

Abstract
Quantitative gene expression analysis in intact single cells can be achieved using single molecule-based fluorescence in situ hybridization (smFISH). This approach relies on fluorescence intensity to distinguish between true signals, emitted from an RNA copy hybridized with multiple oligonucleotide probes, and background noise. Thus, the precision in smFISH is often compromised by partial or nonspecific probe binding and tissue autofluorescence, especially when only a small number of probes can be fitted to the target transcript. Here we provide an accurate approach for setting quantitative thresholds between true and false signals, which relies on on-off duty cycles of photoswitchable dyes. This fluctuation localization imaging-based FISH (fliFISH) uses on-time fractions (measured over a series of exposures) collected from transcripts bound to as low as 8 probes, which are distinct from on-time fractions collected from nonspecifically bound probes or autofluorescence. Using multicolor fliFISH, we identified radial gene expression patterns in mouse pancreatic islets for insulin, the transcription factor, NKX2-2 and their ratio (Nkx2-2/Ins2). These radial patterns, showing higher values in β cells at the islet core and lower values in peripheral cells, were lost in diabetic mouse islets. In summary, fliFISH provides an accurate, quantitative approach for detecting and counting true RNA copies and rejecting false signals by their distinct on-time fractions, laying the foundation for reliable single-cell transcriptomics.
INTRODUCTION
Single-cell analysis techniques provide effective avenues to probe molecular processes and cellular heterogeneities with unprecedented resolution and accuracy (1–3). Imaging-based single-cell transcriptomics is a powerful approach to dissect the identity and functional state of individual cells as it directly provides the exact copy number of RNA species as well as their subcellular location (4). Single-molecule-based fluorescence in situ hybridization (smFISH) has laid the foundation for utilizing a programmable set of oligonucleotide probes to barcode the transcripts of interest (Figure 1A and B) (5–7). Together with combinatorial multicolor labeling and optimized hybridization protocols, smFISH has substantially improved the throughput of single-cell RNA counting (8–10). However, nearly all the smFISH methods rely on a large number of fluorescent probes targeting a single RNA molecule to increase the signal-to-noise ratio (SNR) and separate the true signal, emitted from a single RNA copy, from nonspecific fluorescence background. Furthermore, the fluorescence threshold for distinguishing true signals from background often relies on qualitative assessments and include nonspecific or unbound probes, as well as autofluorescence. This drawback inevitably compromises the reliability of smFISH in providing accurate gene expression values in single cells.
Figure 1.

Illustration of the fliFISH concept. (A) Multiple single-labeled probes (blue) are hybridized to a target RNA molecule (red), enabling the quantification and subcellular localization of the transcripts. (B) In conventional smFISH, integrated fluorescence intensity of multiple probes, acquired in a single image, is used to distinguish the target molecule from background noise. (C) In fliFISH, photoswitchable dye molecules attached to individual probes are stochastically turned on and off to generate photoblinking patterns as illustrated in the corresponding single-probe blinking traces. With each additional probe that is hybridized to the RNA molecule, the ensemble on-time fraction is increased in a predictable way. Knowing the number of probes that are designed to target the RNA molecule, the ensemble on-time fraction expected within a diffraction-limited area can be calculated and used as a threshold for a true signal.
Super-resolution localization microscopy makes use of the temporal fluctuation in fluorescence emission of certain fluorophores to achieve spatial separation of individual molecules within a diffraction-limited spot (11,12). Thus, densely localized molecules can be distinguished by sequentially turning only a few of them on during each exposure. The first demonstration of super-resolution localization microscopy, including stochastic optical reconstruction microscopy (STORM) and photoactivated localization microscopy (PALM), used at least two excitation wavelengths to control the ‘blinking’ behaviors of the fluorophores (13) More recently, it has been observed that certain organic dyes, such as Alexa Fluor and Atto dyes, can be spontaneously switched on and off by a single high-power excitation under low-oxygen, thiol-rich environment (14,15). Moreover, these dyes possess a short ‘on-off’ duty cycle—the time window during which photons are emitted—but yield a robust photon burst per cycle, which makes them ideal for super-resolution localization microscopy (16,17).
Here, we take advantage of these photoswitchable dyes to address the shortcomings and limitations of smFISH. We provide a new quantitative approach for distinguishing between true signals, emitted from single RNA copies, and background noise. Our fluctuation localization imaging-based fluorescence in situ hybridization approach, dubbed fliFISH, increases the accuracy and reliability of single-cell transcriptomic analysis. Our approach relies on the principle that the on-time fraction (the times when a fluorophore emits photons over a series of exposures) of a single FISH probe, tagged with one dye molecule, is distinguished from the ensemble on-time fraction of multiple hybridized probes (Figure 1C). When attached to an RNA molecule in cells, the set of single-labeled oligonucleotide probes is expected to span an area that is within the diffraction limit. Thus, by measuring the on-time fraction of single probes within a diffraction-limited area, it is possible to predict the expected ensemble on-time fraction of multiple probes bound to an RNA molecule. Since the on-time fraction of a single (less blinking) and multiple (more blinking) dye molecules within a sub-diffraction area are significantly different (within a given number of exposure frames, e.g. >5000), it allows us to set a quantitative threshold based on the on-off duty cycle of the specific fluorophores, which clearly separates between true binding to an RNA molecule and autofluorescence or unbound stray probes. Because fliFISH relies on photoblinking patterns, rather than on fluorescence intensity, this approach not only increases the precision of recognizing a true RNA copy, but also decreases the number of required oligonucleotide probes and improves the spatial resolution in the acquired images.
The normal function of tissues and organs relies on a highly ordered structure of cells that assume different roles and harbor distinct patterns of gene expressions. However, conventional approaches cannot reveal the transcriptomic heterogeneity at the single-cell resolution. In contrast, smFISH can measure gene expressions while preserving the structural information within the tissues (18,19). The pancreas is a critical organ involved in digestive and endocrine functions. Degeneration and/or dysfunction of β cells within the pancreatic islets of Langerhans leads to diabetes mellitus. Recently, significant evidence has emerged to suggest the existence of β cell heterogeneity (20–23), which may impact β cell function and/or susceptibility to external environmental stressors that can lead to diabetes. Identifying aberrations in transcriptomic patterns at the cellular and islet levels are therefore critical to understanding the pathophysiology of the disease. Here we demonstrate the applicability of fliFISH in mouse pancreatic β cells in culture and in islet tissue sections, where we quantitatively identify a radial gradient of single-cell gene expression values for insulin and its relevant transcription factor within normal islets.
MATERIALS AND METHODS
Cell culture and probe design
MIN6 cells (mouse pancreatic β cell line) were cultured in DMEM supplemented with 25 mM glucose, 10% fetal bovine serum, 1% l-glutamine, 1% penicillin–streptomycin (Thermo Fisher Scientific), and 0.0005% 2-mercaptoethnol. 266-6 cells (mouse pancreatic acinar cell line) were cultured in DMEM supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin. Cell cultures were maintained in a humid incubator supplied with 5% CO2 at 37°C. Before use, cells were tested to eliminate impact from mycoplasma contamination. Sets of 20 nucleotide (nt) probes were designed to meet the following criteria: 40-60% CG content, no ‘CCC’, ‘GGG’, ‘AAAA’ or ‘TTTT’ repeats, and no stem-loop structure. All probe sequences were confirmed with BLAST to confirm targeting specificity (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Probes were labeled at the 5′ end and synthesized by Integrated DNA Technologies. All probe sequences are listed in Supplementary Table S1.
Mouse and human pancreatic tissues
All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Colorado Denver. 6–8 weeks old female NOD/ShiLtJ (NOD) and NOR/LtJ (NOR) mice were obtained from the Jackson Laboratories (Bar Harbor, ME) and pancreatic tissue was harvested immediately upon arrival. The pancreatic tissues were fixed in 4% PFA overnight, washed in PBS and embedded in carboxymethyl cellulose (CMC) for cryo-sectioning at 8–10 μm thick sections for hybridization. Formalin-fixed paraffin-embedded (FFPE) pancreatic tissue sections (used in Supplementary Figures S8 and S9) were a gift from the UCSF Islet Isolation Core to Dr. Holger A. Russ (University of Colorado, Denver). Before hybridization, these tissue sections were deparaffinized with xylene-ethanol series and the residual formalin was quenched with 0.1% sodium borohydride (NaBH4).
RNA hybridization
Cells for fliFISH were seeded onto 35 mm glass-bottom Petri dishes (No.1 thickness) and grown to 70–80% confluency. The cells were fixed with 4% paraformaldehyde (PFA) for 10 min at room temperature. The PFA residual was quenched with 0.1% NaBH4 after washing with PBS. The cell membranes were permeabilized with 70% ethanol overnight at 4°C. For fliFISH, probes mixture at 4–5 nM (100 nM for tissue sections, equimolar mixture of all probes) was dissolved in hybridization buffer (10% dextran sulfate, 2 mM vanadyl-ribonucleoside complex, 0.02% BSA, 2 × SSC, 10% formamide) and added to the cells or tissue sections for overnight incubation at 37°C. For smFISH using the Stellaris probe set, the standard protocol provided by the manufacturer (Biosearch Technologies) was followed. Before imaging, the cells/tissues were thoroughly rinsed with buffer containing 2 × SSC and 10% formamide. The cell nuclei were stained with 0.05% DAPI.
Microscopy for fliFISH
The fliFISH imaging was performed using Olympus IX-71 inverted microscope base with a 100× oil immersion objective (NA 1.4). Five solid-state lasers were incorporated into the system (405, 488, 542, 594, 640 nm) and the laser power was controlled with electronic drivers and neutral density filters. The fluorescence emission was collected using the same objective and imaged by an electron multiplying CCD camera (Andor iXon Ultra 897). Chromatic aberration was calibrated with multispectral beads and corrected in post-processing. The raw images were initially recorded as 512 × 512 pixels with a pixel size of 160 nm, and stored as 16-bit FITS files. The exposure time and imaging frame rate were controlled with the Andor SOLIS software. Data collection was started when fluorescence blinking became stable (1–3 min after excitation). Based on the literature and our calibration, a 400 s detection window (10 000 frames) was selected in which no significant photobleaching would occur. Imaging was done in buffer containing: 50 mM Tris (pH 8.0), 10 mM NaCl, 10% glucose, 560 μg/ml glucose oxidase (Sigma, G2133), 34 μg/ml catalase (Sigma, C3155) and 1% 2-mercaptoethnol. For quantifying on-time fractions of the different dyes, 0.1 nM probes, tagged with one dye molecule, were diluted in TE buffer and placed for 20 min in a chamber with a glass-bottom that was coated with poly-l-lysine. The solution was then replaced with a fresh imaging buffer to remove unbound probes. Different image acquisition rates and exposure times were tested (results provided in Supplementary Table S2), and 25 Hz acquisition rate and a total exposure of 200-400 s were chosen as the optimal imaging setting.
Image processing
Each raw image frame was first processed with a Laplacian of Gaussian (LoG) filter function to reduce background and the coordinates of all local maxima were identified. Guided by the local maxima coordinates, a Gaussian mask algorithm was used to find the centroid for each blinking spot (24). The intensity threshold of a blinking event was defined as above five times the standard deviation of the sample-specific background fluctuations. A fitted centroid that was 1 pixel away from the original local maxima (e.g. due to strong background interference) was rejected. The size of target mRNAs and the probe spanning area upon hybridization were used to set the limit for the area from which blinking events were considered to come from one mRNA copy. For example, the mouse Ins2 mRNA targeted in this study is about 480 nt long (equivalent to ∼163 nm in space) and the designed eight probes flank a segment of 220 nt (∼75 nm). Therefore, blinking events localized within 1 pixel area (e.g. 160 × 160 nm2) could be grouped to count RNA copies. Since multiple transcripts could exist within a diffraction-limited region, especially for highly expressed genes, the DBSCAN algorithm (25) was used in this study to distinguish multiple transcripts in a diffraction-limited area. This was done by linking clustered blinking events to a discrete RNA molecule based on the actual probe spanning area and error of localization. Here, the ε value in DBSCAN – the distance threshold that determines if two localized events belong to the same cluster – was set to 30 nm (approximation of the distance between two adjacent fluorophores on hybridized probes plus the error of localization in cells). This way, clusters with the expected number of blinking events (frame number × ensemble on-time fraction) could be identified within a diffraction-limited area and each cluster was counted as an individual transcript. Clusters of excessive blinking events (e.g. >10% ensemble on-time fraction) and clusters spanning an area larger than the target hybridization segment were rejected. For all the fluorophores used in our study, the standard error of localization (x) was calculated according to the following equation:
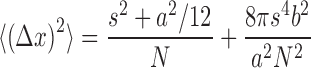 |
(1) |
where s is the standard deviation of the Gaussian point spread function, a is the pixel size, b is the background noise, and N is the collected photons (24). The obtained errors of localization were determined to be ∼20 nm in cultured cells, ∼30 nm in frozen tissue sections, and 40–50 nm in FFPE tissue sections. These values guided the setting of a reasonable ε value in the DBSCAN algorithm for the different conditions. The ensemble on-time fraction thresholds (Fensemble-on) were calculated based on the single-probe on-time fraction (Fsingle-on) that was quantified for each fluorophore and the number of probes (Nprobe) targeting each transcript using the following equation:
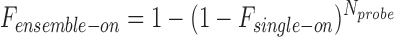 |
(2) |
All image processing was performed with customized C and MATLAB scripts that are available upon request.
Immunofluorescence staining
Pancreatic tissue sections were stained with insulin antibody to identify the location of islets of Langerhans. The tissue sections were permeabilized with 0.5% Triton X-100 for 30 min, followed by blocking with 1 × PBS containing 5% goat serum and 0.3% Triton X-100 for 1 h at room temperature. The sections were incubated in 1:500 dilution monoclonal rabbit anti-insulin IgG (Cell Signaling Technology, #3014) overnight at 4°C. The primary antibody was thoroughly washed with PBS followed with incubation in 1:1000 dilution Alexa 546-goat anti-rabbit F(ab’)2 (Thermo Fisher Scientific). The tissue sections were then subjected to the fliFISH treatment and imaging.
qRT-PCR
Single-cell qRT-PCR was used to validate the expression level of Ins2 in MIN6 cells. The number of input cells (e.g. 200 cells) was counted by flow cytometry (Influx, BD Biosciences) before RNA extraction. The RNA extraction and reverse transcription were conducted using the RNeasy Mini Kit (Qiagen) and iScript cDNA Synthesis Kit (Bio-Rad), respectively. The PCR amplification was performed in a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific). The PCR parameters were: 94°C 30 s, 58°C 1 min, 72°C 1 min for 40 cycles. The segment of Ins2 DNA and the primers that were used to generate the standard curve for quantification are shown in Supplementary Table S3. The generated standard curve is shown in Supplementary Figure S1.
RESULTS AND DISCUSSION
Setting parameters for a quantitative threshold between true signals, emitted from RNA copies and background noise
Our approach relies on the following principle. Given a fluorophore with on-time fraction F and a transcript targeted with N such fluorophores, the ensemble on-time fraction for the transcript would be 1 – (1 – F)N within a diffraction-limited pixel area (160 × 160 nm2). We chose to measure blinking events in a sub-pixel area because the distance spanned by a set of oligonucleotide probes bound to an RNA molecule is expected to be within the diffraction limit. To quantify the on-time fraction for a single Alexa 647-bound probe, single-labeled oligonucleotide FISH probes were diluted in fresh imaging buffer and placed in a glass coverslip chamber for imaging. Using a CW laser with the excitation power of 0.4 kW/cm2, stable photoblinking could be observed with no significant photobleaching for up to 400–600 s. An example for such a trace is shown in Figure 2A. Considering the low, pM-level concentration of probes used in these measurements, the possibility for having multiple probes within a single pixel is negligible. We collected ∼10 000 pixels with detectable blinking events and summarized the single-probe on-state duty cycle in a histogram (Figure 2B). The average on-time fraction for single Alexa 647-labeled probes (Fsingle-on) was found to be 0.19%, which is in agreement with a previous report (16). This sparse-blinking rate minimizes signal overlap between multiple probes and makes Alexa 647 a primary choice for our fliFISH application. According to Equation 2 under Materials and Methods, the ensemble on-time fraction for an RNA molecule targeted by eight single-labeled probes is expected to be around 1.6% (Fensemble-on). Consistent with this prediction, on-time fractions measured within a diffraction-limited pixel area in cells treated with 8 distinct probes, targeting the insulin mRNA, showed a higher number of blinking events (Figure 2C, lower trace). Blinking values in the cell showed values ∼1.7%, indicating the presence of a transcript hybridized to multiple probes, as well as lower values, suggesting non-specific binding, as demonstrated in Figure 2D. Notably, the position of each blinking event can be localized with a precision of about 20 nm, enabling the differentiation of multiple transcripts within a single pixel (Figure 3B).The large difference between the on-time fraction values for one probe and multiple probes, measured within a diffraction-limited area over many exposures (>5000), allows us to set a clear threshold between true binding of multiple probes to an RNA molecule and nonspecific or unbound single probes. Although no other quantitative method for setting a threshold between true and false signals in smFISH is currently available other than using SNR in fluorescence intensity values, a general approach for counting molecules in complexes, such as number of bound FISH probes to a transcript, has been recently demonstrated by qPAINT (26), using 64 oligonucleotide probes. This approach is built on DNA-PAINT (27–29), where blinking events are generated by the transient binding of free-floating dye-labeled oligonucleotide strands, named ‘imagers’, to complementary target-bound or ‘docking’ strands. The number of target molecules (or bound probes) is then quantified by the predicted binding/unbinding kinetics between the fluorescent ‘imagers’ and the ‘docking’ strands. Thus some of the challenges that qPAINT faces are the ability to achieve fine control over probe influx, theoretical calculations of binding kinetics, and elimination of signal interference from a large number of unbound probes. The detection of two fluorescent colors would also be required. In comparison, fliFISH employs single wavelength-dependent intrinsic photoblinking of organic dyes that are directly bound to the FISH probe with no additional colors, oligonucleotides or interactions needed.
Figure 2.

Characterization of on-time fraction for Alexa 647-labeled probes. (A) A representative fluorescence trace from a single probe on glass coverslip. (B) The average on-time fraction of single probes, Fsingle-on, under 0.4 kW/cm2 excitation was measured using a low concentration of probes adhered to a cover glass. The ensemble on-time fraction of N probes, Fensemble-on, can then be determined using Equation (2). (C) Representative fluorescence traces from a single stray probe (upper), and multiple probes bound to a transcript (lower) in the cell, recorded from a diffraction-limited area (single pixel, 160 × 160 nm2). The traces show a significant difference in the number of blinking events to support a clear threshold between truly bound probes and nonspecific background. (D) Upper panel: Fluorescence image showing edge of cells treated with multiple probes targeting Ins2 mRNA. Lower panel: Blinking events, marked by red crosses, are counted within diffraction-limited areas (individual pixels, black squares), showing low (0.3%) and high (1.7%) on-time fraction values that indicate the presence of only one or two probes versus multiple (∼8) probes, respectively.
Figure 3.

fliFISH improves the resolution and reliability of counting RNA copies, especially when using a small number of probes. (A) Comparison between conventional smFISH (left image) and fliFISH (right image) using eight probes to target Ins2 mRNA in pancreatic β cells (MIN6) in culture. (B) fliFISH enables accurate localization of individual blinking events and the distinction between multiple transcripts within a diffraction-limited area.
Demonstrating fliFISH in cultured pancreatic β cells
Figure 3A demonstrates the challenge in quantifying gene expression using conventional smFISH when only a limited number of probes can be fitted to the target RNA and how fliFISH overcomes this challenge to provide reliable transcript counts with high accuracy. We used 8 single-labeled probes to quantify insulin-2 (Ins2) mRNA in pancreatic β cells (MIN6) grown in culture. As shown in Figure 3A, left image, the low SNR makes it difficult to unambiguously identify the location and determine the number of all transcripts within a single cell using conventional smFISH. In comparison, using fliFISH to set the threshold at the expected ensemble on-time fraction for 8 Alexa 647-tagged probes (Supplementary Movie S1), it was possible to precisely locate and quantify Ins2 mRNA copies in these cells (Figure 3A, right image). The low SNR in smFISH images could be attributed to: (i) autofluorescence from cellular structures and tissue components at a comparable intensity to hybridized probes; (ii) signals from nonspecifically-bound stray probes, or from bound probes at out-of-focus planes, contributing to the overall background; (iii) the actual fluorescence intensity at the hybridization site is not linearly proportional to the number of fluorophores; (iv) during the one-time exposure used for smFISH, a considerable number of fluorophores may not be in the on-state due to the stochastic emission of organic dyes. In conventional smFISH, the primary approach to improve SNR is either by increasing the number of probes or by increasing the exposure time. However, when only a limited number of probes can be accommodated, such as the case when targeting a short transcript as Ins2, longer exposure does not always warrant better SNR due to photobleaching and imbalanced accumulation of probe signals and background noise (Supplementary Figure S2A). Rather than fluorescence intensity, fliFISH relies on the on-off duty cycle, determined over many short exposures, for setting the on-time fraction threshold between true and false signals. Since on-off duty cycles are not critically impacted by the points listed above, the preset on-time fraction threshold in fliFISH can accurately determine a true hybridization location even in the presence of background noise. In fliFISH, where the blinking of probes is sparsely distributed and uncorrelated, shorter exposures (i.e. higher frame rates) could in fact improve SNR, as demonstrated in Supplementary Figure S2B. With a higher frame rate, each photon burst is still recoded and the blinking pattern is minimally impacted since the burst of photon emission always exceeds the background fluctuation (Supplementary Figure S2B). Furthermore, in fliFISH the location of each blinking event is precisely recorded and could reach a spatial resolution of 20 nm. Therefore, probes hybridized to the same transcript can be better resolved. As shown in Figure 3B, multiple transcripts are identified by fliFISH in a diffraction-limited area, which could not be resolved using smFISH. It is noteworthy that smFISH and fliFISH yielded the same values for RNA copies per cell when enough probes (34 probes) could be applied in smFISH (Supplementary Figure S3).
During image processing, counts from nonspecific or unbound probes (false-positives) can be substantially reduced by setting the ensemble blinking threshold at the level of expected bound probes (Figure 4A). Applying eight probes, each tagged with one Alexa 647 molecule, for targeting Ins2 in β cells, we observed a fast decay in the number of detected counts when the blinking threshold is increased from 0.2% (on-time fraction of a single probe) to 1.0% (on-time fraction of five probes), when fitting the histogram with a double exponential decay function (dotted line). The faster decay in counts over this threshold range, indicated by the yellow shaded area in Figure 4A, suggests counting bias due to nonspecific or unbound probes (30). Therefore, we set a conservative lower limit cutoff at 1.2% (on-time fraction of six probes), which corresponds to ∼70% hybridization efficiency as reported earlier (8,26). We noted that some counts remained at a certain value when the threshold was set to above 10%, which could potentially result from the presence of multiple mRNAs (>8 copies) in a sub-pixel area, but more likely from the presence of aggregated probes or strongly autofluorescing structures. Therefore, we also set a higher limit cutoff to eliminate these possible false-positive counts. In practice, we rarely found spots that reached above 10% ensemble on-time fraction, partially because we used the DBSCAN algorithm (see Materials and Methods) to distinguish multiple transcripts within a diffraction-limited area, as demonstrated in Figure 3B.
Figure 4.

Quantification of Ins2 gene expression by fliFISH in mouse pancreatic β cell line (MIN6). (A) Removal of counts originating from nonspecifically bound stray probes or aggregated probes can be achieved by presetting the on-time fraction threshold calculated according to the number of probes used. The histogram is fitted with a double exponential decay curve (blue dotted line). (B) Single-cell quantification of Ins2 mRNA using fliFISH. The cells are outlined by the green dotted line and the number of mRNA copies per cell is shown in the center for each cell. (C) Histogram summarizing the distribution of Ins2 copy number per cell. The histogram is fitted with a single exponential decay curve (blue dotted line). (D) Comparison of Ins2 copy number quantified by fliFISH and qRT-PCR shows high agreement.
Essentially, fliFISH relies on an imaging principle that sacrifices the speed of data acquisition for high specificity and spatial resolution, while relying on a lower number of FISH probes (<20) than those needed for smFISH. Using fliFISH, the copy number of Ins2 mRNA in single β cells was precisely counted (Figure 4B). We observed a typical pattern of transcriptional bursting, where at a given point in time, only few β cells show high abundance of Ins2 transcripts, while the majority of cells maintain relatively low transcript levels (Figure 4C) (31). Such heterogeneity in time and space can be uncovered only by single-cell analysis approaches (32,33) and is reflected in the exponential decay in the mRNA lifetime (34,35). To evaluate the accuracy of fliFISH, we quantified Ins2 mRNA in a given number of cells (determined by flow cytometry) using quantitative real-time polymerase chain reaction (qRT-PCR). As shown in Figure 4D, the single-cell expression levels quantified by fliFISH and by single-cell qRT-PCR were in high agreement. As negative control, mouse pancreatic acinar cell line (266-6), which does not express insulin, was used. As shown in Supplementary Figure S4A, no transcription of Ins2 was detected by fliFISH in these cells. In contrast, Elastase mRNA copies were detected at different levels in these cells as expected (Supplementary Figure S4B).
Multiplexed RNA counting in single cells using fliFISH
Over the past few years, the throughput of smFISH has been greatly increased owing to a variety of multiplexing and barcoding strategies (4,8–10,36,37). These approaches support gene expression analysis for multiple genes in single cells simultaneously. To test the potential and compatibility of fliFISH with these techniques, we first targeted multiple genes in single cells using different fluorescent colors to tag each set of probes. We characterized the photoblinking behaviors of additional organic dyes, including Atto 488 and TAMRA, which were found to have the required optical properties (i.e. low on-time fraction and high photon yield) (Supplementary Figures S5 and S6). For instance, the average on-time fraction of Atto 488 was about 0.12% under excitation power of 1 kW/cm2. Using eight Atto 488-labeled probes, fliFISH was applied to quantify mRNA copy number of the Nkx2-2 gene in β cells (Supplementary Figure S6, right). NKX2-2 is a homeodomain transcription factor that is critical for the maintenance and function of β cells, and its downregulation is closely related to reduction in insulin production in diabetic mice (38–40). In addition to probes targeting Ins2 and Nkx2-2, eight TAMRA-labeled probes were used to target the mRNA for glyceraldehyde 3-phosphate dehydrogenase (Gapdh), a housekeeping gene expressed at relatively high levels in most cells. The three genes were targeted simultaneously in single cells, as demonstrated in Figure 5A. The ensemble on-time fraction thresholds set by fliFISH (1.2% for Ins2, 0.8% for Nkx2-2 and 0.6% for Gapdh), allowed a clear distinction between true and false signals for all three colors simultaneously. A significant cell-to-cell heterogeneity in transcript levels was observed for the three genes. Next, we designed a set of probes targeting Ins2, where eight probes were tagged with Alexa 647 and additional eight were tagged with Alexa 594 (Figure 5B). We found a relatively high overlapping rate (up to ∼86%) between the two colors, indicating the detection of RNA copies with high accuracy for both colors, and demonstrating the compatibility of fliFISH with multiplexing by spectral barcoding.
Figure 5.

fliFISH compatibility with multiplexed transcript quantifications in single cells. (A) Simultaneous detection of Ins2, Nkx2-2, and Gapdh mRNA copies, each targeted with 8 probes, tagged with Alexa 647, Atto 488 and TAMRA, respectively. Raw fluorescence images for each channel (left) and fliFISH image (right) of β cells in culture. (B) Detection of Ins2 by fliFISH using a two-color barcode, where probes tagged with Alexa 594 (left) or Alexa 649 (middle) were used simultaneously. The overlap of the two sets is shown on the right.
As mentioned above, the imaging speed is the major technical challenge for fliFISH to achieve high-throughput screening. To capture one image, smFISH requires several seconds while fliFISH requires hundreds of seconds to acquire a sufficient number of frames for precise localization. Using a standard wide-field microscope, a limited number of images along the z-direction (4–6 z-stacks) can be taken due to photobleaching. Therefore, increasing the photon collection efficiency becomes a pressing task to further the application of fliFISH. To tackle this problem, two methodological aspects of fliFISH can be optimized, including combining fliFISH with advanced microscopy platforms such as light-sheet microscopy or multifocus microscopy (41,42), and utilizing faster-blinking dyes (e.g. an ideal F single-on would be 1–2% under the 25 Hz frame rate) to enable the acquisition of sufficient number of frames within a shorter time window. With these advances, the difference in the temporal resolution between smFISH and fliFISH can be reduced to within one order of magnitude. In addition, as fliFISH is mainly a technical improvement in imaging and data processing rather than in hybridization protocols, it can be readily extended to work with MERFISH (10) or smiFISH (43) where primary probes carry orthologous sequence overhangs to which secondary probes coupled to a fluorophore are hybridized, such that the cost for multiplexed gene quantification can be greatly reduced.
Overcoming the inherently high background in tissue samples by fliFISH
One of the challenges for smFISH in tissue sections is the intense autofluorescence background due to the presence of molecular components with complex compositions. To address this challenge, various methods have been developed to either reduce the source of background (e.g. optical clearing (44–46) and expansion microscopy (47)) or enhance the collection efficiency for on-target signals (e.g. light-sheet microscopy (36,48)). However, only few of the techniques have shown full compatibility with smFISH in tissues without special treatments (49,50). In comparison, using fliFISH it is possible to resolve and eliminate different types of background noise based on specific photoblinking patterns to improve specificity, as demonstrated in Supplementary Figure S7. The application of fliFISH is further demonstrated in FFPE pancreatic tissue sections, which are known to have high background, where blinking events were clearly distinguished from background (Supplementary Figures S8 and S9). In Figure 6, we demonstrate the application of fliFISH by targeting Nkx2-2 in pancreatic islets of Langerhans. Using pancreatic tissue cryo-sections, insulin antibody was used to locate the islets, while Nkx2-2 mRNA copies were identified by fliFISH. As expected, the Nkx2-2 mRNA molecules were found predominantly in the endocrine cells of the islet, including insulin-producing β cells, supporting its role in regulating β cell maturation and insulin production.
Figure 6.

Implementation of fliFISH in mouse pancreatic tissue section. (A) An islet of Langerhans, located by an antibody against insulin (red), is highlighted by the dashed line. The nuclei are shown in blue. (B) Nkx2-2 mRNA copies detected by fliFISH in the enlarged area indicated by the square in A.
The role of NKX2-2 in regulating insulin expression can be quantitatively evaluated by targeting Ins2 and Nkx2-2 mRNA copies in pancreatic islets simultaneously. Probes tagged with Atto 488 and Alexa 647were applied to quantify Nkx2-2 and Ins2 mRNA copies, respectively (Figure 7A). Using fliFISH, the transcriptional heterogeneity for both genes was clearly observed in the islets, showing a radial pattern with higher expression levels for both genes in the center of the islets and lower levels at the periphery (Figure 7B and C). This radial architecture is in agreement with the mouse islet architecture, where mature and polarized β cells are found at the core of the islet while proliferative β cells and non-β cells are mostly found at the periphery (51). fliFISH also allowed accurate quantification and mapping of the Nkx2-2/Ins2 copy number ratio, showing a radial pattern as well (Figure 7). While Ins2 transcript numbers were found to be higher than the number of Nkx2-2 transcripts in these cells, Nkx2-2 copy number relative to Ins2 copy number increased toward the center of the islet, supporting a more profound role for the transcription factor in mature and polarized β cells compared with proliferative β cells. Cells in islets of non-obese diabetic (NOD) mice showed a significant decrease in the expression levels of both Nkx2-2 and Ins2 compared to cells in control non-obese resistant (NOR) mice (Supplementary Figure S10A and B). Interestingly, the radial pattern of Nkx2-2/Ins2 gene expression ratio was clearly disrupted in NOD mice islets (Supplementary Figure S10C). Together, fliFISH quantitatively showed a correlative expression of the two genes and the ratio between them, supporting a functional link with a structural pattern between Nkx2-2, insulin production, and maturation of β cells in normal mice and its alteration in NOD mice.
Figure 7.

Spatial profiling of single islet cells by the ratio of gene expression for Nkx2-2 and Ins2. (A) Left: Pancreatic islet is identified by immunostaining targeting insulin (red). Nuclei are shown in blue. Middle: mRNA cope number for each gene is quantified in each cell (outlined) by fliFISH. Right: mRNA copy number ratio of Nkx2-2 to Ins2 is calculated and presented in color-map for each cell. (B) Left: Part of another islet (outlined) showing insulin expression by immunoassay (red). Right: The ratio of Nkx2-2 to Ins2 copy number in each cell is color-mapped, showing a radial decrease from the islet core to the periphery. (C) Upper: Illustration of mouse islet architecture. Lower: Normalized Nkx2-2 and Ins2 copy number per cell as a function of distance from the islet's core, showing radial pattern of gene expression and decrease in Nkx2-2/Ins2 ratio toward the periphery, supporting a role for Nkx2-2 in β cell maturation and insulin expression (data summarized from three different mouse samples, n >100 cells).
In summary, fliFISH is particularly useful when targeting RNA molecules that can accommodate only a small number of oligonucleotide probes (as low as eight probes) and where background noise (from nonspecific binding or autofluorescence) is high. Furthermore, by recording blinking events from each single probe, fliFISH can resolve multiple transcripts localized within a diffraction-limited spot, making it useful for accurate quantification of highly expressed genes. Considering the low number of probes required for fliFISH, it can also be applied to study the tertiary structure of nucleic acids and in situ DNA-RNA-protein interactions. In practice, as fliFISH relies on the SNR of single dye molecules, rather than integrated SNR of multiple molecules, bright dyes with SNR of at least 3 (ideally >5) is required, such as the dyes used in this study. Key parameters including single-probe on-time fraction, localization error, and camera performance should be carefully quantified for each experiment.
CONCLUSION
In this work, we report a quantitative fliFISH approach for reliable detection and accurate counting of RNA copies in single cells, especially when only a small number of probes can be fitted to the target transcript. Instead of relying on fluorescence intensities, fliFISH relies on on-off duty cycles of photoswitchable dyes to distinguish true signals emitted from an RNA copy hybridized with FISH probes, from false signals coming from nonspecifically bound stray probes or autofluorescence. Knowing the on-time fraction of a probe tagged with one dye molecule, the ensemble on-time fraction for a given number of probes that are targeting a specific transcript can be calculated and used as a threshold for distinguishing true from false signals. Current smFISH approaches rely on fluorescence intensities to differentiate hybridized targets from background and are strongly dependent on the SNR in fluorescence intensity acquired in a single image. Therefore, the accuracy of smFISH is strongly impacted by factors, such as insufficient number of probes targeting or binding a specific transcript, dye molecules found in their off-state during the single exposure, out-of-focus emission, or intense autofluorescence. In contrast, fliFISH can provide accurate gene expression values even in the presence of low SNR because it relies on a preset on-time fraction to eliminate autofluorescence and nonspecific binding, which are unlikely to reach the blinking threshold range. In addition to high spatial resolution and superior detection specificity, fliFISH is capable of quantifying short RNA species such as non-coding RNAs, which can accommodate only a limited number of probes, often insufficient for detection by smFISH. We show that fliFISH can be combined with multicolor labeling and barcoding to increase throughput. Using multiplexed fliFISH, we targeted transcripts for both ins2 and the transcription factor, Nkx2-2, in pancreatic islet β cells in tissue sections from control and NOD mice. In control mice islets, we identified a radial single-cell gene expression pattern for both genes, as well as their copy number ratio (Nkx2-2/Ins2), with higher values in cells at the core of the islet and lower values in cells at the periphery. This radial expression pattern was lost in NOD mice. While it has been known that mature, insulin-producing β cells are located at the core of the islet and that NKX2-2 plays a critical role in insulin production, here we provide the first quantitative relationships between the expression levels of the two genes at the single cell level, showing a radial pattern that is disrupted in NOD mice. Together, fliFISH provides quantitative gene expression analysis in single cells with high accuracy even when only a small number of probes are used to target the transcripts and has the potential to significantly advance reliable single cell transcriptomics.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Dr Holger A. Russ for providing FFPE tissue sections.
FUNDING
National Institute of Health (NIDDK)—Human Islet Research Network [UC4DK108101]; Pacific Northwest National Laboratory (PNNL) Microbiome in Transition Initiative. The research was performed using the Environmental Molecular Sciences Laboratory (EMSL), a national scientific user facility sponsored by the Department of Energy's Office of Biological and Environmental Research and located at PNNL. Funding for open access charge: NIH grant.
Conflict of interest statement. None declared.
REFERENCES
- 1. Cui Y., Irudayaraj J.. Inside single cells: quantitative analysis with advanced optics and nanomaterials. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015; 7:387–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dey S.S., Kester L., Spanjaard B., Bienko M., van Oudenaarden A.. Integrated genome and transcriptome sequencing of the same cell. Nat. Biotechnol. 2015; 33:285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mitchell H.D., Markillie L.M., Chrisler W.B., Gaffrey M.J., Hu D., Szymanski C.J., Xie Y., Melby E.S., Dohnalkova A., Taylor R.C. et al. Cells respond to distinct nanoparticle properties with multiple strategies as revealed by single-cell RNA-seq. ACS Nano. 2016; 10:10173–10185. [DOI] [PubMed] [Google Scholar]
- 4. Cui Y., Liu J., Irudayaraj J.. Beyond quantification: in situ analysis of transcriptome and pre-mRNA alternative splicing at the nanoscale. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017; 9:e1443. [DOI] [PubMed] [Google Scholar]
- 5. Femino A.M., Fay F.S., Fogarty K., Singer R.H.. Visualization of single RNA transcripts in situ. Science. 1998; 280:585–590. [DOI] [PubMed] [Google Scholar]
- 6. Levsky J.M., Shenoy S.M., Pezo R.C., Singer R.H.. Single-cell gene expression profiling. Science. 2002; 297:836–840. [DOI] [PubMed] [Google Scholar]
- 7. Raj A., van den Bogaard P., Rifkin S.A., van Oudenaarden A., Tyagi S.. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods. 2008; 5:877–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lubeck E., Cai L.. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat. Methods. 2012; 9:743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lubeck E., Coskun A.F., Zhiyentayev T., Ahmad M., Cai L.. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods. 2014; 11:360–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen K.H., Boettiger A.N., Moffitt J.R., Wang S., Zhuang X.. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015; 348:aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rust M.J., Bates M., Zhuang X.. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods. 2006; 3:793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Betzig E., Patterson G.H., Sougrat R., Lindwasser O.W., Olenych S., Bonifacino J.S., Davidson M.W., Lippincott-Schwartz J., Hess H.F.. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006; 313:1642–1645. [DOI] [PubMed] [Google Scholar]
- 13. Huang B., Bates M., Zhuang X.. Super-resolution fluorescence microscopy. Annu. Rev. Biochem. 2009; 78:993–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vogelsang J., Cordes T., Forthmann C., Steinhauer C., Tinnefeld P.. Controlling the fluorescence of ordinary oxazine dyes for single-molecule switching and superresolution microscopy. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:8107–8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heilemann M., van de Linde S., Mukherjee A., Sauer M.. Super-resolution imaging with small organic fluorophores. Angew. Chem. Int. Ed. Engl. 2009; 48:6903–6908. [DOI] [PubMed] [Google Scholar]
- 16. Dempsey G.T., Vaughan J.C., Chen K.H., Bates M., Zhuang X.. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat. Methods. 2011; 8:1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van de Linde S., Loschberger A., Klein T., Heidbreder M., Wolter S., Heilemann M., Sauer M.. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat. Protoc. 2011; 6:991–1009. [DOI] [PubMed] [Google Scholar]
- 18. Itzkovitz S., Lyubimova A., Blat I.C., Maynard M., van Es J., Lees J., Jacks T., Clevers H., van Oudenaarden A.. Single-molecule transcript counting of stem-cell markers in the mouse intestine. Nat. Cell Biol. 2011; 14:106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bahar Halpern K., Shenhav R., Matcovitch-Natan O., Toth B., Lemze D., Golan M., Massasa E.E., Baydatch S., Landen S., Moor A.E. et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature. 2017; 542:352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dorrell C., Schug J., Canaday P.S., Russ H.A., Tarlow B.D., Grompe M.T., Horton T., Hebrok M., Streeter P.R., Kaestner K.H. et al. Human islets contain four distinct subtypes of beta cells. Nat. Commun. 2016; 7:11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Y.J., Golson M.L., Schug J., Traum D., Liu C., Vivek K., Dorrell C., Naji A., Powers A.C., Chang K.M. et al. Single-cell mass cytometry analysis of the human endocrine pancreas. Cell Metab. 2016; 24:616–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bader E., Migliorini A., Gegg M., Moruzzi N., Gerdes J., Roscioni S.S., Bakhti M., Brandl E., Irmler M., Beckers J. et al. Identification of proliferative and mature beta-cells in the islets of Langerhans. Nature. 2016; 535:430–434. [DOI] [PubMed] [Google Scholar]
- 23. Gutierrez G.D., Gromada J., Sussel L.. Heterogeneity of the pancreatic beta cell. Front. Genet. 2017; 8:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thompson R.E., Larson D.R., Webb W.W.. Precise nanometer localization analysis for individual fluorescent probes. Biophys J. 2002; 82:2775–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ester M., Kriegel H.-P., Sander J., Xu X.. A density-based algorithm for discovering clusters in large spatial databases with noise. Proc. of the Second International Conference on Knowledge Discovery and Data Mining. 1996; 96:AAAI Press; 226–231. [Google Scholar]
- 26. Jungmann R., Avendano M.S., Dai M., Woehrstein J.B., Agasti S.S., Feiger Z., Rodal A., Yin P.. Quantitative super-resolution imaging with qPAINT. Nat. Methods. 2016; 13:439–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jungmann R., Steinhauer C., Scheible M., Kuzyk A., Tinnefeld P., Simmel F.C.. Single-molecule kinetics and super-resolution microscopy by fluorescence imaging of transient binding on DNA origami. Nano Lett. 2010; 10:4756–4761. [DOI] [PubMed] [Google Scholar]
- 28. Jungmann R., Avendano M.S., Woehrstein J.B., Dai M., Shih W.M., Yin P.. Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat. Methods. 2014; 11:313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dai M., Jungmann R., Yin P.. Optical imaging of individual biomolecules in densely packed clusters. Nat. Nanotechnol. 2016; 11:798–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Raj A., Tyagi S.. Detection of individual endogenous RNA transcripts in situ using multiple singly labeled probes. Methods Enzymol. 2010; 472:365–386. [DOI] [PubMed] [Google Scholar]
- 31. Bengtsson M., Stahlberg A., Rorsman P., Kubista M.. Gene expression profiling in single cells from the pancreatic islets of Langerhans reveals lognormal distribution of mRNA levels. Genome Res. 2005; 15:1388–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Raj A., Peskin C.S., Tranchina D., Vargas D.Y., Tyagi S.. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006; 4:e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Raj A., van Oudenaarden A.. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008; 135:216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kennell D., Riezman H.. Transcription and translation initiation frequencies of the Escherichia coli lac operon. J. Mol. Biol. 1977; 114:1–21. [DOI] [PubMed] [Google Scholar]
- 35. Cai L., Friedman N., Xie X.S.. Stochastic protein expression in individual cells at the single molecule level. Nature. 2006; 440:358–362. [DOI] [PubMed] [Google Scholar]
- 36. Shah S., Lubeck E., Zhou W., Cai L.. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron. 2016; 92:342–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moffitt J.R., Hao J., Wang G., Chen K.H., Babcock H.P., Zhuang X.. High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:11046–11051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Doyle M.J., Sussel L.. Nkx2.2 regulates beta-cell function in the mature islet. Diabetes. 2007; 56:1999–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gutierrez G.D., Bender A.S., Cirulli V., Mastracci T.L., Kelly S.M., Tsirigos A., Kaestner K.H., Sussel L.. Pancreatic beta cell identity requires continual repression of non-beta cell programs. J. Clin. Invest. 2017; 127:244–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Churchill A.J., Gutierrez G.D., Singer R.A., Lorberbaum D.S., Fischer K.A., Sussel L.. Genetic evidence that Nkx2.2 acts primarily downstream of Neurog3 in pancreatic endocrine lineage development. Elife. 2017; 6:e20010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Abrahamsson S., Chen J., Hajj B., Stallinga S., Katsov A.Y., Wisniewski J., Mizuguchi G., Soule P., Mueller F., Dugast Darzacq C. et al. Fast multicolor 3D imaging using aberration-corrected multifocus microscopy. Nat. Methods. 2013; 10:60–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Abrahamsson S., Ilic R., Wisniewski J., Mehl B., Yu L., Chen L., Davanco M., Oudjedi L., Fiche J.B., Hajj B. et al. Multifocus microscopy with precise color multi-phase diffractive optics applied in functional neuronal imaging. Biomed. Opt. Express. 2016; 7:855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tsanov N., Samacoits A., Chouaib R., Traboulsi A.M., Gostan T., Weber C., Zimmer C., Zibara K., Walter T., Peter M. et al. smiFISH and FISH-quant - a flexible single RNA detection approach with super-resolution capability. Nucleic Acids Res. 2016; 44:e165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cui Y., Wang X., Ren W., Liu J., Irudayaraj J.. Optical clearing delivers ultrasensitive hyperspectral dark-field imaging for single-cell evaluation. ACS Nano. 2016; 10:3132–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Moffitt J.R., Hao J., Bambah-Mukku D., Lu T., Dulac C., Zhuang X.. High-performance multiplexed fluorescence in situ hybridization in culture and tissue with matrix imprinting and clearing. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:14456–14461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sylwestrak E.L., Rajasethupathy P., Wright M.A., Jaffe A., Deisseroth K.. Multiplexed intact-tissue transcriptional analysis at cellular resolution. Cell. 2016; 164:792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen F., Wassie A.T., Cote A.J., Sinha A., Alon S., Asano S., Daugharthy E.R., Chang J.B., Marblestone A., Church G.M. et al. Nanoscale imaging of RNA with expansion microscopy. Nat. Methods. 2016; 13:679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Long X., Colonell J., Wong A.M., Singer R.H., Lionnet T.. Quantitative mRNA imaging throughout the entire Drosophila brain. Nat. Methods. 2017; 14:703–706. [DOI] [PubMed] [Google Scholar]
- 49. Itzkovitz S., van Oudenaarden A.. Validating transcripts with probes and imaging technology. Nat. Methods. 2011; 8:S12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lyubimova A., Itzkovitz S., Junker J.P., Fan Z.P., Wu X., van Oudenaarden A.. Single-molecule mRNA detection and counting in mammalian tissue. Nat. Protoc. 2013; 8:1743–1758. [DOI] [PubMed] [Google Scholar]
- 51. Roscioni S.S., Migliorini A., Gegg M., Lickert H.. Impact of islet architecture on beta-cell heterogeneity, plasticity and function. Nat. Rev. Endocrinol. 2016; 12:695–709. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.