

Abstract
Background
Ischemic preconditioning (IPC) and ischemic postconditioning (IPoC) are currently among the most efficient strategies protecting the heart against ischemia/reperfusion injury. However, the effect of IPC and IPoC on functional recovery following ischemia/reperfusion is less clear, particularly with regard to the specific receptor‐mediated signaling of the postischemic heart. The current article examines the effect of IPC or IPoC on the regulation and coupling of β‐adrenergic receptors and their effects on postischemic left ventricular function.
Methods and Results
The β‐adrenergic signal transduction was analyzed in 3‐month‐old Wistar rats for each of the intervention strategies (Sham, ischemia/reperfusion, IPC, IPoC) immediately and 7 days after myocardial infarction. Directly after the infarction a cardioprotective potential was demonstrated for both IPC and IPoC: the infarct size was reduced, apoptosis and production of reactive oxygen species were lowered, and the myocardial tissue was preserved. Seven days after myocardial ischemia, only IPC hearts showed significant functional improvement. Along with a deterioration in fractional shortening, IPoC hearts no longer responded adequately to β‐adrenergic stimulation. The stabilization of β‐adrenergic receptor kinase‐2 via increased phosphorylation of Mdm2 (an E3‐ubiquitin ligase) was responsible for desensitization of β‐adrenergic receptors and identified as a characteristic specific to IPoC hearts.
Conclusions
Immediately after myocardial infarction, rapid and transient activation of β‐adrenergic receptor kinase‐2 may be an appropriate means to protect the injured heart from excessive stress. In the long term, however, induction and stabilization of β‐adrenergic receptor kinase‐2, with the resultant loss of positive inotropic function, leads to the functional picture of heart failure.
Keywords: cardiac remodeling, ischemic postconditioning, ischemic preconditioning, myocardial infarction, β‐adrenergic receptor, β‐adrenergic receptor kinase‐2
Subject Categories: Myocardial Infarction, Remodeling, Heart Failure, Contractile function
Clinical Perspective
What Is New?
Ischemic preconditioning and ischemic postconditioning are very efficient clinically applied strategies that protect the heart against ischemia/reperfusion injury.
What remains unknown, however, is their effect on the function of cardiac receptors, which are significantly involved in the functional and structural remodeling of the postischemic myocardium.
Here, we have been able to demonstrate that, unlike ischemic preconditioning, the application of ischemic postconditioning by inducing β‐adrenergic receptor kinase‐2 leads to desensitization of β‐adrenergic receptors, which in turn induces cardiac dysfunction with restricted inotropic functional reserve.
What Are the Clinical Implications?
The findings of this study not only provide a possible explanation for the previously disappointing long‐term clinical trials of ischemic postconditioning but also identify the postconditioned heart as another indication for the therapeutic use of prospective β‐adrenergic receptor kinase‐2 inhibitors.
Introduction
The long‐term prognosis of an acute myocardial infarction is determined both by the extent of the resultant irreversible tissue damage and by the reconstruction processes that are immediately triggered, which are referred to as cardiac remodeling. Short series of repetitive ischemia/reperfusion cycles (IR), which are described as ischemic preconditioning (IPC) and ischemic postconditioning (IPoC), are currently among the most efficient strategies for myocardial protection.1, 2 Both interventions share some signaling pathways, although they are applied at opposing time points of the ischemic period.3 To what extent this protective intervention also leads to a better prognosis over the long term depends fundamentally on its effect on the remodeling of the postischemic myocardium.
In the classical in vivo IPC and IPoC models, a reduction in the size of the infarct is chosen as the relevant end point in most cases even though this was often not accompanied by a corresponding functional recovery.4, 5 However, no information can be gleaned about long‐term functional improvements or positive remodeling using these short‐term models.
Therapeutically, application at the time of reperfusion is an appropriate intervention option, meaning that in routine clinical practice IPoC is the preferred protective strategy.6, 7
However, the results from longitudinal human studies have not provided a uniform picture of the efficacy and safety of IPoC.8 In 2013 a single‐center study by Freixa et al showed rather a significant deterioration in the myocardial salvage and the myocardial salvage index along with reduced pump function 6 months after myocardial ischemia.9 These results correspond to data published by Hahn et al from a multicenter study of 700 patients (POST [Effects of Postconditioning On Myocardial Reperfusion] trial, https://clinicaltrials.gov/show/NCT00942500) that was conducted between 2009 and 2012, also showing no cardiprotective effect.10 Moreover, recently published results of the DANAMI‐3–iPOST (DANish Study of Optimal Acute Treatment of Patients With ST‐elevation Myocardial Infarction, https://clinicaltrials.gov/show/NCT01435408) trial have also demonstrated that IPoC in addition to primary percutaneous coronary intervention failed to significantly reduce death from any cause or hospitalization during a median follow‐up of 38 months.11
It has already been shown for both IPC and IPoC that both prior pharmacological interventions and preexisting diseases (diabetes mellitus, chronic heart failure) can contribute to a reduction in their cardioprotective potential. 12, 13, 14
What is not currently known, however, is the effect of both intervention strategies on cardiac receptors, which, as important components of the functional and structural remodeling, are significantly involved in the functional preservation of the postischemic myocardium and thus play a critical role in the efficiency of IPC or IPoC.15
The β‐adrenergic receptor (β‐AR) is primarily involved in the maintenance and, where necessary, adaptation of cardiac function. The density and the signaling of the cardiac β‐ARs change over the course of many cardiac diseases. In the postischemic failing myocardium these changes are often associated with significant downregulation of the β‐ARs. The activation and intracellular coupling of the β1‐AR, on the other hand, can be inadequately elevated in the early postischemic phase.16, 17, 18
The primary aim of this research was to study the effect of IPC or IPoC on the remodeling of the β‐adrenergic system and their effects on the function of the postischemic myocardium.
A new signaling mechanism for the IPoC has been identified that considerably reduces the coupling of β‐ARs independently of a reduction in the size of the infarction. The resulting restriction in the positive inotropic functional reserve induced a persistent cardiac dysfunction in the postconditioned myocardium.
Methods
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85‐23, revised 1996). The data and analytic methods that support the findings of this study are available from the corresponding author on request.
Langendorff Perfusion
Using the Langendorff technique, Krebs‐perfused hearts from 3‐month‐old female Wistar rats were subject to 45 minutes of global ischemia and then reperfused for 120 minutes (IR). The IR protocol was either supplemented by IPC (3×3 minutes of no flow—5 minutes of perfusion) or IPoC (3×30 seconds of perfusion—30 seconds of no flow), and the corresponding functional recovery and the extent of the resultant tissue damage were compared to the values for a normoxic control group (Figure 1A). For this purpose, rats were anesthetized by isoflurane and killed by cervical dislocation. Their hearts were rapidly excised, and the aortas were cannulated for retrograde perfusion with a 16‐gauge needle connected to a Langendorff perfusion system. A polyvinyl chloride balloon was inserted into the left ventricle (LV) through the mitral valve and held in place by a suture tied around the left atrium. The other end of the tubing was connected to a pressure transducer for continuous measurement of LV pressure. A second transducer connected to the perfusion line just before the heart was used to measure coronary perfusion pressure. Hearts were perfused with an oxygenated saline medium containing (in mmol/L) 140 NaCl, 24 NaHCO3, 2.7 KCl, 0.4 NaH2PO4, 1 MgCl2, 1.8 CaCl2 and 5 glucose, gassed with 95% O2 and 5% CO2, pH 7.4. After attachment to the Langendorff system, hearts were allowed to stabilize for at least 20 minutes. The intraventricular balloon was inflated to give a diastolic pressure of 10 mm Hg. The flow was adjusted to give a perfusion pressure of 50 mm Hg. To avoid differences in oxygen supply due to changes in the coronary resistance, hearts were perfused at a constant flow. The perfusion system was constantly kept at a temperature of 37°C throughout the whole experiment.15 As a pharmacological inhibitor of protein kinase C (PKC), chelerythrine chloride (1 μmol/L, Merck Millipore, Darmstadt, Germany; product #220285) was applied from the onset of reperfusion.
Figure 1.
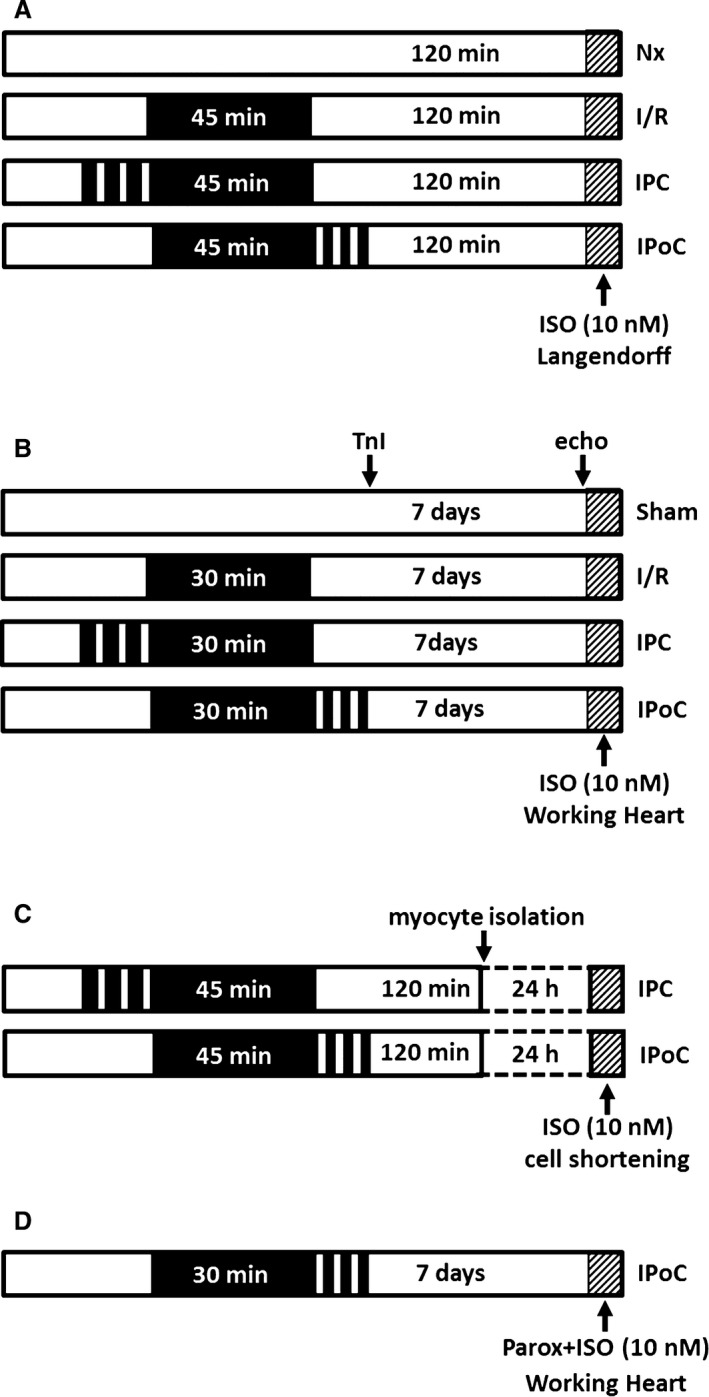
Study designs and treatment protocols. A, Stimulation of β‐ARs at the end of a 2‐hour reperfusion in the Langendorff model. B, Stimulation of β‐ARs 7 days after infarction in the working heart model. C, Cardiomyocytes were isolated from IPC or IPoC hearts and cultured for 24 hours. Cell shortening was determined before and after stimulation with ISO using a cell‐edge detection system. D, Perfusion of IPoC hearts 7 days after an infarction. After 10 minutes’ infusion with paroxetine, the CO was measured continuously for 5 minutes with additional stimulation using ISO. β‐AR indicates β‐adrenergic receptor; CO, cardiac output; IPC, ischemic preconditioning; IPoC, ischemic postconditioning; ISO, isoproterenol.
Measurement of Lactate Dehydrogenase Activity
The lactate dehydrogenase (LDH) release of Langendorff hearts was measured in the perfusate, which was collected for 60 seconds after 5 minutes of reperfusion. LDH activity was determined using a colorimetric LDH assay kit (Abcam, Cambridge, UK). In brief, LDH reduces NAD to NADH, which then interacts with a specific probe to generate color at 450 nm. The intensity of the color is directly proportional to the LDH activity (U/L) in each sample. Fifty‐microliter samples were incubated with LDH substrate mix and measured immediately at OD 450 nm on a microplate reader in a kinetic mode for 45 minutes at 37°C. LDH activity in each sample was calculated according to a standard curve.
Measurement of Superoxide
Fifteen minutes after the start of reperfusion, left ventricular tissue was prepared for cryosectioning by freezing in Tissue‐Tek OCT Compound (Sakura Finetek Germany GmbH, Staufen, Germany). To perform dihydroethidium staining, cryoslices of the LV were incubated with the fluorescent probe dihydroethidium (dissolved in 1× PBS) for 10 minutes at 37°C in a light‐protected humidity chamber, then fixed with Dako Fluorescent Mounting Medium (Dako North America Inc, Carpinteria, CA). Sections were imaged by fluorescence microscopy (LSM 510 META; Carl Zeiss, Jena, Germany) using an excitation wavelength of 488 nm; emission was recorded at 540 nm.19
Analysis was performed by digital image analysis using Leica Confocal Software Lite Version (LCS Lite; Leica, Wetzlar, Germany). The mean fluorescence intensity of n=6 ventricles was used to quantify the extent of superoxide.
Immunohistochemistry Analysis of Active Caspase‐3 and Connexin 43
Subsequent to the particular reperfusion time, Langendorff‐perfused hearts were fixed after snap‐freezing and sectioned using a cryostat (section thickness 10 μm). Briefly, slides were rehydrated in PBS for 15 minutes and then blocked with bovine serum albumin (5%) for 1 hour. Primary antibodies against active caspase‐3 or connexin 43 were diluted 1:100 in PBS/Tween containing 2.5% bovine serum albumin and incubated for 1 hour at room temperature. Slides were washed 20 minutes with PBS/Tween and incubated with an Alexa Fluor‐488 coupled secondary antibody (Molecular Probes, Eugene, OR) for 1 hour. The visualization and quantification of the florescence were carried out using a florescence microscope (Till Photonics, Gräfelfing, Germany).
Rat Model of Ischemia and Reperfusion In Vivo
In brief, female Wistar‐Hannover rats weighing 210 to 265 g were anesthetized by intraperitoneal injection of 60 mg/kg sodium pentobarbital (Euthasol, Produlab Pharma b.v., Raamsdonksveer, the Netherlands). Animals were intubated and mechanically ventilated (Model 683, Harvard Apparatus, Holliston, MA) with room air in a volume of 6.2 mL/kg and a frequency of 72±5 breaths/min according to body weight. Rats were placed in a supine position on a heating pad to maintain body core temperature in physiological range (37.0±1.0°C). Body surface ECG was monitored throughout the experiments (Haemosys, Experimetria Inc, Budapest, Hungary). Left anterior descending coronary artery occlusion was induced by a left thoracotomy. A 5‐0 Prolene suture (Ethicon, Johnson & Johnson Kft Hungary, Budapest, Hungary) was placed around the left anterior descending artery and a small plastic knob, which was threaded through the ligature and placed in contact with the heart, was used for making occlusion for 30 minutes. Animals for the IPC group were subjected to 3 cycles of 3 minutes of ischemia followed by 5 minutes of reperfusion before a 30‐minute test ischemia; IPoC was performed after test ischemia by using 3 cycles of 30 seconds of reperfusion/30 seconds of ischemia. The presence of ischemia was confirmed by the appearance of ST segment elevation and ventricular arrhythmias. After 30 minutes of ischemia or the postconditioning protocol, the occlusion was released, and the suture was removed from the heart. Restoration of blood flow was confirmed by the appearance of arrhythmias observed in the first minutes after the onset of reperfusion. Then the chest was closed in layers with a 4‐0 Monocryl suture (Ethicon, Johnson & Johnson Kft Hungary, Budapest, Hungary), and 0.3 mg/kg nalbuphin (Nalbuphin Orpha, Torrex Chiesi Pharma GmbH, Vienna, Austria) was given subcutaneously to alleviate postoperative pain. Rats were allowed to recover for 7 days after coronary occlusion. Sham‐operated animals were treated with the same procedure except occlusion of the coronary artery (Figure 1B). The surgical procedure was performed according to previous publications.20, 21
Transthoracic Echocardiography
Cardiac morphology and function were also assessed by transthoracic echocardiography 7 days after the induction of ischemia/reperfusion injury or sham operation. Echocardiography was performed as described previously.22 Briefly, rats were anesthetized with sodium pentobarbital (Euthasol, 40 mg/kg body weight, intraperitoneally), the chest was shaved, and the animal was placed in supine position onto a heating pad. Two‐dimensional and M‐mode echocardiographic examinations were performed in accordance with the criteria of the American Society of Echocardiography with a Vivid 7 Dimension ultrasound system (General Electric Medical Systems, Waukesha, WI) using a phased‐array 5.5‐ to 12‐MHz transducer (10S probe). Data of 3 consecutive heart cycles were analyzed (EchoPac Dimension Software, General Electric Medical Systems) and then the mean values of the 3 measurements were calculated and used for statistical evaluation. Systolic and diastolic wall thicknesses were obtained from parasternal short‐axis view at the level of the papillary muscles. The left ventricular diameters were measured by means of M‐mode echocardiography from short‐axis views between the endocardial borders. Functional parameters including fractional shortening and ejection fraction were calculated on short‐axis view images.
Measurement of Functional Parameters of Hearts: Working Heart Perfusion
After echocardiographic examinations, rat hearts were isolated and prepared for working heart perfusion according to Neely et al.23, 24, 25 Hearts were perfused at 37°C with freshly prepared Krebs‐Henseleit bicarbonate buffer containing (in mmol/L) NaCl 118, KCl 4.3, CaCl2 2.4, NaHCO3 25, KH2PO4 1.2, MgSO4 1.2 and glucose 11.1, gassed with 95% O2 and 5% CO2. Preload (17 cm H2O) and afterload (100 cm H2O) were kept constant throughout the experiments. Coronary flow was measured by collecting effluent from the right atrium by a measuring cylinder for 1 minute, aortic flow was measured by a calibrated rotameter (KDG Flowmeters, Sussex, England), and cardiac output (CO) was calculated as the sum of aortic flow and coronary flow. Ventricular pressure was measured by means of a pressure transducer (B. Braun, Melsungen, Germany) connected to a small polyethylene catheter inserted into the LV through the left atrial cannula. Left ventricular developed pressure (LVDP) was calculated as peak systolic pressure minus LV end‐diastolic pressure, positive and negative first derivatives of left ventricular pressure (±dp/dt), and LV end‐diastolic pressure were also defined. Heart rate was derived from the LV pressure curve.
Measurement of Cardiac Troponin I Release in Plasma
Plasma samples were collected into heparinized tubes (Sarstedt, Nümbrecht, Germany) from the femoral vein at the 60th minute of reperfusion, and plasma was separated to determine cardiac troponin I (TnI) release after acute myocardial infarction. Plasma TnI concentration was determined by a conventional ELISA kit (Life Diagnostics, Inc, West Chester, PA) according to the recommendations of the manufacturer. Briefly, plasma samples were diluted 4 to 40 times according to the treatment protocol (ie, sham or ischemic) and to previous preanalyses to get absorbances in the range of standard absorbances. Diluted samples were allowed to react simultaneously with 2 antibodies against rat TnI (1 is immobilized on the microtiter wells, and the other is conjugated to horseradish peroxidase [HRP] in soluble phase), resulting in TnI being sandwiched between the solid phase and HRP‐conjugated antibodies. After 1 hour of incubation at room temperature on a plate shaker, the wells were washed with wash solution to remove unbound HRP‐conjugated antibodies. A solution of tetramethylbenzidine, a HRP substrate, was then added and incubated for 20 minutes, resulting in the development of a blue color. The color development was stopped by addition of 1N HCl, which changed the color to yellow. The concentration of TnI was proportional to the absorbance at 450 nm.
Determination of Infarct Size
After 120 minutes of reperfusion, hearts were isolated for infarct size measurements. Hearts were perfused in a Langendorff perfusion system with 37°C Krebs‐Henseleit buffer to remove blood from the coronary vessels. After 5 minutes of perfusion, the risk area was reoccluded, and hearts were perfused with 4 mL of 0.1% Evans blue dye through the ascending aorta. Following Evans staining, hearts were cut into 5 transverse slices and incubated in 1% triphenyltetrazolium chloride for 10 minutes at 37°C followed by formalin fixation for 10 minutes. Digital images were taken from both surfaces of heart slices by a Nikon DSLR camera (Nikon Corporation, Tokyo, Japan). Planimetric evaluation was carried out to determine infarct size using InfarctSize™ software version 2.5, (Pharmahungary, Szeged, Hungary26).
Western Blot
Total protein was extracted from the LV using Cell Lysis Buffer (10×) (Cell Signaling, Danvers, MA) according to the manufacturer's protocol. Briefly, the homogenate was centrifuged at 14 000g at 4°C for 10 minutes, and supernatant was treated with Laemmli buffer. Protein samples were loaded on NuPAGE Bis‐Tris Precast gels (8%, 10%; Life Technology, Darmstadt, Germany) and subsequently transferred onto nitrocellulose membranes. Blots were incubated with 1 of the following antibodies: anti‐β1‐AR (V‐19) (Santa Cruz Biotechnology, Inc, Heidelberg, Germany; product sc‐568), anti‐β2‐AR (H‐73) (Santa Cruz Biotechnology, Inc, Heidelberg, Germany; product sc‐9042), anti‐β‐adrenergic receptor kinase‐2 (anti‐GRK2; Sigma‐Aldrich, Taufkirchen, Germany; product G0296), anti‐GRK2 phospho S29 (Abcam plc, Cambridge, UK; product ab58520), anti‐β‐arrestin1/2 (New England Biolabs GmbH, Frankfurt am Main, Germany; product #4674), anti‐MDM2/HDM2 (R&D Systems, Wiesbaden, Germany; AF1244), anti‐MDM2 phospho Ser166 (New England Biolabs GmbH, Frankfurt am Main, Germany; product #3521), anti‐PI3 kinase p110α (Becton Dickinson GmbH, Heidelberg, Germany; product #611398), and anti‐GAPDH (Merck Millipore, Darmstadt, Germany; product CB1001). Secondary antibodies (HRP‐conjugated) directed against rabbit IgG or mouse IgG were purchased from Affinity Biologicals (Ancaster, ON, Canada; product SAR‐APHRP, GAM‐APHRP). In order to detect β1‐AR dimers, samples were treated with nonreducing Laemmli buffer and loaded on a 10% NuPAGE Bis‐Tris Precast gel.
Densitometry of immunoblot bands was carried out using Quantity One software from Bio‐Rad Laboratories (Hercules, CA). The intensities of the target proteins were normalized to that of GAPDH of the same sample.
RNA Isolation and Real‐Time Reverse‐Transcriptase Polymerase Chain Reaction
Total RNA was isolated from the LV using peqGold TriFast (peqlab, Biotechnologie GmbH, Erlangen, Germany) according to the manufacturer's protocol. To remove genomic DNA contamination, RNA samples were treated with 1 U DNase/μg RNA (Invitrogen, Karlsruhe, Germany) for 15 minutes at 37°C. One microgram of total RNA was used in a 10‐μL reaction to synthesize cDNA using Superscript RNaseH Reverse Transcriptase (200 U/μg RNA, Invitrogen, Karlsruhe, Germany) and oligo‐dTs as primers. Reverse transcriptase reactions were performed for 50 minutes at 37°C. Real‐time quantitative polymerase chain reaction was performed using MyiQ® detection system (Bio‐Rad, Munich, Germany) in combination with the iTaq Universal SYBR Green Real‐Time PCR Supermix (Bio‐Rad, Munich, Germany). The thermal cycling program consisted of initial denaturation in 1 cycle of 3 minutes at 95°C, followed by 45 cycles of 30 seconds at 95°C, 30 seconds at the individual annealing temperature for each primer, and 30 seconds at 72°C. Primers used for determination had the following sequences: HPRT forward CCA GCG TCG TGA TTA GTG AT, HPRT reverse CAA GTC TTT CAG TCC TGT CC, β1‐AR forward GGC GCT CAT CGT GCT GCT CA, β1‐AR reverse AGG CAC CAC CAG CAG TCC CA, β2‐AR forward GCT TCT GTG CCT TCG CCG GT, β2‐AR reverse AGC CTT CCA TGC CAG GG GCT, β3‐AR forward GGT TGG GCT ATG CCA ACT CT, β3‐AR reverse CCT GTT GAG CGG TGA GTT CT, GRK2 forward GCT CTT CAA GTT GTT GCG GG, GRK2 reverse AAA CCT TCC AGC AGG GAT CG. Quantification was performed as described before.27
Isolation and Culture of Cardiomyocytes From the LV
Heart muscle cells from the LV were isolated from 3‐month‐old Wistar rats. Briefly, hearts were excised and transferred rapidly to ice‐cold saline and mounted on the cannula of a Langendorff perfusion system. Heart perfusion and subsequent steps were all performed at 37°C. First, hearts were perfused in a noncirculating manner for 5 minutes at 10 mL/min. Thereafter, perfusion was continued with recirculation using 50 mL perfusate supplemented with 0.06% (w/v) crude collagenase and 25 μmol/L CaCl2 at 5 mL/min for 25 minutes. Then, the LV was dissected from the RV, minced, and incubated for 5 minutes in medium with 1% (w/v) bovine serum albumin under 5% CO2 and 95% O2. The resulting cell suspension was filtered through a 200‐μm nylon mesh. Filtered cells were washed twice by centrifugation and resuspended in perfusate with a stepwise increase in CaCl2 to 0.2 and 0.5 mmol/L. After further centrifugation, cells were resuspended in serum‐free culture medium and plated in 35‐mm culture dishes (Falcon, type 3001).28
Isolated cardiomyocytes were then incubated with siRNA against GRK2 (Qiagen GmbH, Hilden, Germany, product Rn_Adrbk1_1), siRNA against β‐arrestin1 (Qiagen GmbH, Hilden, Germany, product Rn_Arrb1_2), or siRNA against β‐arrestin2 (Qiagen GmbH, Hilden, Germany, product Rn_Arrb2_1) for 24 hours. Paroxetine (LKT Laboratories, St. Paul, MN) was used as a pharmacological inhibitor of GRK2.
Determination of Cell Contraction
Cell shortening was measured as described before in greater detail.28 Briefly, isolated cardiomyocytes were allowed to contract at room temperature and analyzed using a cell‐edge detection system. Cells were stimulated via 2 AgCl electrodes with biphasic electrical stimuli composed of 2 equal but opposite rectangular 50‐V stimuli of 0.5 milliseconds duration. Each cell was stimulated for 1 minute at a frequency of 2.0 Hz. Every 15 seconds contractions were recorded. The mean of these 4 measurements was used to define the cell shortening of a given cell. Cell lengths were measured via a line camera (data recording at 500 Hz). Data are expressed as cell shortening normalized to diastolic cell length (dL/L [%]).
Statistical Analysis
Data are presented as mean±SD as indicated in the legends of the figures and tables. All data sets were initially analyzed by a Shapiro‐Wilk test to analyze whether the data are normally distributed. If this was the case, data were analyzed either by paired or unpaired t‐test depending on the study design. If the 2 groups had different variances (as analyzed by the Levene test), we calculated the P‐value by a Welch test. In the case that the data were not normally distributed, we performed a Mann‐Whitney test for unpaired samples or a Friedman test for paired samples. In those figures in which more than 2 groups were compared, we performed a 1‐way ANOVA with a subsequent Student‐Newman‐Keuls test for post hoc analysis as long as the data were normally distributed. If data were not normally distributed, we used a Kruskal‐Wallis test to decide whether differences between groups occurred. A P‐value of ≤0.05 is always indicated by an asterisk to indicate that this was used as the threshold to reject the null hypothesis. Statistical analysis was performed by SPSS (IBM, Armonk, NY) version 22.0.
Results
Evidence of Cardiac Protection for IPC and IPoC Algorithms Used on the Ex Vivo Perfused Heart
The efficiency of IPC and IPoC was analyzed in Langendorff perfused hearts using the experimental protocol in Figure 1A. LDH release, the formation of superoxide, and the proportion of activated caspase‐3 were significantly elevated in the hearts in the IR group compared with those in the normoxic group (Figure 2A through 2C). All parameters were improved by both IPC and IPoC. The cardioprotective potential of both protocols was also reflected in the structural preservation (connexin 43), whereas the short‐term functional recovery within the 120‐minute reperfusion period did not improve significantly (Figure 2D and 2E).
Figure 2.
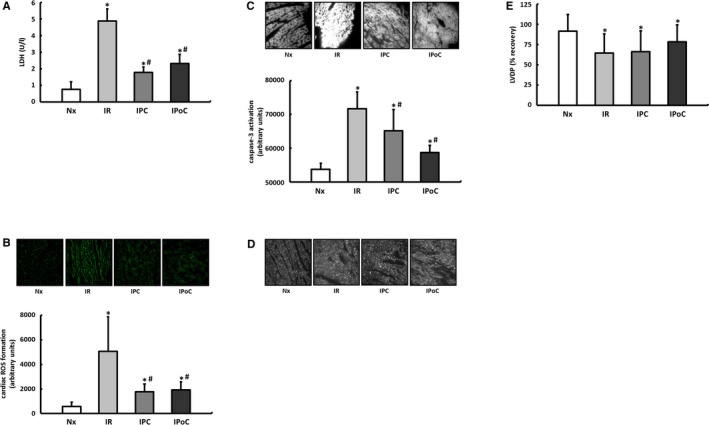
Proof of cardiac protection for IPC and IPoC algorithms in a Langendorff model of myocardial ischemia and reperfusion. A, Five minutes after the onset of reperfusion, the LDH level was determined in the perfusate as a surrogate parameter for tissue damage. B, After 15 minutes of reperfusion reactive oxygen species (ROS) were determined in the left ventricular myocardium using DHE staining. C, Activation of caspase‐3 was determined by immunohistochemistry after 120 minutes of reperfusion. D, The structural preservation of the myocardium was qualitatively assessed using connexin 43 staining. E, Functional recovery was expressed as a percentage of the preischemic LVDP. Data are means±SD of n=6 to 8 hearts. *P≤0.05 vs Nx, # P≤0.05 vs IR. DHE indicates dihydroethidium; IPC, ischemic preconditioning; IPoC, ischemic postconditioning; IR, ischemia/reperfusion; LDH, lactate dehydrogenase; LVDP, left ventricular developed pressure; Nx, normoxia.
Stimulation of β‐ARs 2 Hours After Infarction in the Langendorff Model
In another series of experiments the previously described perfusion protocols (normoxia, IR, IPC, IPoC) were supplemented by a final stimulation with isoproterenol (ISO, 10 nmol/L) for 5 minutes (Figure 1A). Both the ISO‐induced increase in the heart rate and the reduction in the coronary resistance were equally pronounced in the 4 treatment groups. A significant increase in the LVDP was observed only in the hearts in the normoxia, IR, and IPC groups but, unexpectedly, not in IPoC hearts (Table 1).
Table 1.
Influence of IR, IPC, and IPoC on β‐AR–Mediated Effects 2 Hours After Myocardial Infarction
| Heart Rate (beats/min) | ||
|---|---|---|
| Baseline | ISO | |
| Nx | 244±44 | 318±26a |
| IR | 234±42 | 305±48a |
| IPC | 246±24 | 314±52a |
| IPoC | 230±60 | 303±36a |
| Perfusion Pressure (mm Hg) | ||
|---|---|---|
| Baseline | ISO | |
| Nx | 57±8 | 46±3a |
| IR | 64±8 | 55±7a |
| IPC | 62±13 | 50±7a |
| IPoC | 72±18 | 62±14a |
The heart rate increased and the perfusion pressure fell uniformly under ISO stimulation in all 4 treatment groups. A significant increase in the LVDP was observed only in hearts of the Nx, IR, and IPC groups. Data are means±SD of n=6 to 8 hearts.
IPC indicates ischemic preconditioning; IPoC, ischemic postconditioning; IR, ischemia/reperfusion; ISO, isoproterenol; LVDP, left ventricular developed pressure; Nx, normoxia.
P≤0.05 vs baseline.
There were no differences observed in the LV expression of the β1‐AR in the Western blot among the comparator groups. However, compared with normoxic control hearts, the proportion of β1‐AR dimers was significantly reduced in the myocardium of reperfused hearts regardless of the particular protocol used (Figure 3A through 3C).
Figure 3.
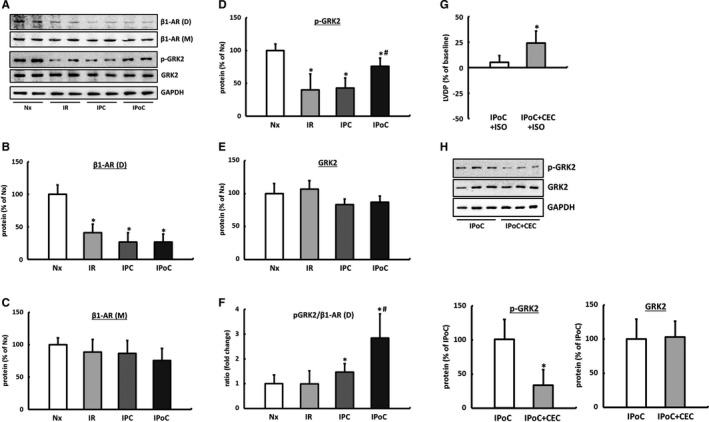
Influence of IR, IPC, and IPoC on β‐adrenergic signaling 2 hours after starting reperfusion. A, Representative immunoblots and quantification by densitometric analysis of (B) β1‐AR dimers (D, MW 95 kDa) and (C) β1‐AR monomers (M, MW 50 kDa). Quantification of the (D) phosphorylated (p‐GRK2) and (E) total GRK2 protein. F, The p‐GRK2 to β1‐AR (D) ratio was calculated for each animal, and group means were expressed as fold change of Nx. Data are means±SD of n=6 hearts. *P≤0.05 vs Nx, # P≤0.05 vs IPC. The significance of PKC for GRK2 phosphorylation. G, ISO‐induced inotropic response of IPoC hearts perfused with the PKC inhibitor CEC. H, The effect of PKC inhibition on GRK2 phosphorylation in IPoC hearts. Immunoblot bands were normalized to GAPDH and expressed as a percentage of Nx. Data are means±SD of n=6 hearts. *P≤0.05 vs IPoC. CEC indicates chelerythrine chloride; D, dimer; GAPDH, Glyceraldehyde 3‐phosphate dehydrogenase; GRK2, G protein‐coupled receptor kinase 2; IPC, ischemic preconditioning; IPoC, ischemic postconditioning; IR, ischemia/reperfusion; ISO, isoproterenol; M, monomer; Nx, normoxic; p‐GRK2, phosphorylated GRK2; PKC, protein kinase C; β1‐AR, β1‐adrenergic receptor.
The β‐adrenergic receptor kinases (GRKs), which mediate the accumulation of β‐arrestin by phosphorylating serine and threonine residues on the intracellular domains of the receptor, are critically involved in the regulatory mechanisms for intracellular coupling of the β‐ARs and, in turn, can prevent activation of the Gs subunit or induce internalization of the receptor.
The phosphorylation, and thus the activation, of GRK2 was significantly reduced in the hearts in the IR and IPC groups. Consequently, the ratio of receptor dimers to phosphorylated GRK2 did not differ from that of the hearts in the control group. In contrast, in postconditioned hearts, the increased proportion of phosphorylated GRK2 combined with the significant reduction in the β‐AR dimers led to an imbalance, which may be the underlying cause of the missing increase in the LVDP following β‐adrenergic stimulation (Figure 3A, 3D through 3F, Table 1).
Significance of PKC for GRK2 Phosphorylation in the Postconditioned Heart
It has already been shown for PKC that it can effectively contribute to the desensitization of G‐protein‐coupled receptors via phosphorylation of GRK2.29 By inhibiting PKC using chelerythrine chloride, the IPoC‐induced phosphorylation of GRK2 is significantly reduced, and consequently, the ISO‐induced increase in the LVDP at the end of a 2‐hour reperfusion can be reestablished (Figure 3G and 3H).
Effect of IR, IPC, and IPoC on the Size of the Infarct and the Cardiac Function In Vivo
IPoC has already induced a specific remodeling of the β‐adrenergic signaling during the short‐term reperfusion ex vivo. It is certainly an important question whether IPoC also affects the coupling of β‐ARs over the long term in vivo (see experimental protocol in Figure 1B). One hour after the onset of reperfusion, the TnI plasma level was determined. Compared with the sham group (1.1±0.8 ng/mL), the plasma samples from the IR group had the highest concentrations of TnI at 104.3±33.6 ng/mL and were significantly reduced by IPC to 32.8±33.9 ng/mL or by IPoC to 41.9±41.1 ng/mL (Figure 4A). The validity of TnI as a marker to determine the size of the infarct in the IR model used here was demonstrated in a separate group of animals. Plasma TnI values measured 1 hour after infarction confirm a reduction in the size of the infarct in conditioned animals of the same magnitude as that determined using triphenyltetrazolium chloride staining in the corresponding hearts 2 hours after starting reperfusion (Figure S1).
Figure 4.
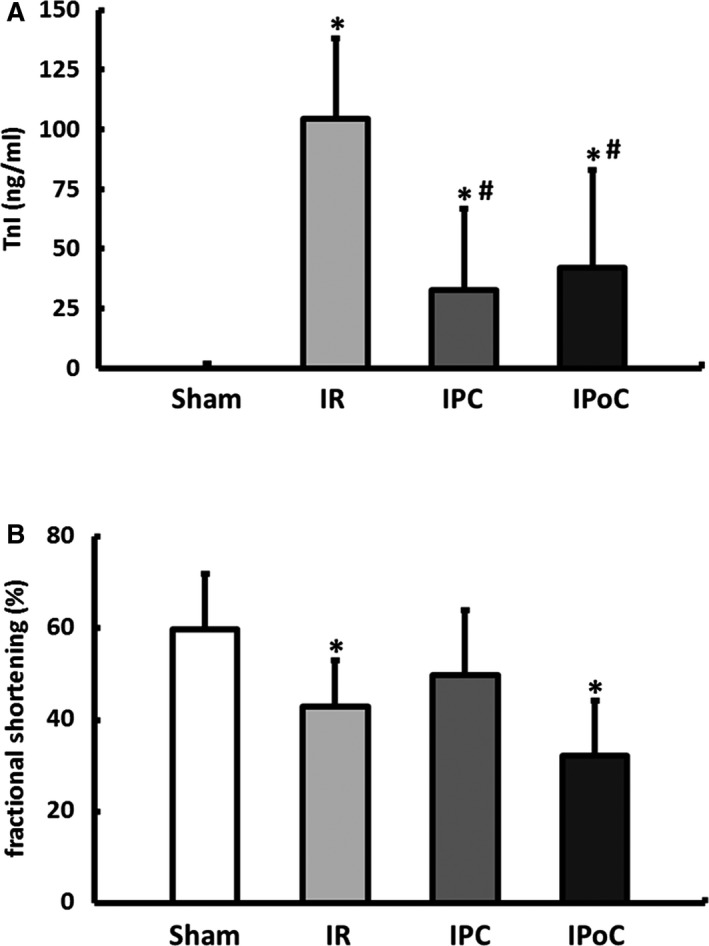
Influence of IR, IPC, and IPoC on infarct size and cardiac function in vivo. A, Plasma levels of cardiac troponin I (TnI) were determined 1 hour after the onset of reperfusion as an early index of myocardial infarct size. B, Left ventricular systolic function, represented as fractional shortening (%), was determined 7 days following the reperfusion. Data are means±SD of n=6 to 10 animals. *P≤0.05 vs Sham, # P≤0.05 vs IR. IPC indicates ischemic preconditioning; IPoC, ischemic postconditioning; IR, ischemia/reperfusion.
After 7 days the left ventricular morphology and function were assessed using echocardiography. Fractional shortening was 59.8±12% in the sham group, which was significantly reduced (42.9±10%) in the IR group. The application of IPC improved the fractional shortening to 49.8±14%, whereas the hearts in the IPoC group only achieved a value of 32.2±12%, which did not differ statistically from the IR group (Figure 4B).
There were no significant differences in the ventricular geometry determined by echocardiography (Table S1) and the wet weight analyzed postmortem (data not shown) between the groups at the end of the experimental period.
Stimulation of β‐ARs 7 Days After Infarction in the Working Heart Model
After the 7 days had elapsed, the pump function and hemodynamics of the hearts were recorded ex vivo in the working heart model, first at a basal level and again immediately after stimulation with ISO (10 nmol/L) (Figure 1B). The basal CO, respectively the LVDP and dP/dtmax, confirmed the values for the function previously determined by echocardiography and also identified the IPC as the only cardioprotective application in terms of a medium‐term preservation of function. The 4 groups did not differ in terms of their heart rates or the coronary flow (Table 2).
Table 2.
Functional Data Were Assessed Ex Vivo in a Working Heart Model
| Heart Rate (beats/min) | ||
|---|---|---|
| Baseline | ISO | |
| Sham | 272±29 | 341±34a |
| IR | 276±48 | 364±32a |
| IPC | 263±22 | 352±72a |
| IPoC | 273±25 | 347±41a |
| Cardiac Output (mL/min) | ||
|---|---|---|
| Baseline | ISO | |
| Sham | 48±7 | 66±6a |
| IR | 36±8b | 51±12a |
| IPC | 42±9 | 54±11a |
| IPoC | 33±10b | 38±15 |
| dP/dtmax (kPa/s) | ||
|---|---|---|
| Baseline | ISO | |
| Sham | 612±58 | 986±251a |
| IR | 468±148b | 876±214a |
| IPC | 515±134 | 872±207a |
| IPoC | 445±114b | 607±186a |
Seven days after the myocardial infarction, cardiac function and hemodynamics were recorded first at a basal level and then 5 minutes after stimulation with ISO (10 nmol/L). dP/dtmax indicates rate of pressure rise; IPC, ischemic preconditioning; IPoC, ischemic postconditioning; IR, ischemia/reperfusion; ISO, isoproterenol; LVDP, left ventricular developed pressure. Data are means±SD of n=6 to 10 hearts.
P≤0.05 vs baseline.
P≤0.05 vs Sham.
After stimulation with ISO, a significant increase was recorded for the CO and the LVDP only for the sham‐operated hearts and the hearts in the IR and the IPC groups. IPoC hearts were no longer able to adequately respond to the β‐adrenergic stimulus in terms of all contraction parameters (Table 2).
On the other hand, the heart rate increased uniformly by 30±4% on average under ISO stimulation in all 4 treatment groups. The coronary flow also rose with the increase in the hearts of the sham group (63%) about one and a half times as high as that in the hearts of the IR, IPC, and IPoC groups, which had a mean rise in the flow of 42±3% and did not differ from each another (Table 2).
Effect of IR, IPC, and IPoC on the Remodeling and the Intracellular Coupling of Cardiac β‐Receptors
The expression (mRNA and protein) of the β1‐ and β2‐ARs was significantly reduced in the hearts in the IR, IPC, and IPoC groups compared with the sham group 7 days after the infarction with no differences seen among the groups. Receptor dimers can no longer be detected under these conditions. The expression of the β3‐AR is stable postischemia and was unaffected by either IPC or IPoC (Figure 5A through 5D, for mRNA data; please see Figure S2). A selective influence of IPoC on the expression of cardiac β‐ARs can therefore be excluded. This result was also verified by measuring sodium‐potassium‐ATPase as a surrogate marker for membrane density, which in the postconditioned myocardium does not differ from IR or IPC hearts (Figure 5K).
Figure 5.
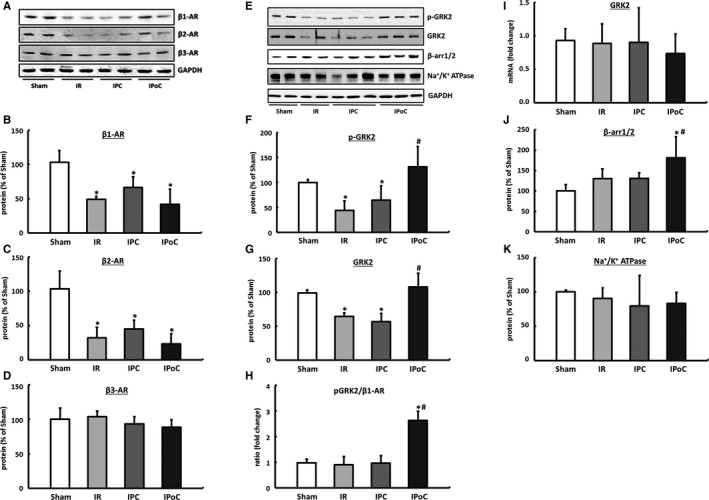
Expression pattern of β‐ARs. A, Representative immunoblots and densitometric analysis of (B) the β1‐AR, (C) the β2‐AR and (D) the β3‐AR. Remodeling and intracellular coupling of cardiac β‐ARs. E, Representative blots and densitometric data of the (F) phosphorylated and (G) total GRK2 protein. H, The pGRK2 to β1‐AR ratio was calculated for each animal, and group means were expressed as fold change of Sham. I, The mRNA expression of GRK2 was analyzed in left ventricular tissue and normalized to HPRT. J, β‐Arrestin1/2, also critically involved in β‐AR coupling, was exclusively upregulated in IPoC hearts. K, Na+/K+ ATPase was determined as a surrogate marker for the membrane density. Immunoblot bands were normalized to GAPDH and expressed as a percentage of Sham. Data are means±SD of n=6 hearts. *P≤0.05 vs Sham, # P≤0.05 vs IR. AR indicates adrenergic receptor; arr, arrestin; GAPDH, Glyceraldehyde 3‐phosphate dehydrogenase; GRK2, G protein‐coupled receptor kinase 2; HPRT, hypoxanthine‐guanine phosphoribosyltransferase; IPC, ischemic preconditioning; IPoC, ischemic postconditioning; IR, ischemia/reperfusion; p‐GRK2, phosphorylated GRK2.
In hearts of the IR and IPC groups, GRK2 was significantly downregulated, meaning that the ratio of β‐ARs to GRK2 was unchanged compared to the hearts of the control group. In IPoC hearts, on the other hand, significant increases in GRK2 as well as its phosphorylated fraction were recorded. The receptor/pGRK2 ratio was shifted 2.6 times toward GRK2, which thus explains the reduced response of IPoC hearts to β‐adrenergic stimuli (Figure 5E through 5H).
The expression of GRK2 was positively correlated with β‐arrestin1/2 in all groups, meaning that desensitization of the β‐receptors in the IPoC hearts was synergistically amplified (Figure 5J).
The high levels of GRK2 in the IPoC hearts cannot be attributed to an increase in the mRNA expression but are instead due to stabilization of the protein (Figure 5I).
Cardiomyocyte as the Cause of Dysfunction in the IPoC Heart
Using the isolated cardiomyocyte model, the previously described molecular and functional remodeling of the postconditioned myocardium can be confirmed at a cellular level (Figure 1C). Cardiomyocytes isolated from postconditioned hearts showed no difference in basal cell shortening after 24‐hour culture compared to IPC myocytes, but their responsivity to ISO was significantly reduced with a simultaneous induction of GRK2 (Figure 6A and 6B). This finding suggests that the limited inotropy of postconditioned hearts can be attributed exclusively to remodeling of the β‐adrenergic signaling and not to structural remodeling processes of the cardiomyocytes or the myocardium. Consequently, postconditioned hearts with simultaneous inhibition of GRK2 should be able to achieve a positive inotropic response to ISO.
Figure 6.
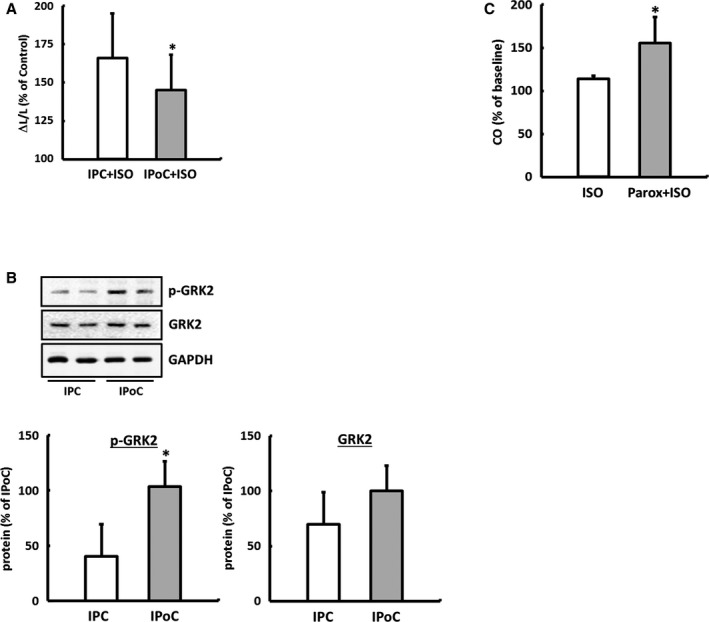
Remodeling of β‐ARs as the main cause for the loss of the inotropic reserve in IPoC hearts. A, Responsivity of cardiomyocytes isolated from IPC or IPoC hearts to ISO was analyzed using a cell‐edge detection system. B, Induction of GRK2 varied depending on whether IPC or IPoC was applied. Data are means±SD of n=4 hearts/216 cells. *P≤0.05 vs IPC. IPoC‐induced loss of the β‐adrenergic functional reserve. C, IPoC hearts 7 days after an infarction were perfused using the working heart technique. After 10 minutes’ infusion with paroxetine, the CO was measured continuously for 5 minutes with additional stimulation using ISO. Data are means±SD of n=4 hearts. *P≤0.05 vs ISO. AR indicates adrenergic receptor; CO, cardiac output; GAPDH, Glyceraldehyde 3‐phosphate dehydrogenase; GRK2, G protein‐coupled receptor kinase 2; IPC, ischemic preconditioning; IPoC, ischemic postconditioning; ISO, isoprenaline; Parox, paroxetine; p‐GRK2, phosphorylated GRK2.
This hypothesis was tested experimentally using the working heart model by perfusing postconditioned hearts after 7 days’ reperfusion first with the GRK2 inhibitor paroxetine for 10 minutes and then stimulating them with ISO (Figure 1D).
With inhibition of the GRK2, the CO of IPoC hearts rose by 55±30% on average during the β‐adrenergic stimulation. In the direct comparison, ISO induced only a moderate increase of 14±4% in the CO of IPoC hearts, which were not perfused with paroxetine (Figure 6C).
The functional relevance of GRK2 (and β‐arrestin) was confirmed in all experimental models (isolated cardiomyocytes, isolated perfused hearts) using selective knockdown with siRNA and/or pharmacological inhibition. The ISO concentration used is based on the dose‐response curves determined on isolated normoxic cardiomyocytes (Figures S3 through S6).
Signaling Mechanisms Underlying GRK2 Induction
GRK2 is regulated both at the transcription level and to a considerable degree via its degradation, in which the E3‐ubiquitin ligase Mdm2 is primarily involved.
The Mdm2 expression did not differ across the 4 treatment groups, but its phosphorylated fraction was significantly reduced in the IR and IPC groups compared with the sham group. In the IPoC hearts the phosphorylated fraction was significantly higher than that in the IR and IPC groups and did not differ from the hearts of the sham group (Figure 7A through 7C).
Figure 7.
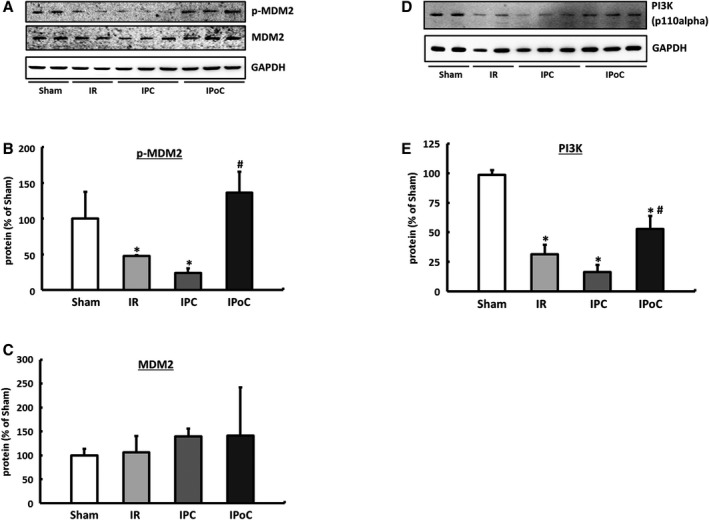
Regulatory mechanisms for GRK2. A, Representative immunoblots and densitometric data of the (B) phosphorylated and (C) total MDM2 protein. D, Western blot image and (E) analysis of PI3 kinase expression. Immunoblot bands were normalized to GAPDH and expressed as a percentage of Sham. Data are means±SD of n=6 hearts. *P≤0.05 vs Sham, # P≤0.05 vs IR. GAPDH indicates Glyceraldehyde 3‐phosphate dehydrogenase; GRK2, G protein–coupled receptor kinase 2; IPC, ischemic preconditioning; IPoC, ischemic postconditioning; IR, ischemia/reperfusion; MDM2, mouse double minute 2 homolog/E3 ubiquitin‐protein ligase Mdm2; p‐GRK2, phosphorylated GRK2; PI3K, phosphoinositide 3‐kinase; p‐MDM2, phosphorylated MDM2.
In the current study, induction or normalization of the phosphoinositide 3‐kinase (PI3 kinase) (Figure 7D and 7E) was detected in IPoC hearts. Because PI3 kinase and Mdm2 have already been described as directly interacting partners, the PI3 kinase/Akt/Mdm2 signaling pathway should be primarily responsible for stabilizing the GRK2 in the hearts of the IPoC group.30
Discussion
The development of contractile dysfunction as a consequence of a myocardial infarction is often unavoidable, even after successful reperfusion. The reduction in the size of the infarct and metabolic disorder by applying IPC or IPoC should also enable positive functional and structural remodeling over the long term based on an improved initial short‐term situation.
The key finding of this study is that, unlike ischemic preconditioning, ischemic postconditioning does not lead to functional preservation of the postischemic myocardium over the long term, despite having short‐term cardioprotective potential.
Just 2 hours after the start of the reperfusion, the intracellular coupling of the β1 receptor via amplified phosphorylation of GRK2 was weakened by application of IPoC. In the long term the specific induction/phosphorylation of GRK2 combined with the reduced expression of β‐ARs caused molecular remodeling in the postconditioned heart, which, as a pathophysiological correlate of a contractile functional disorder, characterizes the start of systolic heart failure. IPoC‐induced desensitization of the β‐receptors only affected the myocardium, however, and thus only reduced the rate of pressure rise and the positive inotropic potential of the ventricle. The pacemaker cells were not affected by the application, meaning that a (desirable) frequency control was not observed. Apart from a desensitization, the coronary vessels were also able to continue ensuring myocardial perfusion as required thanks to the preservation of the β‐adrenergic‐receptor‐mediated vasodilation.
At the moment it is largely unclear to what extent the conditioned heart also benefits over the long term from 1 of the 2 protective strategies and to what extent receptor‐mediated signaling is affected by the 2 strategies. However, effective use of therapy/medication following IPC or IPoC assumes a well‐founded understanding of the receptor status of the conditioned postischemic heart.31
The ratio of β1 to β2 receptors is about 70:30 in the heart. Functionally, their effect is primarily synergistic, but the β1 receptor has a greater effect on the myocardium, whereas the β2 receptor has a greater effect on the smooth muscle cells of the vessels. Quantitatively, the β3‐AR plays a subordinate role in the heart, but protective effects are attributed to it, particularly in the insufficient myocardium.32
No change in the protein concentration of the β1 receptor was recorded during the 2‐hour reperfusion ex vivo. The significantly reduced proportion of receptor dimers indicates, however, that remodeling of the β‐adrenergic signaling has already occurred during this short period independent of the 2 intervention strategies. A reduction in the receptor dimers in turn leads to a lessening of the signal transduction and to accelerated receptor internalization.33, 34, 35, 36
The in vivo protocol used led to a significant reduction in the β1‐ and β2‐AR subtypes, with the expression of the β3‐AR remaining stable, in the left ventricular myocardium in the IR, IPC, and IPoC hearts 7 days after the infarction.
The effect on the intracellular coupling varied depending on whether IPC or IPoC was used. The response of the postconditioned left ventricular myocardium to a β‐adrenergic stimulus was significantly reduced toward all comparator groups. Selective increases in the GRK2 protein and its phosphorylated fraction in the heart of the IPoC group were identified as the cause.
It was originally postulated that an elevation in the GRK2 level during the progression of heart failure may be a sensible protective mechanism that protects the heart against excessive exposure to catecholamines.37 For the postinfarcted heart, rapid and transient elevation or activation of the GRK2 could reduce the reperfusion injury and protect the myocardium against hypercontractions. This mechanism may be causally involved in the initial trend toward improved recovery of postconditioned hearts compared with the IR and IPC groups in the Langendorff model. At the start of the reperfusion the IPoC‐induced phosphorylation of GRK2 by CEC could be significantly reduced, and consequently the ISO‐induced increase in the LVDP can be reestablished. Consequently, the activation of GRK2 should also be a PKC‐dependent process in the reperfused myocardium as well.
A sustained and long‐term upregulation of GRK2 with the resultant desensitization or uncoupling of cardiac receptors leads inevitably to impaired cardiac function and loss of the positive inotropic reserve.38, 39, 40 In a transgenic mouse model, a cardiac‐specific overexpression of the GRK2 inhibitor βARKct not only improved the basal and catecholamine‐stimulated heart function, it also led to a significant reduction in cardiac dysfunction and mortality in various models of heart failure.41, 42, 43 These results were confirmed by using cardiac‐specific heterozygous GRK2 knockout mice that also had a significantly better prognosis after induction of heart failure.44
Particularly when considering the early cardioprotective effects that can be achieved by the postconditioning protocol used here, the importance of desensitized or uncoupled β‐ARs becomes clear because, along with the loss of their β‐adrenergic contraction reserve, IPoC hearts have the worst heart function at rest measured using echocardiography after 7 days have elapsed.
At this point there are only a few verified findings for the precise regulatory mechanisms for GRK2 within the cardiovascular system. The expression is controlled both at the level of the transcription and by affecting the degradation and thus the stabilization of the protein.45 The precise interplay between synthesis and degradation under physiological and pathophysiological conditions is largely unknown, but ubiquitination with subsequent proteasomal degradation is considered to play a significant role in the degradation of GRK2.46 Both β‐arrestin and the E3‐ubiquitin ligase Mdm2 are also involved in this process, initiating the degradation of GRK2 by the proteasome by forming a complex.47 Various functions are attributed to β‐arrestin as part of the regulatory mechanisms that underlie the receptor coupling or desensitization. Together with GRK2, it is directly involved in the uncoupling and internalization of G‐protein‐coupled receptors. Furthermore, in the absence of receptor stimulation, it can inhibit GRK2‐Mdm2 interaction and thus the degradation of GRK2, but if the receptor is stimulated, it can boost complex formation and thus the degradation of GRK2.48
Based on the present results, the significant increase in GRK2 levels in IPoC hearts can be ascribed to impaired degradation that is caused by increased phosphorylation of Mdm2. Phosphorylated Mdm2 translocates increasingly into the nucleus and is thus no longer available for ubiquitination of GRK2 in the cytosol. It is also postulated that the interaction between phosphorylated Mdm2 and GRK2 is impaired, and subsequently, the complex cannot be transported to the proteasome.47
PI3 kinase and Akt have already been identified as direct upstream elements of Mdm2 and are considered responsible for its phosphorylation.30, 49, 50 The induction of both PI3 kinase and Akt plays a crucial role in cardioprotection during IPoC and is classified as a key element of the prosurvival RISK pathway.51, 52
The functional effects of a pharmacological GRK2 inhibition could be demonstrated in both isolated cardiomyocytes and in the isolated, perfused heart. In the IPoC heart, the CO could only be significantly elevated by β‐adrenergic stimulation with selective inhibition of GRK2. These findings emphasize not only the importance of GRK2 for cardiac function, they also identify GRK2 as the reason for the loss of inotropic functional reserve of the postconditioned myocardium. GRK2 is currently being considered as a therapeutic target for chronic heart failure.53 Along with the cardioprotective potential immediately after the start of reperfusion, selective inhibition of GRK2 could also achieve significant functional improvement in the postconditioned myocardium over the long term (Figure 8).
Figure 8.
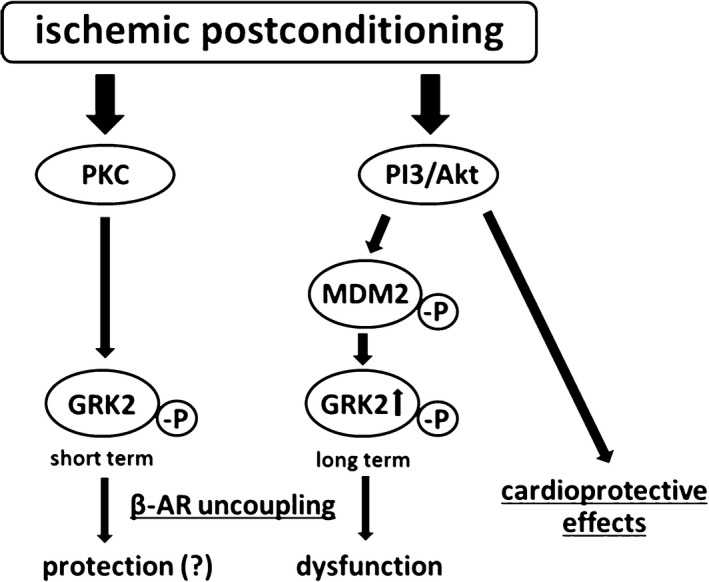
IPoC‐induced desensitization of β‐ARs. In the short term, rapid and transient activation of the GRK2 could reduce the reperfusion injury and protect the myocardium against hypercontractions. In the long term, however, stabilization of GRK2 and its phosphorylated fraction leads to a desensitization or decoupling of β‐ARs with a resultant loss of the positive inotropic functional reserve. AR indicates adrenergic receptor; GRK2, G protein‐coupled receptor kinase 2; MDM2, mouse double minute 2 homolog/E3 ubiquitin‐protein ligase Mdm2; PI3, phosphoinositide 3‐kinase; PKC, protein kinase C.
In summary, the results of this study demonstrate for the first time that the application of IPoC induces desensitization or uncoupling of β‐ARs, which leads to induction of cardiac dysfunction with a restricted inotropic functional reserve. With comparable cardioprotective properties immediately after acute myocardial infarction, IPC also ensures that the function of the postischemic myocardium is preserved over the long term without exerting a negative effect on the β‐adrenergic system. The present results must be considered a possible reason behind the disappointing results obtained in longitudinal clinical studies to date and should lead to a critical review of the clinical use of IPoC.
Sources of Funding
The study was supported by the Deutsche Stiftung für Herzforschung and by the “János Bolyai Scholarship” of the Hungarian Academy of Sciences (Bencsik, Csonka); Ferdinandy was a Szentágothai Fellow of the National Program of Excellence (Grant TAMOP 4.2.4.A/2‐11‐1‐2012‐0001).
Disclosures
None.
Supporting information
Table S1. Functional and Morphological Data Assessed by Echocardiography
Figure S1. The validity of TnI as a marker to determine the infarct size.
Figure S2. Expression pattern of β‐ARs in left ventricular tissue.
Figure S3. The effect of GRK2 and β‐arrestin on the contraction of isolated ventricular cardiomyocytes.
Figure S4. The functional relevance of GRK2 in ex‐vivo perfused hearts.
Figure S5. Concentration‐response curves to isoproterenol in isolated cardiomyocytes.
Figure S6. Western blot analysis to assess the efficiency of the siRNA‐mediated knockdown.
(J Am Heart Assoc. 2017;6:e006809 DOI: 10.1161/JAHA.117.006809.)29273639
References
- 1. Hausenloy DJ, Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res. 2006;70:240–253. [DOI] [PubMed] [Google Scholar]
- 2. Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten‐Johansen J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–H588. [DOI] [PubMed] [Google Scholar]
- 3. Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre‐, post‐, and remote conditioning. Circ Res. 2015;116:674–699. [DOI] [PubMed] [Google Scholar]
- 4. van Vuuren D, Genis A, Genade S, Lochner A. Postconditioning the isolated working rat heart. Cardiovasc Drugs Ther. 2008;22:391–397. [DOI] [PubMed] [Google Scholar]
- 5. Couvreur N, Lucats L, Tissier R, Bize A, Berdeaux A, Ghaleh B. Differential effects of postconditioning on myocardial stunning and infarction: a study in conscious dogs and anesthetized rabbits. Am J Physiol Heart Circ Physiol. 2006;291:H1345–H1350. [DOI] [PubMed] [Google Scholar]
- 6. Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L'Huillier I, Aupetit JF, Bonnefoy E, Finet G, André‐Fouët X, Ovize M. Postconditioning the human heart. Circulation. 2005;112:2143–2148. [DOI] [PubMed] [Google Scholar]
- 7. Granfeldt A, Lefer DJ, Vinten‐Johansen J. Protective ischaemia in patients: preconditioning and postconditioning. Cardiovasc Res. 2009;83:234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hausenloy DJ, Erik Bøtker H, Condorelli G, Ferdinandy P, Garcia‐Dorado D, Heusch G, Lecour S, van Laake LW, Madonna R, Ruiz‐Meana M, Schulz R, Sluijter JP, Yellon DM, Ovize M. Translating cardioprotection for patient benefit: position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res. 2013;98:7–27. [DOI] [PubMed] [Google Scholar]
- 9. Freixa X, Bellera N, Ortiz‐Pérez JT, Jiménez M, Paré C, Bosch X, De Caralt TM, Betriu A, Masotti M. Ischaemic postconditioning revisited: lack of effects on infarct size following primary percutaneous coronary intervention. Eur Heart J. 2012;33:103–112. [DOI] [PubMed] [Google Scholar]
- 10. Hahn JY, Yu CW, Park HS, Song YB, Kim EK, Lee HJ, Bae JW, Chung WY, Choi SH, Choi JH, Bae JH, An KJ, Park JS, Oh JH, Kim SW, Hwang JY, Ryu JK, Lim DS, Gwon HC. Long‐term effects of ischemic postconditioning on clinical outcomes: 1‐year follow‐up of the POST randomized trial. Am Heart J. 2015;169:639–646. [DOI] [PubMed] [Google Scholar]
- 11. Engstrøm T, Kelbæk H, Helqvist S, Høfsten DE, Kløvgaard L, Clemmensen P, Holmvang L, Jørgensen E, Pedersen F, Saunamaki K, Ravkilde J, Tilsted HH, Villadsen A, Aarøe J, Jensen SE, Raungaard B, Bøtker HE, Terkelsen CJ, Maeng M, Kaltoft A, Krusell LR, Jensen LO, Veien KT, Kofoed KF, Torp‐Pedersen C, Kyhl K, Nepper‐Christensen L, Treiman M, Vejlstrup N, Ahtarovski K, Lønborg J, Køber L; Third Danish Study of Optimal Acute Treatment of Patients With ST Elevation Myocardial Infarction–Ischemic Postconditioning (DANAMI‐3–iPOST) Investigators . Effect of ischemic postconditioning during primary percutaneous coronary intervention for patients with ST‐segment elevation myocardial infarction: a randomized clinical trial. JAMA Cardiol. 2017;2:490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007;59:418–458. [DOI] [PubMed] [Google Scholar]
- 13. Lecour S, Bøtker HE, Condorelli G, Davidson SM, Garcia‐Dorado D, Engel FB, Ferdinandy P, Heusch G, Madonna R, Ovize M, Ruiz‐Meana M, Schulz R, Sluijter JP, Van Laake LW, Yellon DM, Hausenloy DJ. ESC working group cellular biology of the heart: position paper: improving the preclinical assessment of novel cardioprotective therapies. Cardiovasc Res. 2014;104:399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferdinandy P, Hausenloy DJ, Heusch G, Baxter GF, Schulz R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol Rev. 2014;66:1142–1174. [DOI] [PubMed] [Google Scholar]
- 15. Schreckenberg R, Maier T, Schlüter KD. Post‐conditioning restores pre‐ischaemic receptor coupling in rat isolated hearts. Br J Pharmacol. 2009;156:901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. White DC, Hata JA, Shah AS, Glower DD, Lefkowitz RJ, Koch WJ. Preservation of myocardial β‐adrenergic receptor signaling delays the development of heart failure after myocardial infarction. Proc Natl Acad Sci USA. 2000;97:5428–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Anthonio RL, Brodde OE, van Veldhuisen DJ, Scholtens E, Crijns HJ, van Gilst WH. β‐Adrenoceptor density in chronic infarcted myocardium: a subtype specific decrease of β1‐adrenoceptor density. Int J Cardiol. 2000;72:137–141. [DOI] [PubMed] [Google Scholar]
- 18. Strasser RH, Marquetant R. Sensitization of the beta‐adrenergic system in acute myocardial ischaemia by a protein kinase C‐dependent mechanism. Eur Heart J. 1991;12:48–53. [DOI] [PubMed] [Google Scholar]
- 19. Nazarewicz RR, Bikineyeva A, Dikalov SI. Rapid and specific measurements of superoxide using fluorescence spectroscopy. J Biomol Screen. 2013;18:498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bencsik P, Kupai K, Giricz Z, Görbe A, Pipis J, Murlasits Z, Kocsis GF, Varga‐Orvos Z, Puskás LG, Csonka C, Csont T, Ferdinandy P. Role of iNOS and peroxynitrite‐matrix metalloproteinase‐2 signaling in myocardial late preconditioning in rats. Am J Physiol Heart Circ Physiol. 2010;299:H512–H518. [DOI] [PubMed] [Google Scholar]
- 21. Bencsik P, Pálóczi J, Kocsis GF, Pipis J, Belecz I, Varga ZV, Csonka C, Görbe A, Csont T, Ferdinandy P. Moderate inhibition of myocardial matrix metalloproteinase‐2 by ilomastat is cardioprotective. Pharmacol Res. 2014;80:36–42. [DOI] [PubMed] [Google Scholar]
- 22. Kocsis GF, Sárközy M, Bencsik P, Pipicz M, Varga ZV, Pálóczi J, Csonka C, Ferdinandy P, Csont T. Preconditioning protects the heart in a prolonged uremic condition. Am J Physiol Heart Circ Physiol. 2012;303:H1229–H1236. [DOI] [PubMed] [Google Scholar]
- 23. Neely JR, Liebermeister H, Battersby EJ, Morgan HE. Effect of pressure development on oxygen consumption by isolated rat heart. Am J Physiol. 1967;212:804–814. [DOI] [PubMed] [Google Scholar]
- 24. Ferdinandy P, Szilvássy Z, Csont T, Csonka C, Nagy E, Koltai M, Dux L. Nitroglycerin‐induced direct protection of the ischaemic myocardium in isolated working hearts of rats with vascular tolerance to nitroglycerin. Br J Pharmacol. 1995;115:1129–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ferdinandy P, Csonka C, Csont T, Szilvássy Z, Dux L. Rapid pacing‐induced preconditioning is recaptured by farnesol treatment in hearts of cholesterol‐fed rats: role of polyprenyl derivatives and nitric oxide. Mol Cell Biochem. 1998;186:27–34. [PubMed] [Google Scholar]
- 26. Csonka C, Kupai K, Kocsis GF, Novák G, Fekete V, Bencsik P, Csont T, Ferdinandy P. Measurement of myocardial infarct size in preclinical studies. J Pharmacol Toxicol Methods. 2010;61:163–170. [DOI] [PubMed] [Google Scholar]
- 27. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(–ΔΔC(T)). Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 28. Tastan I, Schreckenberg R, Mufti S, Abdallah Y, Piper HM, Schlüter KD. Parathyroid hormone improves contractile performance of adult rat ventricular cardiomyocytes at low concentrations in a non‐acute way. Cardiovasc Res. 2009;82:77–83. [DOI] [PubMed] [Google Scholar]
- 29. Krasel C, Dammeier S, Winstel R, Brockmann J, Mischak H, Lohse MJ. Phosphorylation of GRK2 by protein kinase C abolishes its inhibition by calmodulin. J Biol Chem. 2001;276:1911–1915. [DOI] [PubMed] [Google Scholar]
- 30. Mayo LD, Donner DB. A phosphatidylinositol 3‐kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA. 2001;98:11598–11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ovize M, Baxter GF, Di Lisa F, Ferdinandy P, Garcia‐Dorado D, Hausenloy DJ, Heusch G, Vinten‐Johansen J, Yellon DM, Schulz R. Postconditioning and protection from reperfusion injury: where do we stand? Cardiovasc Res. 2010;87:406–423. [DOI] [PubMed] [Google Scholar]
- 32. Niu X, Watts VL, Cingolani OH, Sivakumaran V, Leyton‐Mange JS, Ellis CL, Miller KL, Vandegaer K, Bedja D, Gabrielson KL, Paolocci N, Kass DA, Barouch LA. Cardioprotective effect of beta‐3 adrenergic receptor agonism: role of neuronal nitric oxide synthase. J Am Coll Cardiol. 2012;59:1979–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lan TH, Kuravi S, Lambert NA. Internalization dissociates β2‐adrenergic receptors. PLoS One. 2011;6:e17361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. He J, Xu J, Castleberry AM, Lau AG, Hall RA. Glycosylation of beta(1)‐adrenergic receptors regulates receptor surface expression and dimerization. Biochem Biophys Res Commun. 2002;297:565–572. [DOI] [PubMed] [Google Scholar]
- 35. Hebert TE, Moffett S, Morello JP, Loisel TP, Bichet DG, Barret C, Bouvier M. A peptide derived from a beta2‐adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J Biol Chem. 1996;271:16384–16392. [DOI] [PubMed] [Google Scholar]
- 36. Kobayashi H, Ogawa K, Yao R, Lichtarge O, Bouvier M. Functional rescue of β1‐adrenoceptor dimerization and trafficking by pharmacological chaperones. Traffic. 2009;10:1019–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Matkovich SJ, Diwan A, Klanke JL, Hammer DJ, Marreez Y, Odley AM, Brunskill EW, Koch WJ, Schwartz RJ, Dorn GW II. Cardiac‐specific ablation of G‐protein receptor kinase 2 redefines its roles in heart development and β‐adrenergic signaling. Circ Res. 2006;99:996–1003. [DOI] [PubMed] [Google Scholar]
- 38. Koch WJ, Rockman HA, Samama P, Hamilton RA, Bond RA, Milano CA, Lefkowitz RJ. Cardiac function in mice overexpressing the beta‐adrenergic receptor kinase or a beta ARK inhibitor. Science. 1995;268:1350–1353. [DOI] [PubMed] [Google Scholar]
- 39. Lymperopoulos A. GRK2 and β‐arrestins in cardiovascular disease: something old, something new. Am J Cardiovasc Dis. 2011;1:126–237. [PMC free article] [PubMed] [Google Scholar]
- 40. Raake PW, Zhang X, Vinge LE, Brinks H, Gao E, Jaleel N, Li Y, Tang M, Most P, Dorn GW II, Houser SR, Katus HA, Chen X, Koch WJ. Cardiac G‐protein‐coupled receptor kinase 2 ablation induces a novel Ca2+ handling phenotype resistant to adverse alterations and remodeling after myocardial infarction. Circulation. 2012;125:2108–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tevaearai HT, Walton GB, Keys JR, Koch WJ, Eckhart AD. Acute ischemic cardiac dysfunction is attenuated via gene transfer of a peptide inhibitor of the β‐adrenergic receptor kinase (βARK1). J Gene Med. 2005;7:1172–1177. [DOI] [PubMed] [Google Scholar]
- 42. Harding VB, Jones LR, Lefkowitz RJ, Koch WJ, Rockman HA. Cardiac βARK1 inhibition prolongs survival and augments β‐blocker therapy in a mouse model of severe heart failure. Proc Natl Acad Sci USA. 2001;98:5809–5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Siryk‐Bathgate A, Dabul S, Lymperopoulos A. Current and future G protein‐coupled receptor signaling targets for heart failure therapy. Drug Des Devel Ther. 2013;7:1209–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rockman HA, Choi DJ, Akhter SA, Jaber M, Giros B, Lefkowitz RJ, Caron MG, Koch WJ. Control of myocardial contractile function by the level of beta‐adrenergic receptor kinase 1 in gene‐targeted mice. J Biol Chem. 1998;273:18180–18184. [DOI] [PubMed] [Google Scholar]
- 45. Penela P, Murga C, Ribas C, Tutor AS, Peregrín S, Mayor F Jr. Mechanisms of regulation of G protein‐coupled receptor kinases (GRKs) and cardiovascular disease. Cardiovasc Res. 2006;69:46–56. [DOI] [PubMed] [Google Scholar]
- 46. Penela P, Ruiz‐Gómez A, Castaño JG, Mayor F Jr. Degradation of the G protein‐coupled receptor kinase 2 by the proteasome pathway. J Biol Chem. 1998;273:35238–35244. [DOI] [PubMed] [Google Scholar]
- 47. Salcedo A, Mayor F Jr, Penela P. Mdm2 is involved in the ubiquitination and degradation of G‐protein‐coupled receptor kinase 2. EMBO J. 2006;25:4752–4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nogués L, Salcedo A, Mayor F Jr, Penela P. Multiple scaffolding functions of {beta}‐arrestins in the degradation of G protein‐coupled receptor kinase 2. J Biol Chem. 2011;286:1165–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Feng J, Tamaskovic R, Yang Z, Brazil DP, Merlo A, Hess D, Hemmings BA. Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt‐dependent phosphorylation. J Biol Chem. 2004;279:35510–35517. [DOI] [PubMed] [Google Scholar]
- 50. Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K, Masuyama N, Gotoh Y. Akt enhances Mdm2‐mediated ubiquitination and degradation of p53. J Biol Chem. 2002;277:21843–21850. [DOI] [PubMed] [Google Scholar]
- 51. Tsang A, Hausenloy DJ, Mocanu MM, Yellon DM. Postconditioning: a form of “modified reperfusion” protects the myocardium by activating the phosphatidylinositol 3‐kinase‐Akt pathway. Circ Res. 2004;95:230–232. [DOI] [PubMed] [Google Scholar]
- 52. Zhu M, Feng J, Lucchinetti E, Fischer G, Xu L, Pedrazzini T, Schaub MC, Zaugg M. Ischemic postconditioning protects remodeled myocardium via the PI3K‐PKB/Akt reperfusion injury salvage kinase pathway. Cardiovasc Res. 2006;72:152–162. [DOI] [PubMed] [Google Scholar]
- 53. Schumacher‐Bass SM, Traynham CJ, Koch WJ. G protein‐coupled receptor kinase 2 as a therapeutic target for heart failure. Drug Discov Today Ther Strateg. 2012;9:155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Functional and Morphological Data Assessed by Echocardiography
Figure S1. The validity of TnI as a marker to determine the infarct size.
Figure S2. Expression pattern of β‐ARs in left ventricular tissue.
Figure S3. The effect of GRK2 and β‐arrestin on the contraction of isolated ventricular cardiomyocytes.
Figure S4. The functional relevance of GRK2 in ex‐vivo perfused hearts.
Figure S5. Concentration‐response curves to isoproterenol in isolated cardiomyocytes.
Figure S6. Western blot analysis to assess the efficiency of the siRNA‐mediated knockdown.