 labeling along two of the MtQ microtubules nearest the HC shank was also visible, as was seen with TbMORN1 labeling of isolated flagella (Esson et al., 2012). We also performed dual TbMORN1-TbSmee1 negative-stain immuno-EM using the cell line containing HA-tagged TbSmee1 (Figure S2). The signal for TbMORN1 was present along the full length of the HC and the two proximal MtQ microtubules, with sparse labeling on the centrin arm, while TbSmee1 was found predominantly on the HC shank and the two proximal MtQ microtubules.
Figure 2.
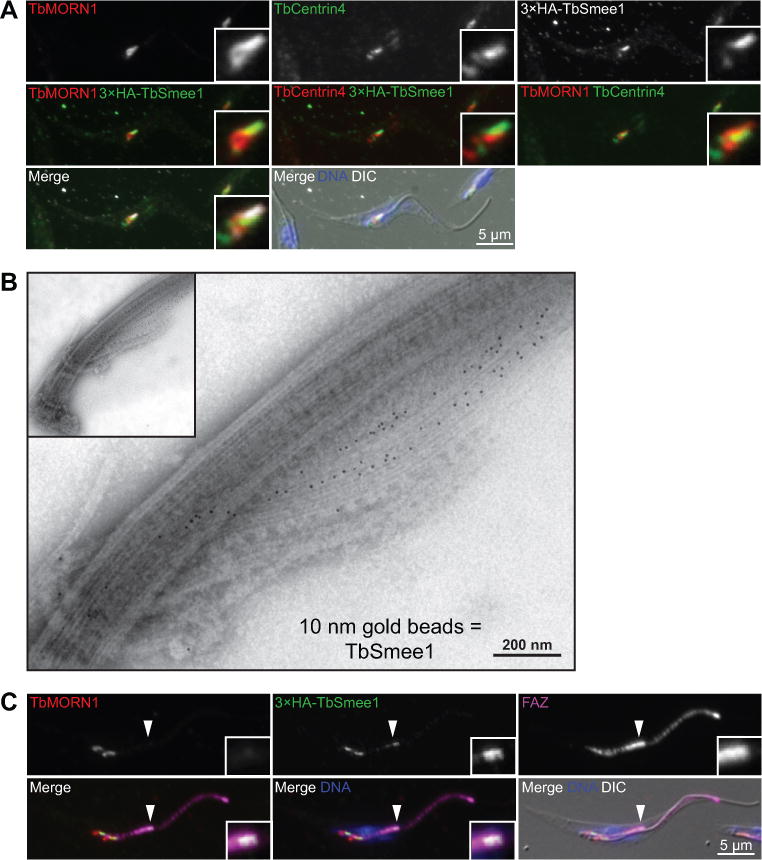
TbSmee1 localizes to the hook complex and the tip of the new FAZ. (A) Cells expressing 3×HA-TbSmee1 fixed and labeled with antibodies against TbMORN1 (TbMORN1; red), TbCentrin4 (TbCentrin4; green, red), and anti-HA (3×HA-TbSmee1; white, green) and DAPI to label the DNA (DNA; blue). The cells were visualized using fluorescence and DIC microscopy. (B) Flagella were isolated from the same cells as in A and labeled with antibodies against TbSmee1 and a secondary antibody conjugated to 10 nm gold particles. The labeled isolated flagella were negatively stained and examined by transmission electron microscopy. (C) The same cells as in A were fixed and labeled with antibodies against TbMORN1 (TbMORN1; red), an antibody against the FAZ (FAZ; magenta), anti-HA (3×HA-TbSmee1; green), and DAPI to label DNA (DNA; blue). The filled arrowhead shows the new FAZ tip in a dividing cell. Insets show threefold magnifications of the area around the tip of the new FAZ.
During our immunofluorescence experiments, we noticed that in dividing cells TbSmee1 showed an additional localization that coincided with the tip of the extending FAZ, which is visible after the duplication of the HC and centrin arm. Several proteins are known to localize to the new FAZ tip, including TbPLK and TOEFAZ1 (also known as CIF1), which are important regulators of cytokinesis in trypanosomes (de Graffenried et al., 2008; McAllaster et al., 2015; Zhou, Gu, et al., 2016). The bilobe component TBCCD1 is also present on the FAZ tip, but it is a constitutive component (André et al., 2013). To confirm this localization, we stained cells containing HA-tagged TbSmee1 with antibodies against TbMORN1 to label the HC, the monoclonal antibody 1B41 that labels the FAZ, and anti-HA to label TbSmee1 (Figure 2C). In cells containing two HCs, we were able to localize the additional TbSmee1 signal to the tip of the new FAZ, as identified by the bright puncta at the tip of 1B41 signal. To confirm this localization, we integrated a triple-Ty1 tag into one endogenous allele of TOEFAZ1 and labeled these cells with anti-Ty1 and antibodies against TbSmee1 (Figure S2). In dividing cells, we were able to see colocalization between TOEFAZ1 and TbSmee1 at the tip of the new FAZ. TbSmee1 signal at the new FAZ tip was also present in cells at later points of the cell cycle that had already undergone nuclear duplication, although the signal follows a pattern similar to that of TbPLK, which declines prior to the initiation of cytokinesis (Ikeda and de Graffenried, 2012).
TbSmee1 depletion leads to slowed growth and defective cytokinesis
We created a TbSmee1 conditional knockout (cKO) cell line in PCFs to investigate protein function. This approach removes both endogenous alleles using homologous recombination and introduces an epitope-tagged, doxycycline-inducible copy of the gene of interest at an ectopic locus (Figure S3) (Ochatt et al., 1999; Urbaniak et al., 2006; de Graffenried et al., 2013). Removing doxycycline from the media blocks expression of the ectopic copy. We generated a cell line containing a doxycycline-inducible copy of TbSmee1 at the rDNA locus tagged with three copies of the Ty1 epitope; the endogenous copies of TbSmee1 were then replaced with puromycin and blasticidin selection markers (Bastin et al., 1996). Titration with antibodies raised against TbSmee1 showed that endogenous levels of protein expression could be maintained with 35 ng mL−1 of doxycycline, which was used for all the subsequent experiments (Figure S4). Cells grown with doxycycline divided at a rate similar to the parental cell line (Wirtz et al., 1999). Upon washout of doxycycline, TbSmee1 expression levels rapidly decreased, as assessed by anti-Ty1 western blotting, and the rate of cell division decreased by approximately 40% when measured at 6 days after washout (Figure 3A; Figure S5). The TbSmee1-depleted cells maintained this slow-growth phenotype over the course of eight days of monitoring, suggesting that the protein is not absolutely essential for the viability of PCFs but is necessary for maximal growth rates.
Figure 3.
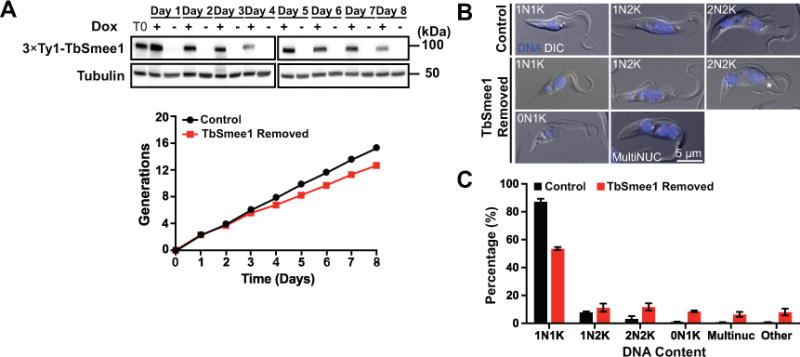
Depletion of TbSmee1 leads to a slow-growth phenotype and causes a cytokinetic delay. (A) TbSmee1 cKO cells were grown for 8 days in either the presence (+; Control) or absence (-; TbSmee1 Removed) of doxycycline. Cells from each culture were monitored by cell count and collected daily to monitor for TbSmee1 depletion by Ty1 western blotting, using tubulin as a loading control. T0 represents the culture at the start of each experiment. (B) Cells expressing TbSmee1 (Control) and cells depleted of TbSmee1 (TbSmee1 Removed) for 6 days were fixed with PFA and treated with DAPI to label DNA. These cells were evaluated by fluorescence and DIC imaging. Asterisk shows a detached flagellum tip. (C) Quantification of DNA content of TbSmee1-depleted cells (TbSmee1 Removed) and control cells (Control) after 6 days. The “other” category contains a mixed population of multi-kinetoplast cells and nucleus-only cells. Data are means ± SD of three independent experiments.
We fixed control- and TbSmee1-depleted cells at various time points and stained them with DAPI to identify cell cycle progression defects (Figure 3B). Trypanosomes maintain their mitochondrial DNA in a structure known as the kinetoplast, which is linked to the basal body. Early in the cell cycle, parasites have one nucleus and one kinetoplast (1N1K). Among the earliest events in cell division is the maturation of the probasal body and extension of the new flagellum (Sherwin and Gull, 1989). The FP duplicates and the two flagella are partitioned into individual pockets, with the new flagellum always present in the more posterior pocket (Lacomble et al., 2010). At this point, the basal bodies begin to separate, which segregates the replicated kinetoplast DNA, producing a 1N2K cell (Robinson and Gull, 1991). Karyokinesis then occurs as the new flagellum continues to elongate, producing a 2N2K cell, which undergoes cytokinesis to produce two 1N1K cells (Sherwin and Gull, 1989; Wheeler et al., 2013). Cells lacking TbSmee1 showed diminished numbers of 1N1K cells (87% to 54%) and an increase in 2N2K cells (3.3% to 12%) (Figure 3C). Anucleate (0N1K) cells and cells containing more than two nuclei were also evident. We also noted elevated numbers of 2N2K cells where the two flagella were not linked by the FC (8.8% ± 7.9 in control cells, versus 34.4% ± 5.6 in cells lacking TbSmee1), which should not occur until after the cleavage furrow has ingressed considerably (Wheeler et al., 2013; Varga et al., 2017). This phenotype is likely due to a delay in furrow ingression, as we have seen previously in cells depleted of the essential cytokinetic protein TOEFAZ1 (McAllaster et al., 2015). Taken together, depletion of TbSmee1 causes a moderate delay in cytokinesis that slows cell growth and leads to the production of a small population of cells lacking the correct complement of DNA.
Loss of TbSmee1 causes a loss of TbPLK from the new FAZ tip
Control and cells that had been depleted of TbSmee1 for 6 days were stained with antibodies against TbPLK and the FAZ marker 1B41 to establish if there were alterations in the localization of the kinase (Figure 4A, B). Under wild-type conditions TbPLK is first visible at the basal body and centrin arm/HC, followed by migration to the tip of the new FAZ and FC (Ikeda and de Graffenried, 2012). The kinase remains on the tip of the new FAZ, although with diminished signal intensity as the cell cycle progresses, until it disappears just prior to cytokinetic furrow ingression. In cells lacking TbSmee1, TbPLK was still present on the basal body and centrin arm/HC during the early stages of cell division, but the kinase did not appear to migrate to the tip of the new FAZ at later time points. The loss of TbPLK localization in TbSmee1-depleted cells did not lead to a substantial decrease TbPLK expression, as measured by TbPLK western blotting (Figure S6A, B). A similar phenotype was previously seen in cells lacking TOEFAZ1, however in that case cytokinesis was compromised (McAllaster et al., 2015; Zhou, Gu, et al., 2016). To confirm that the alteration in TbPLK localization was due to loss of TbSmee1 and not a secondary effect, we analyzed cells after two days of TbSmee1 depletion (Figure S6C). We noted a similar loss of TbPLK localization to the new FAZ tip at this time point, which argues that what we are observing at later time points is the result of a chronic phenotype, which is consistent with the persistent slow-growth of the cells lacking TbSmee1.
Figure 4.
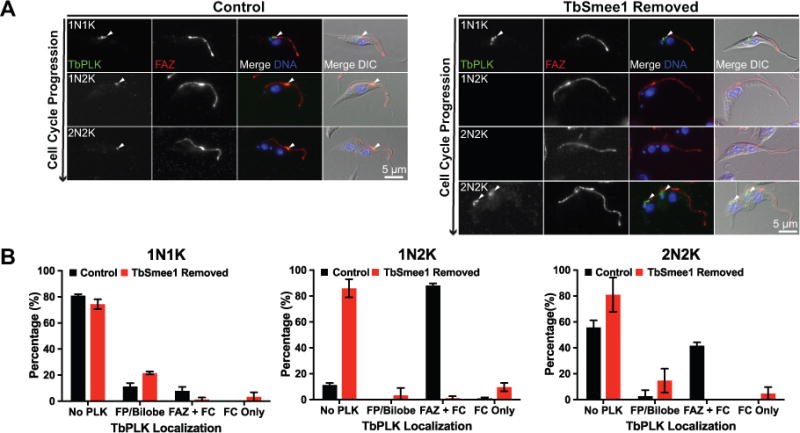
TbPLK localization is absent from the new FAZ tip when TbSmee1 is depleted. (A) Control cells (Control) and cells depleted of TbSmee1 for 6 days were fixed and labeled with antibodies against TbPLK (TbPLK; green) and FAZ (FAZ; red). DAPI was used to visualize DNA (DNA; blue) and the samples were imaged using fluorescence and DIC microscopy. Arrowheads denote PLK localization. (B) Quantitation of the localization of TbPLK in control and TbSmee1-depleted cells (TbSmee1 Removed) in A, sorted by DNA content. Data are means ± SD of three independent experiments.
Cells lacking TbSmee1 show a delay in HC biogenesis
We determined if depletion of TbSmee1 had any effect on morphology or HC number. The duplication of the HC and centrin arm have not been investigated in great detail, so prior to looking at TbSmee1-depleted cells we established distinct morphological categories and put them in temporal order using centrin arm and basal body number to mark cell cycle progression (Figure 5A). We employed antibodies against TbMORN1 to label the HC and a monoclonal antibody against TbCentrin4 to label the centrin arm and the basal body. Most 1N cells contained one basal body complex (comprising the mature- and pro- basal body), one centrin arm, and one HC, which we defined as category I. A subset of 1N cells, termed category II, contained two basal body complexes and two centrin arms, but had only one HC, suggesting that production of a new centrin arm precedes HC duplication. Category III cells contain two centrin arms and two HCs that are paired into two sets containing one of each structure; the pairs were still in close proximity to one another. In category IV cells, the paired structures had begun to separate, which coincided with nuclear duplication.
Figure 5.
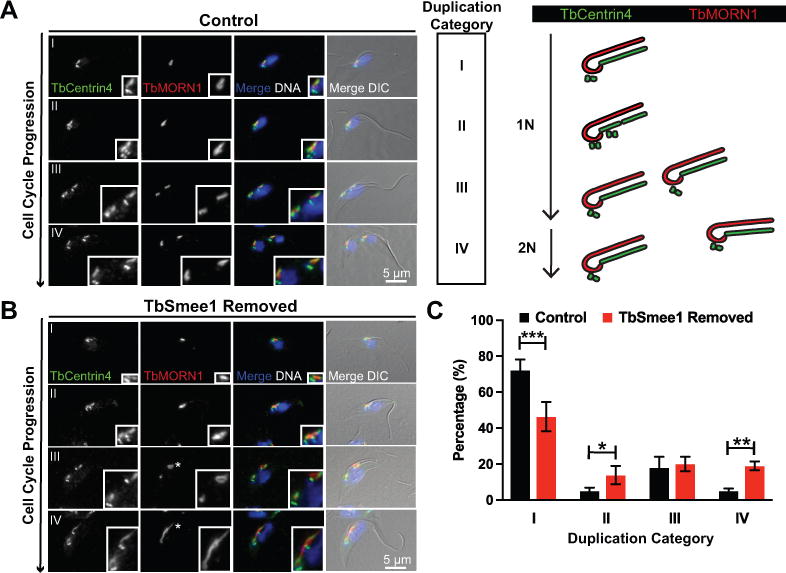
TbSmee1 depletion leads to delayed hook complex replication. (A) Control cells were fixed and labeled with antibodies against TbMORN1 (TbMORN1; red) and TbCentrin4 (TbCentrin4; green); DAPI was used to visualize DNA (DNA; blue). Fluorescence and DIC microscopy was used to analyze the cells. Rows depict cells progressing through the cell cycle and indicate stage of hook complex-centrin arm replication as depicted in the schematic representation. (B) Cells depleted of TbSmee1 for 6 days (TbSmee1 Removed) were fixed, antibody labeled, and visualized as above. Asterisk highlights hook complexes that have altered morphology. (C) Quantitation of hook complex-centrin arms from control and TbSmee1-depleted cells (TbSmee1 Removed) found in each duplication stage. Data are the means ±SD of three independent experiments. *, P ≤ 0.05; **, P ≤ 0.01; ***, P≤ 0.001 (multiple unpaired two-tailed Student’s t test).
With separate categories established to identify HC and centrin arm duplication intermediates, we next evaluated these structures in TbSmee1-depleted cells. Centrin arm duplication appeared unaffected by TbSmee1 depletion, so we separated cells into the previously-defined categories based exclusively upon this unperturbed structure. Cells lacking TbSmee1 showed a decrease in category I cells and a concomitant increase in category II and IV cells (Figure 5B, C). It is likely that the increase in category IV cells is caused by the delay in cytokinesis that we observed in our DAPI/DIC experiments. The increase in category II cells argues that cells lacking TbSmee1 have delayed the assembly of the HC.
TbSmee1-depleted cells have altered HC morphologies
Cells lacking TbSmee1 had substantial defects in HC duplication and developed structures with complex morphologies. We categorized the defects in HCs lacking TbSmee1, confining our initial analysis to cells containing a single copy of the HC and centrin arm to minimize contributions from cells undergoing division because the morphology of the HC can vary during its replication (Figure 6A). We established several different morphological categories for cells containing a single HC (Figure 6B). In control cells the majority of HCs, as labeled by TbMORN1, were positioned alongside the centrin arm and wrapped around the flagellum (Figure 6A, C). Approximately 15% of cells contained a “branched” complex, which may mark an early duplication intermediate. In a majority of cells depleted of TbSmee1, the HC lost its normal morphology and formed a more oval-like shape, which we termed “teardrop” (Figure 6A, C). There was also a slight elevation in the number of branched complexes. Additionally, we observed small numbers of cells with shortened, bar-like HCs and a similar number of cells with complexes where the shank region was substantially elongated. Cells analyzed after two days of TbSmee1 depletion had a similar distribution of HC morphologies, showing that the phenotypes arise quickly and are not due to secondary effects (Figure S7).
Figure 6.
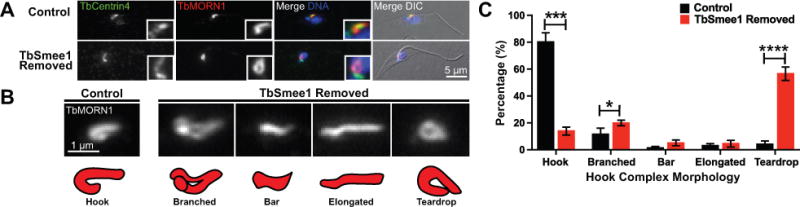
Depletion of TbSmee1 leads to altered hook complex morphologies. (A) Control cells and TbSmee1-depleted cells (TbSmee1 Removed) were fixed and labeled with an antibody against TbCentrin4 (TbCentrin4; green), antibodies against TbMORN1 (TbMORN1; red), and DAPI to visualize DNA (DNA; blue). A non-dividing 1N1K cell is shown. Insets are threefold magnifications. (B) Six-fold magnifications of TbMORN1-labeled (TbMORN; white) hook complexes with varying morphologies from non-dividing 1N1K control and TbSmee1-depleted cells (TbSmee1 Removed) categorized as shown in the schematic. (C) Quantification of hook complex morphologies in non-dividing 1N1K control and TbSmee1-depleted (TbSmee1 Removed) cells. Data are means ± SD of three independent experiments. *, P ≤ 0.05; ***, P≤ 0.001; ****, P ≤ 0.0001(multiple unpaired two-tailed Student’s t test).
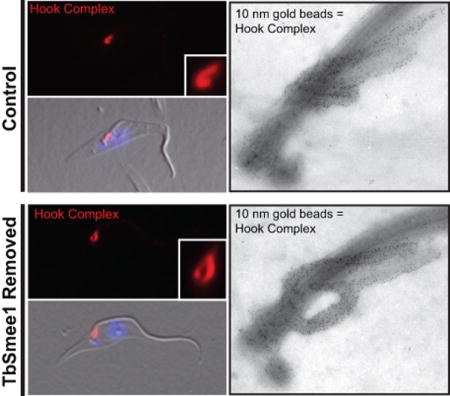
Our immunofluorescence experiments showed substantial alterations in HC morphology in cells lacking TbSmee1, but the small size (~2 µm) of the structure makes it difficult to study these alterations in more detail at the light level. To improve our resolution, we performed iEM on flagella from both control and TbSmee1-depleted cells that were isolated via detergent extraction and corset microtubule depolymerization (Esson et al., 2012). The isolated flagella were first labeled with antibodies against TbMORN1 to identify the HC and centrin arm, followed by secondary antibodies conjugated to 10 nm gold particles. The samples were then labeled with aurothioglucose as a negative stain and imaged using transmission electron microscopy. The shank and hook regions of the HC were heavily decorated with gold particles, while the centrin arm, which sits opposite to the shank, had lower levels of TbMORN1 labeling. In cells lacking TbSmee1, we observed flagella with smaller HCs (Figure 7A, top right), similar to the bar-like structures we observed by immunofluorescence. HCs where the hook segment was no longer wrapped tightly around the flagellum were also visible; these altered HCs have similarity to the teardrop morphology (Figure 7A, bottom left). We also observed flagella where the TbMORN1 labeling decorated an elongated structure on the centrin arm-side of the flagellum, which could be an extension of the HC or additional TbMORN1 labeling on the centrin arm (Figure 7A, bottom right). This altered morphology is most similar to the “elongated” HCs that were observed by immunofluorescence. In general, we noted elevated TbMORN1 labeling on the centrin arm and that the overall pattern of TbMORN1 labeling appeared to extend beyond its normal endpoint in cells lacking TbSmee1 (Figure 7A, B). We decided to characterize these changes in more detail using quantitative methods.
Figure 7.
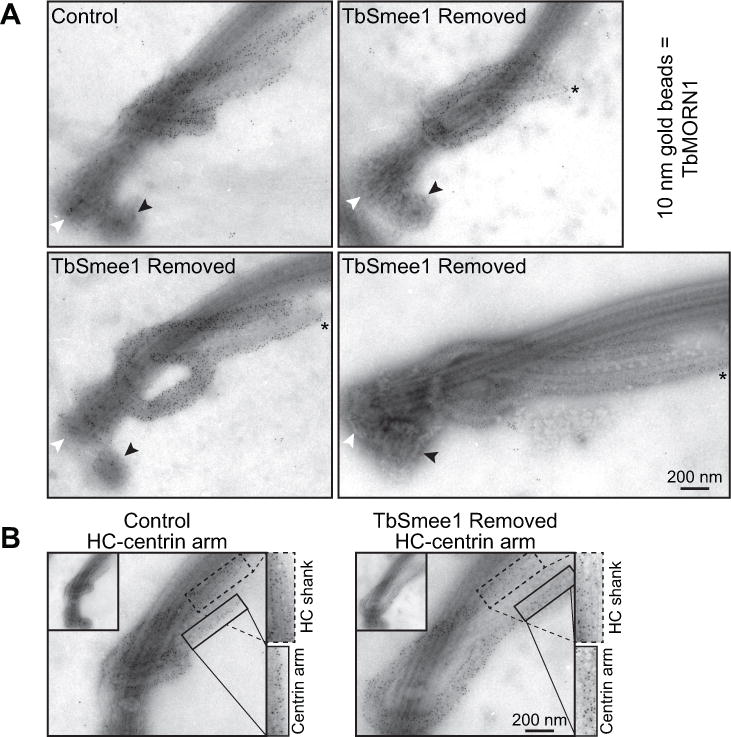
Ultrastructural examination of hook complex and centrin arm by immuno-electron microscopy. (A) Flagella were isolated from control and cells depleted of TbSmee1 (TbSmee1 Removed) for 6 days, and then labeled with antibodies against TbMORN1 and a secondary antibody conjugated to 10 nm gold beads. Black arrowhead denotes the probasal body, while white arrowhead denotes the mature basal body. Asterisk demarcates the elongation of the centrin arm. (B) Isolated flagella from control and TbSmee1-depleted cells (TbSmee1 Removed) on day 6. Boxes highlight density of TbMORN1 10 nm gold particles on HC shank (dashed box) and centrin arm (solid box).
TbSmee1-depleted cells have altered TbMORN1 distribution
We designed a graphical user interface (GUI) in MATLAB using functions found in the Image Processing Toolbox™ to allow for semi-automated quantitation of gold particles on present on HC-centrin arm pairs. In addition to quantitating gold particles on the whole structure, this GUI allows the user to segment each of the structures into 200 nm regions, leading to the quantitation of the relative density of gold particles per 200 nm region (Figure 8A). Using isolated flagella labeled with anti-TbMORN1 antibodies and secondary antibodies conjugated to 10 nm gold particles, we segmented a total of 75 HC-centrin arms from three independent experiments of both control and TbSmee1-removed cells. We used the total number of 200 nm segments per HC-centrin arm as a measure of combined length for the two structures. In control cells, more than half the combined structures contained 15 or 16 segments, with 90% falling within 13 and 17 segments (Figure 8B). Removal of TbSmee1 resulted in structures with significant variability in size, with more than 30% of cells containing combined structures of more than 18 segments, including 15% that were composed of more than 20 segments.
Figure 8.
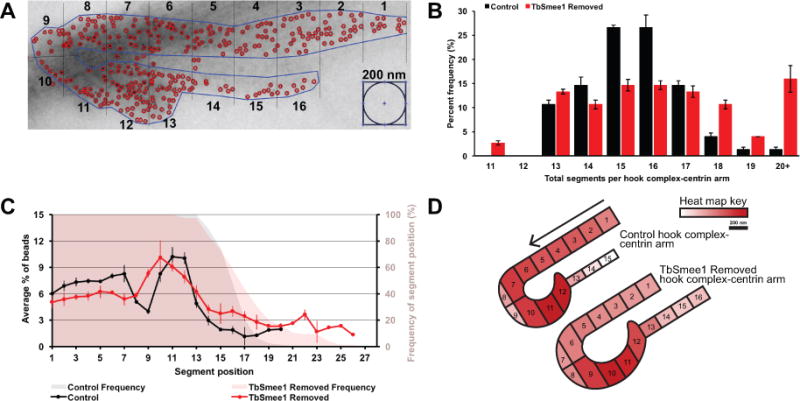
Loss of TbSmee1 leads to HC-centrin arms with varying sizes and redistribution of TbMORN1 labeling. (A) Representative output of MATLAB GUI created to segment HC-centrin arms into 200 nm regions and count TbMORN1 immunogold within each region. (B) Quantitation of HC-centrin arm segments from control and TbSmee1 removed isolated flagella on day 6. Data is mean ± SEM of three independent experiments. (C) Quantitation of the percentage of TbMORN1 immunogold particles in each segment position and the frequency of the presence of the segment position on day 6 in control and TbSmee1-removed cells. Data is mean ± SD of three independent experiments. (D) Heat map of the percentage of TbMORN1 immunogold particles per segment in representative illustrations of median-length HC-centrin arms in control and TbSmee1-removed cells.
Given the increased number of 200 nm segments in TbSmee1-removed HC-centrin arms, we quantified the distribution of gold particles along the combined structure. This was accomplished by using the percent density of gold particles per 200 nm segment. In control cells, the segments representing the shank of the HC each contained between 6% and 8% of the total gold particles (Figure 8C). At the most posterior region of the HC, which starts to wrap around the flagellum, the number of gold particles per segment decreases because this curved region leaves part of the 200 nm segments empty. The final segments of the HC that lead to the centrin arm contain the highest concentration of TbMORN1, after which the particle density declines substantially on the segments comprising the centrin arm. In cells lacking TbSmee1, the distribution of TbMORN1 is altered along the combined structures. We created a heat map schematic to represent the alteration in TbMORN1 gold particle distribution on median-length HC-centrin arm structures in TbSmee1-depleted cells (Figure 8D). Color representation of bead distribution along the median length of the HC-centrin arm pair shows that the concentration of gold particles on the shank of the HC from TbSmee1-depleted cells is lower than in control cells, while there is a concomitant increase in the concentration of beads on the centrin arm. While the distribution of gold particles and the median length of the combined HC-centrin arm structure were altered in cells lacking TbSmee1, the total number of beads on control and experimental conditions was not significantly different (Figure S8A). When control and TbSmee1-depleted structures are plotted in terms of segment length and total number of particles, there is no correlation between the longer structures and increased number of gold particles (Figure S8B). These results strongly argue that the additional gold particle labeling seen on the centrin arm structure in TbSmee1-depleted cells does not arise from elevated levels of TbMORN1 incorporation. To confirm this result, we made lysates from control cells and cells lacking TbSmee1 over the course of 8 days and blotted them with anti-TbMORN1 antibodies. We observed no significant difference in TbMORN1 levels at any time point (Figure S8C).
Re-expression of TbSmee1 restores cytokinesis and HC morphology
We sought to determine if cells could regenerate HCs with the correct morphology if TbSmee1 expression was restored. Many cytoskeletal structures, such as the basal body array found in Paramecium, rely on previously-assembled structures for structural and positional information to direct their assembly (Beisson and Sonneborn, 1965; Beisson et al., 2001). We took cells that had been depleted of TbSmee1 for 6 days, which showed the expected 40% growth defect compared to control cells, and restored protein expression by adding doxycycline back to the media (Figure 9A, Figure S9). The treated cells rapidly turned on TbSmee1 expression and began to grow at a similar rate to control cells. We assessed morphology and DNA content using DAPI/DIC in cells that had constitutively expressed TbSmee1, cells lacking TbSmee1, and rescued cells (Figure 9B, Figure S10A–C). The rescued cells re-established normal cellular morphology. Moreover, rescued cells restored HC morphology by anti-TbMORN1 antibody labeling at day 7 and day 9 (1 or 3 days after adding-back doxycycline). Remarkably, within 1 day of TbSmee1 rescue, 65% of cells had HCs with normal morphology, compared to 15% in cells lacking TbSmee1 expression (Figure S5A, B). After 3 days of TbSmee1 rescue the morphology of the HC was fully restored in all cells.
Figure 9.
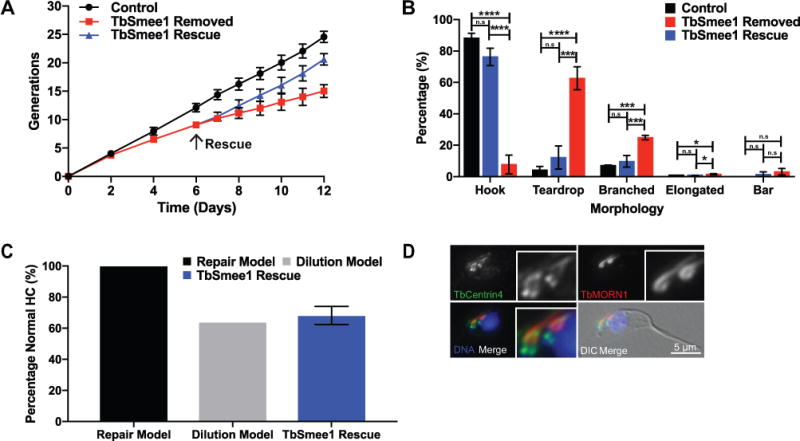
Restoring TbSmee1 in depleted cells leads to recovery of cell growth and hook complex morphology. (A) Growth of control and TbSmee1-depleted cells (TbSmee1 Removed) was monitored for 6 days by cell growth before the addition of doxycycline (arrow) to rescue the TbSmee1-depleted cells (TbSmee1 Rescue). Monitoring continued for an additional 6 days. Data are means ± SD of three independent experiments. (B) Quantification of hook complex morphologies on day 9 in non-dividing 1N1K control, TbSmee1-depleted (TbSmee1 Removed) and TbSmee1-rescued (TbSmee1 Rescue) cells. (C) Modeling of hook complex morphology restoration after 24 hours of doxycycline re-addition. (D) A dividing TbSmee1-rescued cell detergent extracted, fixed, and labeled with antibodies against TbCentrin4 (green) and antibodies against TbMORN1 (red). DAPI was used to label DNA (DNA; blue). DIC and fluorescence microscopy were used for imaging. Data are means ± SD of three independent experiments. n.s, not significant; *, P ≤ 0.05, ***, P≤ 0.001; ****, P ≤ 0.0001(One-way ANOVA).
There are two possible pathways for HC restoration upon TbSmee1 re-expression: newly-formed HCs are produced with the correct morphology while the aberrant ones remain, or both old and new HCs are restored once TbSmee1 is available. If the first model is correct, we would expect morphologically aberrant HCs to remain in the population until they are removed by dilution during cell passaging. We should also be able to observe dividing cells with an aberrant HC in the anterior of the cell and a newly-formed, restored HC in the posterior position, which reflects the inheritance pattern of the structure. If the second model is correct, all the HCs in the population should be rapidly restored regardless of when they were formed.
To distinguish between these two models, we depleted TbSmee1 expression for 6 days, then restored expression for 24 hours, followed by fixation and staining with antibodies against TbMORN1 and TbCentrin4 to label the HC and centrin arm, respectively. Tetracycline-inducible expression in trypanosomes has been previously shown to reach steady-state levels within 5 hours of tetracycline addition (Wirtz and Clayton, 1995; Wirtz et al., 1999). Since the TbSmee1-rescued cells underwent 1.44 cell divisions within the 24-hour period of TbSmee1 re-expression, we would expect every cell in the culture to have undergone at least one round of division with TbSmee1 expression turned on. In contrast to the model where HC morphology is restored upon re-expression of TbSmee1, we observed that approximately 30% of the HCs in the culture still had aberrant morphology, which is consistent with the model where old HCs cannot be repaired and must be lost from the culture via dilution (Figure 9C). We also observed that in the subset of dividing cells containing two HCs, aberrant HCs were present on the anterior side of the cell, marking them as the older structure, while the newly-formed structure had the correct morphology (Figure 9D). These results argue that restoring HC morphology upon TbSmee1 re-expression can only occur on newly-formed HCs and that the old HCs cannot be repaired.
Uptake of fluid-phase and membrane markers is delayed in cells lacking TbSmee1
The substantial defect in HC morphology observed in cells lacking TbSmee1 led us to investigate potential alterations to rates of entry into the FP. In BSFs, depletion of TbMORN1 causes rapid lethality, possibly due to defects in endocytosis, but little is known about changes in trafficking in procyclic cells lacking HC components (Morriswood and Schmidt, 2015). To test membrane uptake, we employed the lipid-intercalating dye FM4-64FX, which is rapidly internalized into the FP (Figure 10A) (Hall et al., 2005; Bonhivers et al., 2008; Absalon et al., 2008). Both control and TbSmee1-depleted cells were labeled with dye at 4 °C, then warmed to 27 °C to allow uptake, followed by washing at 4 °C to remove excess dye. The cells were then treated with Hoechst to label DNA and imaged live to determine the distribution of FM4-64FX. We chose to avoid fixation of the cells because we noted that cells lacking TbSmee1 were somewhat fragile and did not fix well after the multiple incubation and centrifugation steps necessary for the FM4-64FX labeling. We confined our analysis to 1N1K cells to limit complications from dividing cells. In control samples, cells that had been held at 4 °C showed labeling primarily at the cell surface, with limited pocket labeling (Figure 10B). Control cells that were incubated at 27 °C had labeling primarily at the flagellar pocket, with some residual labeling of the cell surface. Cells lacking TbSmee1 showed a substantial reduction in flagellar pocket labeling (Figure 10C).
Figure 10.
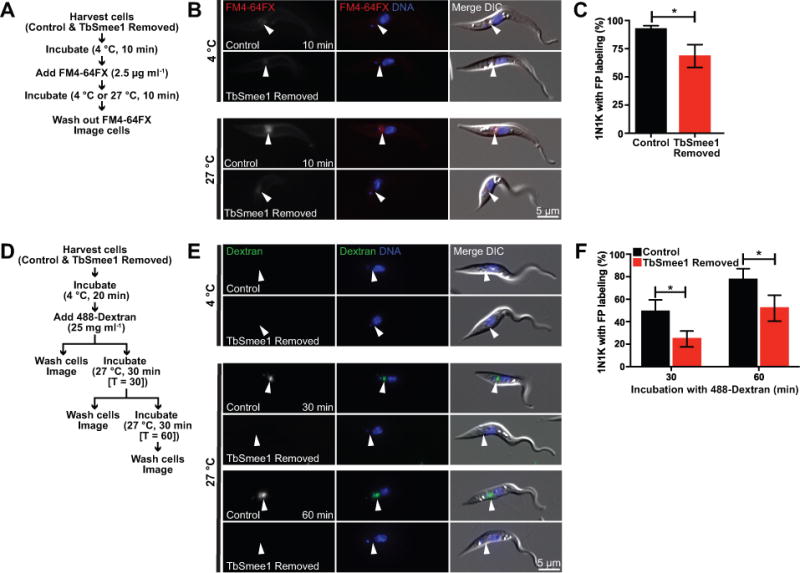
Depletion of TbSmee1 results in delayed uptake of membrane and fluid-phase markers into the flagellar pocket. (A) Workflow of FM4-64FX uptake protocol. (B) Live cell DIC and fluorescence microscopy of representative control cells treated with FM4-64FX (FM4-64FX; red) at either 4 °C or 27 °C and Hoechst to label DNA (DNA; blue). Closed arrowhead indicates FP region. (C) Quantitation of 1N1K control and TbSmee1-removed cells with FM4-64FX labeling in the flagellar pocket. Data are means ± SD of three independent experiments. *, P ≤ 0.05 (Student’s t test). (D) Workflow of 488-Dextran uptake protocol. (E) Live cell DIC and fluorescence microscopy of representative control cells treated with fluorescent dextran (Dextran; green) at either 4°C or 27°C and Hoechst to label DNA (DNA; blue). Closed arrowhead indicates FP region. (F) Quantitation of 1N1K control and TbSmee1-removed cells with fluorescent dextran in FP region. Data are means ± SD of three independent experiments. *, P ≤ 0.05 (multiple two-tailed unpaired Student’s t test).
We tested the ability of cells lacking TbSmee1 to internalize fluorescently-labeled dextran into the FP to determine if the rate of fluid-phase uptake was also altered. Alexa 488-conjugated 10 kDa dextran has been previously shown to enter the FP in both PCFs and BSFs (Figure 10D) (Engstler et al., 2004; Hall et al., 2004; Hall et al., 2005; Morriswood and Schmidt, 2015). We cooled control and TbSmee1-depleted cells to 4 °C to block endocytosis, followed by labeling with Alexa-488 dextran. A portion of the cells were then washed, labeled with Hoechst, and then imaged live to determine dextran localization. The rest of the cells were warmed to 27 °C for either 30 or 60 minutes, followed by washing, labeling with Hoechst, and live cell imaging of 1N1K cells (Figure 10E). At 4 °C, neither control nor TbSmee1-depleted cells acquired any FP-specific dextran labeling. In the control sample, 30 min of incubation with dextran at 27 °C yielded approximately 50% of control cells with dextran labeling in their FPs, while after 60 mins this number increased to 80% (Figure 10F). Cells lacking TbSmee1 had fewer FPs that were labeled with Alexa 488 dextran, with only 22% after a 30-min incubation and 50% after a 60-min incubation.
While depletion of TbSmee1 led to a clear difference in the rate of dextran uptake, it was unclear if there was a difference in the amount of dextran within the FPs. To test this, we selected all the 1N1K cells that had clear dextran labeling in their FPs from both control and TbSmee1-depleted cells and measured the fluorescence intensity of each labeled flagellar pocket (Figure S11). There was no significant difference in the distribution of dextran signal intensities among the two sets of FPs, which suggests that the capacity of the TbSmee1-depleted FPs was not altered.
Defects in the endo-lysosomal pathway could cause alterations in the rate of uptake from the FP by influencing the rate of endocytosis. While TbSmee1 does not localize to any endomembrane compartments, it is possible that the protein influences these structures indirectly, which could contribute to the delayed uptake phenotype. The LAMP-like protein p67 is a lysosomal marker in T. brucei that has been used to assess the size and morphology of post-Golgi secretory compartments, which are frequently altered if rates of flux from the FP change (Kelley et al., 1999; Peck et al., 2008). We stained control cells and cells lacking TbSmee1 with an antibody against p67 to determine the size and morphology of the lysosomal compartment (Figure S12). In control cells, the p67 signal in a single focus between the nucleus and kinetoplast, as has been reported by others (Brickman and Balber, 1993; Alexander et al., 2002). In the absence of TbSmee1, the p67 appeared very similar to control cells, forming a single, well-defined puncta. This contrasts with cases such as the depletion of µ1 subunit of the adaptor complex, where p67 is mislocalized to the cell surface, or the dominant-negative form of RAB4 and ARF1 RNAi, where the lysosomal complex increased in size significantly due to trafficking defects (Hall et al., 2005; Price et al., 2007; Tazeh et al., 2009).
Discussion
In this work, we have characterized the HC component TbSmee1 as a means to understand the function of this enigmatic cytoskeletal structure. TbSmee1 localizes to the shank portion of the HC and is necessary for maintaining the correct distribution of TbMORN1 on the HC (Morriswood et al., 2013; McAllaster et al., 2015). In the absence of TbSmee1, cells divide at a slower rate and show evidence of cytokinetic defects such as the appearance of anucleate and multinucleated cells. TbPLK, which is necessary for the duplication of many essential cytoskeletal structures, no longer localizes to the tip of the new FAZ. The overall morphology of the HC is also disrupted, leading to structures with extended shanks or diminished association with the flagellum. These morphological defects correlate with a decreased rate of cargo entry into the FP, as shown by our FM4-64FX and Alexa-488 dextran assays, and may indicate that the shape of the HC affects the rate at which cells uptake material from their surroundings.
In the absence of TbSmee1, TbMORN1 is present on the centrin arm at higher levels, along with an increase in the total length of the combined HC-centrin arm complex. The most direct explanation for this observation is that TbSmee1 is responsible for maintaining the morphology of the HC and that in its absence the distribution of HC components is perturbed, although the hierarchy of these effects has not been established. The morphological defects appear to arise during the duplication of the HC, which is significantly delayed in the absence of TbSmee1, producing asymmetric intermediate structures that could result in the large variation in total HC-centrin arm structure size that we observe by EM and IF. There is no apparent defect in the duplication or morphology of the centrin arm, which suggests that the duplication of the two structures is separable and can occur autonomously. This autonomy may extend to function, as RNAi of primarily HC proteins such as TbMORN1 appears to cause defects in entry of material into the FP, while the absence of primarily centrin arm proteins such as TbCentrin2 and TbCentrin4 causes defects in new FAZ filament assembly, leading to detached flagella (Selvapandiyan et al., 2007; Shi et al., 2008; Morriswood et al., 2009; de Graffenried et al., 2013).
The decreased growth observed in the absence of TbSmee1 may be due to defects in multiple aspects of cell division. Delayed assembly of the HC may retard cell cycle progression, either by blocking the assembly of a new FP or the segregation of replicated FPs. It is not known if HC duplication occurs de novo or if it relies on the old structure as a template, but cells that inherit a HC with altered morphology are still viable and can produce normal structures once TbSmee1 expression is restored. Since endocytosis and general flux through the FP is less vital to PCFs, they are more likely to tolerate defects in the HC structure (Langreth and Balber, 1975; Natesan et al., 2007). Depletion of TbMORN1 in PCFs leads to a slow-growth phenotype, but depletion in BSFs rapidly produces cells with highly enlarged FPs and cell death via lysis (Morriswood et al., 2009). The speed and severity of the cell lysis in BSFs makes it very difficult to assess alterations to the HC, which may take one or two cell divisions to manifest.
The localization of TbSmee1 to the HC shank and the tip of the new FAZ suggests interplay between the trypanosome cytokinetic complex and the cytoskeletal structures surrounding the FP. The function of TbSmee1 at the tip of the extending FAZ appears to involve the recruitment of TbPLK, which initially localizes to the centrin arm/HC region before loading onto the FAZ tip later in the cell cycle. The specific localization of the kinase to the HC or centrin arm has not been established, although TbPLK does phosphorylate TbCentrin2 and TbSmee1 is a potential substrate (de Graffenried et al., 2013; McAllaster et al., 2015). TbPLK recruitment to the centrin arm/HC remains in the absence of TbSmee1, so the interaction between TbPLK and TbSmee1 appears confined to the FAZ tip. A set of other proteins including TOEFAZ1, CIF2, and TbAUK1 reside on the new FAZ tip and contribute to the positioning of the cytokinetic furrow (McAllaster et al., 2015; Zhou, Gu, et al., 2016; Zhou, Gu, et al., 2016). Depletion of TOEFAZ1 causes significant defects in cytokinesis and blocks TbPLK recruitment to the FAZ, suggesting that kinase activity at the point of furrow ingression may be important for completion of cell division (McAllaster et al., 2015; Zhou, Gu, et al., 2016). However, previous work shows that TbPLK inhibition late in the cell cycle, when it is present on the FAZ tip, has minimal effect on growth (Li et al., 2010; Lozano-Nunez et al., 2013). Similarly, TbSmee1 depletion blocks TbPLK recruitment to the new FAZ but has only a limited effect on cell division, which is consistent with the kinase having limited function late in the cell cycle. We have recently identified the domain within TOEFAZ1 that is necessary to retain TbPLK at the tip of the new FAZ and showed that this domain is dispensable trypanosome growth, emphasizing that the kinase does not have an essential role at this location at later points of the cell cycle (Sinclair-Davis et al., 2017). It is more likely that TbPLK inhibition blocks cytokinesis indirectly by inhibiting new FAZ formation early in the cell cycle, which makes it impossible to correctly position the complex that triggers cytokinesis. Based on localization data, it is possible that the cytokinetic complex is assembled at the anterior end of the duplicating HC-centrin arm complex and is then loaded onto the new FAZ tip. This may explain how TbPLK and TbSmee1 end up as part of the cytokinetic complex, even if they are dispensable components.
TbSmee1 depletion causes a delay in the entry of both membrane and fluid phase markers into the flagellar pocket. The constriction at the top of FP adds another point for controlling entry and exit from the cell, possibly as a way to help protect against detection by the host. Tracking entry into the pocket with gold-conjugated cargo has shown that material accumulates at a small channel created by the MtQ as it traverses the FPC, suggesting that this path constitutes the primary mode of entry into the FP (Gadelha et al., 2009). It is remarkable that this small deformation can handle the levels of flux present in BSFs, considering the high rate of internalization necessary to clear bound antibody. Depletion of TbMORN1 in BSFs appeared to block the entry of the lectin ConA, which binds to surface glycoproteins, and diminished the entry of the fluid phase marker BSA. Fluorescent dextran, which has a significantly smaller hydrodynamic radius than either BSA or ConA, was able to access the pocket normally but was not endocytosed (Morriswood and Schmidt, 2015). While the morphology of the HC was not assessed in BSFs, these results suggest that TbMORN1 is necessary for allowing access to the FP and that in its absence the FPC may form a tighter seal than it should, which limits the ability of larger cargo to enter the pocket effectively. It is possible that the diminished rate of uptake we have observed in PCFs lacking TbSmee1 is due to the alterations in HC shape and distribution of HC components along the structure. If the HC does not wrap around the flagellar neck tightly it may not be able to maintain the channel properly, which would slow down the rate of uptake significantly. The HC and FPC may play combating roles at the top of the FP, with the FPC forming a seal and limiting entry, while the HC deforms the seal at the point where the MtQ is present so that material can enter. Inhibiting the function of the HC by altering its shape may allow the FPC to seal the top of the pocket too tightly, which then limits uptake. In PCFs this delay in uptake may not have a strong effect because the endocytic rate is quite slow, so a delay in FP entry would not affect the overall rate of endocytosis (Langreth and Balber, 1975; Natesan et al., 2007). This would be quite different in BSFs, where the endocytic rate is very high (Engstler et al., 2004). Even a small delay in uptake may alter finely-tuned equilibria and disrupt the trafficking rates of essential receptors.
Experimental Procedures
Cell Culture
All experiments were performed using Lister 427 T. brucei brucei cells and the 427 derived 29-13 cell line, which contains the machinery for doxycycline-mediated gene expression (Wirtz et al., 1999). Cell growth was monitored using a particle counter (Z2 Coulter Counter; Beckmann Coulter).
Antibodies
Antibodies were obtained from the following sources: anti-Ty1 from Cynthia He (National University of Singapore, Singapore), 1B41 from Linda Kohl (Centre National de la Recherché Scientifique, Paris, France), anti-TbMORN1 and anti-TbSmee1 from Brooke Morriswood (University of Würzburg, Würzburg, Germany), and anti-alpha tubulin (Sigma). The TbPLK antibodies and TbCentrin4 monoclonal antibody have been described previously (de Graffenried et al., 2008; Ikeda and de Graffenried, 2012). The goat anti-HA and biotin-conjugated donkey anti-goat antibodies are from Jackson ImmunoResearch (West Grove, PA). Gold conjugated secondary antibodies and streptavidin are from BBInternational (Cardiff, UK). The specificity of the TbSmee1 antibody is shown in Figure S13.
Immunofluorescence
Cells were harvested, washed once in phosphate-buffered saline (PBS), then adhered to coverslips by centrifugation. For DNA and DIC analysis, cells were then fixed in 4% paraformadehyde in PBS for 15 minutes at room temperature, followed by three washes in PBS before mounting. For direct methanol fixation, the cells were immersed in methanol cooled to −20 °C for 20 minutes, air dried, then rehydrated in PBS. For cytoskeletal extractions, cells were incubated in extraction buffer (PEME-NP-40; 0.1 M PIPES, pH 6.9, 2m M EGTA, 1 mM MgSO4, 0.1 mM EDTA, 0.5% NP-40) for 5 minutes at room temperature, washed three times in PBS, then fixed in −20 °C methanol for 10 minutes and rehydrated in PBS. Staining with primary and secondary antibodies was performed as described previously (McAllaster et al., 2015).
Flagellar isolation and immuno-gold electron microscopy
For negatively stained isolated flagella, cells were pelleted at 800 × g, washed twice with PBS, and spun onto glow-discharged Formvar- and carbon-coated grids (EMS). Grids were then inverted onto a drop of PEME buffer plus 1% NP-40 (5 minutes, RT), then washed three times in PEME buffer plus 1% NP-40 (RT) and incubated on a drop of PEME buffer plus 1% NP-40 and 1 M KCl (20 minutes, 4 °C). Cells were then washed four times in PEME buffer plus 1% NP-40 and 1 M KCl (4 °C). Finally, the grids were washed four times in PEME buffer (without KCl, RT), fixed in 2.5% glutaraldehyde, rinsed in ultra-pure water, and stained with 1% aurothioglucose (Sigma-Aldrich).
For isolated flagella that were labeled with immunogold particles, grids were treated as above, except after the final four PEME washes without KCl at RT, grids were blocked by washing in five drops of 2% BSA in PBS and incubated in primary antibody diluted in blocking buffer (1 hr, RT). The grids were then moved through seven drops of blocking solution and incubated in secondary antibody for 1 hour at RT (BBInternational). Grids were then incubated in blocking solution (5 min, RT), fixed, and stained with aurothioglucose, as above (Sigma-Aldrich). Images were taken on a Phillips 410 transmission electron microscope at 100kV equipped with a 1k × 1k Advantage HR CCD camera from Advanced Microscopy Techniques (AMT) using AMT imaging software.
For the dual immunogold labeling, isolated flagella from a cell line in which TbSmee1 was labeled with a 3X-HA epitope tag were prepared and blocked as above. Grids were then incubated with goat anti-HA primary (1 hr, RT) (Jackson ImmunoResearch) and moved through seven drops of blocking solution. Grids were incubated with biotin-conjugated donkey anti-goat secondary antibody (Jackson ImmunoResearch), as well as rabbit anti-TbMORN1 primary (1 hr, RT). Finally, grids were moved through five drops of blocking solution and incubated with 15 nm gold-conjugated streptavidin and 10 nm goat anti-rabbit secondary antibodies for 1 hr RT (BBInternational). Grids were stained and imaged as above.
Cloning and cell line assembly
All DNA constructs were made as described previously using Gibson Assembly® (McAllaster et al., 2016). Briefly, the endogenous tagging of TbSmee1 (927.10.8820) with a triple HA epitope tag was targeted by the 500 bp of UTR proximal to the open reading frame (ORF), along with the first 500 bp of the ORF (McAllaster et al., 2015). Targeting constructs were assembled in a sequencing plasmid (PCR4Blunt), then linearized by restriction digest before electroporation with a GenePulser Xcell (Bio-Rad) into 427 cells. Clonal cell lines were selected for using 1 µg mL−1 of puromycin, and putative clones were screened by loci PCR, western blotting, and immunofluorescence. A similar scheme was used for the triple Ty1-TOEFAZ1 and the triple Ty1-HaloTag® TbSmee1.
The TbSmee1 cKO cell line created as described in Figure S3 and was maintained with 35 ng mL−1 of doxycycline (Figure S4).
Induction and recovery of TbSmee1 conditional knockout
The induction of the TbSmee1 cKO was performed as described previously, with the exception of washing three times with complete Beck’s medium to remove the doxycycline used to maintain expression (de Graffenried et al., 2013). For recovery of the TbSmee1 cKO, an additional flask of TbSmee1-depleted cells was seeded at 1×106 cells mL−1 and received 35 ng mL−1 of doxycycline on day 6 of the induction. This flask was then treated as described above.
Uptake of FM4-64FX
Microscopic analysis of FM4-64 uptake was carried out by a modification of the assay described by Bonhivers and coworkers (Bonhivers et al., 2008). The modifications included a total of 2.4 × 107 induced (6 days) and non-induced TbSmee1 cKOs harvested by centrifugation, washed in PBS, then resuspended in 3 mL of PBS, 0.1 mM adenosine, and 10 mM glucose. The cells also either remained at 4 °C or were shifted to 27 °C for 10 minutes. To prepare the cells for live imaging, they were washed twice in ice-cold PBS containing 0.1 mM adenosine, 10 mM glucose, and 8 µM of Hoechst 33342 (Thermo Fisher). Then, the cells were resuspended in 100 µL the above buffer without the addition of Hoechst 33342 and were kept on ice until ready to image.
For live imaging, 10 µl of cell suspension was spotted onto a glass slide and a coverslip was laid on top prior to visualization with a Zeiss Observer Z1 equipped with a Hamamatsu Orca Flash 4.0 camera (Hammamatsu) and a Plan-Apochromat 100×/1.4 oil immersion lens (Zeiss).
Uptake of fluorescent dextran
Analysis of fluorescent uptake was performed by a modification of the assay done by Hall and coworkers (Hall et al., 2005). Modifications of the assay were that induced (6 days) and non-induced TbSmee1 cKOs were harvested and resuspended in ice-cold complete media containing 25 mg mL−1 of Alexa Fluor-488 dextran 10,000 (Thermo Fisher) for a final concentration of 6.7 × 108 cells mL−1 in 75 µL. One-third of the volume was taken for a T = 0 min sample, while the remainder was shifted to 27 °C for either 30 min or 1 h. To prepare the cells for live imaging, cells were washed a total of five times: twice with ice-cold complete media then three times with ice-cold complete media containing 8 µM of Hoechst 33342 (Thermo Fisher). Cells were resuspended in 100 µL of PBS containing 0.1 mM adenosine and 10 mM glucose and kept on ice until ready to image. Live cell imaging was performed as described above.
Measurements
For quantitation of TbPLK localization and hook complex morphology, maximum intensity Z-projections of the TbPLK and TbMORN1 channels were generated to ensure localization of TbPLK and the entirety of the hook complex structure before assigning phenotype. Quantitation of 488-dextran and FM4-64FX uptake was performed using “sum slices” projection and “Analyze Particles” function after selecting regions of interest. Measurements were conducted in ImageJ version 1.51n (National Institutes of Health) and exported to Excel (Microsoft) for analysis.
Quantitation of immuno-gold electron microscopy
Semi-automated quantitation of immuno-gold electron microscopy was performed with a custom MATLAB GUI (The MathWorks, Inc) containing the “Polycenter” function (http://www.biomecardio.com/matlab/polycenter.html). The data was exported to Excel (Microsoft) for analysis.
Statistics
All error bars represent the standard deviation (SD) from the mean for three independent experiments, except for measurements of pixel intensity and hook complex and centrin arm segment frequency distribution where the error bars represent standard error of the mean (SEM). For quantitation of DNA content for both depletion and rescue of the TbSmee1 cKOs, a total of 200 cells per condition were counted. Quantitation of PLK localization the following numbers of cells were counted for each DNA stage from a population of 400 cells: 200–350 1N1K cells, 30 – 50 1N2K cells, and 10–50 2N2K cells. Characterization of HC-centrin arm duplication used a population size of 400 cells. Analysis of hook morphology upon depletion was based on a population of 200 cells, where morphologies from only 1N1K cells were used. For the rescue experiment, a total of 150 - 1N1K cells were quantitated, while modeling of rescue of HC morphology versus dilution was performed using 75–150 1N1K cells. For analysis of HC-centrin arm by iEM, micrographs from 25 non-dividing cells were used. For experiments looking at uptake of FM4-64FX and 488-Dextran, 95 and 100 - 1N1K cells were quantitated, respectively. All statistical analyses were performed in Prism7 (GraphPad Software, Inc.)
Supplementary Material
Figure S1 Dual immuno-electron microscopy with TbMORN1 confirms TbSmee1 localization to the HC shank. Flagella were isolated from 3×HA-TbSmee1 cells and then labeled with antibodies against HA, a biotin-conjugated secondary antibody, and 15 nm gold-conjugated streptavidin. TbMORN1 was labeled with anti-TbMORN1 followed by a 10 nm gold-conjugated secondary antibody. The labeled isolated flagella were negatively stained and examined by transmission electron microscopy.
Figure S2 TbSmee1 colocalizes with TOEFAZ1 at the new FAZ tip. Cells expressing endogenously tagged 3 × Ty1- TOEFAZ1 were fixed and labeled with antibodies against TbSmee1 (TbSmee1; green) and Ty1 (3 × Ty1- TOEFAZ1; red), along with DAPI to label the DNA (DNA; blue). The cells were visualized using fluorescence microscopy and DIC. The filled arrowheads depict the location of the new FAZ tip in a dividing cell and insets are three-fold magnification of the area surrounding new FAZ tip.
Figure S3 Schematic of TbSmee1 conditional knockout creation. Steps 1,2: 3×Ty1-TbSmee1 was integrated into an ectopic rDNA locus and is under doxycycline control. Step 3: At the endogenous locus, one allele of TbSmee1 was replaced with a blasticidin resistance cassette. Step 4: Expression of the ectopic copy of 3×Ty1-TbSmee1 was induced through the addition of doxycycline and the remaining endogenous allele of TbSmee1 was replaced with a puromycin resistance cassette. To observe the resultant phenotypes of TbSmee1 depletion, doxycycline is removed from the culture medium, which turns off the expression of the ectopic 3×Ty1-TbSmee1.
Figure S4 Titration of doxycycline to match expression of ectopic 3×Ty1-TbSmee1 to endogenous levels. The TbSmee1 cKO cell line was grown in a variety of doxycycline concentrations before being collected for western blot analysis. The TbSmee1 cKO lysates and control T. brucei 29-13 lysates were separated by SDS-PAGE, transferred to nitrocellulose, and probed with rabbit anti-TbSmee1 and anti-tubulin as a loading control. The blot was analyzed semi-quantitatively to determine that 30 ng mL−1 of doxycycline approximated endogenous levels of expression, so 35 ng mL−1 was used in all following experiments to slightly overexpress TbSmee1 to ensure normal growth.
Figure S5 TbSmee1-depletion leads to a 40% decrease in cell growth. TbSmee1 cKO cells were grown for 8 days in either the presence (Control) or absence (TbSmee1 Removed) of doxycycline. Cells from each culture were monitored by cell count, and cultures were re-seeded to starting densities every two days using either doxycycline- or vehicle- containing media. T0 represents the culture at the start of each experiment.
Figure S6 TbPLK mislocalization is not due to change in protein expression. (A) TbSmee1 cKO cells were grown for 8 days in either the presence (+) or absence (-) of doxycycline. Cells from each culture were monitored by cell count and collected daily to monitor for TbPLK expression by anti-TbPLK western blotting, using tubulin as a loading control. T0 represents the culture at the start of each experiment. (B) Semi-quantitative analysis of western blot for TbPLK expression in TbSmee1 cKOs. Values are normalized against anti-tubulin loading control and are relative to TbPLK expression at T0. Data are means ± SD of three independent experiments. (C) Quantitation of TbPLK localization at 48 hours of TbSmee1 depletion. Data are means ± SD of three independent experiments.
Figure S7 TbSmee1 depletion for 2 days leads to altered HC morphology. Quantitation of HC morphology in non-dividing 1N1K control (Control) and TbSmee1-depleted cells (TbSmee1 Removed) for 2 days. Data are means ± SD of three independent experiments.
Figure S8 Amount of immunogold particles remains the same between control and TbSmee1-depleted cells independent of HC-centrin arm size. (A) Quantitation of total number of TbMORN1 immunogold particles on HC-centrin arms of control and TbSmee1-removed cells. Each marker represents one HC-centrin arm and the error bars indicate quartiles. n.s; not significant (two-tailed unpaired Student’s t test). (B) Correlation of total TbMORN1 immunogold particles on HC-centrin arm to total number of HC-centrin arm segments. Dotted lines indicate linear regressions. (C) TbSmee1 cKO cells were grown for 8 days in either the presence (+) or absence (-) of doxycycline. Cells from each culture were monitored by cell count and collected daily to monitor for TbMORN1 expression by anti-TbMORN1 western blotting, using tubulin as a loading control. T0 represents the culture at the start of each experiment. The TbMORN1 western blot was semi-quantitatively analyzed with the TbMORN1 values being normalized against the anti-tubulin loading control and are relative to TbMORN1 expression at T0. Data are means ± SD of three independent experiments.
Figure S9 Addition of doxycycline to TbSmee1- depleted cells restores expression of the ectopic 3×Ty1- TbSmee1 allele and leads to restored cell growth. TbSmee1 cKO cells were treated with either doxycycline (Control; +) or vehicle control (TbSmee1 Removed; -) for 6 days before treating TbSmee1-depleted cells with doxycycline (AB; TbSmee1 Rescue) for an additional 6 days. Cells were monitored for cell growth, collected at the indicated intervals, and lysed for western blotting. The lysates were separated by SDS-PAGE, transferred to nitrocellulose, and blotted with anti-Ty1 and anti-tubulin (loading control) antibodies. Molecular weights in kDa are indicated on the right.
Figure S10 Restoring TbSmee1 expression leads to recovery of normal DNA content and hook complex morphology. (A) Quantitation of DNA content in control, TbSmee1-rescued, and TbSmee1 – depleted (TbSmee1 Removed) cells on day 7 (one day after doxycycline addition) of experiment. Data are means ± SD of three independent experiments. (B) Quantitation of hook complex morphologies in non-dividing 1N1K control, depleted TbSmee1 (TbSmee1 Removed) and rescued TbSmee1 (TbSmee1 Rescue) cells on day 7. Data are means ± SD of three independent experiments. (C) Quantitation of DNA content in control, TbSmee1-rescued, and TbSmee1-depleted (TbSmee1 Removed) cells on day 9 (three days post addition of doxycycline to rescue). Data are means ± SD of three independent experiments.
Figure S11 TbSmee1-depleted cells take up the same amount of fluorescent dextran as control cells. (A) Quantitation of raw integrated density of fluorescence intensity in non-dividing 1N1K control and TbSmee1-removed cells after 30 minutes of incubation with dextran. Fluorescence intensity is plotted on a logarithmic scale and each marker represents one cell. Error bars are mean ± SEM. (B) Quantitation of raw integrated density of fluorescence intensity in non-dividing 1N1K control and TbSmee1-removed cells after 60 minutes of incubation with dextran. Fluorescence intensity was plotted on a logarithmic scale and each marker represents one cell. Error bars are mean ± SEM.
Figure S12 Lysosomal marker p67 is not altered upon depletion of TbSmee1. Control cells (Control) and cells depleted of TbSmee1 for 6 days were fixed and labeled with anti-p67 (p67; red). DAPI was used to visualize DNA (DNA; blue) and the samples were imaged using fluorescence and DIC microscopy.
Figure S13 Specificity of the rabbit anti-TbSmee1 polyclonal. Three cell lines were used to validate the anti-TbSmee1 polyclonal: T. brucei Lister 427 (427), a cell line with TbSmee1 endogenously tagged with HaloTag® (TbSmee1 3×Ty1-HaloTag ET), and a cell line where the expression of TbSmee1 is under doxycycline control (TbSmee1 cKO). TbSmee1 cKO cell line was treated with either doxycycline (+) or vehicle control (-) for 6 days before lysates for western blotting were made. The lysates for all cell lines were separated using SDS-PAGE, transferred to nitrocellulose, then blotted with rabbit anti-TbSmee1. The membrane was then stripped and reprobed with anti-Ty1. Two exposures of the anti-TbSmee1 blot are shown to better display the HaloTagged protein (open arrowhead) and the endogenous protein (asterisk). Molecular weights are shown in kDa on the left.
Acknowledgments
We thank Brooke Morriswood and Daja Schichler for providing the TbSmee1 antisera and for critical evaluation of the manuscript. We would like to thank Tom Sladewski and Nick Hilton for helpful advice and engaging conversations. We would also like to thank Richard Bennett for the use of his microscope, along with Geoff Williams and Sue Vaughan for assistance with TEM. This work was funded by the NIH (RO1 AI112953-01 to CLdG; ANS-D was supported by 5 T32 GM 7601-37). The content is solely the responsibility of the authors and does not necessarily reflect the official views of the NIH.
Footnotes
Author Contributions
JAP, MRM, and CLdG performed the conception and design of the study; JAP and ANS-D performed the experiments; JAP, ANS-D, and CLdG performed analysis or interpretation of the data; JAP, ANS-D, and CLdG wrote the manuscript. CLdG functioned in a supervisory role and acquired the funding to conduct the work.
References
- Absalon S, Blisnick T, Bonhivers M, Kohl L, Cayet N, Toutirais G, et al. Flagellum elongation is required for correct structure, orientation and function of the flagellar pocket in Trypanosoma brucei. Journal of Cell Science. 2008;121:3704–3716. doi: 10.1242/jcs.035626. [DOI] [PubMed] [Google Scholar]
- Acosta-Serrano A, Vassella E, Liniger M, Kunz Renggli C, Brun R, Roditi I, Englund PT. The surface coat of procyclic Trypanosoma brucei: programmed expression and proteolytic cleavage of procyclin in the tsetse fly. Proceedings of the National Academy of Sciences. 2001;98:1513–1518. doi: 10.1073/pnas.041611698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander DL, Schwartz KJ, Balber AE, Bangs JD. Developmentally regulated trafficking of the lysosomal membrane protein p67 in Trypanosoma brucei. J Cell Sci. 2002;115:3253–3263. doi: 10.1242/jcs.115.16.3253. [DOI] [PubMed] [Google Scholar]
- Allen CL, Goulding D, Field MC. Clathrin-mediated endocytosis is essential in Trypanosoma brucei. EMBO J. 2003;22:4991–5002. doi: 10.1093/emboj/cdg481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- André J, Harrison S, Towers K, Qi X, Vaughan S, McKean PG, Ginger ML. The tubulin cofactor C family member TBCCD1 orchestrates cytoskeletal filament formation. Journal of Cell Science. 2013;126:5350–5356. doi: 10.1242/jcs.136515. [DOI] [PubMed] [Google Scholar]
- Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, et al. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010;38:D457–62. doi: 10.1093/nar/gkp851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastin P, Bagherzadeh Z, Matthews KR, Gull K. A novel epitope tag system to study protein targeting and organelle biogenesis in Trypanosoma brucei. Molecular & Biochemical Parasitology. 1996;77:235–239. doi: 10.1016/0166-6851(96)02598-4. [DOI] [PubMed] [Google Scholar]
- Beisson J, Sonneborn TM. Cytoplasmic Inheritance of the Organization of the Cell Cortex in Paramecium aurelia. Proceedings of the National Academy of Sciences. 1965;53:275–282. doi: 10.1073/pnas.53.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisson J, Clérot JC, Fleury-Aubusson A, Garreau de Loubresse N, Ruiz F, Klotz C. Basal body-associated nucleation center for the centrin-based cortical cytoskeletal network in Paramecium. Protist. 2001;152:339–354. doi: 10.1078/1434-4610-00072. [DOI] [PubMed] [Google Scholar]
- Bonhivers M, Nowacki S, Landrein N, Robinson DR. Biogenesis of the Trypanosome Endo-Exocytotic Organelle Is Cytoskeleton Mediated. PLoS Biology. 2008;6:e105. doi: 10.1371/journal.pbio.0060105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickman MJ, Balber AE. Trypanosoma brucei rhodesiense: membrane glycoproteins localized primarily in endosomes and lysosomes of bloodstream forms. Exp Parasitol. 1993;76:329–344. doi: 10.1006/expr.1993.1041. [DOI] [PubMed] [Google Scholar]
- Büscher P, Cecchi G, Jamonneau V, Priotto G. Human African trypanosomiasis. Lancet. 2017 doi: 10.1016/S0140-6736(17)31510-6. [DOI] [PubMed] [Google Scholar]
- de Graffenried CL, Anrather D, Von Raußendorf F, Warren G. Polo-like kinase phosphorylation of bilobe-resident TbCentrin2 facilitates flagellar inheritance in Trypanosoma brucei. Mol Biol Cell. 2013;24:1947–1963. doi: 10.1091/mbc.E12-12-0911. http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-12-0911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graffenried CL, Ho HH, Warren G. Polo-like kinase is required for Golgi and bilobe biogenesis in Trypanosoma brucei. The Journal of Cell Biology. 2008;181:431–438. doi: 10.1083/jcb.200708082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean S, Marchetti R, Kirk K, Matthews KR. A surface transporter family conveys the trypanosome differentiation signal. Nature. 2009;459:213–217. doi: 10.1038/nature07997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean S, Sunter JD, Wheeler RJ. TrypTag.org: A Trypanosome Genome-wide Protein Localisation Resource. Trends Parasitol. 2017;33:80–82. doi: 10.1016/j.pt.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstler M, Pfohl T, Herminghaus S, Boshart M, Wiegertjes G, Heddergott N, Overath P. Hydrodynamic flow-mediated protein sorting on the cell surface of trypanosomes. Cell. 2007;131:505–515. doi: 10.1016/j.cell.2007.08.046. [DOI] [PubMed] [Google Scholar]
- Engstler M, Thilo L, Weise F, Grünfelder CG, Schwarz H, Boshart M, Overath P. Kinetics of endocytosis and recycling of the GPI-anchored variant surface glycoprotein in Trypanosoma brucei. Journal of Cell Science. 2004;117:1105–1115. doi: 10.1242/jcs.00938. [DOI] [PubMed] [Google Scholar]
- Esson HJ, Morriswood B, Yavuz S, Vidilaseris K, Dong G, Warren G. Morphology of the trypanosome bilobe, a novel cytoskeletal structure. Eukaryotic Cell. 2012;11:761–772. doi: 10.1128/EC.05287-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field MC, Carrington M. The trypanosome flagellar pocket. Nat Rev Micro. 2009;7:775–786. doi: 10.1038/nrmicro2221. [DOI] [PubMed] [Google Scholar]
- Freeman JC. The penetration of the peritrophic membrane of the tsetse flies by trypanosomes. Acta Tropica. 1973;30:347–355. [PubMed] [Google Scholar]
- Gadelha C, Rothery S, Morphew M, McIntosh JR, Severs NJ, Gull K. Membrane domains and flagellar pocket boundaries are influenced by the cytoskeleton in African trypanosomes. PNAS. 2009;106:17425–17430. doi: 10.1073/pnas.0909289106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheiratmand L, He CY. A Comparative Proteomic Analysis Reveals a New Bi-Lobe Protein Required for Bi-Lobe Duplication and Cell Division in Trypanosoma brucei. PLoS ONE. 2010;5:e9660. doi: 10.1371/journal.pone.0009660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson W, Bailey M. The development of Trypanosoma brucei within the tsetse fly midgut observed using green fluorescent trypanosomes. Kinetoplastid Biol Dis. 2003;2:1. doi: 10.1186/1475-9292-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünfelder CG, Engstler M, Weise F, Schwarz H, Stierhof YD, Morgan GW, et al. Endocytosis of a glycosylphosphatidylinositol-anchored protein via clathrin-coated vesicles, sorting by default in endosomes, and exocytosis via RAB11-positive carriers. Mol Biol Cell. 2003;14:2029–2040. doi: 10.1091/mbc.E02-10-0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BS, Pal A, Goulding D, Acosta-Serrano A, Field MC. Trypanosoma brucei: TbRAB4 regulates membrane recycling and expression of surface proteins in procyclic forms. Exp Parasitol. 2005;111:160–171. doi: 10.1016/j.exppara.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Hall BS, Pal A, Goulding D, Field MC. Rab4 is an essential regulator of lysosomal trafficking in trypanosomes. J Biol Chem. 2004;279:45047–45056. doi: 10.1074/jbc.M407271200. http://www.jbc.org/lookup/doi/10.1074/jbc.M407271200. [DOI] [PubMed] [Google Scholar]
- He CY, Pypaert M, Warren G. Golgi duplication in Trypanosoma brucei requires Centrin2. Science. 2005;310:1196–1198. doi: 10.1126/science.1119969. [DOI] [PubMed] [Google Scholar]
- Höög JL, Lacomble S, Bouchet-Marquis C, Briggs L, Park K, Hoenger A, Gull K. 3D Architecture of the Trypanosoma brucei Flagella Connector, a Mobile Transmembrane Junction. PLoS Negl Trop Dis. 2016;10:e0004312. doi: 10.1371/journal.pntd.0004312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Zhou Q, Han X, Li Z. CRL4WDR1 Controls Polo-like Kinase Protein Abundance to Promote Bilobe Duplication, Basal Body Segregation and Flagellum Attachment in Trypanosoma brucei. PLoS Pathog. 2017;13:e1006146. doi: 10.1371/journal.ppat.1006146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda KN, de Graffenried CL. Polo-like kinase is necessary for flagellum inheritance in Trypanosoma brucei. Journal of Cell Science. 2012;125:3173–3184. doi: 10.1242/jcs.101162. [DOI] [PubMed] [Google Scholar]
- Kelley RJ, Alexander DL, Cowan C, Balber AE, Bangs JD. Molecular cloning of p67, a lysosomal membrane glycoprotein from Trypanosoma brucei. Molecular & Biochemical Parasitology. 1999;98:17–28. doi: 10.1016/s0166-6851(98)00155-8. [DOI] [PubMed] [Google Scholar]
- Lacomble S, Vaughan S, Gadelha C, Morphew MK, Shaw MK, McIntosh JR, Gull K. Three-dimensional cellular architecture of the flagellar pocket and associated cytoskeleton in trypanosomes revealed by electron microscope tomography. Journal of Cell Science. 2009;122:1081–1090. doi: 10.1242/jcs.045740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacomble S, Vaughan S, Gadelha C, Morphew MK, Shaw MK, McIntosh JR, Gull K. Basal body movements orchestrate membrane organelle division and cell morphogenesis in Trypanosoma brucei. Journal of Cell Science. 2010;123:2884–2891. doi: 10.1242/jcs.074161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langreth SG, Balber AE. Protein uptake and digestion in bloodstream and culture forms of Trypanosoma brucei. The Journal of Protozoology. 1975;22:40–53. doi: 10.1111/j.1550-7408.1975.tb00943.x. [DOI] [PubMed] [Google Scholar]
- Li Z, Umeyama T, Li Z, Li Z, Wang CC. Polo-like kinase guides cytokinesis in Trypanosoma brucei through an indirect means. Eukaryotic Cell. 2010;9:705–716. doi: 10.1128/EC.00330-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano-Nunez A, Ikeda KN, Sauer T, de Graffenried CL. An analogue-sensitive approach identifies basal body rotation and flagellum attachment zone elongation as key functions of PLK in Trypanosoma brucei. Mol Biol Cell. 2013;24:1321–1333. doi: 10.1091/mbc.E12-12-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllaster MR, Ikeda KN, Lozano-Nunez A, Anrather D, Unterwurzacher V, Gossenreiter T, et al. Proteomic identification of novel cytoskeletal proteins associated with TbPLK, an essential regulator of cell morphogenesis in Trypanosoma brucei. Mol Biol Cell. 2015;26:3013–3029. doi: 10.1091/mbc.E15-04-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllaster MR, Sinclair-Davis AN, Hilton NA, de Graffenried CL. A unified approach towards Trypanosoma brucei functional genomics using Gibson assembly. Molecular & Biochemical Parasitology. 2016;210:13–21. doi: 10.1016/j.molbiopara.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira-Leite FF. A Trypanosome Structure Involved in Transmitting Cytoplasmic Information During Cell Division. Science. 2001;294:610–612. doi: 10.1126/science.1063775. [DOI] [PubMed] [Google Scholar]
- Morriswood B. Form, Fabric, and Function of a Flagellum-Associated Cytoskeletal Structure. Cells. 2015;4:726–747. doi: 10.3390/cells4040726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morriswood B, Schmidt K. A MORN Repeat Protein Facilitates Protein Entry into the Flagellar Pocket of Trypanosoma brucei. Eukaryotic Cell. 2015;14:1081–1093. doi: 10.1128/EC.00094-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morriswood B, Havlicek K, Demmel L, Yavuz S, Sealey-Cardona M, Vidilaseris K, et al. Novel bilobe components in Trypanosoma brucei identified using proximity-dependent biotinylation. Eukaryotic Cell. 2013;12:356–367. doi: 10.1128/EC.00326-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morriswood B, He CY, Sealey-Cardona M, Yelinek J, Pypaert M, Warren G. The bilobe structure of Trypanosoma brucei contains a MORN-repeat protein. Molecular & Biochemical Parasitology. 2009;167:95–103. doi: 10.1016/j.molbiopara.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Mugnier MR, Cross GAM, Papavasiliou FN. The in vivo dynamics of antigenic variation in Trypanosoma brucei. Science. 2015;347:1470–1473. doi: 10.1126/science.aaa4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natesan SKA, Peacock L, Matthews K, Gibson W, Field MC. Activation of endocytosis as an adaptation to the mammalian host by trypanosomes. Eukaryotic Cell. 2007;6:2029–2037. doi: 10.1128/EC.00213-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochatt CM, Bütikofer P, Navarro M, Wirtz E, Boschung M, Armah D, Cross GA. Conditional expression of glycosylphosphatidylinositol phospholipase C in Trypanosoma brucei. Molecular & Biochemical Parasitology. 1999;103:35–48. doi: 10.1016/s0166-6851(99)00111-5. [DOI] [PubMed] [Google Scholar]
- Overath P, Engstler M. Endocytosis, membrane recycling and sorting of GPI-anchored proteins: Trypanosoma brucei as a model system. Molecular Microbiology. 2004;53:735–744. doi: 10.1111/j.1365-2958.2004.04224.x. [DOI] [PubMed] [Google Scholar]
- Peck RF, Shiflett AM, Schwartz KJ, McCann A, Hajduk SL, Bangs JD. The LAMP-like protein p67 plays an essential role in the lysosome of African trypanosomes. Molecular Microbiology. 2008;68:933–946. doi: 10.1111/j.1365-2958.2008.06195.x. [DOI] [PubMed] [Google Scholar]
- Price HP, Stark M, Smith B, Smith DF. TbARF1 influences lysosomal function but not endocytosis in procyclic stage Trypanosoma brucei. Molecular & Biochemical Parasitology. 2007;155:123–127. doi: 10.1016/j.molbiopara.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DR, Gull K. Basal body movements as a mechanism for mitochondrial genome segregation in the trypanosome cell cycle. Nature. 1991;352:731–733. doi: 10.1038/352731a0. [DOI] [PubMed] [Google Scholar]
- Robinson DR, Sherwin T, Ploubidou A, Byard EH, Gull K. Microtubule polarity and dynamics in the control of organelle positioning, segregation, and cytokinesis in the trypanosome cell cycle. The Journal of Cell Biology. 1995;128:1163–1172. doi: 10.1083/jcb.128.6.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roditi I, Schwarz H, Pearson TW, Beecroft RP, Liu MK, Richardson JP, et al. Procyclin gene expression and loss of the variant surface glycoprotein during differentiation of Trypanosoma brucei. The Journal of Cell Biology. 1989;108:737–746. doi: 10.1083/jcb.108.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotureau B, Subota I, Buisson J, Bastin P. A new asymmetric division contributes to the continuous production of infective trypanosomes in the tsetse fly. Development. 2012;139:1842–1850. doi: 10.1242/dev.072611. [DOI] [PubMed] [Google Scholar]
- Rudenko G. African trypanosomes: the genome and adaptations for immune evasion. Essays Biochem. 2011;51:47–62. doi: 10.1042/bse0510047. [DOI] [PubMed] [Google Scholar]
- Salmon D, Geuskens M, Hanocq F, Hanocq-Quertier J, Nolan D, Ruben L, Pays E. A novel heterodimeric transferrin receptor encoded by a pair of VSG expression site-associated genes in T. brucei. Cell. 1994;78:75–86. doi: 10.1016/0092-8674(94)90574-6. [DOI] [PubMed] [Google Scholar]
- Selvapandiyan A, Kumar P, Morris JC, Salisbury JL, Wang CC, Nakhasi HL. Centrin1 is required for organelle segregation and cytokinesis in Trypanosoma brucei. Mol Biol Cell. 2007;18:3290–3301. doi: 10.1091/mbc.E07-01-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwin T, Gull K. The Cell Division Cycle of Trypanosoma brucei brucei: Timing of Event Markers and Cytoskeletal Modulations. Philosophical Transactions of the Royal Society B: Biological Sciences. 1989;323:573–588. doi: 10.1098/rstb.1989.0037. [DOI] [PubMed] [Google Scholar]
- Shi J, Franklin JB, Yelinek JT, Ebersberger I, Warren G, He CY. Centrin4 coordinates cell and nuclear division in T. brucei. Journal of Cell Science. 2008;121:3062–3070. doi: 10.1242/jcs.030643. [DOI] [PubMed] [Google Scholar]
- Sinclair-Davis AN, McAllaster MR, de Graffenried CL. A functional analysis of TOEFAZ1 uncovers protein domains essential for cytokinesis in Trypanosoma brucei. J Cell Sci. 2017;130:3918–3932. doi: 10.1242/jcs.207209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunter JD, Gull K. The Flagellum Attachment Zone: “The Cellular Ruler” of Trypanosome Morphology. Trends Parasitol. 2016;32:309–324. doi: 10.1016/j.pt.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AE, Godfrey DG. A new organelle of bloodstream salivarian trypanosomes. The Journal of Protozoology. 1969;16:466–470. doi: 10.1111/j.1550-7408.1969.tb02302.x. [DOI] [PubMed] [Google Scholar]
- Tazeh NN, Silverman JS, Schwartz KJ, Sevova ES, Sutterwala SS, Bangs JD. Role of AP-1 in Developmentally Regulated Lysosomal Trafficking in Trypanosoma brucei. Eukaryotic Cell. 2009;8:1352–1361. doi: 10.1128/EC.00156-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetley L, Turner CM, Barry JD, Crowe JS, Vickerman K. Onset of expression of the variant surface glycoproteins of Trypanosoma brucei in the tsetse fly studied using immunoelectron microscopy. Journal of Cell Science. 1987;87(Pt 2):363–372. doi: 10.1242/jcs.87.2.363. [DOI] [PubMed] [Google Scholar]
- Tiengwe C, Bush PJ, Bangs JD. Controlling transferrin receptor trafficking with GPI-valence in bloodstream stage African trypanosomes. PLoS Pathog. 2017;13:e1006366. doi: 10.1371/journal.ppat.1006366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeyama T, Wang CC. Polo-like kinase is expressed in S/G2/M phase and associated with the flagellum attachment zone in both procyclic and bloodstream forms of Trypanosoma brucei. Eukaryotic Cell. 2008;7:1582–1590. doi: 10.1128/EC.00150-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbaniak MD, Turnock DC, Ferguson MAJ. Galactose starvation in a bloodstream form Trypanosoma brucei UDP-glucose 4′-epimerase conditional null mutant. Eukaryotic Cell. 2006;5:1906–1913. doi: 10.1128/EC.00156-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urwyler S, Studer E, Renggli CK, Roditi I. A family of stage-specific alanine-rich proteins on the surface of epimastigote forms of Trypanosoma brucei. Molecular Microbiology. 2007;63:218–228. doi: 10.1111/j.1365-2958.2006.05492.x. [DOI] [PubMed] [Google Scholar]
- Varga V, Moreira-Leite F, Portman N, Gull K. Protein diversity in discrete structures at the distal tip of the trypanosome flagellum. Proc Natl Acad Sci USA. 2017;114:E6546–E6555. doi: 10.1073/pnas.1703553114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan S, Kohl L, Ngai I, Wheeler RJ, Gull K. A repetitive protein essential for the flagellum attachment zone filament structure and function in Trypanosoma brucei. Protist. 2008;159:127–136. doi: 10.1016/j.protis.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Vickerman K. On the surface coat and flagellar adhesion in trypanosomes. Journal of Cell Science. 1969;5:163–193. doi: 10.1242/jcs.5.1.163. [DOI] [PubMed] [Google Scholar]
- Wang M, Gheiratmand L, He CY. An interplay between Centrin2 and Centrin4 on the bi-lobed structure in Trypanosoma brucei. Molecular Microbiology. 2012;83:1153–1161. doi: 10.1111/j.1365-2958.2012.07998.x. [DOI] [PubMed] [Google Scholar]
- Wheeler RJ, Scheumann N, Wickstead B, Gull K, Vaughan S. Cytokinesis in Trypanosoma bruceidiffers between bloodstream and tsetse trypomastigote forms: implications for microtubule-based morphogenesis and mutant analysis. Molecular Microbiology. 2013;90:1339–1355. doi: 10.1111/mmi.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz E, Clayton C. Inducible Gene Expression in Trypanosomes Mediated by a Prokaryotic Repressor. Science. 1995;268:1179–1183. doi: 10.1126/science.7761835. [DOI] [PubMed] [Google Scholar]
- Wirtz E, Leal S, Ochatt C, Cross GM. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Molecular & Biochemical Parasitology. 1999;99:89–101. doi: 10.1016/s0166-6851(99)00002-x. [DOI] [PubMed] [Google Scholar]
- Yaro M, Munyard KA, Stear MJ, Groth DM. Combatting African Animal Trypanosomiasis (AAT) in livestock: The potential role of trypanotolerance. Vet Parasitol. 2016;225:43–52. doi: 10.1016/j.vetpar.2016.05.003. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Gu J, Lun ZR, Ayala FJ, Li Z. Two distinct cytokinesis pathways drive trypanosome cell division initiation from opposite cell ends. Proceedings of the National Academy of Sciences. 2016;113:3287–3292. doi: 10.1073/pnas.1601596113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Hu H, Li Z. An EF-hand-containing Protein in Trypanosoma brucei Regulates Cytokinesis Initiation by Maintaining the Stability of the Cytokinesis Initiation Factor CIF1. J Biol Chem. 2016;291:14395–14409. doi: 10.1074/jbc.M116.726133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Dual immuno-electron microscopy with TbMORN1 confirms TbSmee1 localization to the HC shank. Flagella were isolated from 3×HA-TbSmee1 cells and then labeled with antibodies against HA, a biotin-conjugated secondary antibody, and 15 nm gold-conjugated streptavidin. TbMORN1 was labeled with anti-TbMORN1 followed by a 10 nm gold-conjugated secondary antibody. The labeled isolated flagella were negatively stained and examined by transmission electron microscopy.
Figure S2 TbSmee1 colocalizes with TOEFAZ1 at the new FAZ tip. Cells expressing endogenously tagged 3 × Ty1- TOEFAZ1 were fixed and labeled with antibodies against TbSmee1 (TbSmee1; green) and Ty1 (3 × Ty1- TOEFAZ1; red), along with DAPI to label the DNA (DNA; blue). The cells were visualized using fluorescence microscopy and DIC. The filled arrowheads depict the location of the new FAZ tip in a dividing cell and insets are three-fold magnification of the area surrounding new FAZ tip.
Figure S3 Schematic of TbSmee1 conditional knockout creation. Steps 1,2: 3×Ty1-TbSmee1 was integrated into an ectopic rDNA locus and is under doxycycline control. Step 3: At the endogenous locus, one allele of TbSmee1 was replaced with a blasticidin resistance cassette. Step 4: Expression of the ectopic copy of 3×Ty1-TbSmee1 was induced through the addition of doxycycline and the remaining endogenous allele of TbSmee1 was replaced with a puromycin resistance cassette. To observe the resultant phenotypes of TbSmee1 depletion, doxycycline is removed from the culture medium, which turns off the expression of the ectopic 3×Ty1-TbSmee1.
Figure S4 Titration of doxycycline to match expression of ectopic 3×Ty1-TbSmee1 to endogenous levels. The TbSmee1 cKO cell line was grown in a variety of doxycycline concentrations before being collected for western blot analysis. The TbSmee1 cKO lysates and control T. brucei 29-13 lysates were separated by SDS-PAGE, transferred to nitrocellulose, and probed with rabbit anti-TbSmee1 and anti-tubulin as a loading control. The blot was analyzed semi-quantitatively to determine that 30 ng mL−1 of doxycycline approximated endogenous levels of expression, so 35 ng mL−1 was used in all following experiments to slightly overexpress TbSmee1 to ensure normal growth.
Figure S5 TbSmee1-depletion leads to a 40% decrease in cell growth. TbSmee1 cKO cells were grown for 8 days in either the presence (Control) or absence (TbSmee1 Removed) of doxycycline. Cells from each culture were monitored by cell count, and cultures were re-seeded to starting densities every two days using either doxycycline- or vehicle- containing media. T0 represents the culture at the start of each experiment.
Figure S6 TbPLK mislocalization is not due to change in protein expression. (A) TbSmee1 cKO cells were grown for 8 days in either the presence (+) or absence (-) of doxycycline. Cells from each culture were monitored by cell count and collected daily to monitor for TbPLK expression by anti-TbPLK western blotting, using tubulin as a loading control. T0 represents the culture at the start of each experiment. (B) Semi-quantitative analysis of western blot for TbPLK expression in TbSmee1 cKOs. Values are normalized against anti-tubulin loading control and are relative to TbPLK expression at T0. Data are means ± SD of three independent experiments. (C) Quantitation of TbPLK localization at 48 hours of TbSmee1 depletion. Data are means ± SD of three independent experiments.
Figure S7 TbSmee1 depletion for 2 days leads to altered HC morphology. Quantitation of HC morphology in non-dividing 1N1K control (Control) and TbSmee1-depleted cells (TbSmee1 Removed) for 2 days. Data are means ± SD of three independent experiments.
Figure S8 Amount of immunogold particles remains the same between control and TbSmee1-depleted cells independent of HC-centrin arm size. (A) Quantitation of total number of TbMORN1 immunogold particles on HC-centrin arms of control and TbSmee1-removed cells. Each marker represents one HC-centrin arm and the error bars indicate quartiles. n.s; not significant (two-tailed unpaired Student’s t test). (B) Correlation of total TbMORN1 immunogold particles on HC-centrin arm to total number of HC-centrin arm segments. Dotted lines indicate linear regressions. (C) TbSmee1 cKO cells were grown for 8 days in either the presence (+) or absence (-) of doxycycline. Cells from each culture were monitored by cell count and collected daily to monitor for TbMORN1 expression by anti-TbMORN1 western blotting, using tubulin as a loading control. T0 represents the culture at the start of each experiment. The TbMORN1 western blot was semi-quantitatively analyzed with the TbMORN1 values being normalized against the anti-tubulin loading control and are relative to TbMORN1 expression at T0. Data are means ± SD of three independent experiments.
Figure S9 Addition of doxycycline to TbSmee1- depleted cells restores expression of the ectopic 3×Ty1- TbSmee1 allele and leads to restored cell growth. TbSmee1 cKO cells were treated with either doxycycline (Control; +) or vehicle control (TbSmee1 Removed; -) for 6 days before treating TbSmee1-depleted cells with doxycycline (AB; TbSmee1 Rescue) for an additional 6 days. Cells were monitored for cell growth, collected at the indicated intervals, and lysed for western blotting. The lysates were separated by SDS-PAGE, transferred to nitrocellulose, and blotted with anti-Ty1 and anti-tubulin (loading control) antibodies. Molecular weights in kDa are indicated on the right.
Figure S10 Restoring TbSmee1 expression leads to recovery of normal DNA content and hook complex morphology. (A) Quantitation of DNA content in control, TbSmee1-rescued, and TbSmee1 – depleted (TbSmee1 Removed) cells on day 7 (one day after doxycycline addition) of experiment. Data are means ± SD of three independent experiments. (B) Quantitation of hook complex morphologies in non-dividing 1N1K control, depleted TbSmee1 (TbSmee1 Removed) and rescued TbSmee1 (TbSmee1 Rescue) cells on day 7. Data are means ± SD of three independent experiments. (C) Quantitation of DNA content in control, TbSmee1-rescued, and TbSmee1-depleted (TbSmee1 Removed) cells on day 9 (three days post addition of doxycycline to rescue). Data are means ± SD of three independent experiments.
Figure S11 TbSmee1-depleted cells take up the same amount of fluorescent dextran as control cells. (A) Quantitation of raw integrated density of fluorescence intensity in non-dividing 1N1K control and TbSmee1-removed cells after 30 minutes of incubation with dextran. Fluorescence intensity is plotted on a logarithmic scale and each marker represents one cell. Error bars are mean ± SEM. (B) Quantitation of raw integrated density of fluorescence intensity in non-dividing 1N1K control and TbSmee1-removed cells after 60 minutes of incubation with dextran. Fluorescence intensity was plotted on a logarithmic scale and each marker represents one cell. Error bars are mean ± SEM.
Figure S12 Lysosomal marker p67 is not altered upon depletion of TbSmee1. Control cells (Control) and cells depleted of TbSmee1 for 6 days were fixed and labeled with anti-p67 (p67; red). DAPI was used to visualize DNA (DNA; blue) and the samples were imaged using fluorescence and DIC microscopy.
Figure S13 Specificity of the rabbit anti-TbSmee1 polyclonal. Three cell lines were used to validate the anti-TbSmee1 polyclonal: T. brucei Lister 427 (427), a cell line with TbSmee1 endogenously tagged with HaloTag® (TbSmee1 3×Ty1-HaloTag ET), and a cell line where the expression of TbSmee1 is under doxycycline control (TbSmee1 cKO). TbSmee1 cKO cell line was treated with either doxycycline (+) or vehicle control (-) for 6 days before lysates for western blotting were made. The lysates for all cell lines were separated using SDS-PAGE, transferred to nitrocellulose, then blotted with rabbit anti-TbSmee1. The membrane was then stripped and reprobed with anti-Ty1. Two exposures of the anti-TbSmee1 blot are shown to better display the HaloTagged protein (open arrowhead) and the endogenous protein (asterisk). Molecular weights are shown in kDa on the left.