

Abstract
Apis mellifera capensis Eschscholtz and A.m. scutellata Lepeletier are subspecies of western honey bees that are indigenous to the Republic of South Africa (RSA). Both subspecies have invasive potential and are organisms of concern for areas outside their native range, though they are important bees to beekeepers, agriculture, and the environment where they are native. The aim of the present study was to examine genetic differentiation among these subspecies and estimate their phylogenetic relationships using complete mitochondrial genomes sequences. We used 25 individuals that were either assigned to one of the subspecies or designated hybrids using morphometric analyses. Phylogenetic analyses of mitogenome sequences by maximum likelihood (ML) and Bayesian inference identified a monophyletic RSA clade, subdivided into two clades. A haplotype network was consistent with the phylogenetic trees. However, members of both subspecies occurred in both clades, indicating that A.m. capensis and A.m. scutellata are neither reciprocally monophyletic nor do they exhibit paraphyly with one subspecies nested within the other subspecies. Furthermore, no mitogenomic features were diagnostic to either subspecies. All bees analyzed from the RSA expressed a substantial level of haplotype diversity (most samples had unique haplotypes) but limited nucleotide diversity. The number of variable codons across protein-coding genes (PCGs) differed among loci, with CO3 exhibiting the most variation and ATP6 the least.
Introduction
The western honeybee, Apis mellifera L. (Hymenoptera, Apidae) is a eusocial species and a major pollinator that is ecologically and economically important1,2. A global analysis of the modern diversity of A. mellifera using morphometric and genetic approaches suggests that there are approximately 26 subspecies classified into at least six different evolutionary groups: (A) African subspecies, (M) northern and western European subspecies, (C) north Mediterranean subspecies, (O and Z) Middle Eastern subspecies, and (Y) in Ethiopia3–7. This great diversity may have arisen due to regional adaptation to the distinctive ecological conditions in each geographic region8.
Eleven A. mellifera subspecies have been identified throughout Africa based on discrete behavioral, morphological, and regional adaptation to different ecological conditions9,10. There are two honey bee subspecies that are indigenous to the Republic of South Africa (RSA), A.m. scutellata Lepeletier 1836 and A.m. capensis Eschscholtz 1821. Apis mellifera capensis (the Cape honey bee) is exclusively distributed in the RSA, whereas the distribution of A.m. scutellata extends north, out of the RSA, into the middle of the continent9. Both of these subspecies differ reproductively and behaviorally and both exhibit traits that confer strong invasive potential.
Apis mellifera capensis can be socially parasitic due to several specific reproductive characteristics that some Cape worker bees exhibit: thelytokous parthenogenesis (the ability of workers to lay diploid, female-destined eggs without mating)11, the possession of spermathecae12, and high ovariole numbers13. Furthermore, Cape honey bee workers have short latency periods before becoming reproductive, and produce queen-like pheromones rapidly after queen loss in the colony14,15. In the early 1990’s, A.m. capensis colonies were moved by migratory beekeepers into regions of the RSA where A.m. scutellata was endemic but that historically lacked A.m. capensis16. There, A.m. capensis has become a social parasite of A.m. scutellata colonies. This happens when some A.m. capensis worker bees drift into A.m. scutellata colonies and replace the existing queen. The parasitic A.m. capensis worker(s) begin producing offspring, though they cannot reproduce at the rate a normal queen does. Thus, the colony dwindles and ultimately dies. This problem became known as “capensis calamity”17.
Apis mellifera scutellata (the African savannah honey bee) is known for being defensive, having a high swarming rate, being migratory, and possessing other phenotypes many beekeepers view negatively9,18,19. This is the bee that was introduced into Brazil in 195720,21 and later became known as the “African,” “Africanized,” or “killer” bee of the Americas as it expanded its range throughout South, Central, and Southern North America22–24.
A stable hybrid zone separates the populations of A.m. capensis and A.m. scutellata in the RSA, with A.m. scutellata to the north and A.m. capensis to the south9. Neither subspecies has naturally expanded into the other subspecies’ geographic region despite their parasitic (A.m. capensis) or invasive (A.m. scutellata) capabilities25,26. In addition to the wide variety of behavioral and reproductive differences between the two subspecies, morphometric comparisons demonstrate that the subspecies have undergone sufficient morphological differentiation to be recognized as distinct subspecies9. However, recent studies have revealed multiple diagnostic markers to differentiate different subspecies but A.m. capensis and A.m. scutellata clusters in a single monophyletic RSA clade27–29.
Given their unique biologies, it is reasonable to assume that A.m. capensis and A.m. scutellata might have distinct genetic structures. The arrangement of A.m. capensis and scutellata based on morphological characters suggests two distinct subspecies, but further examination is required to find reliable and robust genetic markers that are diagnostic for the two subspecies. Given the more rapid evolutionary rate and coalescence of the mitochondrial genome30–32, the mitochondrion may yield diagnostic markers even in the absence of diagnostic nuclear markers.
The rapid development of Next Generation Sequencing (NGS)33,34 methods has provided opportunities to extract large amounts of data from organisms rapidly, thus improving our understanding of patterns of divergence at multiple taxonomic levels35,36. Use of NGS data, combined with the availability of a published mitochondrial genomes makes it possible to assemble the complete mitogenomes of A.m. capensis, A.m. scutellata and hybrid honey bees to examine differentiation among them. Here, we generated complete mitogenome sequences for 25 honey bees sampled from 25 discrete geographical regions across RSA. Given the morphological differentiation between A.m. capensis and A.m. scutellata, we sought to determine if the complete mitogenome sequences could be used to differentiate between the two honey bee subspecies accurately. The results of this study provide new insights regarding the mitogenome diversity of honey bee maternal lineages in RSA, but also clearly underscore the need for large-scale genomic studies in biogeography and evolutionary ecology of Apis.
Results
Genome structure, organization and composition
We collected complete mitogenomes from 25 honey bees (including two published in37,38 (Table 1). The assembled mitogenome sequences of all 25 honey bees were circular molecules that ranged in length from 16,338–16,513 bp (Table 2). On average, we mapped over 70,000 Illumina HiSeq. 3000 paired-end reads (2 × 100 bp) per honey bee sample (range from 33,469–390,359), corresponding to a sequence depth that averaged 479× (range from 204–2,392×). The number of ambiguous sites in final assemblies was low (0.01%). These ambiguous sites were largely in non-coding regions (particularly the AT-rich region). Only one sequence had ambiguities in any of the protein-coding genes (PCGs) or rRNAs. Thus, our results demonstrate the effectiveness of genome skimming for the assembly of complete mitochondrial sequences.
Table 1.
Summary information for honey bee samples collected in the Republic of South Africa. Bees were sampled from 25 apiaries.
| Apiary No. | geographical region | abbreviations | Apis mellifera subspecies | geographical coordinates | GenBank Accession Number |
|---|---|---|---|---|---|
| 1 | Bredasdorp | BD | A.m. capensis | 34°50ʹS–20°35ʹE | MG552681 |
| 2 | Citrusdaal | CD | A.m. capensis | 32°84ʹS–19°24ʹE | MG552682 |
| 3 | Cape Town | CT | A.m. capensis | 33°96ʹS–18°45ʹE | MG552683 |
| 4 | George | GE | A.m. capensis | 33°98ʹS–22°47ʹE | MG552684 |
| 5 | Grahamstown | GT | A.m. capensis | 33°31ʹS–26°49ʹE | MG552685 |
| 6 | Knysna | KN | A.m. capensis | 34°05ʹS–22°99ʹE | KX870183 |
| 7 | Langebaan | LA | A.m. capensis | 33°00ʹS–18°31ʹE | MG552686 |
| 8 | Laingsburg | LB | A.m. capensis | 33°27ʹS–20°85ʹE | MG552687 |
| 9 | Moorreesburg | MB | A.m. capensis | 33°10ʹS–18°74ʹE | MG552688 |
| 10 | Modderfontein | MF | A.m. capensis | 33°18ʹS–25°80ʹE | MG552689 |
| 11 | Plettenburg Bay | PB | A.m. capensis | 34°05ʹS–23°36ʹE | MG552690 |
| 12 | Port Elizabeth | PE | A.m. capensis | 33°87ʹS–25°39ʹE | MG552691 |
| 13 | Riversdale | RD | A.m. capensis | 34°10ʹS–21°20ʹE | MG552692 |
| 14 | St. Francis | SF | A.m. capensis | 34°17ʹS–24°81ʹE | MG552693 |
| 15 | Stellenbosch | ST | A.m. capensis | 33°85ʹS–18°82ʹE | MG552694 |
| 16 | Swellendam | SW | A.m. capensis | 34°05ʹS–20°65ʹE | MG552695 |
| 17 | Worcester | WD | A.m. capensis | 33°52ʹS–19°49ʹE | MG552696 |
| 18 | Bloemfontein | BL | A.m. scutellata | 29°20ʹS–27°20ʹE | MG552698 |
| 19 | Kroonstad | KR | A.m. scutellata | 27°27ʹS–27°50ʹE | MG552699 |
| 20 | Pretoria | PT | A.m. scutellata | 25°70ʹS–28°10ʹE | MG552700 |
| 21 | Springbok | SP | A.m. scutellata | 29°65ʹS–17°83ʹE | MG552701 |
| 22 | Upington | UP | A.m. scutellata | 28°52ʹS–21°24ʹE | MG552702 |
| 23 | Vryburg | VR | A.m. scutellata | 26°96ʹS–24°76ʹE | MG552703 |
| 24 | Beaufort West | BW | Hybrid | 32°34ʹS–22°62ʹE | KX943034 |
| 25 | Klawer | KL | Hybrid | 32°02ʹS–18°78ʹE | MG552697 |
Geographical region is identified by the main city/town closest to the sampled apiary and is abbreviated for use elsewhere in the manuscript. Subspecies identity was confirmed morphometrically following9. The GPS location of each apiary is noted (geographical coordinates). Finally, we include the GenBank accession numbers for the sequenced mitogenome of each bee.
Table 2.
Data on the 39 sequenced mitogenomes used in the study.
| Gene regions | Aligned length | Parsimony Informative sites | Variable sites | Variable sites (%) | Variable amino acid sites | Variable amino acid sites (%) | Verage AT (%) |
|---|---|---|---|---|---|---|---|
| ND2 | 1002 | 23 | 42 | 4.2 | 7 | 2.1 | 86.3 |
| COI | 1566 | 34 | 73 | 4.7 | 6 | 1.1 | 76.1 |
| CO2 | 676 | 11 | 30 | 4.4 | 3 | 1.3 | 80.2 |
| ATP8 | 159 | 3 | 7 | 4.4 | 1 | 1.9 | 88.1 |
| ATP6 | 681 | 5 | 13 | 1.9 | 2 | 0.9 | 84.5 |
| CO3 | 848 | 19 | 41 | 4.8 | 11 | 4.2 | 82.7 |
| ND3 | 354 | 12 | 15 | 4.2 | 2 | 1.7 | 85.5 |
| ND5 | 1668 | 38 | 64 | 3.8 | 13 | 2.3 | 85.5 |
| ND4 | 1319 | 29 | 57 | 4.3 | 15 | 3.4 | 86.5 |
| ND4L | 264 | 3 | 15 | 5.7 | 2 | 2.3 | 85.9 |
| ND6 | 504 | 16 | 27 | 5.4 | 5 | 3.0 | 86.9 |
| CYB | 1152 | 21 | 42 | 3.6 | 7 | 1.8 | 80.3 |
| ND1 | 918 | 21 | 49 | 5.3 | 12 | 3.9 | 82.9 |
| tRNAs – 22 genes | 1526 | 14 | 26 | 1.7 | N/A | N/A | 87.0 |
| *L-rRNA | 1381 | 17 | 41 | 3.0 | N/A | N/A | 84.3 |
| †S-rRNA | 787 | 7 | 21 | 2.7 | N/A | N/A | 81.0 |
| AT-rich region | 1053 | 75 | 170 | 16.1 | N/A | N/A | 96.1 |
| Intergenic sites | 982 | 24 | 62 | 6.3 | N/A | N/A | 94.3 |
| Complete mitogenome | 16768 | 371 | 789 | 4.7 | 86 | 2.3 | 84.7 |
*L-rRNA: Large subunit of ribosomal RNA.
†S-rRNA: Small subunit of ribosomal RNA.
Mitogenome content and organization was consistent with published data37,38 with 38 regions: 13 protein-coding genes (PCGs), two ribosomal RNAs (lrRNA and srRNA), 22 tRNAs, and an AT-rich non-coding region. The mitochondrial gene orders and arrangements were identical to those of other published A. mellifera mitochondria37–40. The genes encoded on heavy and light strands and the initiation and termination codons for all individuals are identical to those from our previous publications37,38. There were not cases where PCGs appeared to have an unusual start or stop codon; all PCGs initiated with ATA, ATC, ATG, or ATT and all ended with TAA or with a T that can be polyadenylated to form a TAA stop codon. All subspecies exhibited the same patterns of start and stop codon usage, such that the same start codon was found in all samples of all subspecies for a given gene. If the T was polyadenylated to form a stop codon, it was the same across all samples.
The nucleotide composition of the mitogenomes of A.m. capensis, A.m. scutellata and hybrids were strongly biased toward A and T with an average AT content of ~85% across the entire genome (Tables 3 and S1). Base composition did vary among regions of the mitochondrial genomes, with the highest AT content being found in the AT-rich region, as expected, and the lowest being found in COI (cytochrome oxidase I; Table 2). The number of variable codons differed among the PCGs (Table 2), with Cytochrome c oxidase subunit 3 (CO3) exhibiting the highest degree variation (over 4% of amino acids sites varied among samples) while ATP Synthase 6 (ATP6) showed much greater conservation (0.9%). Both NADH dehydrogenase 4 (ND4) and NADH dehydrogenase 5 (ND5) exhibited variation in the number of codons, with most variation observed in A.m. capensis (Table 2). There were no amino acid substitutions that presented synapomorphies uniting any of the subspecies for which multiple individuals were sampled. Instead, substitutions were either found in just a few individuals of a subspecies, were unique to one sample, or were shared among subspecies. The levels of variation differed among gene regions (Table 2), with the PCGs averaging just under 5% of variable sites. While the AT-rich region showed the greatest variation, some of this may be due to errors in alignment since the extreme base composition (over 95% AT) made identification of homology within this region challenging.
Table 3.
Genome size and nucleotide compositions among the mitogenomes of Apis mellifera capensis, A.m. scutellata and hybrid honey bees.
| Apis mellifera subspecies | whole mtDNA genome | *PCGs | †L-rRNA | ∫S-rRNA | AT-rich region | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| size range and avg bp | AT % | size range and avg bp | AT % | size range and avg bp | AT % | size range and avg bp | AT % | size range and avg bp | AT % | |
| A.m. capensis | 16,343–16,513 16,444.9 |
84.8 | 3,662–3,665 3,663.1 |
83.1 | 1,325–1,328 1,326.7 |
88 | 780–786 784.4 |
81 | 797–925 862.7 |
96 |
| A.m. scutellata | 16,338–16,479 16,422.4 |
84.7 | 3,662–3,663 3,662.1 |
83.1 | 1,326–1,328 1,326.4 |
84.1 | 785 785 |
81 | 767–931 858 |
96 |
| Hybrids | 16,340–16,455 16,397.5 |
84.7 | 3,662–3,663 3,662.5 |
83.1 | 1,326 1,326 |
84.1 | 780–785 782.5 |
81 | 798–874 836 |
95.8 |
*PCGs: protein-coding genes, includes stop codons.
†L-rRNA: Large subunit of ribosomal RNA.
∫S-rRNA: Small subunit of ribosomal RNA.
When comparing the RSA subspecies and hybrids, there was little variation and the subspecies did not appear distinct in overall characteristics of the mitogenome (Table 3). While the length of the mitogenome, the PCGs, the rRNAs and the AT-rich region varied, this variation occurred within subspecies and did not define subspecies (Table 3).
Variation among taxa
The multiple sequence alignment of all 39 mitogenomes produced 789 variable sites, of which 371 were parsimony informative. When only the 13 PCGs and two rRNAs were considered, this was reduced to 559 variables sites, of which 278 were parsimony informative. Using the concatenated data set (13 PCGs and two rRNAs), the A.m. capensis mitogenome sequences generated 124 variable positions, of which 44 were parsimony informative. There were 83 variable sites within A.m. scutellata mitogenome sequences, of which 15 were parsimony informative sites (the lower variation in A.m. scutellata is likely due to the smaller number of A.m. scutellata individuals sequenced). The multiple sequence alignment of hybrid mitogenomes included 34 variable sites and no parsimony informative sites.
Phylogenetic trees and distances among taxa
The phylogenetic trees using either the complete mitogenome or just the 13 PCGs and two rRNAs showed that the RSA honey bees constituted a well-supported clade, divided into two subclades (Figs 1, 2, 3, and 4). Each subclade consisted of honey bees from different regions and both subspecies. Although this demonstrated a lack of genetic structure between the subspecies, one clade is primarily comprised of A.m. capensis (with only one A.m. scutellata), while the other includes the remaining A.m. scutellata samples (and only three A.m. capensis). Both subclades within the RSA group included one of the hybrid samples. In addition, the other well-sampled subspecies, A.m. mellifera, was also not monophyletic. For both the complete and concatenated datasets, our Bayesian analysis produced a concordant tree topology to the ML tree (Figs 2 and 4).
Figure 1.

Maximum likelihood phylogenetic tree constructed with RAxML approach for the 39 Apis mellifera mitogenomes using concatenated sequences (13 PCGs + two rRNAs). The number represents the bootstrap values which are shown behind each node. The uppermost clade shows the identity of each bee as S for A.m. scutellata, C for A.m. capensis, or H for hybrid. The geographic origin of each bee follows its identity (S, C, or H) and is reported as the abbreviation noted in Table 1.
Figure 2.

Bayesian inference (BI) phylogenetic tree constructed with MrBayes approach for the 39 Apis mellifera mitogenomes using concatenated sequences (13 PCGs + two rRNAs). The number indicates BI posterior probability values which are shown behind each node. The uppermost clade shows the identity of each bee as S for A.m. scutellata, C for A.m. capensis, or H for hybrid. The geographic origin of each bee follows its identity (S, C, or H) and is reported as the abbreviation noted in Table 1.
Figure 3.

Maximum likelihood phylogenetic tree constructed with RAxML approach for the 39 Apis mellifera mitogenomes using complete mitochondrial genome sequences. The number represents the bootstrap values which are shown behind each node. The uppermost clade shows the identity of each bee as S for A.m. scutellata, C for A.m. capensis, or H for hybrid. The geographic origin of each bee follows its identity (S, C, or H) and is reported as the abbreviation noted in Table 1.
Figure 4.

Bayesian inference (BI) phylogenetic tree constructed with MrBayes approach for the 39 Apis mellifera mitogenomes using complete mitochondrial genome sequences. The number indicates BI posterior probability values which are shown behind each node. The uppermost clade shows the identity of each bee as S for A.m. scutellata, C for A.m. capensis, or H for hybrid. The geographic origin of each bee follows its identity (S, C, or H) and is reported as the abbreviation noted in Table 1.
The two datasets (complete mitogenome or 13 PCGs and two rRNAs) largely yielded similar results (e.g., compare Figs 1 to 2, 3 and 4), except that the placement of A.m. intermissa is in a different phylogenetic position when using the two data sets. Apis mellifera intermissa clusters with the RSA clade when complete mitogenomes are analyzed (Figs 3 and 4), whereas it is nested within a larger A.m. mellifera clade when using the concatenated data set (Figs 1 and 2). Thus, the inclusion of the harder-to-align data does have some impact on phylogenetic reconstruction. The inclusion of more sites, especially more variable sites, is expected to result in higher support values. However, there is no consistent pattern with some relationships showing higher support with the complete mitogenome, and others with the concatenated dataset. This may reflect that some sites in the non-coding regions are misaligned, so the increased number of variable sites in complete mitogenome dataset may be offset by greater noise in this alignment. This emphasizes the importance of focusing on regions than can be aligned with confidence.
Using the concatenated data set, the maximum value of the uncorrected genetic distance (p-distance) for all pairs of taxa that included one of the 25 RSA mitogenomes was between A.m. ligustica (0.015). The minimum value (0, indicating identical sequences) was only found in nine pairwise comparisons among the 25 South African mitogenomes (data not shown). The overall mean value of pairwise p-distance across all 39 mitogenomes was 0.006. The Nei genetic distances within groups of A.m. capensis and A.m. scutellata were 0.002 and 0.001, with an overall distance 0.002.
Population comparisons and haplotype network
The level of haplotype diversity was extremely high for the subspecies for which we had two or more samples. Each honey bee contained a unique haplotype with the exception of samples from hybrid region (KL) and A.m. scutellata (S_NJ601784) which shared a single haplotype (Fig. 5, Table S2). Although haplotype diversity was high among all subspecies, nucleotide diversity was lower among the RSA subspecies (and hybrids) than among A.m. mellifera samples (Table 4). Similarly, the average number of nucleotide differences (K), is much higher for A.m. mellifera than for the RSA subspecies (Table 4). The haplotype network of the concatenated data set generated 38 distinct haplotypes, supporting the results of the phylogeny that showed no population differentiation at the subspecies level (Fig. 5, Table S2).
Figure 5.
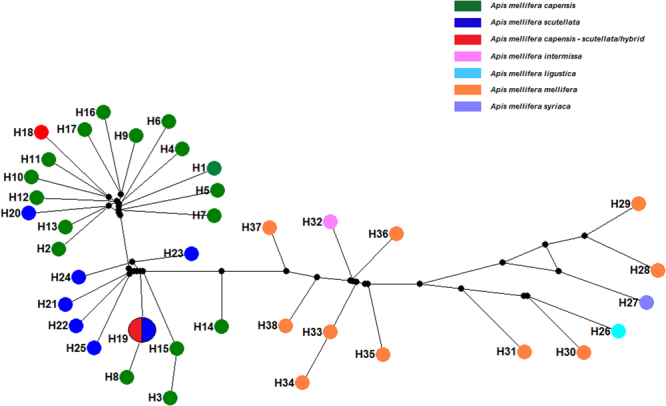
Median-joining parsimony haplotype network constructed for the concatenated sequences of 39 Apis mellifera mitogenomes. H1 – H38 refers to the distinct haplotypes. The size of each circle is proportional to haplotype frequency. Each taxa is marked with different colors as shown in the key. H19 was found in two individuals.
Table 4.
Population genetic indices calculated based on concatenated sequences over examined mitogenomes.
| Apis mellifera subspecies | *N | V | M | H | HD | π | K | Tajima D | Fu’s Fs |
|---|---|---|---|---|---|---|---|---|---|
| A.m. capensis | 17 | 124 | 125 | 17 | 1 ± 0.02 | 0.002 ± 0.0002 | 24.1 | −1.5–P > 0.1 | −4.6–P < 0.05 |
| A.m. scutellata | 7 | 83 | 84 | 7 | 1 ± 0.07 | 0.002 ± 0.0004 | 27.1 | −1.2–P > 0.1 | −0.05–P = 0.5 |
| Hybrids | 2 | 34 | 34 | 2 | 1 ± 0.5 | 0.002 ± 0.001 | 34 | — | 3.5–P = 1 |
| A.m. mellifera | 13 | 424 | 433 | 13 | 1 ± 0.03 | 0.009 ± 0.001 | 113 | −0.85–P > 0.1 | 0.064–P = 0.5 |
*N: number of individuals; V: number of polymorphic sites; M: number of mutations; H: number of haplotypes; HD: haplotype diversity; π: nucleotide diversity; K: average number of nucleotide differences.
Discussion
In this study, we compared whole mitogenomes from A.m. capensis, A.m. scutellata and hybrids in the RSA, along with published mitogenomes from three other subspecies to infer phylogenetic relationships and genetic differentiation among them. Our phylogenetic analyses suggested that the RSA honey bees are genetically closer to one another than to other taxa included in the tree. Although A.m. capensis and A.m. scutellata are genetically indistinguishable when using mitochondrial genomes, morphological analyses do separate these into two distinct clusters9. Furthermore, A.m. capensis and A.m. scutellata have distinctive physiological and behavioral differences8,9,11. Thus, the two subspecies are quite distinct and should not be considered as a single subspecies, even if currently no genetic diagnostic markers have been identified for either subspecies.
Although mitogenomes show significant rearrangement of gene orders among Insecta a putative ancestral gene order has been inferred, based in part on studies of Hemiptera41,42. There have been rearrangements on the branch leading to Apis42 but arrangement and gene orders observed in the examined mitogenomes for honey bees from the RSA were identical to those reported for other honey bees, supporting conservation of mitogenome gene order within this genus40,43,44. It is possible that there will not be major rearrangements within Apis but it is likely that A. mellifera needs to be studied more broadly across the complete modern diversity of Apis37,38 to establish this with confidence.
The consensus mitogenomes obtained in this study varied slightly in size: ±170 bp in A.m. capensis; ±141 bp in A.m. scutellata; and ±115 bp for the hybrids. Variation in mitogenome size is typically the consequence of variation in the non-coding regions in insects45, which is consistent with our observations of A. mellifera mitogenomes. Only limited size variation was detected in the coding and rRNA regions. The mitogenomes of A. mellifera has an AT-bias (84.9%), with guanine as the rarest nucleotide43, which is consistent with other observations that the mitochondrial genomes of insects in general have very strong A+T base composition in the non-coding control region46,47.
We observed different levels of variation in the PCGs genes across all datasets. The greatest variation looking at nucleotides was in ND4L (followed by ND4), though at the amino acid level the greatest variation was in the CO3 gene. For both metrics, ATP6 exhibited the highest level of conservation. De Jager et al.45 found that the greatest variation in the Diuraphis noxia (Hemiptera: Aphididae) was observed in the ND5 nucleotide gene but the causal effects of variation in ND5 was not fully understood. These results suggest that the degree of stabilizing selection differs on different genes within the mitochondria, and that this likely also differs among different taxonomic groups. However, too little information currently exists on insect mitochondria to elucidate why there may be different levels of selection among genes, and how universally there are differences among different taxonomic groups.
Using the complete mitochondrial genome sequences, we found that A.m. intermissa clustered at an intermediate position (Figs 3 and 4). The phylogenetic position of A.m. intermissa has been reported by previous investigators and supported by the present study, indicating that A.m. intermissa might be a hybrid subspecies or that the sampled bee was misidentified as a true representative of the subspecies48. We support sequencing of additional samples to confirm the phylogenetic position of this subspecies.
Mitochondrial DNA has been widely used as a marker in population genetic, biogeographic, phylogenetic and DNA barcoding studies49. Due to their relative rapid coalescence, one should be able to distinguish reciprocally monophyletic groups even when analyses of nuclear markers fail to do so. However, based on our results, mitogenomes appear to be poor diagnostic phylogenetic markers in Apis, as has also been found in some other taxa50. Our results are not likely due to specific mitochondrial inheritance patterns since similar results have been reported for these subspecies using nuclear SNP loci27,28. The absence of genetic differentiation, even with mitochondrial DNA sequences, in taxa where there are clear phenotypic differences among populations has been found in a variety of invertebrate and vertebrate taxa51–53, and may be due to the retention of ancestral genetic variation in recently diverged lineages.
An alternative explanation for the lack of genetic differentiation between A.m. capensis and A.m. scutellata could also be due to the impact of beekeeping activities, which can involve the exchange of queens and colonies. These are known to contribute to the admixture pattern among honey bee populations8,54. This, in fact, led to a major problem in RSA known as the “capensis calamity.” This occurred because A.m. capensis workers were moved from southern portions of RSA into northern portions outside their native range55. The A.m. capensis workers became social parasites of A.m. scutellata colonies there, which is a possible reason that their mitogenomes are represented in bees that were morphometrically identified as A.m. scutellata56. This problem has slowed or nearly eliminated the movement of managed hives between southern and northern RSA, so the problem should be diminishing. That said, in spite of the movement of bees, the morphological and physiological differences between A.m. capensis and A.m. scutellata persist.
The high genetic diversity in A.m. capensis, A.m. scutellata, hybrids, and other subspecies could be due to large population sizes within their natural habitats9. The observed low nucleotide diversity with high haplotype diversity observed in both RSA subspecies might suggest a genetic bottleneck occurred in the past, followed by rapid population expansion for both subspecies. This hypothesis could be investigated via the sampling of more sites and more individuals throughout RSA.
While SNP analyses can differentiate other honey bee subspecies belonging to different evolutionary groups29,57,58, mitogenome sequences are not able to differentiate A.m. capensis and A.m. scutellata. There are several reasons this could occur. First, while some hybrids can be identified morphologically, there may be others (particularly if hybrids backcross to one of the parentals) that are morphologically like one subspecies, but may retain the mitogenome of the second subspecies. Second, there could be incomplete lineage sorting, resulting in a mitochondrial gene tree that does not match the subspecies tree59. Finally, a small number of genes may be responsible for the phenotypic differences among subspecies, while the remainder of the nuclear genome (as well as the mitogenome) have not yet become reciprocally monophyletic60. If this is the case, then the small number of genes responsible for this differentiation would be expected to provide diagnostic markers. Exploration of complete or mostly complete nuclear genomes may be necessary to understand patterns of genetic differentiation, gene flow and divergence among A.m. capensis and A.m. scutellata. Such analyses may eventually identify diagnostic markers, thereby allowing more rapid identification of invasive species. Such methods would enhance our ability to detect hidden genetic structure among regional populations of honey bees in RSA.
Methods
Honey bee samples, DNA extraction and sequencing
Honey bee samples were collected from managed colonies located across 25 geographical regions in RSA (Table 1, Fig. 6) in April/May 2013 and April 201461. During the field collections, 50–70 adult worker bees were collected from the brood nest of 1–10 colonies located in 1–3 apiaries sampled per each of the 25 geographic regions. In total, 1,000+ colonies were sampled. All bees were collected into 50 ml tubes containing ≥ 98% ethanol. After collection, the samples were imported into the U.S. per USDA APHIS regulations and approval and stored at −80 °C. Bustamante et al.62 conducted a morphometric analysis on 10 bees from a subset of sampled colonies (N = 240) to classify colonies as A.m. capensis, A.m. scutellata, or hybrids according to standard procedures9,63. The thorax of each bee that was identified morphometrically was preserved in 95% ethanol and stored at −80 °C until molecular processing.
Figure 6.

Origin of 25 honey bees collected from managed hives in the Republic of South Africa. The numbers coincide with the sampling regions discussed in Table 1. The map of South Africa was downloaded from this link (http://www.conceptdraw.com/How-To-Guide/geo-map-africa-south-africawas) and re-designed using Adobe Indesign graphical program version CS6 from this link (http://www.adobe.com/products/indesign.html?sdid=KKQLR&mv=search&s_kwcid=AL!3085!3!155829415173!b!!!!indesign%20cs6&ef_id=V2Cx9wAABOu6AgoK:20170830124634:s).
One bee from each geographic region was selected for use in the present study. In total, 17 A.m. capensis, six A.m. scutellata, and two hybrid bees were used (Table 1 and Fig. 6), including two whose complete mitogenomes had been published previously37,38. Total genomic DNA was extracted from the thoracic tissue of each bee using a Wizard® Genomic DNA Purification kit (Promega, USA) according to the manufacturer’s instructions. The quality of DNA was checked using a 1% agarose gel and quantified using a Quant-iT PicoGreen dsDNA Assay Kit (Life Technologies). From this, 500 ng of DNA was utilized for the construction of paired-end libraries comparable with Illumina sequencing by RAPiD Genomics (Gainesville, Florida, USA). The constructed libraries were multiplexed and sequenced with PE-100 cycle runs (2 × 100 bp) using Next Generation Sequencing on an Illumina HiSeq. 3000 platform (San Diego, California, USA).
Sequence quality control, assembly and annotation
Mitochondrial data were obtained using the genome skimming method64. Short reads were filtered by quality in two steps using the fastx-toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). The first removed bases from the 3′ end of the sequence that had a phred score below 20 and removed the entire read if the resulting length was <50 nt (fastq_quality_trimmer -Q 33 -t 20 -l 50). In the second step, reads that passed the first criteria, but that had ≥10% of the bases with a phred score <20 (fastq_quality_filter -Q 33 -q 20 -p 90) were removed. The resulting sequences were aligned to the bee mitochondria reference genome (NCBI accession NC_0015661) using Mosaik Aligner65 version 2.1.33 (parameters -mmp 0.05, -ls 500 -m all -a all).
Illumina sequence reads were mapped to A.m. capensis and A.m. scutellata (KX870183 and KJ601784.1) mitogenomes using Geneious R9.166, and the assembly that exhibited the fewest conflicts (e.g., that mapped to A.m. capensis or to A.m. scutellata) was used. The mapping procedures are described in37.
The locations and orientation of protein-coding genes (PCGs), transfer RNA genes (tRNAs) and ribosomal RNA genes (rRNA) genes were identified by multiple alignments to the reference mitogenome using Geneious R9.166. All PCGs for each honey bee mitogenome were translated into amino acids and were manually checked to ensure that each could encode a functional protein (i.g., we examined putative PCGs to determine whether they lacked start or stop codons or exhibited frame shifts and/or premature stop codons.
Sequence alignment
We aligned 23 novel complete mitogenome sequences with two additional mitogenomes that we published previously37,38, as well as 14 published mitogenomes primarily from other A. mellifera subspecies (Table S3). All mitogenomes were aligned using Muscle 3.8.3167 implemented in Mesquite 3.0468 with default parameters. The alignment was manually adjusted to maintain reading frame integrity in the protein coding genes.
Our alignment included the complete mitogenome nucleotide sequence. However, since homology was harder to assign in some regions (particularly intergenic regions and the AT-rich region), we also analyzed a concatenated alignment that included all 13 PCGs and two rRNAs to represent a dataset in which we had higher confidence in the alignment. This allowed us to determine if our conclusions depended upon alignment or were robust to inclusion of specific regions and homology assessment in non-coding regions. We estimated p-distance between taxa using MEGA7.
Phylogenetic analyses
Phylogenetic analyses of the complete (entire mitogenome) and concatenated datasets (13 PCGs and two rRNAs) were performed using two different tree-building methods, maximum likelihood (ML) and Bayesian Inference (BI). Maximum Likelihood analyses were performed using RAxML version 8.0.269 with the GTRGAMMA model and 1,000 rapid bootstraps to assess nodal support. BI was performed based on the concatenated data set using MrBayes version 3.2.470. Before running MrBayes, the complete and concatenated datasets were run through jModelTest 3.771 and the AIC was used to select the best model. The best model for the complete dataset was GTR+I+G (shape = 0.7050, pinvar = 0.81), and the best model for the 13 PCGs and two rRNAs was TVM+I+G (shape = 0.8920, pinvar = 0.834). The BI used two independent runs with 8,000,000 generations in each and four chains. Each chain was sampled every 1,000 generations with a burn-in of 25%. The remaining trees were condensed using a 50% majority-rule consensus tree with posterior probabilities.
We also assessed the evolutionary distance between A.m. capensis and A.m. scutellata using uncorrected p-distances among unique haplotypes of the 13 PCG and two rRNA data set with MEGA772.
Population genetic analyses
Our initial results suggested there could be some bias due to mis-alignment in the analysis of the complete dataset. Therefore, we focused population genetic analyses on the 13 PCGs and two rRNA dataset. A haplotype network was generated using the median-joining method with default settings in NETWORK v. 4.6.1.373 for the concatenated dataset (13 PCGs and two rRNAs) from the 39 samples (25 sequenced from the RSA, and 14 previously published mitogenomes from Africa and Europe). All networks were visualized and manually adjusted to ensure sufficient resolution. We computed the haplotype diversity (HD), nucleotide diversity (π), number of haplotypes (H), number of variable sites (V), number of mutations (M), average number of nucleotide differences (K) and neutrality indices using DnaSP v574.
Electronic supplementary material
Acknowledgements
The authors would like to thank the current and former members of the University of Florida Honey Bee Research and Extension Laboratory who collected honey bee samples across the Republic of South Africa. This includes Tomas Bustamante, Mark Dykes, Ashley Mortensen, and Daniel Schmehl. We also thank Mathias Ellis for his assistance with sample collection. We graciously acknowledge Mike Allsopp (ARC-Plant Protection Research Institute, South Africa), Christian Pirk (University of Pretoria, South Africa), and Garth Cambray for the assistance they provided in coordinating field sample collections and/or providing samples. We also thank the South African beekeepers who allowed us to sample their colonies. This project was financed through a cooperative agreement provided by the United States Department of Agriculture, Animal and Plant Health Inspection Service (USDA-APHIS) and by the Florida Department of Agriculture and Consumer Services through the guidance of the Honey Bee Technical Council.
Author Contributions
A.E. and J.D.E. designed and coordinated the research. J.D.E. collected the samples. A.E. conducted library construction and sequencing. A.E., R.T.K. and E.L.B. analyzed the data. A.E. drafted the paper with the input of J.D.E., R.T.K. and E.L.B. All authors read and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-19759-3.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Esther E, et al. Detoxification mechanisms of honey bees (Apis mellifera) resulting in tolerance of dietary nicotine. Scientific reports. 2015;5:11779. doi: 10.1038/srep11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haddad N, et al. Next generation sequencing of Apis mellifera syriaca identifies genes for Varroa resistance and beneficial bee keeping traits. Insect Science. 2015;23(4):579–590. doi: 10.1111/1744-7917.12205. [DOI] [PubMed] [Google Scholar]
- 3.Ruttner, F. Biogeography and taxonomy of honeybees. Springer Verlag, Heidelberg, Berlin, New York. 284 (1988).
- 4.Garnery L, Cornuet JM, Solignac M. Evolutionary history of the honey bee Apis mellifera inferred from mitochondrial DNA analysis. Molecular Ecology. 1992;1:145–154. doi: 10.1111/j.1365-294X.1992.tb00170.x. [DOI] [PubMed] [Google Scholar]
- 5.Franck P, Garnery L, Solignac M, Cornuet J-M. Molecular confirmation of a fourth lineage in honeybees from the Near East. Apidologie. 2000;31:167–180. doi: 10.1051/apido:2000114. [DOI] [Google Scholar]
- 6.Franck P, et al. Genetic diversity of the honeybee in Africa: microsatellite and mitochondrial data. Heredity. 2001;86(4):420–430. doi: 10.1046/j.1365-2540.2001.00842.x. [DOI] [PubMed] [Google Scholar]
- 7.Alburaki M, et al. A fifth major genetic group among honeybees revealed in Syria. BMC Genetics. 2013;14(1):1. doi: 10.1186/1471-2156-14-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gupta, R. K., Reybroeck, W., van Veen, J. W. & Gupta, A. Beekeeping for Poverty Alleviation and Livelihood Security. 63 (2014).
- 9.Hepburn, H. R. & Radloff, S. E. Honeybees of Africa. Springer Science & Business Media (1998).
- 10.Sheppard, W. Honey Bee Diversity – Races, Ecotypes and Strains. In Joe Graham (Eds.), The Hive and the Honey Bee. 53–67 (2015).
- 11.Onions GW. South African ‘fertile worker bees’. Agric J Union S Afr. 1912;1:720–728. [Google Scholar]
- 12.Ruttner F. The problem of the Cape bee (Apis mellifera capensis Escholtz): parthenogenesis-size of population-evolution. Apidologie. 1977;8(3):281–294. doi: 10.1051/apido:19770305. [DOI] [Google Scholar]
- 13.Phiancharoen M, Pirk CWW, Radloff SE, Hepburn R. Clinal nature of the frequencies of ovarioles and spermathecae in Cape worker honeybees, Apis mellifera capensis. Apidologie. 2010;41:129–134. doi: 10.1051/apido/2009054. [DOI] [Google Scholar]
- 14.Crewe RM, Velthuis HHW. False queens: a consequence of mandibular gland signals in worker honeybees. Natur wissenschaften. 1980;67:467. doi: 10.1007/BF00405650. [DOI] [Google Scholar]
- 15.Velthuis, H. H. W., Ruttner, F. & Crewe, R. M. Differentiation in reproductive physiology and behaviour during the development of laying worker honey bees. Social Insects Springer Berlin Heidelberg 231–243 (1990).
- 16.Hepburn HR, Crewe RM. Portrait of the Cape honeybee, Apis mellifera capensis. Apidologie. 1991;22:567–580. doi: 10.1051/apido:19910601. [DOI] [Google Scholar]
- 17.Allsopp MH. The capensis calamity. S Afr Bee J. 1992;64:52–55. [Google Scholar]
- 18.Winston ML, Taylor OR, Otis GW. Some differences between temperate and tropical African and South American honeybees. Bee World. 1983;64:12–21. doi: 10.1080/0005772X.1983.11097902. [DOI] [Google Scholar]
- 19.Neumann P, Radloff SE, Moritz RFA, Hepburn HR, Reece SL. Social parasitism by honeybee workers (Apis mellifera capensis Esch.): Host finding and resistance of hybrid host colonies. Behav Ecol. 2001;12:419–428. doi: 10.1093/beheco/12.4.419. [DOI] [Google Scholar]
- 20.Kerr WE. Introducao de abelhas africanas no Brasil. Brasil Apícola. 1957;3:211–213. [Google Scholar]
- 21.Kerr WE. The history of the introduction of African bees in Brazil. Apiculture in Western Australia. 1967;2:53–55. [Google Scholar]
- 22.Moritz RF, Härtel S, Neumann P. Global invasions of the western honeybee (Apis mellifera) and the consequences for biodiversity. Ecoscience. 2005;12(3):289–301. doi: 10.2980/i1195-6860-12-3-289.1. [DOI] [Google Scholar]
- 23.Harrison JF, Fewell JH, Anderson KE, Loper GM. Environmental physiology of the invasion of the Americas by Africanized honeybees. Integrative and Comparative Biology. 2006;46(6):1110–1122. doi: 10.1093/icb/icl046. [DOI] [PubMed] [Google Scholar]
- 24.Vital MVC, Hepburn R, Radloff S, Fuchs S. Geographic distribution of Africanized honeybees (Apis mellifera) reflects niche characteristics of ancestral African subspecies. Braz J Nat Conserv. 2012;10:184–190. [Google Scholar]
- 25.Beekman M, Allsopp MH, Wossler TC, Oldroyd BP. Factors affecting the dynamics of the honeybee (Apis mellifera) hybrid zone of South Africa. Heredity. 2008;100:13–18. doi: 10.1038/sj.hdy.6801058. [DOI] [PubMed] [Google Scholar]
- 26.Scott Schneider S, DeGrandi-Hoffman G, Smith DR. The African honey bee: factors contributing to a successful biological invasion. Annual Reviews in Entomology. 2004;49(1):351–376. doi: 10.1146/annurev.ento.49.061802.123359. [DOI] [PubMed] [Google Scholar]
- 27.Chapman NC, et al. A SNP test to identify Africanized honeybees via proportion of ‘African’ ancestry. Molecular Ecology Resources. 2015;15(6):1346–1355. doi: 10.1111/1755-0998.12411. [DOI] [PubMed] [Google Scholar]
- 28.Harpur BA, et al. Assessing patterns of admixture and ancestry in Canadian honey bees. Insectes Sociaux. 2015;62(4):479–489. doi: 10.1007/s00040-015-0427-1. [DOI] [Google Scholar]
- 29.Wallberg A, et al. A worldwide survey of genome sequence variation provides insight into the evolutionary history of the honeybee Apis mellifera. Nature genetics. 2014;46(10):1081–1088. doi: 10.1038/ng.3077. [DOI] [PubMed] [Google Scholar]
- 30.Moore WS. Inferring phylogenies from mtDNA variation: mitochondrial‐gene trees versus nuclear‐gene trees. Evolution. 1995;49:718–726. doi: 10.1111/j.1558-5646.1995.tb02308.x. [DOI] [PubMed] [Google Scholar]
- 31.Oliveira DCSG, Raychoudhury R, Lavrov DV, Werren JH. Rapidly evolving mitochondrial genome and directional selection in mitochondrial genes in the parasitic wasp Nasonia (Hymenoptera: Pteromalidae) Molecular Biology and Evolution. 2008;25(10):2167–2180. doi: 10.1093/molbev/msn159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaltenpoth, M. et al. Accelerated evolution of mitochondrial but not nuclear genomes of Hymenoptera: new evidence from crabronid wasps. PLoS one7(3) (2012). [DOI] [PMC free article] [PubMed]
- 33.Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 34.Mardis ER. A decade’s perspective on DNA sequencing technology. Nature. 2011;470:198–203. doi: 10.1038/nature09796. [DOI] [PubMed] [Google Scholar]
- 35.Duchene, S., Archer, F. I., Vilstrup, J., Caballero, S. & Morin, P. A. Mitogenome phylogenetics: The Impact of Using Single Regions and Partitioning Schemes on Topology, Substitution Rate and Divergence Time Estimation. Plos One6(11) (2011). [DOI] [PMC free article] [PubMed]
- 36.Vilstrup JT, et al. Mitogenomic phylogenetic analyses of the Delphinidae with an emphasis on the Globicephalinae. BMC Evol Biol. 2011;11:65. doi: 10.1186/1471-2148-11-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eimanifar A, Kimball RT, Braun EL, Ellis JD. The complete mitochondrial genome of the Cape honey bee Esch., Apis mellifera capensis (Insecta: Hymenoptera: Apidae) Mitochondrial DNA Part B. 2016;1:817–819. doi: 10.1080/23802359.2016.1241682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eimanifar A, Kimball RT, Braun EL, Ellis JD. The complete mitochondrial genome of the hybrid honey bee, Apis mellifera capensis × Apis mellifera scutellata, from South Africa. Mitochondrial DNA Part B. 2016;1(1):856–857. doi: 10.1080/23802359.2016.1250132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gibson JD, Hunt GJ. The complete mitochondrial genome of the invasive Africanized Honey Bee, Apis mellifera scutellata (Insecta: Hymenoptera: Apidae) Mitochondrial DNA Part A. 2016;27(1):561–562. doi: 10.3109/19401736.2014.905858. [DOI] [PubMed] [Google Scholar]
- 40.Hu P, et al. Complete mitochondrial genome of the Algerian honeybee, Apis mellifera intermissa (Hymenoptera: Apidae) Mitochondrial DNA Part A. 2016;27(3):1791–1792. doi: 10.3109/19401736.2014.963815. [DOI] [PubMed] [Google Scholar]
- 41.Shao R, Baker SC. The highly rearranged mitochondrial genome of plague thrips, Thrips imagines (Insecta: Thysanoptera): convergence of two novel gene boundaries and an extraordinary rearrangement of rRNA genes. Molecular Biology and Evolution. 2003;20:362–370. doi: 10.1093/molbev/msg045. [DOI] [PubMed] [Google Scholar]
- 42.Dowton M. Assessing the relative rate of (mitochondrial) genomic change. Genetics. 2004;167(2):1027–1030. doi: 10.1534/genetics.103.024885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crozier RH, Crozier YC. The mitochondrial genome of the honeybee Apis mellifera: complete sequence and genome organization. Genetics. 1993;133(1):97–117. doi: 10.1093/genetics/133.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haddad NJ. Mitochondrial genome of the Levant Region honeybee, Apis mellifera syriaca (Hymenoptera: Apidae) Mitochondrial DNA Part A. 2016;27(6):4067–4068. doi: 10.3109/19401736.2014.1003846. [DOI] [PubMed] [Google Scholar]
- 45.De Jager L, Burger NFV, Botha AM. Complete mitochondrial genome of Diuraphis noxia (Hemiptera: Aphididae) from nine populations, SNP variation between populations, and comparison with other Aphididae species. African Entomology. 2014;22(4):847–862. doi: 10.4001/003.022.0409. [DOI] [Google Scholar]
- 46.Chai HN, Du YZ, Zhai BP. Characterization of the complete mitochondrial genome of Cnaphalocrocis medinalis and Chilo suppressalis (Lepidoptera: Pyralidae) International Journal of Biological Sciences. 2012;8:561–579. doi: 10.7150/ijbs.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gonçalves R, Freitas AI, Jesus J, De la Rúa P, Brehm A. Structure and genetic variation of the mitochondrial control region in the honey bee Apis mellifera. Apidologie. 2015;46(4):515–526. doi: 10.1007/s13592-014-0341-y. [DOI] [Google Scholar]
- 48.Han F, Wallberg A, Webster MT. From where did the Western honeybee (Apis mellifera) originate? Ecology and Evolution. 2012;2(8):1949–1957. doi: 10.1002/ece3.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fourdrilis, S. et al. Mitochondrial DNA hyperdiversity and its potential causes in the marine periwinkle Melarhaphe neritoides (Mollusca: Gastropoda). PeerJ4 (2016). [DOI] [PMC free article] [PubMed]
- 50.Wu J, Krutovskii KV, Strauss SH. Abundant mitochondrial genome diversity, population differentiation and convergent evolution in pines. Genetics. 1998;150(4):1605–1614. doi: 10.1093/genetics/150.4.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buerkle CA. The historical pattern of gene flow among migratory and nonmigratory populations of prairie warblers (Aves: Parulinae) Evolution. 1999;53:1915–1924. doi: 10.1111/j.1558-5646.1999.tb04572.x. [DOI] [PubMed] [Google Scholar]
- 52.Bulgin NL, Gibbs HL, Vickery P, Baker AJ. Ancestral polymorphisms in genetic markers obscure detection of evolutionarily distinct populations in the endangered Florida grasshopper sparrow (Ammodramus savannarum floridanus) Mol Ecol. 2003;12:831–844. doi: 10.1046/j.1365-294X.2003.01774.x. [DOI] [PubMed] [Google Scholar]
- 53.Lee CF, Tsai CL, Konstantinov A, Yeh WB. Revision of Mandarella Duvivier from Taiwan, with a new species, new synonymies and identities of highly variable species (Insecta, Chrysomelidae, Galerucinae, Alticini) Zookeys. 2016;568:23–49. doi: 10.3897/zookeys.568.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Techer MA, et al. Genetic characterization of the honeybee (Apis mellifera) population of Rodrigues Island, based on microsatellite and mitochondrial DNA. Apidologie. 2015;46(4):445–454. doi: 10.1007/s13592-014-0335-9. [DOI] [Google Scholar]
- 55.Neumann P, Moritz RFA. The Cape honeybee phenomenon: The sympatric evolution of a social parasite in real time? Behav Ecol Sociobiol. 2002;52:271–281. doi: 10.1007/s00265-002-0518-7. [DOI] [Google Scholar]
- 56.Neumann P, Hepburn R. Behavioural basis for social parasitism of Cape honeybees (Apis mellifera capensis) Apidologie. 2002;33(2):165–192. doi: 10.1051/apido:2002008. [DOI] [Google Scholar]
- 57.Ilyasov RA, Poskryakov AV, Nikolenko AG. New SNP markers of the honeybee vitellogenin gene (Vg) used for identification of subspecies Apis mellifera mellifera L. Russian Journal of Genetics. 2015;51(2):163–168. doi: 10.1134/S1022795415020088. [DOI] [PubMed] [Google Scholar]
- 58.Muñoz, I. et al. Reduced SNP panels for genetic identification and introgression analysis in the dark honey bee (Apis mellifera mellifera). PloS one10(4) (2015). [DOI] [PMC free article] [PubMed]
- 59.Maddison WP. Gene trees in species trees. Systematic Biology. 1997;46(3):523–536. doi: 10.1093/sysbio/46.3.523. [DOI] [Google Scholar]
- 60.Toews DP, et al. Plumage genes and little else distinguish the genomes of hybridizing warblers. Current Biology. 2016;26(17):2313–2318. doi: 10.1016/j.cub.2016.06.034. [DOI] [PubMed] [Google Scholar]
- 61.Mortensen AN, et al. Differences in Varroa destructor infestation rates of two indigenous subspecies of Apis mellifera in the Republic of South Africa. Experimental and Applied Acarology. 2016;68(4):509–515. doi: 10.1007/s10493-015-9999-8. [DOI] [PubMed] [Google Scholar]
- 62.Bustamante, T., Baiser, B. & Ellis, J. Determining if wing geometry can be used to discriminate between the South African honey bee subspecies Apis mellifera scutellata and Apis mellifera capensis (Unpublished, 2017).
- 63.Meixner MD, et al. Standard methods for characterising subspecies and ecotypes of Apis mellifera. Journal of Apicultural Research. 2013;52(4):1–28. doi: 10.3896/IBRA.1.52.4.05. [DOI] [Google Scholar]
- 64.Straub SCK, et al. Navigating the tip of the genomic iceberg: Next-generation sequencing for plant systematics. American Journal of Botany. 2012;99:349–364. doi: 10.3732/ajb.1100335. [DOI] [PubMed] [Google Scholar]
- 65.Lee, W. P. et al. MOSAIK: a hash-based algorithm for accurate next-generation sequencing short-read mapping. (CK Hsiao, Ed.). PLoS one9(3) (2014). [DOI] [PMC free article] [PubMed]
- 66.Kearse M, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maddison, W. P. & Maddison, D. R. Mesquite: a modular system for evolutionary analysis. Version 3.10, http://mesquiteproject.org (2016).
- 69.Stamatakis A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 70.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 71.Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14(9):817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 72.Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Molecular Biology and Evolution. 2016;33(7):1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bandelt HJ, Forster P, Rohl A. Median joining networks or inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 74.Librado P, Rozas J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.