

ABSTRACT
The Lyme disease-causing organism Borrelia burgdorferi is transmitted into the mammalian host by an infected-tick bite. Successful infection relies on the ability of this extracellular pathogen to persist and colonize different tissues. B. burgdorferi encodes a large number of adhesins that are able to interact with host ligands to facilitate adherence and tissue colonization. Multiple glycosaminoglycan binding proteins present in B. burgdorferi offer a degree of redundancy of function during infection, and this highlights the importance of glycosaminoglycans as host cell receptors for spirochete adherence. Of particular interest in this study is Borrelia glycosaminoglycan binding protein (Bgp), which binds to heparin-related glycosaminoglycans. The properties of a bgp transposon mutant and a trans-complemented derivative were compared to those of the wild-type B. burgdorferi in the in vitro binding assays and in infection studies using a C3H/HeJ mouse infection model. We determined that the loss of Bgp impairs spirochete adherence, infectivity, and tissue colonization, resulting in a reduction of inflammatory manifestations of Lyme disease. Although Bgp is not essential for infectivity, it is an important virulence factor of B. burgdorferi that allows adherence and tissue colonization and contributes to disease severity.
KEYWORDS: adhesin, B. burgdorferi, Bgp, glycosaminoglycan, infectivity, Lyme disease, qPCR
INTRODUCTION
Lyme disease is the most common vector-borne illness in the United States, with an estimated 300,000 new cases occurring each year (1, 2). Symptoms of the disease range from acute skin involvement with the characteristic bull's eye rash known as erythema migrans and generalized flu-like symptoms to chronic and debilitating inflammatory manifestations such as Lyme carditis and arthritis (3, 4). Infection with Borrelia burgdorferi or other Lyme spirochetes triggers an inflammatory response due to the expression of a large number of immunogenic lipoproteins on the surface of these pathogens (5, 6). Many of these lipoproteins, as well as other proteins, function as surface adhesins and play a critical role in the ability of this extracellular pathogen to infect and persist within the host (7). The relatively small, 1.52-Mbp, genome of extracellular B. burgdorferi encodes a large repertoire of adhesion mechanisms, highlighting the importance of this function in Lyme disease pathogenesis.
Investigations of host-pathogen interactions between B. burgdorferi adhesins and components of the host extracellular matrix (ECM) and other receptors, such as collagen, fibronectin, glycosaminoglycans (GAGs), and integrins, have been an area of active research for many years. The ubiquitous expression of GAGs, particularly in the ECM of skin, neuronal, and joint tissues, which are commonly affected in patients with Lyme disease, has prompted investigations to identify the spirochetal proteins that bind to these important host molecules (8–10). GAGs are long carbohydrate chains composed of disaccharide repeats covalently linked to a protein core; the composition of the disaccharide repeats and the sulfation patterns generate different types of GAGs, including heparan sulfate, dermatan sulfate, and chondroitin sulfate, which have been investigated to determine recognition by B. burgdorferi strains (11).
The ability of Lyme disease-causing spirochete strains to bind to GAGs (including purified heparin, heparan sulfate, and dermatan sulfate on host cells) and proteoglycans was recognized quite early (11–14). Concerted efforts were made to identify and characterize GAG-binding adhesins using biochemical approaches that led to the identification of Borrelia glycosaminoglycan-binding protein (Bgp) (12, 15). Bgp is a 26-kDa protein expressed on the outer membrane of the spirochete and has been classified as a GAG-binding adhesin since it was purified using heparin affinity chromatography and possesses hemagglutination activity. Binding of recombinant Bgp to immobilized heparin and the observation of partial inhibition of B. burgdorferi attachment to heparin following preincubation with recombinant Bgp further validated the role of Bgp as a heparin-binding hemagglutinin (15). The relative contribution of Bgp-mediated GAG binding to the virulence of Lyme spirochetes has not been assessed systematically due to the unavailability of appropriate mutant and complemented strains. The gene encoding Bgp was suggested to be the first gene in a two-gene operon, with the downstream essential gene encoding phosphate acetyltransferase (Pta), thus making genetic manipulations difficult (16, 17).
In 2012, a genome-wide transposon mutagenesis study of B. burgdorferi performed by Lin et al. identified approximately 4,800 single-insertion events, from which over 400 mutants were characterized for infectivity (18). A bgp mutant strain, T06TC117, which harbors a single transposon insertion within the BB_0588 gene, was identified. The study utilized two methods to investigate the infectivity of a mutant library of 502 signature-tagged mutants (STM) with disruption in 422 genes (18). A Luminex/PCR-based system was used for detection of transposon insertion in spirochete DNA recovered from organs of the infected mice directly or after tissue cultures. The median fluorescence intensity (MFI) value correlated with the quantities of spirochete DNA present. T06TC117 had a mean MFI of 33 and 0% positive cultures, indicating the importance of Bgp as a virulence factor of B. burgdorferi (18). The retention of viability of the bgp mutants suggested that expression of the essential Pta protein of B. burgdorferi was not perturbed in this mutant strain. Furthermore, this transposon mutagenesis screen did not identify any insertions in the pta gene, validating that the protein is essential for B. burgdorferi survival (18).
Two of the most commonly studied B. burgdorferi GAG-binding adhesins are decorin binding protein A (DbpA) and DbpB, which have been shown to bind to decorin, a proteoglycan prevalent in the ECM that binds to collagen via conserved lysine residues (19, 20). Adherence of B. burgdorferi to immobilized decorin was inhibited in the presence of recombinant DbpA (21). Overexpression of these proteins in a high-passage, nonadherent B. burgdorferi strain lacking the plasmid encoding DbpA/B restored binding to immobilized decorin (22). In decorin-deficient mice, B. burgdorferi dissemination, tissue colonization, and inflammatory manifestations of disease were diminished (23–25). Several reports have demonstrated the affinity of DbpA/B for decorin and dermatan sulfate and showed correlation of this binding with joint colonization and Lyme arthritis development and persistence within the host. These results suggest that the expression of decorin-binding proteins and other GAG-binding adhesins enhances the virulence of these spirochetes and contributes to the pathogenesis of Lyme disease (22, 24, 26–32).
Another B. burgdorferi protein, BBK32, originally described as a fibronectin binding protein, was also shown to have affinity for GAGs, specifically heparin and heparan sulfate (33, 34). An observation that BBK32 was able to bind to HEp-2 epithelial cells, which lack fibronectin, prompted further investigation into other potential receptors (35). The finding that enzymatic digestion with heparinase effectively removed adherence, coupled with the ability of BBK32 to bind to immobilized heparin as efficiently as fibronectin, confirmed the proposed affinity of this protein for GAGs (34). Studies comparing a BBK32 mutant strain in which the gene was disrupted by allelic exchange revealed impaired infectivity and tissue colonization of mice infected with the mutant strain (36). Multiple truncations and deletions in the bbk32 gene showed that the fibronectin and GAG-binding domains of the encoded protein were independent of one another. Furthermore, removal of the GAG-binding activity of BBK32, and not its fibronectin domain, abolished joint colonization, demonstrating the importance of spirochete adherence to GAGs in inflammatory manifestations of disease (35).
Unlike DbpA/B and BBK32, the GAG-binding contributions of Bgp have not been characterized thoroughly yet, in part because the mutagenesis assays required to elucidate the role of this protein during infection are complicated by the organization of the gene, and also because the protein has dual function both as a GAG-binding adhesin and 5′ methylthioadenosine/S-adenosyl homocysteine (MTA/SAH) nucleosidase. In addition to hydrolysis of MTA and SAH, this class of enzymes has several critical functions, such as regulation of methylation reactions, production of quorum-sensing autoinducer compounds, and regulation of the synthesis of polyamines (37, 38). Due to the extreme auxotrophic nature of B. burgdorferi, these nucleosidases have been suggested to be important for the recycling of nutrients, particularly the purine adenine by hydrolysis of MTA and SAH (38, 39).
The use of the specific mutants is the ideal method to directly demonstrate the role of an adhesin in attachment to the host cells in vitro and in colonization of the specific tissues in vivo. The purpose of this study was to characterize the bgp mutant of B. burgdorferi strain B31 using in vitro and in vivo experimental techniques. We sought to investigate how loss of this protein affects spirochete adherence, infectivity, tissue colonization, and disseminated inflammatory manifestations of Lyme disease. Our findings demonstrate that even though Bgp is not essential for infectivity, the protein is an important virulence factor employed by the spirochete for tissue colonization and, ultimately, Lyme disease pathogenesis.
RESULTS
Characterization of a bgp mutant of B. burgdorferi strain B31.
We used the derivatives of low-passage, infectious B. burgdorferi strain B31 in this study. Strain B31 5A4 included in this study retains lp56 and lacks only lp5, which appears to possess only one open reading frame other than those required for plasmid maintenance (18). The bbe02 mutant of the wild-type (WT) strain B31 5A18, B31 5A18NP1, was the strain used for generation of our bgp mutant strain, bgpmut. The bgp mutant possesses a single transposon in the BB_0588 gene (18). Thus, the difference between these strains is primarily the presence of a defective bgp gene in the mutant. Both strains B31 5A4 and B31 5A18 were included in parallel in different experiments. To restore Bgp expression in the mutant strain, we generated a trans-complemented strain that carries a wild-type bgp-pta operon on the pJSB175 shuttle vector of B. burgdorferi. Sequencing of the B. burgdorferi genome suggested that the bgp-pta genes are under the control of a single promoter, with no additional promoter sequences identified in the interspace between the two genes. To confirm if the bgp-pta genes are indeed present in an operon, we designed a single set of primers that were at the N-terminal region of BB_0588 and toward the 3′ end of BB_0589 for reverse transcriptase (RT)-PCR. The presence of a single band of 1.9 kB in strain B31 5A4 (WT) indicates that both genes are transcribed as a single mRNA transcript (Fig. 1A). We did not detect any bgp transcript in our mutant, suggesting disruption of the gene expression. Our complemented strain restored the transcript to a size comparable to that of the WT strain B31 5A4 (Fig. 1A).
FIG 1.

Growth and bgp expression in strains B31 5A4 (WT), bgpmut, and bgpcomp. (A) Reverse transcriptase PCR (RT-PCR) with bgp-pta operon primers produced a 1.9-kb product in WT and bgpcomp strains. No transcript was detected in the bgpmut strain. RT-PCR using pta primers produced a 1-kb transcript, indicating that transcription of the downstream gene was not affected by the bgp mutagenesis. A control without reverse transcriptase indicates that all RNA samples were free from DNA contamination. (B) Growth rates of all B. burgdorferi strains were comparable. (C) Western blot analysis using anti-Bgp antibodies showed the absence of Bgp expression in the mutant strain. Anti-Pta antibodies showed that this protein is expressed uniformly, albeit at low levels, in all strains. Flagellin protein detected by anti-FlaB antibodies was included as a loading control.
The organization of bgp-pta into an operon raises concern that disruption of the bgp gene may interfere with the expression of Pta and thus affect the phenotype of the mutant. Pta is considered to be an essential gene for B. burgdorferi cell wall biogenesis, and supplementation with mevalonate is required for spirochete survival if the expression of this protein is perturbed (16). To determine if mutagenesis of bgp gene alters Pta, thus resulting in a defect in growth, we conducted growth assays. There was no impairment of growth in our bgpmut and bgpcomp strains compared to WT strain B31 5A4 (Fig. 1B), suggesting that Pta expression was not altered in our mutant strain.
Based upon our growth assay results, we hypothesized that the insertion of the transposon in the same direction as the bgp gene allows expression of the downstream pta gene through the flgB promoter of the gentamicin cassette within the transposon. In addition, there is also growing evidence to support that an alternative transcriptional start site upstream of the pta gene may allow for gene transcription and Pta expression in the absence of Bgp (16, 40). To test this hypothesis, we performed an RT-PCR using primers spanning the pta gene and found that the bgpmut strain had pta transcript present (Fig. 1A). We observed elevated levels of transcription of pta in our bgp mutant and complemented strains, suggesting that the strong flgB promoter of gentamicin cassette may be driving increased expression. We performed RT-PCR using the same RNA samples and a combination of gentamicin- and pta-specific primers. Although we failed to obtain a product by RT-PCR, an ∼3.5-kb product was obtained by PCR using the same primers and genomic DNA of the mutant (data not shown). The absence of transcript initiated by the bgp promoter or by the flgB promoter of the gentamicin cassette resulting in a product as a read-through suggests that a second transcription start site lies within the bgp gene and may be used for pta transcription in bgpmut. Our Western blot analysis detected higher levels of Bgp in the complemented strain and also slightly higher levels of Pta protein expression in the bgpcomp strain (Fig. 1C). Thus, the elevated levels of pta transcript in our bgpmut and bgpcomp strains were reflected by a moderate increase in Pta protein expression, likely due to posttranscriptional regulation of pta as described previously (17, 41).
It is also important to note that the highest Pta levels occur under growth conditions different from those for optimum Bgp expression under in vitro conditions. Bgp is expressed at 33°C, while Pta is expressed at 23°C (41, 42). Western blot analysis of B. burgdorferi grown at physiological temperature showed lower levels of Pta expression (41). Therefore, we evaluated Pta expression after incubation of cultures for a week at 23°C by Western blotting to compare different strains when protein is expressed at a maximum level. Furthermore, we expected that any regulatory mechanisms for pta expression would remain intact in our bgpmut strains. The observation of more pta transcript with moderate increase in protein expression is likely a testament to the posttranscriptional regulation of the gene (17, 41).
Using anti-Bgp antisera, we show that Bgp is absent only in the bgpmut strain while its expression is restored in the bgpcomp strain (Fig. 1C). Higher expression of Bgp was observed in our bgpcomp strain, as evidenced by Western blotting. This result is consistent with the employment of the trans-complementation strategy, where copy number may affect the amount of protein produced. Such increased expression in B. burgdorferi strains has been reported in other studies that utilized a similar genetic approach (34, 43). Thus, all changes observed for our mutant strain can be attributed to the lack of expression of Bgp.
Disruption of the bgp gene impairs spirochete adherence in vitro.
Our laboratory has previously shown that Bgp has the highest affinity for heparin/heparan sulfate and little affinity for chondroitin sulfate C. We first confirmed these results using adherence assays to quantify binding of Bgp to GAGs. Our results showed that Bgp bound to heparin at significantly higher levels (P ≤ 0.0001) than our negative control, OspC, and only slightly to chondroitin sulfate C (Fig. 2). The binding pattern of the recombinant Bgp to GAGs was consistent with the trend reported previously (15). Binding of OspC to both GAGs was comparable to that seen in the buffer-only and no-protein control (NPC), showing the specific interaction of Bgp to GAGs.
FIG 2.
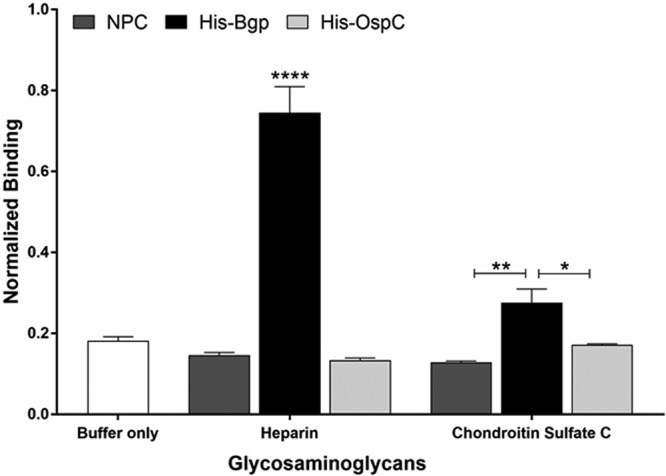
Bgp is a GAG-binding adhesin with affinity for heparin. Biotinylated GAGs were able to bind to recombinant Bgp protein (20 μg/ml), with the most-significant adherence to heparin. Bgp binds poorly to chondroitin sulfate C. Recombinant OspC was included as a negative control. The no-protein control (NPC) shows that interaction of Bgp was specific for GAGs. Each bar represents the mean ± standard deviation (SD) for quadruplicate samples. Data were normalized by dividing the total absorbance obtained in each well by the coated protein concentration in parallel control wells as determined by ELISA. The adjusted binding was then averaged for four replicates per treatment, and the percentage of biotinylated GAGs bound to the respective proteins is shown. Statistical significance (****, P ≤ 0.0001; **, P ≤ 0.01; *, P ≤ 0.05) was determined by the Student t test for comparison of samples with unequal variances.
We further investigated if binding to GAGs is affected by disruption of the bgp gene in B. burgdorferi using 35S-labeled intact bacterial strains. No-GAG (phosphate-buffered saline [PBS]) control wells demonstrate that B. burgdorferi strains do not bind to the empty wells appreciably (Fig. 3A). We found a highly significant (P ≤ 0.001) reduction in binding of the mutant strain to heparin compared to the B31 5A4 (WT) strain (Fig. 3B), while binding to chondroitin sulfate C was not affected (Fig. 3C). The bgpcomp strain was able to restore binding to heparin. Although the binding level of the complemented strain was slightly lower, it was not statistically different from that of the WT strain (Fig. 3B). Binding to chondroitin sulfate C remained relatively unchanged irrespective of Bgp expression (Fig. 3C). Taken together, these results suggest that adherence to heparin is impaired in the absence of Bgp despite retention of other spirochete GAG-binding adhesins.
FIG 3.

Binding of the bgp mutant to heparin is significantly reduced. Plate wells were coated with purified heparin or chondroitin sulfate C (5 mg/ml) or PBS. 35S-labeled B31 5A4 (WT), bgpmut, and bgpcomp strains were added, and binding was quantified by scintillation counting. (A) Buffer-only-treated (control) wells showed low nonspecific binding by each strain. (B) Adherence to heparin was significantly reduced in the mutant strain compared to WT and bgpcomp strains. (C) Overall binding of all strains to chondroitin sulfate C was unaffected by Bgp expression and was comparable to that seen in buffer control wells as shown in panel A. After subtracting the background count for empty well from all treated-well radiolabel counts determined by scintillation counting, the adjusted binding was divided by the total bacterial input label for each strain, and the percent binding was calculated. Each bar represents the mean ± SD for quadruplicate samples. Statistical significance (***, P ≤ 0.001) was determined by the Student t test for comparison of samples with unequal variances (ns, not significant).
To assess spirochete adherence to mammalian cells, we used two epithelial cell lines, i.e., human embryonic kidney cells (HEK293) and kidney epithelial cells isolated from African green monkey (Vero), and an endothelial cell line derived from human umbilical vein (EA.hy296). Cell monolayers were grown to confluence and incubated with 35S-labeled bacterial strains. For this set of experiments, only the results for strain B31 5A18 (WT) is shown. We found that the mutant strain was significantly impaired in its ability to bind to both human epithelial and endothelial cell lines. Binding of the bgpmut strain to the Vero cell line was only slightly reduced compared to strain B31 5A18 (Fig. 4), consistent with previous findings showing that significant adherence of B. burgdorferi to Vero cells was facilitated by GAG-independent binding mechanisms (11). Our results demonstrate that in the absence of Bgp, B. burgdorferi adherence to both endothelial and epithelial cells is significantly diminished despite retention of other GAG-binding adhesins in the spirochetes, thus highlighting the importance of Bgp for tissue colonization.
FIG 4.
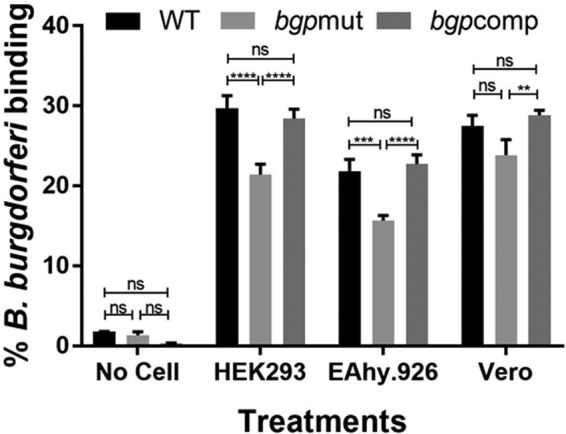
B. burgdorferi mutant lacking Bgp expression is impaired in binding to epithelial and endothelial cell lines. 35S-labeled B31 5A18 (WT), bgpmut, and bgpcomp strains were incubated with epithelial (HEK293 and Vero) and endothelial (EA.hy926) cell lines. The bgpmut strain showed statistically significant reduction in binding to HEK293 and EA.hy926 cells compared to the WT and bgpcomp strains. Binding of the mutant strain to Vero cells was slightly reduced (ns). Binding of the bgpcomp strain, in which Bgp expression is restored, was comparable to that in the WT strain. Wells containing no cells were included as a control for nonspecific binding to wells. After subtracting the mean background (four empty wells) binding levels, the adjusted counts for all treated wells were divided by the total bacterial input for the respective strain, and the percent binding was calculated. Each bar represents the mean ± SD for quadruplicate samples. Statistical significance (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001) was determined by the Student t test for comparison of samples with unequal variances.
The absence of Bgp attenuates infectivity and reduces tissue colonization and inflammatory disease in C3H/HeJ mice.
Our in vitro characterization of Bgp-mediated spirochete adherence led us to investigate the infectivity, tissue colonization, and histopathological changes during infection of susceptible mice. We infected 4-week-old female C3H/HeJ mice with a range of inoculum doses from 100 to 100,000 spirochetes per mouse of the B31 5A4 and B31 5A18 (WT), bgpmut, or bgpcomp strains. Two weeks postinfection, mice were euthanized, and ear, injection site skin, and bladder tissues were harvested for B. burgdorferi recovery in growth medium. Liquid cultures were checked weekly by microscopy for the presence of live spirochetes, indicative of positive infection. We found that the bgpmut strain was somewhat impaired in causing infection in the mouse model of Lyme disease. With an initial inoculum of 102 bacteria from WT B. burgdorferi strains (both B31 5A4 and 5A18), all mice became infected and spirochetes were recovered from different tissues. At the same dose of infection by the bgpmut strain, no mice (0/3) became infected, demonstrating a significant attenuation of infectivity compared to the WT strain at this dose of infection (Table 1). We observed partial restoration of infectivity in our bgpcomp strain with one mouse infected at a dose of 102 spirochetes per mouse. At an inoculum of 103 bgpcomp spirochetes, 2/3 mice for skin and bladder were positive for live spirochetes, while only 1 of 3 mice was positive when ear tissue was cultured. The reduction in infectivity in the bgpmut strain relative to the WT strain and the partial restoration of culture positivity in the complemented strain suggest that Bgp is an important virulence factor needed for colonization of these tissues.
TABLE 1.
Infectivity of WT, bgpmut, and bgpcomp strains in C3H miced
| B. burgdorferi strain | Inoculum | No. of culture-positive mice/no. of mice tested |
|||
|---|---|---|---|---|---|
| Ear | Skin (IS)e | Bladder | Total (%) | ||
| B31 5A4 | 100 | 3/3 | 3/3 | 3/3 | 9/9 (100) |
| 1,000 | 4/4 | 4/4 | 4/4 | 12/12 (100) | |
| 10,000 | 3/3 | 3/3 | 3/3 | 9/9 (100) | |
| B31 5A18 | 100 | 3/3 | 3/3 | 2/3 | 8/9 (90) |
| 1,000 | 4/4 | 4/4 | 4/4 | 12/12 (100) | |
| 10,000 | 3/3 | 3/3 | 3/3 | 9/9 (100) | |
| bgpmut | 100 | 0/5 | 0/5 | 0/5 | 0/15 (0) |
| 1,000 | 5/5 | 5/5 | 5/5 | 15/15 (100) | |
| 10,000 | 5/5 | 5/5 | 5/5 | 15/15 (100) | |
| 100,000 | 5/5 | 5/5 | 5/5 | 15/15 (100) | |
| bgpcomp | 100 | 1b/3 | 1b/3 | 1b/3 | 3/9 (33) |
| 1,000 | 1b/3 | 2b,c/3 | 2a,c/3 | 5/9 (56) | |
| 10,000 | 3/3 | 2a,b/3 | 3/3 | 8/9 (90) | |
Mouse number 1.
Mouse number 2.
Mouse number 3.
Cultures were monitored at 14 days postharvest.
IS, injection site.
Western blot analysis using sera from the mice 14 days postinfection further supported these findings (Fig. 5). Consistent with stimulation of humoral immune response only when spirochetes persist in high numbers, both WT strains showed significantly higher antibody reactivity with B31 lysate than the mutant, and several protein bands were detected when even 100 spirochetes/mouse were injected. Sera from our bgpmut strain-infected mice at this dose failed to show any reactivity, and this lower antibody response also supports the results that show lack of bgpmut culture recovery at this dose. This result is also consistent with the original results obtained with the bgp mutant determined by STM (18). The overall diminished immune response observed for the mutant at this dose improved when 103 to 104 mutant spirochetes were used for infection, as evidenced by the recognition of additional protein bands, albeit the response remained significantly lower than for the WT strains. Our bgpcomp strain showed strong reactivity for only 1/3 mice at inoculums of 100 spirochetes/mouse, consistent with the infection and recovery of spirochetes from the same mouse. Mice with low humoral immune response also retained fewer bacteria as detected by quantitative real-time PCR (qPCR) analysis of various tissues to determine colonization. Reactions of sera from the complemented-strain-infected mice at doses of ≥103 spirochetes/mouse were also significantly higher than in bgpmut-infected mice at the same inoculum.
FIG 5.

Immune response against total B. burgdorferi lysate demonstrates the diminished persistence of the bgpmut strain in mice. Proteins of B. burgdorferi lysate resolved by SDS-PAGE were blotted on an Immobilon-P membrane, and strips cut from the blots were then probed with sera recovered from different mice 14 days postinfection. For each strain, strips cut from the same blot were used, except for the mutant, for which strips from two blots run together in the same setup were used. Anti-mouse-IgG-AP secondary antibodies were used for detection of protein bands. Arrows indicate potential recognition of medium components associated with B. burgdorferi by mice sera, because these bands were also detected when probed with uninfected mouse serum. Mean densitometric readings with sera from all mice infected with each dose of infection with the respective strains are provided at the bottom. Sera recovered from WT strain (B31 5A4 and B31 A18)-infected mice produced a strong reaction (3/3 mice); however, the intensity of bands was not significantly different between doses of infection for the two WT strains. An intense reaction by WT strains observed even at doses as low as 100 spirochetes/mouse was significantly higher than in the bgpmut-infected mice with B. burgdorferi proteins (***, P < 0.001 compared to B31 5A4; **, P < 0.01 compared to B31 5A18) and was not significantly different from the reaction in the complemented strain at the same dose of infection. The difference between immunological responses against B31 5A4 and bgpmut strains was most significant at an inoculum of 103 bacteria/mouse (****, P < 0.0001) and was reduced, albeit remaining significant, at the infection dose of 104 bacteria/mouse (*, P < 0.05). A significant difference between B31 5A18 and bgpmut was also observed at an inoculum of ≥103 bacteria (***, P < 0.001). The reaction of sera from the complemented-strain-infected mice at doses of ≥103 spirochetes/mouse was significantly higher than from bgpmut-infected mice at the same inoculum (**, P < 0.01).
To study changes in tissue colonization, we used DNA isolated from the joints of infected mice to quantitatively determine the spirochete burden in the tissue. Spirochete infiltration of the joint is associated with the common inflammatory Lyme disease complication, Lyme arthritis. Our results showed that inoculums as low as 100 spirochetes of either of the two WT strains (n = 3) produced detectable levels of colonization in the joints (Fig. 6A and B). These results agree with our ability to recover live bacteria from tissues and the strong immune response observed in mice infected with either of the two WT strains. We were able to recover live spirochetes from one mouse infected with B31 5A18 at an inoculum of 100, and a serological response was detected by Western blotting using serum from this mouse; however, surprisingly, joints of this mouse remained qPCR negative. We observed a reduction in spirochete burden in joints of bgpmut-infected animals (n = 5) and failed to detect any spirochete DNA present when mice were inoculated with 100 mutant bacteria (Fig. 6C), again consistent with both the culture recovery results and serology. Interestingly, the ability of the bgpmut strain to colonize joints was impaired only at low inoculums, while inoculation with 104 to 105 mutant bacteria was able to produce colonization of the joint at levels similar to that seen with the WT strain (Fig. 6C). Consistent with our previous results, spirochete DNA was also detected from joints of the majority of bgpcomp-infected mice; 2 mice from each group (n = 3) were positive for colonization by qPCR, while DNA could not be detected from one animal (Fig. 6D).
FIG 6.

Disruption of bgp reduces the efficiency of joint colonization by B. burgdorferi in C3H/HeJ mice. Fourteen days after infection, mice were euthanized, and tissues were collected for DNA isolation and qPCR analysis of spirochete burden within joint tissues. Each point is representative of one mouse sample. Three/four mice for the WT and five mice for the mutant were used. (A) B31 5A4 spirochetes showed colonization of the joint in all mice at an inoculum dose as low as 100 B. burgdorferi spirochetes. (B) B31 A18 was less efficient at colonization as evidenced by 1 mouse remaining PCR negative at an inoculum of 100. (C) We failed to detect any spirochete DNA from mice infected with 100 bgpmut spirochetes, such that the difference between B31 5A4-100 and bgpmut-100 is statistically significant (P ≤ 0.05). At a higher inoculum (104 spirochetes), joint colonization for the bgpmut strain was similar to that of the WT B31 A18-infected mice. (D) Only partial restoration of joint colonization was evident in the complemented strain, with 1/3 mice for all inoculums remaining PCR negative for B. burgdorferi DNA. Colonization by B31 A18 and complemented strains were comparable in the other 2/3 mice.
Lyme disease is primarily an inflammatory disease, particularly during chronic or persistent infections. The manifestation of inflammatory disease in mice was determined by histopathological examination of heart and joint sections stained with hematoxylin and eosin. Scoring of sections from the hearts and joints of infected mice in a blinded manner showed that inflammatory disease is attenuated in mice infected with the mutant strain (Table 2). Scoring was based on infiltration of white blood cells into the pericardial structures and vasculature, as well as changes in the synovial structures of the joint. Inflammation of the knee and tibiotarsus was observed in all mice (3/3, 4/4) infected with WT strain B31 5A4, with higher inflammatory responses observed when mice were infected with increasing doses. The hearts of all mice infected with this strain also showed an inflammatory response. We observed similarly strong inflammatory responses in the majority of the mice infected with B31 5A18; however, one mouse at an inoculum of 100 (1/3) remained equivocal for inflammation at the knee. This is consistent with the poor serological detection of B. burgdorferi proteins and our negative qPCR result for this animal. Thus, strain B31 5A18 may have a slight attenuation in virulence compared to strain B31 5A4 (attributable to the loss of lp56). Pathological changes to the heart tissue were present to similar degrees across higher inoculums in both WT strains. At an inoculum of 100 spirochetes/mouse, none of the mice (0/5) displayed inflammation of the joint when infected with bgpmut, while 2/5 mice were positive for slight inflammation in the heart. These results indicate impairment of the bgpmut strain to colonize heart and joints and induce inflammation, particularly at a low dose of infection. At a dose of 103 mutant bacteria/mouse, differences in the inflammatory response in knees between either of the two WT strains and the mutant strain were less pronounced; however, significant increases in tibiotarsus inflammation by the WT strains reflect an attenuation of the inflammatory response to mutants (++ to +++ versus +). At higher inoculums (104 bacteria), there was no apparent difference between WT and bgpmut strains and similar pathological changes were observed for all strains. It is nonetheless important to note that mice infected at lower-dose inoculums with either WT or complemented strains showed a relatively higher inflammatory response than the mutant strain. The knee and tibiotarsus of complemented-strain-infected animals were positive at all inoculums and showed little difference in severity compared to the WT strain-infected mice. One mouse from each inoculum dose remained equivocal for inflammation of the heart (1/3 in all cases) when infected with the complemented strain. These results agree with other test results indicating that the respective animals showed low antibody response and remained qPCR negative for spirochete DNA. Overall, histopathological examination of the knee and tibiotarsus showed more inflammation in the WT strain-infected mice than in the bgpmut-infected animals, while some inflammation of joints was observed in all animals infected with the complemented strain even at a dose of 100 spirochetes (Table 2). Restoration of increased inflammation in the complemented strain at a higher dose of infection highlights the importance of Bgp in the pathogenesis of Lyme disease, as relevant to heart and joint inflammatory disease.
TABLE 2.
Results of histological examinations for tibiotarsal joint swelling and carditis 14 days after B. burgdorferi infection
| B. burgdorferi strain | Inoculum dose (no. of bacteria) | Inflammation score for mice 1 to 5a |
||
|---|---|---|---|---|
| Knee | Tibiotarsus | Carditis | ||
| B31 5A4 WT | 100 | +/++, +, + | +++, +++, +++ | +, +, + |
| 1,000 | +, +, +, + | +++, +++, ++, +++ | +, +, +, + | |
| 10,000 | ++, +, + | +++, +++, +++ | +, +, + | |
| B31 5A18 WT | 100 | +/−, +, + | ++, ++, +++ | +, +, + |
| 1,000 | +, +, +, + | +++, +++, +++, ++ | ++, ++, +, + | |
| 10,000 | +++, +++, ++ | +++, +++, ++ | ++, ++, +++ | |
| bgpmut | 100 | −, −, −, −, − | −, −, −, −, − | −, −, −, +, + |
| 1,000 | +, +, +, +, + | +, +, +, +, + | +, ++, ++, ++, ++, ++ | |
| 10,000 | +, ++, ++, ++, ++ | +, +, ++, ++, ++ | ++, ++, ++, +++, +++ | |
| 100,000 | ++, ++, ++, +++, +++ | ++, +++, +++, +++, +++ | +++, +++, +++, +++, +++ | |
| bgpcomp | 100 | +, +/++, + | ++, ++, ++ | +/−, +, + |
| 1,000 | +, +, + | +++, +++, +++ | +/−, +, + | |
| 10,000 | +, +, + | +++, +++, +++ | +/−, +, + | |
Three to five mice were infected with each strain, and inflammation scores are listed in sequential order for each mouse (numbered 1 to 5). Scores ranged from − (no inflammation) to +++ (severe arthritis). For carditis, scoring was based upon the number of infiltrating white blood cells between cardiomyocytes, loss of cell structure, and the breakdown of structures surrounding blood vessels/perivascular cuffing. For joints, scoring was based on infiltration of white blood cells in and around periarticular structures, the presence of cells within the synovium, and thickening of articular surfaces. +/−, samples that showed the presence of histopathological changes without a large number of infiltrating immune cells.
The overall histopathological scoring of tibiotarsus (Table 2) was consistent and most reflective of infectivity and tissue colonization levels and also correlates well to the inflammatory response. Thus, the highest severity of arthritis by strain B31 5A4 was observed even when 100 spirochetes were used, while the same level of severity was observed when infection doses of ≥103 bacteria from WT B31 5A18 and complemented strains and 105 bacteria from the bgpmut strain were used. These mice also showed higher infectivity and tissue colonization (Table 1 and Fig. 6) and good antibody response (Fig. 5). The lack of infectivity of bgpmut at a dose of 100 bacteria per mouse both by culture recovery (Table 1) and by qPCR (Fig. 6) also showed high correlation with lack of antibody response and joint inflammation (Table 2). We conclude that even though a bgp mutant remained infectious, the absence of Bgp reduced infectivity and tissue colonization and also diminished inflammatory disease severity of infection by B. burgdorferi.
DISCUSSION
Understanding the genetic organization of bgp (BB_0588)-pta (BB0_589) has been a point of controversy within the field. A 2011 report by Sze and Li mapped the transcriptional start site of pta to the 5′ region upstream of bgp. RT-PCR with primers spanning the predicted operon produced a transcript consistent with the two genes being cotranscribed (17). A subsequent report in 2015 by Richards and coworkers failed to detect a bgp-pta transcript by RT-PCR (16). These two studies utilized slightly different primer pairs and cycling parameters for RT-PCR analysis, which may have contributed to their conflicting results. A more recent report (2017) by Adams et al. utilized in vivo expression technology (IVET) and 5′ end mapping to characterize alternative transcriptional start sites throughout the Borrelia genome and also identified a potential second transcription start site for pta (40). We utilized an RT-PCR approach to determine if the two genes were transcribed together. Primers for the work presented here were designed to begin after the translational start site of bgp and toward the 3′ end of the pta gene to amplify the region spanning the proposed operon. Our results are consistent with the presence of bgp and pta genes in an operon (Fig. 1A). RT-PCR with the same RNA samples using a combination of gentamicin and pta primers failed to result in a product, although the same primers resulted in the expected-size product when genomic DNA was used. Our results support the suggested presence of a second transcriptional start site for pta within BB_0588 (16, 40).
A bgp-pta operon complicates a straightforward analysis of the bgp mutant due to potential disruption of downstream pta gene expression. Pta is considered an essential protein, and perturbation in its expression negatively affects spirochete survival. We selected a bgp mutant strain generated through a genome-wide STM transposon mutagenesis study (18). This strain remained viable, suggesting retention of Pta protein expression. Pta functions in the essential AckA-Pta pathway, in which AckA converts acetate to acetyl phosphate, which is followed by acetyl phosphate conversion to acetyl coenzyme A (acetyl-CoA) by Pta. Due to the absence of enzymes for the tricarboxylic acid (TCA) cycle and oxidative phosphorylation in B. burgdorferi, the acetyl-CoA is fed into the mevalonate pathway for cell wall synthesis (42). Efforts to mutate the pta gene have failed unless the culture medium is supplemented with mevalonate (16, 18, 42). Regulatory functions of Pta have also been proposed but remain controversial and unsubstantiated. Xu and coworkers explored if small high-energy phosphate donors, including the substrate for Pta, acetyl phosphate, could activate the Rrp2-RpoN-RpoS regulon such that Pta was directly involved in modulating the amount of available acetyl phosphate and downstream activation of critical virulence pathways (42). Exogenous sodium acetate was added to the culture, resulting in activation of the pathway. Subsequently, employment of a Pta-overexpressing strain showed the lack of pathway activation. Correlation between acetyl phosphate concentrations and expression of virulence genes was assumed to be due to the effects of Pta; however, the study did not account for the effects of acid stress due to the addition of sodium acetate and failed to accurately draw comparisons between exogenous sodium acetate and endogenous acetyl phosphate levels. These shortcomings were later elaborated by showing that the absence of Pta (mevalonate-supplemented mutant) does not change the activation of the Rrp2-RpoN-RpoS pathway (16). Thus, the essentiality of Pta is most likely due to its role in cell wall biogenesis.
Our sequence analysis demonstrated that the direction of transcription through the flgB promoter, used for the expression of the gentamicin cassette within the transposon, is the same as the bgp open reading frame (data not shown), potentially allowing transcription of the downstream pta gene through the flgB promoter. Read-through from the bgp promoter might also maintain pta expression. Our results indicate that expression of pta in bgpmut might be through a second transcriptional start site upstream of the pta gene. Our growth studies indicated that the mutant is able to survive and grow in a fashion similar to that of the WT strain, providing the first evidence that the essential Pta protein is present in the bgp mutant strain. Furthermore, RT-PCR and Western blot analysis confirmed that only Bgp, and not Pta, is missing in our bgpmut strain. By RT-PCR using the pta-specific primers, we were able to detect the presence of pta transcript in our mutant strain and observed elevated transcript levels in our complemented strain. The increased transcript level in our complemented strain is likely due to a higher operon copy number because of its presence on the shuttle vector. It has been reported that pta transcript is posttranscriptionally regulated by CsrABb, such that pta is repressed when CsrABb is upregulated, which has been noted particularly under growth conditions at 37°C (41). Our efforts to genetically manipulate bgp while retaining pta expression is expected to maintain regulatory regions or systems relating to Pta, and the posttranscriptional regulation of the mRNA is likely responsible for the uniform expression of Pta in the WT and mutant strains and the only slight increase in the level of Pta protein expression in the complemented strain (17, 41). Therefore, changes observed in our mutant are attributable only to the absence of Bgp protein.
It is widely accepted that GAGs are important receptors for spirochete adherence and tissue colonization in the pathogenesis of Lyme disease. Given the ubiquitous expression of heparan sulfate on mammalian tissues, we hypothesized that a significant reduction in spirochete adherence to heparin in vitro would also impair spirochete adherence to the ECM and therefore reduce the spirochete ability to colonize tissues during infection. Our results from in vitro binding experiments validate previous findings and show that recombinant Bgp binds significantly to heparin and less efficiently to chondroitin sulfate C. These observations agree with previous findings in which a different B. burgdorferi strain was used (15). We show for the first time that in the absence of Bgp expression in B. burgdorferi, GAG binding is significantly reduced (Fig. 4). This significant (nearly 4-fold) reduction in binding from 18.6% of the WT strain to only 4.77% of the bgpmut strain indicates that Bgp contributes to GAG-mediated adherence of the Lyme spirochetes.
The B. burgdorferi genome encodes a high number of adhesins; ∼30 adhesins have been identified and extensively investigated to date, and numerous other putative adhesins have yet to be characterized (7, 44, 45). Many B. burgdorferi adhesins have redundant, overlapping functions and affinities allowing for compensation during plasmid loss or expression profile changes between vector and host (7, 45). We have demonstrated Bgp as a GAG-binding adhesin, which binds significantly to heparin (15). At least two other adhesins that recognize heparin have been identified: BBK32, which has been experimentally shown to bind purified heparin (34), and DbpA, which has a confirmed heparin binding consensus sequence (46). Reduction but not elimination of binding to heparin can be attributed to redundancy of function due to remaining B. burgdorferi GAG-binding adhesins (7, 22, 28, 34, 35). Thus, compensation by other heparin binding adhesins, such as BBK32, could be responsible for the remaining 4.77% of spirochete binding to the purified heparin observed in our bgpmut strain. In strains such as N40, which lack BBK32, the effect of a bgp mutation would likely be even more significant because it lacks this protein (47). Binding to chondroitin sulfate C is significantly less than binding of heparin, even by the WT strain, which is consistent with previously published results that showed that different B. burgdorferi strains recognize this GAG poorly (11, 14, 23).
We further report a significant reduction in the ability of the bgpmut strain to adhere to mammalian cells compared to both WT and complemented strains, particularly with respect to epithelial and endothelial cells. Reduced adherence of the bgpmut to purified heparin and mammalian cells in vitro is also reflected in diminished tissue colonization and inflammatory disease manifestations. Although bgp mutant spirochetes retain the ability to infect mice, it is impaired compared to that of the WT strains. Mutagenesis of other GAG-binding adhesin-encoding genes also resulted in retention of infectivity, albeit with demonstration of reduced virulence. For example, bbk32 mutants display variable results, with some mutants exhibiting an increased 50% infective dose (ID50), reduced dissemination, and tissue colonization (36, 48, 49).
It is speculated that in addition to adhesin function, BBK32 may also have a role in modulating the immune response through the formation of superfibronectin and interference in the innate immune response (50). A decrease in infectivity observed for the bgp mutant could also be attributed to the other functional role of this protein. Bgp is a dual-function protein that exhibits both GAG binding and MTA/SAH nucleosidase activity. This nucleosidase is responsible for critical functions such as detoxification by removal of metabolic by-products MTA and SAH, salvage of nutrients, regulation of SAM-mediated methylation reactions, and quorum sensing (37–39). We observed a decrease in spirochete burden within tissues when mice were infected with bgpmut spirochetes compared to what was seen with the WT strain. Consistent with decreased colonization, the inflammatory responses were also reduced in the joints and hearts of mice infected with low doses of mutant spirochetes as determined by histopathological examination (Table 2). Mutagenesis of the bgp gene impacts both activities of Bgp, which may contribute to the attenuation of virulence of the mutant. Our findings here suggest that the contribution of Bgp in cell adherence contributes to tissue colonization; however, more-targeted mutagenesis studies are needed to investigate whether the lack of Bgp-associated nucleosidase activity contributes to the reduced infectivity of the bgp mutant.
Our trans-complemented strain was able to restore binding to purified heparin compared to the WT strain (Fig. 4B). This trans-complemented strain displayed only partial restoration of the WT phenotype during infection of mice as indicated by Western blotting and determination of tissue colonization, because one infected mouse remained negative by both serological test and qPCR at an inoculum of 100 bacteria. One mouse from higher inoculums of 103 to 104 bacteria remained negative by qPCR for colonization, despite testing positive by Western blotting (Fig. 5 and 6). This observation of full restoration of functional activity in vitro and only partial restoration in vivo may be attributed to subtle changes occurring during genetic manipulation, as reported for other B. burgdorferi mutants. Partial restoration of function in the complemented strain has also been reported for the mutants of other GAG-binding adhesins (36).
Collectively, our findings indicate that Bgp is an important virulence factor for B. burgdorferi. Our current work has focused heavily on evaluating the adhesin function of Bgp; however, it is possible that the MTA/SAH nucleosidase functions of the protein also contribute to lower infectivity and poor spirochete survival within the host, thus resulting in decreased colonization and inflammation. Additional studies to characterize the dual function of this protein will provide evidence and mechanistic support for the critical role that Bgp plays in Lyme disease pathogenesis.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Low-passage, infectious B31 Borrelia burgdorferi strains were grown according to standard in vitro laboratory cultivation methods. B31 5A18 and B31 5A4 are the two WT strains carried in parallel for the majority of experiments here. WT strain B31 5A18 is the parental strain, in which mutation of the bbe02 gene generated strain B31 5A18NP1, which was used to generate the strain designated T06TC117 (bgpmut) in the laboratory of Steven Norris (51). The mutant strain harbors a single transposon cassette insertion within the bgp (BB_0588) gene. trans-complementation of T06TC117 was achieved by transformation with plasmid pJSB175Bgp589, such that expression of functional Bgp is restored in strain T06TC117pJSB175Bgp589 (bgpcomp). All clones were cultured in Barbour-Stoenner-Kelly (BSK-II) medium containing 6% rabbit serum at 33°C until the cultures reached mid-logarithmic phase or ∼1 × 108 to 2 × 108 cells/ml. All strains were checked for plasmid loss by previously described methods (52, 53) (see Fig. S1 and Table S1 in the supplemental material). The bgpmut and bgpcomp strains lacked lp28-4, cp9, lp5, and lp56, which are not required for mouse infection (18, 54, 55). The wild-type strain B31 5A4 retained these plasmids except lp5 and cp9.
RT-PCR.
RNA was isolated from B. burgdorferi strains by using kits and as suggested by the manufacturer. Briefly, nucleic acids were isolated from the cells by phase separation using TRIzol reagent (Thermo Scientific, Waltham, MA) followed by RNA purification using the RNeasy kit (Qiagen, Valencia, CA) according to the manufacturers' protocols. Bacterial pellets were washed once with 1× PBS and resuspended in 500 μl of TRIzol reagent. The homogenate was then transferred to a MaXtract high-density tube, and chloroform was added. Following phase separation, the RNA collected from the aqueous phase was transferred to the RNeasy spin column and purified. RNA samples were treated with 1 U/μg RQ01 DNase (Promega, Madison, WI) to remove any contaminating DNA. The pure RNA sample was used as a template for cDNA synthesis for RT-PCR. The cycling parameters for bgp RT-PCR were as follows: cDNA synthesis at 47°C for 30 min, initial denaturation at 94°C for 2 min, 40 cycles of denaturation at 94°C for 15 s, annealing at 50°C for 30 s, extension at 68°C for 2 min, and final extension at 68°C for 5 min. Primers were designed based on the known bgp and pta gene sequences (56). For detection of the bgp-pta operon, primers spanning BB_0588 (5′TATAAGGAGTGTGATTTTATGAATAA3′) and BB_0589 (5′TTAAATGCTTATCATTAAAGCACTT3′) were used. For pta RT-PCR, the following modifications were made: cDNA synthesis was at 53°C for 30 min and annealing at 58°C for 2 min. Primers for BB_0589 were used (5′GCTTTCAATTCAGCCAAAGT3′ and 5′ACCAATAAAGCTACTCTGAGAG3′). In order to confirm that contaminating DNA is not responsible for bgp or pta amplification, control reactions were also performed in parallel using the same parameters in the absence of reverse transcriptase.
SDS-PAGE and Western blotting.
For examination of protein expression in B. burgdorferi strains, ∼2 × 108 cells/ml were centrifuged and washed three times with PBS to remove medium components, and pellets were resuspended in 1× PBS and an equal volume of 2× SDS loading dye. In order to obtain the maximum expression of Pta in all strains, the cultures were moved to 23°C for 1 week prior to centrifugation. Proteins were then analyzed on 12% SDS-polyacrylamide gels and transferred to an Immobilon-P polyvinylidene difluoride membrane (Millipore, Billerica, MA). Anti-Bgp mouse polyclonal antibodies were generated as previously described (15). Immunoblotting was performed with anti-Bgp mouse serum diluted to 1:2,000 or with anti-Pta mouse serum, as generated by Janakiram Seshu, diluted to 1:750 in the blocking buffer, 1× PBS containing 5% skim milk. As a loading control, immunoblotting was conducted in parallel using monoclonal anti-FlaB antibodies diluted to 1:200. Secondary anti-mouse IgG antibodies conjugated with either alkaline phosphatase or horseradish peroxidase (HRP) diluted 1:2,000 were used to detect proteins of interest. A 1:1 ratio of substrate mixture containing 5-bromo-4-chloro-3-indolylphosphate (BCIP) and Nitro Blue Tetrazolium (NBT) in 70% dimethylformamide (DMF; Sigma, Billerica, MA) for alkaline phosphatase or 1× diaminobenzidine tetrahydrochloride (DAB; Thermo Scientific, Waltham, MA) for HRP was added, and the mixture was incubated at room temperature until bands developed.
Determining the genetic organization of the bgp-pta operon in bgpmut.
The bgp gene is disrupted by a gentamicin-containing transposon from pMarGentKan (18). Genomic DNA from the bgp mutant strain was isolated by standard genomic DNA purification, digested with the restriction enzyme Spe1 (New England BioLabs, Ipswich, MA), and cloned in pUC19 (Thermo Scientific, Waltham, MA). Transformation of Top10 competent E. coli cells was followed by selection on gentamicin (15 μg/ml)-containing Luria-Bertani plates to select for the DNA fragment containing the resistance cassette of the transposon. Plasmids were isolated and sequenced using bgp primers to determine the location and direction of the transposon within the gene. Sequencing was performed at Tufts University Core Facility in Boston, MA.
Murine infection.
All animal infection studies were performed in accordance with approved IACUC protocols under Rutgers University standard animal care procedures. Young C3H/HeJ mice have been shown to be highly susceptible to B. burgdorferi (57). Therefore, 4-week-old C3H/HeJ female mice were needle inoculated by subcutaneous injection of a serial dilution of B. burgdorferi WT, bgpmut, or bgpcomp strains containing from 100 to 100,000 spirochetes per mouse. Three/four mice for the WT and complemented strains were used, while five mice were used for the bgp mutant strain. Two weeks postinfection, blood was obtained by cardiac puncture, animals were euthanized, and tissues were harvested for spirochete culture, histopathology, and qPCR. Serum collected from each mouse was diluted 1:100 and used for Western blotting. Detection of primary sera with 1:2,500 dilution of anti-mouse-IgG-AP revealed proteins of interest. Joint and heart tissues were fixed in formalin and sent for serial histological sectioning and hematoxylin and eosin staining at Rutgers Core Facility. Initially, Stephen Barthold scored some samples for inflammation. All samples were rescored in a blinded manner by two researchers with degrees that included studies in veterinary pathology for arthritis and carditis/cardiovascular disease severity.
Our trans-complementation strategy with a plasmid-borne WT bgp gene required maintained selective pressure with streptomycin. For in vitro work, supplementation of culture media with streptomycin was able to positively select for bacteria carrying the complementation plasmid. We maintained antibiotic selective pressure in vivo through supplementation of the animals' water with streptomycin (5 mg/ml) and artificial sweetener (1.5 mg/ml) to make it palatable (58; our unpublished data).
Detection of B. burgdorferi tissue colonization by qPCR.
DNA isolated from joint tissue as previously described (59, 60) was used to determine the spirochete burden by qPCR. Briefly, our multiplex test utilizes gene-specific primers (i) for the Lyme spirochete recA gene (5′GTGGATCTATTGTATTAGATGAGGCTCTCG and 5′GCCAAAGTTCTGCAACATTAACACCTAAAG) to produce a 222-bp amplicon and (ii) for the mouse Nidogen gene (5′CCAGCCACAGAATACCATCC and 5′GGACATACTCTGCTGCCATC) to produce a 154-bp amplicon. Molecular beacon probes tagged to the fluorophore 6-carboxyfluorescein (FAM) (RecA) or Quasar670 (Nidogen) that were complementary to the target sequences of the amplicons produced were designed and detected in real-time PCR. In the absence of target, the molecular beacons remain organized into a hairpin structure that inhibits fluorescence, allowing for sensitive detection of DNA with low background. DNA concentrations were adjusted to 40 ng/μl, and a 5-μl sample was used as the template for each multiplex qPCR in a total reaction volume of 25 μl. The reaction mixture contained 1× AmpliTaq Gold buffer (Applied Biosystems, Carlsbad, CA), 0.5 mg/ml bovine serum albumin (BSA), 3 mM MgCl2, 0.25 mM each deoxynucleotide triphosphate, 0.5 μM recA set of primers, 0.1 μM Nidogen set of primers, 50 ng of each molecular beacon, and 2.5 U AmpliTaq Gold enzyme (Applied Biosystems, Carlsbad, CA). The thermal profile for all reactions was as follows: initial enzyme activation at 95°C for 10 min, followed by 60 cycles of 95°C for 15 s, 60°C for 30 s, and 75°C for 20 s. The spirochete load within the tissue was determined from the standard curve. Overlapping Nidogen curves in the duplex assay confirmed that a consistent concentration of mouse DNA was used, allowing an accurate determination of the B. burgdorferi load.
Recombinant protein production.
Escherichia coli BL21(DE3λ) was transformed with the expression vector pET30a-bgp or with pET30a-ospC for all negative-control experiments and purified as described previously (23). Cultures were induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside; Lab Scientific, Highlands, NJ) overnight at 37°C. Pellets were resuspended in a total volume of 5 ml lysis buffer containing bacterial protein extraction reagent (BPER; Thermo Scientific, Waltham, MA), 100 μl of protease inhibitor cocktail (Sigma, Billerica, MA), lysozyme at a final concentration of 2 mg/ml, and ∼2,500 U of benzonase. Sonication was performed until maximum lysis was achieved. Affinity chromatography using bacterial lysate loaded onto His-Bind resin was performed according to the manufacturer's instructions (Millipore). Protein purity was checked by SDS-PAGE, and concentration was determined using a bicinchoninic acid (BCA) kit (Sigma, Billerica, MA).
Binding of biotinylated GAGs to recombinant Bgp.
Quadruplicate wells of 96-well plates were coated with 50 μl of 20 μg/ml Bgp or with OspC protein as a negative control. After blocking with 1% BSA containing 1× PBS for 1 h, 50 μl of 200-μg/ml biotinylated heparin or chondroitin sulfate C, diluted in 1× PBS, was added. PBS alone was added in control wells. To confirm that the same concentrations of GAGs were used, quadruplicate wells were coated with 50 μl of 5-mg/ml biotinylated GAGs that were detected using 1:200 dilution of anti-biotin-HRP (Sigma, Billerica, MA)/anti-streptavidin-HRP (Calbiogen, San Mateo, CA). For protein controls, 50 μl of recombinant proteins coated in the wells was directly detected using anti-poly-His-HRP. Following washes with 1× PBS to remove unbound antibody, tetramethylbenzidine (TMB) substrate (KPL, Milford, MA) was added to all wells, and quantification of bound GAG was detected by measuring the A670 nm. By dividing the absorbance obtained from each treated well by the average protein absorbance value from quadruplicate control wells, as determined by enzyme-linked immunosorbent assay (ELISA), binding was normalized to account for any differences due to each protein coating level. Mean binding levels from quadruplicate wells were then calculated.
Adherence of radiolabeled bacteria to purified glycosaminoglycans and mammalian cell lines.
To assess adherence of the WT, bgpmut, and bgpcomp strains, we radiolabeled strains with a [35S]methionine-cysteine mixture, prepared aliquots, and stored them at −80°C until use. Bacteria were thawed, resuspended in BSK-H medium, allowed to recover for 2 h at room temperature, and then diluted 1:3 in buffer containing 10 mM glucose, 50 mM NaCl, and 10 mM HEPES at pH 7. Binding of radiolabeled bacterial strains to quadruplicate wells coated with heparin or chondroitin sulfate C (5 mg/ml) was conducted as described previously (15). In addition, cell lines, including HEK293 (human embryonic kidney epithelium), C6 glioma (rat neuronal), EA.hy926 (human endothelial), and Vero (monkey bladder fibroblast), were grown to confluence in their respective standard cell culture media for binding assays in Nunc Maxisorp break-apart plates. Binding assays were conducted as described previously using quadruplicate wells for each cell type (11). Briefly, 50 μl of bacteria prepared as described above was added to the plate containing mammalian cells, centrifuged at 800 × g, and incubated at 37°C in an incubator with 5% CO2 for 1 h. Quadruplicate wells coated with 1× PBS served as a “no-cell” control for binding. The total scintillation count obtained for each strain (50 μl) determined the total input per well. After subtracting the background counts for empty wells from the radiolabel count obtained for each treated well, values were divided by the total input count to calculate the percent binding to both GAGs and mammalian cells. The average percent binding from four replicates for each treatment is documented in the figures.
Supplementary Material
ACKNOWLEDGMENTS
We appreciate Luke Fritzky and Joel Pierre of the histology core of Rutgers Biomedical and Health Sciences for assistance in organ sample preparation, sectioning, and staining for histopathological examination. We are thankful to Stephen Barthold, scientist emeritus at University of California at Davis, for initial histopathological scoring of some joint and heart sections and to Vitomir Djokic and Lavoisier Akoolo, both graduates in Veterinary Medicine, for examining and scoring all samples in a blinded manner.
This work was supported by grant AI089921 to N.P. from the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00667-17.
REFERENCES
- 1.Hinckley AF, Connally NP, Meek JI, Johnson BJ, Kemperman MM, Feldman KA, White JL, Mead PS. 2014. Lyme disease testing by large commercial laboratories in the United States. Clin Infect Dis 59:676–681. doi: 10.1093/cid/ciu397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nelson CA, Saha S, Kugeler KJ, Delorey MJ, Shankar MB, Hinckley AF, Mead PS. 2015. Incidence of clinician-diagnosed Lyme disease, United States, 2005-2010. Emerg Infect Dis 21:1625–1631. doi: 10.3201/eid2109.150417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steere AC. 2001. Lyme disease. N Engl J Med 345:115–125. doi: 10.1056/NEJM200107123450207. [DOI] [PubMed] [Google Scholar]
- 4.Steere AC, Coburn J, Glickstein L. 2004. The emergence of Lyme disease. J Clin Invest 113:1093–1101. doi: 10.1172/JCI21681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oosting M, Buffen K, van der Meer JW, Netea MG, Joosten LA. 2016. Innate immunity networks during infection with Borrelia burgdorferi. Crit Rev Microbiol 42:233–244. doi: 10.3109/1040841X.2014.929563. [DOI] [PubMed] [Google Scholar]
- 6.Skogman BH, Hellberg S, Ekerfelt C, Jenmalm MC, Forsberg P, Ludvigsson J, Bergstrom S, Ernerudh J. 2012. Adaptive and innate immune responsiveness to Borrelia burgdorferi sensu lato in exposed asymptomatic children and children with previous clinical Lyme borreliosis. Clin Dev Immunol 2012:294587. doi: 10.1155/2012/294587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coburn J, Fischer JR, Leong JM. 2005. Solving a sticky problem: new genetic approaches to host cell adhesion by the Lyme disease spirochete. Mol Microbiol 57:1182–1195. doi: 10.1111/j.1365-2958.2005.04759.x. [DOI] [PubMed] [Google Scholar]
- 8.Hook M, Kjellen L, Johansson S. 1984. Cell-surface glycosaminoglycans. Annu Rev Biochem 53:847–869. doi: 10.1146/annurev.bi.53.070184.004215. [DOI] [PubMed] [Google Scholar]
- 9.Isaacs RD. 1994. Borrelia burgdorferi bind to epithelial cell proteoglycans. J Clin Invest 93:809–819. doi: 10.1172/JCI117035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson RL, Busch SJ, Cardin AD. 1991. Glycosaminoglycans: molecular properties, protein interactions, and role in physiological processes. Physiol Rev 71:481–539. [DOI] [PubMed] [Google Scholar]
- 11.Parveen N, Robbins D, Leong JM. 1999. Strain variation in glycosaminoglycan recognition influences cell-type-specific binding by Lyme disease spirochetes. Infect Immun 67:1743–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leong JM, Morrissey PE, Ortega-Barria E, Pereira ME, Coburn J. 1995. Hemagglutination and proteoglycan binding by the Lyme disease spirochete, Borrelia burgdorferi. Infect Immun 63:874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leong JM, Robbins D, Rosenfeld L, Lahiri B, Parveen N. 1998. Structural requirements for glycosaminoglycan recognition by the Lyme disease spirochete, Borrelia burgdorferi. Infect Immun 66:6045–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leong JM, Wang H, Magoun L, Field JA, Morrissey PE, Robbins D, Tatro JB, Coburn J, Parveen N. 1998. Different classes of proteoglycans contribute to the attachment of Borrelia burgdorferi to cultured endothelial and brain cells. Infect Immun 66:994–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parveen N, Leong JM. 2000. Identification of a candidate glycosaminoglycan-binding adhesin of the Lyme disease spirochete Borrelia burgdorferi. Mol Microbiol 35:1220–1234. doi: 10.1046/j.1365-2958.2000.01792.x. [DOI] [PubMed] [Google Scholar]
- 16.Richards CL, Lawrence KA, Su H, Yang Y, Yang XF, Dulebohn DP, Gherardini FC. 2015. Acetyl-phosphate is not a global regulatory bridge between virulence and central metabolism in Borrelia burgdorferi. PLoS One 10:e0144472. doi: 10.1371/journal.pone.0144472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sze CW, Li C. 2011. Inactivation of bb0184, which encodes carbon storage regulator A, represses the infectivity of Borrelia burgdorferi. Infect Immun 79:1270–1279. doi: 10.1128/IAI.00871-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin T, Gao L, Zhang C, Odeh E, Jacobs MB, Coutte L, Chaconas G, Philipp MT, Norris SJ. 2012. Analysis of an ordered, comprehensive STM mutant library in infectious Borrelia burgdorferi: insights into the genes required for mouse infectivity. PLoS One 7:e47532. doi: 10.1371/journal.pone.0047532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salo J, Loimaranta V, Lahdenne P, Viljanen MK, Hytonen J. 2011. Decorin binding by DbpA and B of Borrelia garinii, Borrelia afzelii, and Borrelia burgdorferi sensu stricto. J Infect Dis 204:65–73. doi: 10.1093/infdis/jir207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pikas DS, Brown EL, Gurusiddappa S, Lee LY, Xu Y, Hook M. 2003. Decorin-binding sites in the adhesin DbpA from Borrelia burgdorferi: a synthetic peptide approach. J Biol Chem 278:30920–30926. doi: 10.1074/jbc.M303979200. [DOI] [PubMed] [Google Scholar]
- 21.Guo BP, Brown EL, Dorward DW, Rosenberg LC, Hook M. 1998. Decorin-binding adhesins from Borrelia burgdorferi. Mol Microbiol 30:711–723. doi: 10.1046/j.1365-2958.1998.01103.x. [DOI] [PubMed] [Google Scholar]
- 22.Fischer JR, Parveen N, Magoun L, Leong JM. 2003. Decorin-binding proteins A and B confer distinct mammalian cell type-specific attachment by Borrelia burgdorferi, the Lyme disease spirochete. Proc Natl Acad Sci U S A 100:7307–7312. doi: 10.1073/pnas.1231043100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parveen N, Caimano M, Radolf JD, Leong JM. 2003. Adaptation of the Lyme disease spirochaete to the mammalian host environment results in enhanced glycosaminoglycan and host cell binding. Mol Microbiol 47:1433–1444. doi: 10.1046/j.1365-2958.2003.03388.x. [DOI] [PubMed] [Google Scholar]
- 24.Brown EL, Wooten RM, Johnson BJ, Iozzo RV, Smith A, Dolan MC, Guo BP, Weis JJ, Hook M. 2001. Resistance to Lyme disease in decorin-deficient mice. J Clin Invest 107:845–852. doi: 10.1172/JCI11692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang FT, Brown EL, Wang T, Iozzo RV, Fikrig E. 2004. Protective niche for Borrelia burgdorferi to evade humoral immunity. Am J Pathol 165:977–985. doi: 10.1016/S0002-9440(10)63359-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benoit VM, Fischer JR, Lin YP, Parveen N, Leong JM. 2011. Allelic variation of the Lyme disease spirochete adhesin DbpA influences spirochetal binding to decorin, dermatan sulfate, and mammalian cells. Infect Immun 79:3501–3509. doi: 10.1128/IAI.00163-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hagman KE, Yang X, Wikel SK, Schoeler GB, Caimano MJ, Radolf JD, Norgard MV. 2000. Decorin-binding protein A (DbpA) of Borrelia burgdorferi is not protective when immunized mice are challenged via tick infestation and correlates with the lack of DbpA expression by B. burgdorferi in ticks. Infect Immun 68:4759–4764. doi: 10.1128/IAI.68.8.4759-4764.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin YP, Benoit V, Yang X, Martinez-Herranz R, Pal U, Leong JM. 2014. Strain-specific variation of the decorin-binding adhesin DbpA influences the tissue tropism of the lyme disease spirochete. PLoS Pathog 10:e1004238. doi: 10.1371/journal.ppat.1004238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blevins JS, Hagman KE, Norgard MV. 2008. Assessment of decorin-binding protein A to the infectivity of Borrelia burgdorferi in the murine models of needle and tick infection. BMC Microbiol 8:82–94. doi: 10.1186/1471-2180-8-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo BP, Norris SJ, Rosenberg LC, Hook M. 1995. Adherence of Borrelia burgdorferi to the proteoglycan decorin. Infect Immun 63:3467–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi Y, Xu Q, McShan K, Liang FT. 2008. Both decorin-binding proteins A and B are critical for the overall virulence of Borrelia burgdorferi. Infect Immun 76:1239–1246. doi: 10.1128/IAI.00897-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salo J, Jaatinen A, Soderstrom M, Viljanen MK, Hytonen J. 2015. Decorin binding proteins of Borrelia burgdorferi promote arthritis development and joint specific post-treatment DNA persistence in mice. PLoS One 10:e0121512. doi: 10.1371/journal.pone.0121512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Probert WS, Johnson BJ. 1998. Identification of a 47 kDa fibronectin-binding protein expressed by Borrelia burgdorferi isolate B31. Mol Microbiol 30:1003–1015. doi: 10.1046/j.1365-2958.1998.01127.x. [DOI] [PubMed] [Google Scholar]
- 34.Fischer JR, LeBlanc KT, Leong JM. 2006. Fibronectin binding protein BBK32 of the Lyme disease spirochete promotes bacterial attachment to glycosaminoglycans. Infect Immun 74:435–441. doi: 10.1128/IAI.74.1.435-441.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin YP, Chen Q, Ritchie JA, Dufour NP, Fischer JR, Coburn J, Leong JM. 2015. Glycosaminoglycan binding by Borrelia burgdorferi adhesin BBK32 specifically and uniquely promotes joint colonization. Cell Microbiol 17:860–875. doi: 10.1111/cmi.12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seshu J, Esteve-Gassent MD, Labandeira-Rey M, Kim JH, Trzeciakowski JP, Hook M, Skare JT. 2006. Inactivation of the fibronectin-binding adhesin gene bbk32 significantly attenuates the infectivity potential of Borrelia burgdorferi. Mol Microbiol 59:1591–1601. doi: 10.1111/j.1365-2958.2005.05042.x. [DOI] [PubMed] [Google Scholar]
- 37.Parveen N, Cornell KA, Bono JL, Chamberland C, Rosa P, Leong JM. 2006. Bgp, a secreted glycosaminoglycan-binding protein of Borrelia burgdorferi strain N40, displays nucleosidase activity and is not essential for infection of immunodeficient mice. Infect Immun 74:3016–3020. doi: 10.1128/IAI.74.5.3016-3020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parveen N, Cornell KA. 2011. Methylthioadenosine/S-adenosylhomocysteine nucleosidase, a critical enzyme for bacterial metabolism. Mol Microbiol 79:7–20. doi: 10.1111/j.1365-2958.2010.07455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cornell KA, Primus S, Martinez JA, Parveen N. 2009. Assessment of methylthioadenosine/S-adenosylhomocysteine nucleosidases of Borrelia burgdorferi as targets for novel antimicrobials using a novel high-throughput method. J Antimicrob Chemother 63:1163–1172. doi: 10.1093/jac/dkp129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams PP, Flores Avile C, Popitsch N, Bilusic I, Schroeder R, Lybecker M, Jewett MW. 2017. In vivo expression technology and 5′ end mapping of the Borrelia burgdorferi transcriptome identify novel RNAs expressed during mammalian infection. Nucleic Acids Res 45:775–792. doi: 10.1093/nar/gkw1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karna SL, Prabhu RG, Lin YH, Miller CL, Seshu J. 2013. Contributions of environmental signals and conserved residues to the functions of carbon storage regulator A of Borrelia burgdorferi. Infect Immun 81:2972–2985. doi: 10.1128/IAI.00494-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu H, Caimano MJ, Lin T, He M, Radolf JD, Norris SJ, Gherardini F, Wolfe AJ, Yang XF. 2010. Role of acetyl-phosphate in activation of the Rrp2-RpoN-RpoS pathway in Borrelia burgdorferi. PLoS Pathog 6:e1001104. doi: 10.1371/journal.ppat.1001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaw DK, Hyde JA, Skare JT. 2012. The BB0646 protein demonstrates lipase and hemolytic activity associated with Borrelia burgdorferi, the aetiological agent of Lyme disease. Mol Microbiol 83:319–334. doi: 10.1111/j.1365-2958.2011.07932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Antonara S, Ristow L, Coburn J. 2011. Adhesion mechanisms of Borrelia burgdorferi. Adv Exp Med Biol 715:35–49. doi: 10.1007/978-94-007-0940-9_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brissette CA, Gaultney RA. 2014. That's my story, and I'm sticking to it–an update on B. burgdorferi adhesins. Front Cell Infect Microbiol 4:41. doi: 10.3389/fcimb.2014.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morgan A, Wang X. 2013. The novel heparin-binding motif in decorin-binding protein A from strain B31 of Borrelia burgdorferi explains the higher binding affinity. Biochemistry 52:8237–8245. doi: 10.1021/bi401376u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan K, Awan M, Barthold SW, Parveen N. 2012. Comparative molecular analyses of Borrelia burgdorferi sensu stricto strains B31 and N40D10/E9 and determination of their pathogenicity. BMC Microbiol 12:157. doi: 10.1186/1471-2180-12-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hyde JA, Weening EH, Chang M, Trzeciakowski JP, Hook M, Cirillo JD, Skare JT. 2011. Bioluminescent imaging of Borrelia burgdorferi in vivo demonstrates that the fibronectin-binding protein BBK32 is required for optimal infectivity. Mol Microbiol 82:99–113. doi: 10.1111/j.1365-2958.2011.07801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X, Liu X, Beck DS, Kantor FS, Fikrig E. 2006. Borrelia burgdorferi lacking BBK32, a fibronectin-binding protein, retains full pathogenicity. Infect Immun 74:3305–3313. doi: 10.1128/IAI.02035-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prabhakaran S, Liang X, Skare JT, Potts JR, Höök M. 2009. A novel fibronectin binding motif in MSCRAMMs targets F3 modules. PLoS One 4:e5412. doi: 10.1371/journal.pone.0005412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Norris SJ. 2012. How do Lyme borrelia organisms cause disease? The quest for virulence determinants. Open Neurol J 6:119–123. doi: 10.2174/1874205X01206010119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bunikis I, Kutschan-Bunikis S, Bonde M, Bergstrom S. 2011. Multiplex PCR as a tool for validating plasmid content of Borrelia burgdorferi. J Microbiol Methods 86:243–247. doi: 10.1016/j.mimet.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 53.Chan K, Casjens S, Parveen N. 2012. Detection of established virulence genes and plasmids to differentiate Borrelia burgdorferi strains. Infect Immun 80:1519–1529. doi: 10.1128/IAI.06326-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Purser JE, Norris SJ. 2000. Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc Natl Acad Sci U S A 97:13865–13870. doi: 10.1073/pnas.97.25.13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Labandeira-Rey M, Skare JT. 2001. Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Infect Immun 69:446–455. doi: 10.1128/IAI.69.1.446-455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, White O, Ketchum KA, Dodson R, Hickey EK, Gwinn M, Dougherty B, Tomb J-F, Fleischmann RD, Richardson D, Peterson J, Kerlavage AR, Quackenbush J, Salzberg S, Hanson M, van Vugt R, Palmer N, Adams MD, Gocayne J, Weidman J, Utterback T, Watthey L, McDonald L, Artiach P, Bowman C, Garland S, Fujii C, Cotton MD, Horst K, Roberts K, Hatch B, Smith HO, Venter JC. 1997. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature 390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- 57.Barthold SW, Beck DS, Hansen GM, Terwilliger GA, Moody KD. 1990. Lyme borreliosis in selected strains and ages of laboratory mice. J Infect Dis 162:133–138. doi: 10.1093/infdis/162.1.133. [DOI] [PubMed] [Google Scholar]
- 58.Chan K, Alter L, Barthold SW, Parveen N. 2015. Disruption of bbe02 by insertion of a luciferase gene increases transformation efficiency of Borrelia burgdorferi and allows live imaging in Lyme disease susceptible C3H mice. PLoS One 10(6):e0129532. doi: 10.1371/journal.pone.0129532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chan K, Marras SA, Parveen N. 2013. Sensitive multiplex PCR assay to differentiate Lyme spirochetes and emerging pathogens Anaplasma phagocytophilum and Babesia microti. BMC Microbiol 13:295. doi: 10.1186/1471-2180-13-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schlachter S, Chan K, Marras SAE, Parveen N. 2017. Detection and differentiation of Lyme spirochetes and other tick-borne pathogens from blood using real-time PCR with molecular beacons. Methods Mol Biol 1616:155–170. doi: 10.1007/978-1-4939-7037-7_10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.