

Supplemental Digital Content is available in the text.
Keywords: apolipoprotein E, atherosclerosis, cholesterol, macrophage, monocyte
Abstract
Objective—
The MC1-R (melanocortin 1 receptor) is expressed by monocytes and macrophages where it mediates anti-inflammatory actions. MC1-R also protects against macrophage foam cell formation primarily by promoting cholesterol efflux through the ABCA1 (ATP-binding cassette transporter subfamily A member 1) and ABCG1 (ATP-binding cassette transporter subfamily G member 1). In this study, we aimed to investigate whether global deficiency in MC1-R signaling affects the development of atherosclerosis.
Approach and Results—
Apoe−/− (apolipoprotein E deficient) mice were crossed with recessive yellow (Mc1re/e) mice carrying dysfunctional MC1-R and fed a high-fat diet to induce atherosclerosis. Apoe−/− Mc1re/e mice developed significantly larger atherosclerotic lesions in the aortic sinus and in the whole aorta compared with Apoe−/− controls. In terms of plaque composition, MC1-R deficiency was associated with less collagen and smooth muscle cells and increased necrotic core, indicative of more vulnerable lesions. These changes were accompanied by reduced Abca1 and Abcg1 expression in the aorta. Furthermore, Apoe−/− Mc1re/e mice showed a defect in bile acid metabolism that aggravated high-fat diet–induced hypercholesterolemia and hepatic lipid accumulation. Flow cytometric analysis of leukocyte profile revealed that dysfunctional MC1-R enhanced arterial accumulation of classical Ly6Chigh monocytes and macrophages, effects that were evident in mice fed a normal chow diet but not under high-fat diet conditions. In support of enhanced arterial recruitment of Ly6Chigh monocytes, these cells had increased expression of L-selectin and P-selectin glycoprotein ligand 1.
Conclusions—
The present study highlights the importance of MC1-R in the development of atherosclerosis. Deficiency in MC1-R signaling exacerbates atherosclerosis by disturbing cholesterol handling and by increasing arterial monocyte accumulation.
Atherosclerosis is a chronic inflammatory disease of large- and medium-sized arteries that gives eventually rise to major cardiovascular complications such as myocardial infarction and stroke. The disease is characterized by the gradual accumulation of cholesterol-containing LDL (low-density lipoprotein) particles in arterial walls, which triggers a multitude of inflammatory responses and consequent entry of leukocytes into the intima.1 Monocytes and their descendant macrophages are the most abundant leukocyte subsets in atherosclerotic plaques and also the dominant effector cells in the pathogenesis of the disease.2 In the mouse, lesional macrophages predominantly originate from infiltrating classical Ly6Chigh monocytes,3,4 whereas the nonclassical Ly6Clow monocytes are known to patrol the vasculature but enter the lesions less frequently.5,6 Monocyte-derived macrophages then populate lesions and ingest lipids, primarily in the form of oxidized LDL, which transforms them into foam cells. Eventually, these cells can undergo apoptosis, which, in combination with defective clearance of cellular debris, contributes to the formation of necrotic core.7 This is one of the key features of vulnerable plaques that are prone to rupture and cause life-threatening thrombosis.
The melanocortin system has appeared as an intriguing pharmacological target for modulating immune responses in many inflammatory diseases including atherosclerosis.8,9 The system consists primarily of melanocortin peptides, derived from the post-translational processing of the precursor molecule POMC (proopiomelanocortin), their cellular receptors, and 2 endogenous antagonists, agouti and agouti-related protein. Proteolytic cleavage of the POMC produces melanocortin peptides, namely adrenocorticotropic hormone and α-, β- and γ-melanocyte–stimulating hormones, which bind to a family of G-protein–coupled melanocortin receptors, named from MC1-R to MC5-R. Of these 5 receptors, MC1-R was the first member of the receptor family to be cloned.10 MC1-R stimulates eumelanin synthesis in the skin, being the best-characterized genetic determinant of skin and hair color,11 but it is now clear that its physiological functions extend well beyond the regulation of skin pigmentation. Among other functions, it mediates anti-inflammatory responses in a variety of different leukocytes, but its immunomodulatory role has been studied in most detail in macrophages.9 Furthermore, we recently identified a new regulatory function for MC1-R in macrophage cholesterol transport.12 Activation of MC1-R signaling in macrophages suppressed uptake of oxidized LDL, while concomitantly promoting unloading of intracellular cholesterol that initiates a process called reverse cholesterol transport (RCT). Conversely, dysfunctional MC1-R aggravated cholesterol accumulation and foam cell formation in cultured macrophages. In atherosclerotic Apoe−/− (apolipoprotein E deficient) mice, pharmacological targeting of MC1-R reduced plasma cholesterol level and promoted signs of plaque stability. However, it remains to be determined whether loss-of-function of MC1-R causes a reverse phenotype with enhanced atherosclerosis, thus potentially providing evidence that the integrity of the receptor is a decisive factor for the outcome of the disease.
Thus, we set out to determine whether deficiency in MC1-R signaling contributes to the development of atherosclerosis. To this end, we crossed Apoe−/− mice with the recessive yellow Mc1re/e mouse that carries a nonfunctional MC1-R because of a single-base deletion mutation13 and then characterized the plaque phenotype, cholesterol homeostasis, and tissue leukocyte profiles of this mouse model. We here show that a defect in MC1-R signaling enhances atherogenesis and leads to the formation of more advanced plaques.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
MC1-R Deficiency Accelerates Diet-Induced Atherosclerosis in Apoe−/− Mice
To determine the role of MC1-R in the development of atherosclerosis, we generated Apoe−/− Mc1re/e mice, fed them a high-fat diet (HFD) for 12 weeks, and then analyzed atherosclerotic plaque size and composition using wild-type Apoe−/− mice as controls. We observed that Apoe−/− Mc1re/e mice developed significantly larger atherosclerotic plaques in the aortic sinus (Figure 1A and 1B). Likewise, total lesion area in the aorta, as evidenced by en face staining, was increased by ≈50% in MC1-R–deficient mice (Figure 1C and 1D). Of particular note, quantification of the acellular areas in the aortic sinus plaques revealed markedly increased necrotic cores in these mice (Figure 1E and 1F). This finding was accompanied by reduced α-SMA (α-smooth muscle actin) expression and by lower collagen content in the plaques (Figure 1E and 1G). Plaque macrophage content was increased in Apoe−/− Mc1re/e mice, but the ratio of phenotypic M1- and M2-type macrophage markers was unchanged (Figure I in the online-only Data Supplement). In contrast to HFD conditions, Apoe−/− Mc1re/e mice on a chow diet displayed no change in atherosclerotic lesion area (Figure II in the online-only Data Supplement). However, in line with the phenotype of HFD-fed mice, necrotic core area was increased in the aortic plaques of Apoe−/− Mc1re/e mice (Figure II in the online-only Data Supplement). Plaque collagen content was lower in chow-fed mice compared with HFD mice, but there was no difference between the genotypes in this regard (Figure II in the online-only Data Supplement). Collectively, these data indicate that MC1-R deficiency enhances atherogenesis in HFD-fed mice and leads to the formation of more vulnerable plaques.
Figure 1.
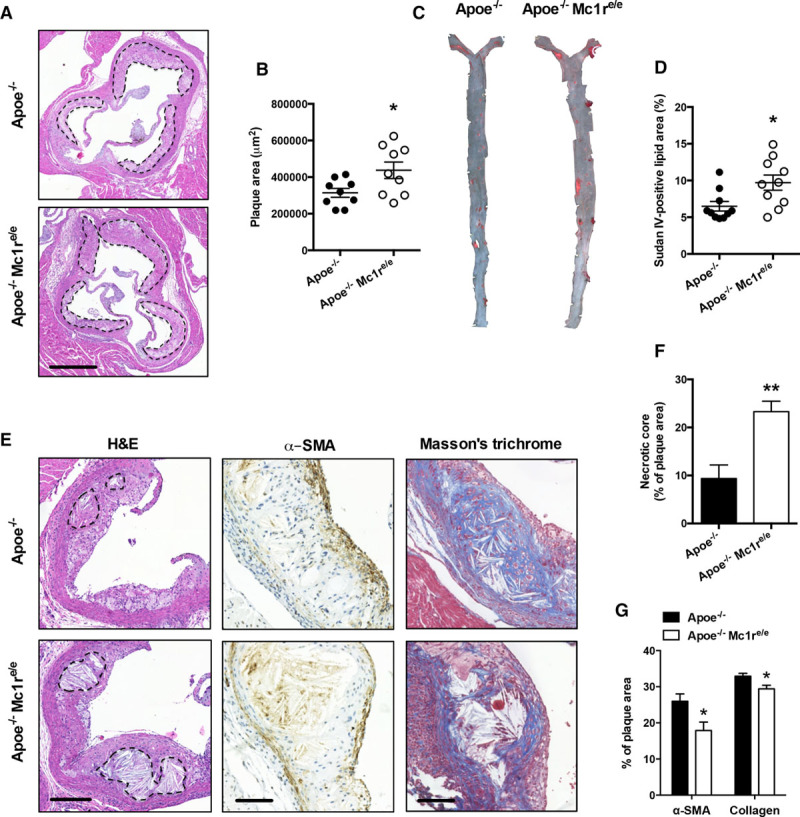
MC1-R (melanocortin 1 receptor) deficiency accelerates atherosclerosis in Apoe−/− mice. A, Representative images of hematoxylin and eosin (H&E) staining of the aortic sinus of Apoe−/− and Apoe−/− Mc1re/e mice fed a high-fat diet (HFD) for 12 wk. Scale bar, 500 µm. B, Quantification of plaque area in aortic sinuses. C, D, Representative en face Sudan IV stainings and quantification of Sudan IV–positive lipid area in the aorta. E, Representative images of necrotic core areas, and α-SMA (α-smooth muscle actin) and Masson trichrome stainings in the aortic sinus. Scale bars, 200 µm (left) and 50 µm (middle, right). Necrotic areas are indicated with dashed lines in H&E-stained images. F, Quantification of acellular necrotic core areas as percentage of total plaque area. G, Quantification of α-SMA–positive area and plaque collagen content as percentage of total plaque area. n=8 to 10 mice per group in each graph. *P<0.05 and **P<0.01 vs Apoe−/− mice. Values are mean±SEM. Apoe indicates apolipoprotein.
We next performed quantitative (real-time)-polymerase chain reaction (qPCR) analysis for aortic lysates and screened the samples for a selection of genes that are involved in cholesterol transport and plaque stability. This was performed in mice that were fed a normal chow diet or HFD for 4 weeks to identify whether MC1-R deficiency per se causes changes in gene expression or whether an interaction between the gene defect and excess of dietary cholesterol is needed to drive these effects. The 4-week HFD time point was chosen because upregulation of inflammatory markers at the mRNA level is detectable at this early time point. First, we turned our attention to RCT-promoting proteins, namely ABCA1 (ATP-binding cassette transporter subfamily A member 1) and ABCG1 (ATP-binding cassette transporter subfamily G member 1), which were previously shown to be increased by MC1-R activation in macrophages.12 These transporters mediate cholesterol efflux and thereby facilitate the clearance of excess cholesterol from macrophages.14 Corroborating our previous results, we found that MC1-R–deficient mice had reduced aortic expression of Abca1 and Abcg1 (Figure III in the online-only Data Supplement), changes that occurred independent of the diet. Second, we quantified the mRNA levels of plaque stability markers and observed that Acta2 (gene encoding α-SMA) and Col3a1 (collagen type III α 1) were downregulated in MC1-R–deficient mice (Figure III in the online-only Data Supplement), further supporting the findings of plaque histology. The downregulation of genes was similar in both diet groups of MC1-R–deficient mice.
MC1-R Deficiency Exacerbates HFD-Induced Hypercholesterolemia and Hepatic Lipid Accumulation in Apoe−/− Mice
We next investigated whether MC1-R deficiency affects plasma cholesterol levels during early phases of atherosclerosis when shifting from normal chow diet to HFD. Cholesterol measurements revealed that Apoe−/− Mc1re/e mice are more prone to HFD-induced hypercholesterolemia compared with Apoe−/− mice, but are able to maintain normal cholesterol levels in the absence of excess dietary cholesterol (Figure 2A). The increase in total cholesterol on HFD was attributable to the elevation in non-HDL cholesterol (Figure 2B), namely LDL and VLDL (very-low-density lipoprotein) fractions, whereas HDL cholesterol showed no genotype difference (Figure 2C). After 4 weeks of HFD, relative liver weight was also increased in Apoe−/− Mc1re/e mice (Figure 2D). Oil red O staining of liver sections indicated aggravated lipid accumulation in these mice (Figure 2E). In support of this finding, quantification of extracted lipids from liver samples showed that Apoe−/− Mc1re/e mice had more tissue total cholesterol in the HFD state compared with controls (Figure 2F).
Figure 2.
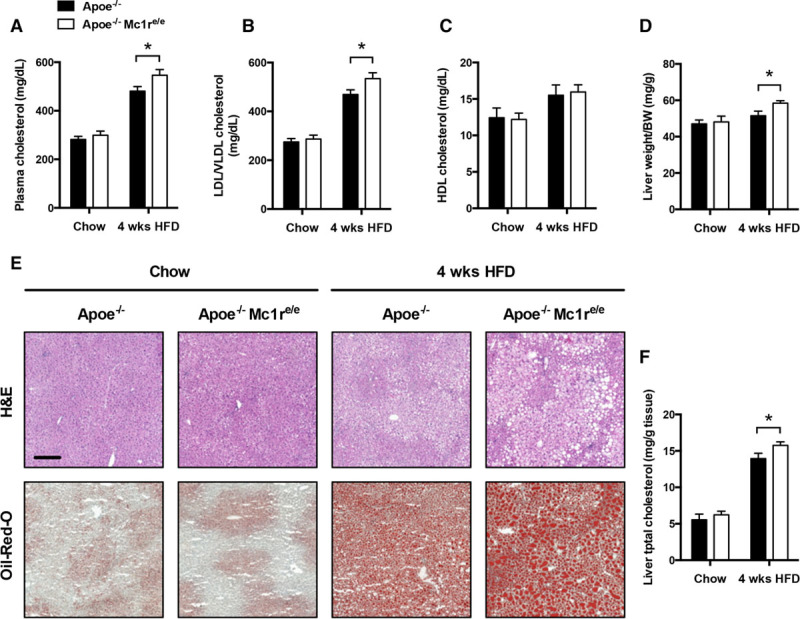
Apoe−/− Mc1re/e mice show increased plasma cholesterol levels and hepatic lipid accumulation after 4 wk of high-fat diet (HFD). A, Total cholesterol; B, LDL (low-density lipoprotein)/VLDL (very-low-density lipoprotein) cholesterol; and C, HDL (high-density lipoprotein) cholesterol levels in the plasma of Apoe−/− and Apoe−/− Mc1re/e fed a normal chow diet or an HFD for 4 wk. D, Liver:body weight ratio in Apoe−/− and Apoe−/− Mc1re/e. E, F, Representative hematoxylin and eosin (H&E-stained and oil red O–stained liver sections and quantification of liver total cholesterol in Apoe−/− and Apoe−/− Mc1re/e. Scale bar, 200 μm. n=8 to 10 mice per group in each graph. *P<0.05 and **P<0.01 vs Apoe−/− mice. Values are mean±SEM. Apoe indicates apolipoprotein; BW, body weight; and Mc1r, melanocortin 1 receptor.
To identify mechanisms contributing to the increased hypercholesterolemia and hepatic lipid accumulation, we determined the expression levels of genes involved in cholesterol synthesis and transport in the liver. Hepatic mRNA levels of cholesterol synthesis genes and their transcriptional regulators were unchanged in Apoe−/− Mc1re/e mice (Figure IV in the online-only Data Supplement). Similarly, the expression of LDL receptor and the HDL receptor Scarb1 or Srb1 was comparable between the genotypes. Interestingly, ABCG5 and ABCG8, which form heterodimers together and mediate the excretion of neutral sterols in the liver, were upregulated in chow-fed Apoe−/− Mc1re/e. Furthermore, Abcg5 and Abcg8 mRNA levels were increased by HFD as expected, but Apoe−/− Mc1re/e mice fell short of this compensation and showed significantly less upregulated Abcg5 and Abcg8 levels in the HFD state (Figure IV in the online-only Data Supplement). We therefore aimed to test whether this reduction has a functional consequence in terms of cholesterol transport efficiency. We performed an in vivo RCT assay by injecting 3H-cholesterol–loaded primary macrophages into both genotypes and measured the transport capacity of cholesterol through the RCT pathway. Because Mc1re/e macrophages have impaired cholesterol efflux,12 we injected macrophages from Apoe−/− mice to both genotypes to control for possible differences in the initiating step of RCT and to specifically investigate the RCT pathway downstream of macrophage cholesterol efflux. However, RCT to plasma, liver or feces was not significantly changed in Apoe−/− Mc1re/e (Figure IV in the online-only Data Supplement). Likewise, total excretion of neutral sterols into the bile and feces was comparable between the genotypes (Figure 3A and 3B), suggesting that the reduced Abcg5 and Abcg8 mRNA levels do not result in impaired cholesterol excretion in the liver.
Figure 3.
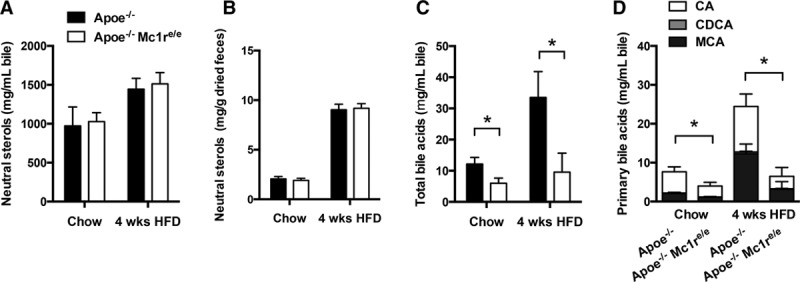
MC1-R (melanocortin 1 receptor) deficiency has no effect on biliary or fecal excretion of neutral sterols, but reduces the amount of biliary bile acids. A, The concentration of neutral sterols (cholesterol) in the gall bladder bile of Apoe−/− and Apoe−/− Mc1re/e fed a normal chow diet or a high-fat diet (HFD) for 4 wk. B, Total neutral sterols in feces excreted over 48 h and expressed as mg/g of dried feces. Fecal neutral sterols were calculated as a sum of cholesterol and coprostanol. Concentration of total bile acids (C) and primary bile acids (D) in the gall bladder bile of Apoe−/− and Apoe−/− Mc1re/e. Primary bile acids consist of cholic acid (CA), chenodeoxycholic acid (CDCA), and muricholic acid (MCA). n=4 to 6 mice per group in each graph. *P<0.05 vs Apoe−/− mice by 2-way ANOVA. Values are mean±SEM. Apoe indicates apolipoprotein E.
To investigate other possible causes of hypercholesterolemia, we measured the size and composition of the bile acid pool in the gall bladder and feces by gas–liquid chromatography. These analyses revealed that total bile acids were reduced in the gall bladder bile (Figure 3C), but not in fecal samples of Apoe−/− Mc1re/e (Figure V in the online-only Data Supplement). Both diet groups of MC1-R–deficient mice showed a reduction in the primary bile acid cholic acid, whereas the amount of secondary bile acids such as deoxycholic acid and lithocholic acid were increased in these mice (Figure 3D; Figure V in the online-only Data Supplement). Furthermore, the fecal ratio of cholic acid:chenodeoxycholic acid increased after HFD as a protective mechanism against excessive cholesterol absorption, but this increase was markedly blunted in Apoe−/− Mc1re/e (Figure V in the online-only Data Supplement). We next quantified the hepatic expression of key enzymes and transcriptional regulators involved in the synthesis and conjugation of bile acid synthesis. However, none of these enzymes or regulators showed altered expression (Figure VI in the online-only Data Supplement), suggesting that the early steps of cholesterol conversion to bile acids and subsequent conjugation are not impaired in MC1-R–deficient mice. Furthermore, transporters responsible for the uptake of bile acids into hepatocytes and for their excretion into bile showed no changes at the mRNA level (Figure VI in the online-only Data Supplement).
Monocyte Dynamics in MC1-R–Deficient Apoe−/− Mice
Because the effects of MC1-R deficiency on leukocyte counts have not been previously explored, we next quantified total leukocytes and different leukocyte subsets in the blood, spleen, and aorta of mice that were fed either a chow diet or HFD for 4 weeks. Given that hypercholesterolemia is known to trigger monocytosis and predominant expansion of Ly6Chigh monocytes, we focused on quantifying Ly6Chigh and Ly6Clow monocyte subsets. Flow cytometric analyses revealed that Apoe−/− and Apoe−/− Mc1re/e mice had equal numbers of circulating total leukocytes, lymphocytes, neutrophils, and monocytes (Figure 4A). The extent of HFD-induced monocytosis was also similar between the genotypes (Figure 4A). Of particular note, MC1-R–deficient mice on chow diet had lower number of blood Ly6Clow monocytes (Figure 4A). On the other hand, bone marrow of these mice was more sensitive to HFD-induced hypercholesterolemia and displayed exaggerated Ly6Chigh monocytosis (Figure VII in the online-only Data Supplement). Considering that ABCA1 and ABCG1 centrally regulate the proliferation of hematopoietic stem and progenitor cells,15–17 we analyzed the levels of these transporters in the bone marrow and found that Abcg1 expression was specifically reduced in HFD-fed Apoe−/− Mc1re/e mice (Figure VII in the online-only Data Supplement).
Figure 4.
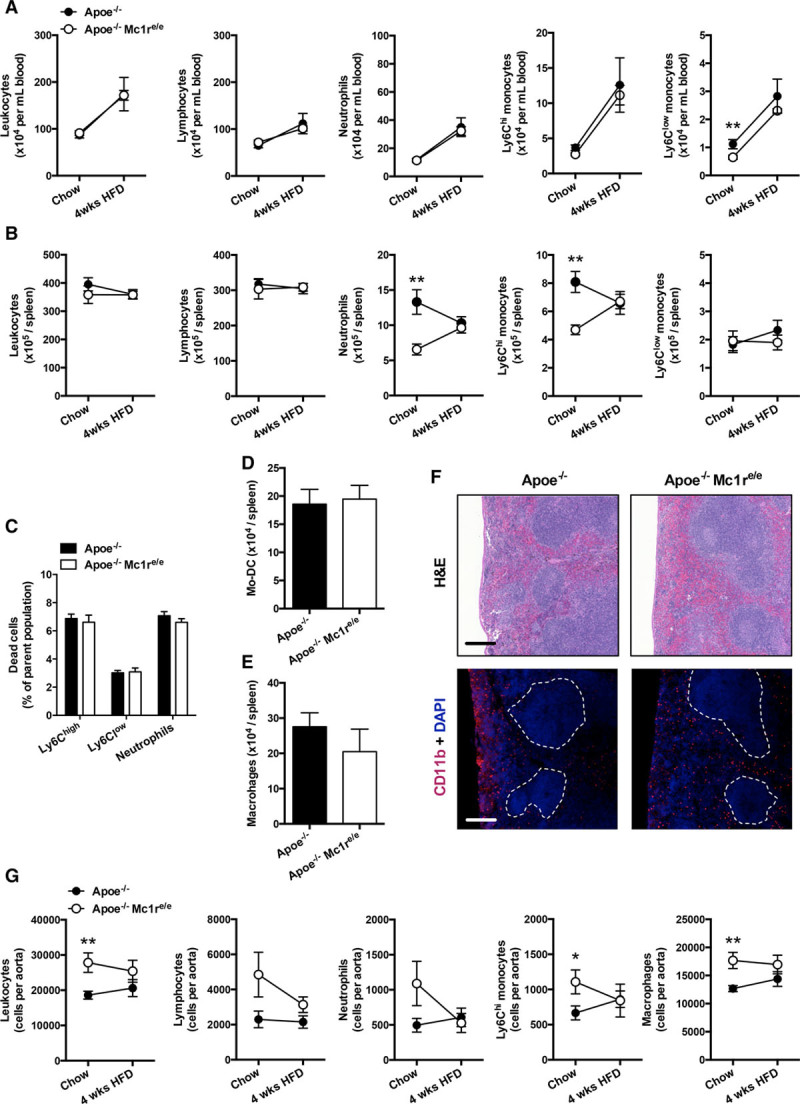
Characterization of leukocyte profiles in Apoe−/− Mc1re/e mice. A, B, Quantification of total leukocytes (CD45+), lymphocytes (CD45+, CD11b−), neutrophils (CD45+, CD11b+, Ly6G+), and Ly6Clow and Ly6Chigh monocytes (CD45+, CD11b+, CD115+, Ly6Clow/high) in the blood and spleen of Apoe−/− and Apoe−/− Mc1re/e mice. C, Percentage of dead monocytes and neutrophils in the spleen of chow-fed mice. D, E, Number of monocyte-derived dendritic cells (CD45+, CD11b+, CD11c+) and macrophages (CD45+, CD11b+, F4/80high) in the spleen of chow-fed mice. F, Representative hematoxylin and eosin (H&E)–stained and CD11b-stained spleen sections from chow-fed Apoe−/− and Apoe−/− Mc1re/e mice. Dashed lines indicate the areas of white pulps. Scale bar, 200 μm. G, Enumeration of total leukocytes, lymphocytes, neutrophils, Ly6Chigh monocytes, and macrophages (CD45+, CD11b+, F4/80high) in the aortic lysates from Apoe−/− and Apoe−/− Mc1re/e mice. n=6 to 10 mice per group in each graph. Samples for chow and 4-wk high-fat diet (HFD) groups were derived from different sets of mice. *P<0.05 and **P<0.01 vs Apoe−/− mice. Values are mean±SEM. Apoe indicates apolipoprotein; and MC1-R, melanocortin 1 receptor.
Characterization of splenic reservoir monocytes showed that Apoe−/− Mc1re/e mice had markedly reduced numbers of Ly6Chigh monocytes during baseline conditions when mice were fed a normal chow diet (Figure 4B). Likewise, the splenic pool of neutrophils was reduced in these mice (Figure 4B). After 4 weeks of HFD, monocyte and neutrophil levels were identical between the genotypes (Figure 4B). To examine whether MC1-R deficiency affects local cell differentiation or death in the spleen, thereby explaining the reduced cell counts, we quantified the percentage of dead cells among neutrophils and monocytes and the total number of monocyte-derived macrophages and dendritic cells in the spleen of chow-fed mice. However, Apoe−/− Mc1re/e mice displayed no changes in the tendency for cell death or monocyte differentiation (Figure 4C through 4E), suggesting that the monocytes are exiting the spleen and get recruited to sites of inflammation. Immunostaining of the spleen further revealed that CD11b+ cells in Apoe−/− Mc1re/e mice were most notably lost from the subcapsular red pulp (Figure 4F), which is the primary site of rapidly recruitable monocytes.18 Supporting the notion of enhanced deployment of splenic monocytes, we observed that MC1-R–deficient mice were accumulating more leukocytes in the aorta during chow-fed conditions. This difference was largely attributable to increases in aortic Ly6Chigh monocyte and macrophage counts (Figure 4G). The phenotype was lost when mice were challenged with HFD (Figure 4G). Cytokine screening of the plasma of chow-fed mice uncovered that Apoe−/− Mc1re/e mice indeed showed signs of increased inflammation with significant peaking of IL-1β (interleukin-1β), IL-5, IFN-γ (interferon-γ), TNF-α (tumor necrosis factor-α), and CCL2 (chemokine (C-C motif) ligand 2; Figure VIII in the online-only Data Supplement). CCL2 conveys chemotactic signal specifically for monocytes and is likely to contribute to the increased infiltration of monocytes into the aorta of Apoe−/− Mc1re/e mice. Taken together, deficiency of MC1-R resulted in lower number of splenic Ly6Chigh monocytes, which were appearing in higher frequency in the aorta and giving probably rise to increased macrophage accumulation during the onset of atherosclerosis.
Deficiency of MC1-R Does Not Modify the Expression of Endothelial Adhesion Molecules in the Aorta
To dissect the underlying mechanism leading to increased monocyte entry to the aorta, we first investigated whether MC1-R deficiency aggravates endothelial inflammation, thus reinforcing monocyte adhesion and migration. Because the immunophenotype of Apoe−/− Mc1re/e mice was more prominent without HFD, we focused on studying mice on chow diet in subsequent experiments. Flow cytometric analysis of aortic endothelial cells indicated no genotype-dependent difference in terms of the expression of VCAM-1 (vascular cell adhesion molecule 1), ICAM-1 (intercellular adhesion molecule 1), PECAM-1 (platelet and endothelial cell adhesion molecule 1), or P-selectin (Figure 5A and 5B). Furthermore, when lysing the whole aorta and performing qPCR analysis of the same adhesion molecules, we observed no significant mRNA changes in the aorta of Apoe−/− Mc1re/e mice compared with control group (Figure 5C). Immunostaining of aortic roots further revealed similar expression of VCAM-1, which mediates rolling and firm adhesion of Ly6Chigh to endothelial cells during atherogenesis19 (Figure 5D). Collectively, these data suggest that the increased accumulation of Ly6Chigh monocytes in MC1-R–deficient mice is not relying on enhanced activation of the aortic endothelium.
Figure 5.
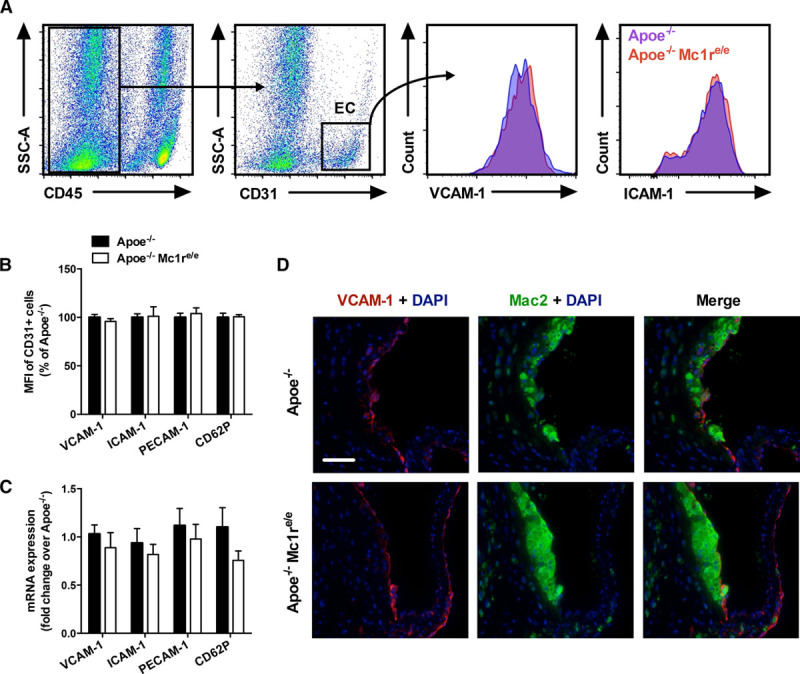
MC1 (melanocortin 1) deficiency does not affect adhesion molecule expression in the aortic endothelium. A, Representative dot plots for the gating of endothelial cells (CD45−, CD31+) in aortic lysates and histograms for VCAM-1 (vascular cell adhesion molecule 1) and ICAM-1 (intercellular adhesion molecule 1) staining. B, Mean fluorescence intensity (MFI, expressed as percentage of Apoe−/− mice) of VCAM-1, ICAM-1, PECAM-1 (platelet and endothelial cell adhesion molecule 1), and CD62P in aortic endothelial cells of chow-fed Apoe−/− and Apoe−/− Mc1re/e mice. C, Quantitative real-time-polymerase chain reaction analysis of Vcam1, Icam1, Pecam1, and Cd62p expression in the whole aorta of chow-fed Apoe−/− and Apoe−/− Mc1re/e mice. D, Representative staining of VCAM-1 and Mac2 in the aortic roots of chow-fed Apoe−/− and Apoe−/− Mc1re/e mice. Scale bar, 50 μm. n=6 to 8 mice per group in each graph. Values are mean±SEM. Apoe indicates apolipoprotein E; EC, endothelial cells; and SSC-A, side scatter area.
Monocytes From Apoe−/− Mc1re/e Mice Express Higher Levels of CD62L and PSGL-1
We next sought to explore whether MC1-R deficiency modifies the characteristics of monocytes because no apparent difference was noted in the endothelial cells. Previous studies have shown that Ly6Chigh and Ly6Clow use different chemokines and their corresponding receptors to access sites of atherosclerosis, among which CCR2 (C-C chemokine receptor type 2), CCR5 (C-C chemokine receptor type 5), and CX3CR1 (C-X3-C motif chemokine receptor 1) are the key receptors because blocking any of these receptors reduces monocyte recruitment and combined receptor blockage almost abolishes atherosclerosis.20,21 We therefore measured the mRNA expression of these receptors in flow-sorted splenic Ly6Chigh and Ly6Clow monocytes (Figure 6A). As expected, Ccr2 expression was upregulated in Ly6Chigh monocytes compared with their Ly6Clow counterparts, whereas Ccr2 mRNA level was significantly lower in Apoe−/− Mc1re/e mice (Figure 6B). No differences were noted in Ccr5 or Cx3cr1 expression (Figure 6B).
Figure 6.
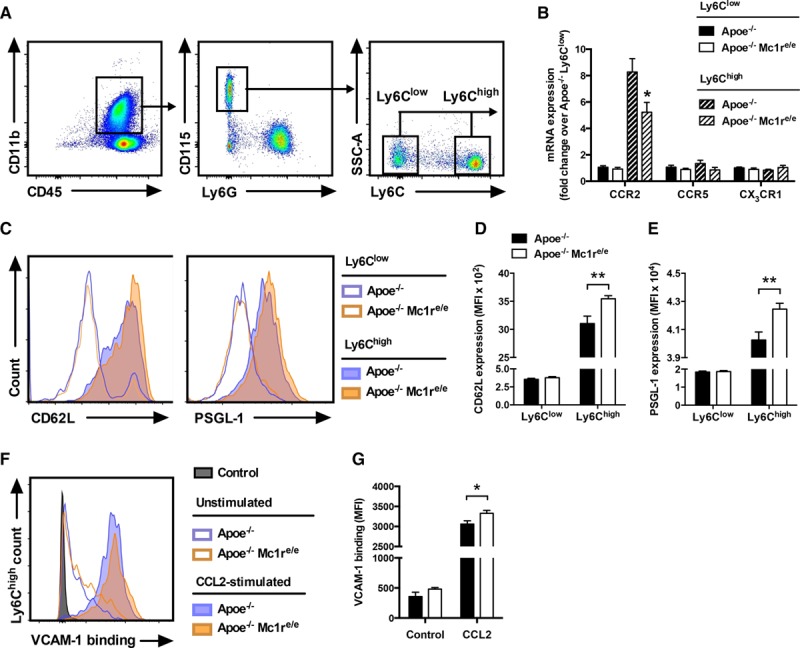
Monocytes from Apoe−/− Mc1re/e mice show increased adhesion molecule expression and binding activity to VCAM-1 (vascular cell adhesion molecule 1). A, Representative dot plots for the gating and sorting of splenic Ly6Clow and Ly6Chigh monocytes (CD45+, CD11b+, CD115+, and Ly6Clow/high). B, Quantification of Ccr2 (C-C chemokine receptor type 2), Ccr5 (C-C chemokine receptor type 5), and Cx3cr1 (C-X3-C motif chemokine receptor 1) mRNA levels in the sorted Ly6Clow and Ly6Chigh monocytes from chow-fed Apoe−/− and Apoe−/− Mc1re/e mice. Representative histograms (C) and mean fluorescence intensity (MFI) of CD62L (D) and PSGL-1 (P-selectin glycoprotein ligand 1; E) in the Ly6Clow- and Ly6Chigh-gated splenic monocytes from Apoe−/− and Apoe−/− Mc1re/e mice. n=6 to 8 mice per group in each graph. F, Representative histogram of binding of blood Ly6Chigh monocytes to soluble VCAM-1-Fc under unstimulated and CCL2 (chemokine (C-C motif) ligand 2)-stimulated (5 μg/mL, 5 min) conditions. The histogram shows also a control sample that was incubated with secondary antibody that was not preconjugated to soluble VCAM-1-Fc. G, Quantification of VCAM-1-Fc binding in Ly6Chigh-gated monocytes from Apoe−/− and Apoe−/− Mc1re/e mice. n=4 to 6 mice per group in each graph. *P<0.05 and **P<0.01 vs Apoe−/− mice. Values are mean±SEM. Apoe indicates apolipoprotein; MC1-R, melanocortin 1 receptor; and SSC-A, side scatter area.
Murine Ly6Chigh monocytes also express L-selectin (CD62L), PSGL-1 (P-selectin glycoprotein ligand 1), LFA-1 (lymphocyte function–associated antigen 1), and VLA-4 (very late antigen 4), which all contribute to monocyte adhesion to activated endothelium.22–24 Quantification of these adhesion molecules by flow cytometry revealed that Ly6Chigh monocytes from MC1-R–deficient mice express higher levels of CD62L and PSGL-1 (Figure 6C and 6D). Conversely, Ly6Clow monocytes from the same mice showed reduced expression of CD18 (Figure IX in the online-only Data Supplement), which together with CD11a forms the integrin LFA-1. CD49d, which is part of the integrin complex of VLA-4, was unaffected by MC1-R deficiency (Figure IX in the online-only Data Supplement). In addition, determination of integrin-binding activity by incubating monocytes with soluble VCAM-1 fragments demonstrated distinct effects for MC1-R deficiency. Specifically, stimulation with CCL2 unveiled that Ly6Chigh monocytes from MC1-R–deficient mice have slightly but significantly increased binding affinity for VCAM-1 (Figure 6E and 6F). Overall, these data indicate that MC1-R deficiency modifies the adhesion properties of classical monocytes mainly by increasing CD62L and PSGL-1 expression.
Discussion
The present study demonstrates a critical role for MC1-R in the development of atherosclerosis. First, we showed that deficiency in MC1-R signaling accelerates atherosclerosis and formation of more vulnerable plaques and worsens diet-induced hypercholesterolemia. Second, we identified that MC1-R regulates arterial accumulation of classical Ly6Chigh monocytes by modulating L-selectin and PSGL-1 expression.
We recently identified that MC1-R enhances cholesterol efflux from macrophages by upregulating Abca1 and Abcg1 expression.12 These effects translated into therapeutic benefits when atherosclerotic Apoe−/− mice were treated with a selective MC1-R agonist. Although no effect was noted in terms of lesion size during early atherosclerosis, MC1-R activation promoted signs of plaque stability with increased α-SMA expression and collagen deposition. Herein, we characterized the plaque phenotype of Apoe−/− Mc1re/e mice and were able to corroborate our previous findings. These mice with deficient MC1-R displayed a reverse phenotype with reduced Abca1 and Abcg1 expression and downregulation of α-SMA and Col3a1. Remarkably, MC1-R deficiency accelerated plaque formation and expanded necrotic core size in the lesions, a central characteristic of vulnerable plaques. The precise mechanisms leading to the exaggerated formation of necrotic cores in Apoe−/− Mc1re/e mice are currently unknown, but reduced Abca1 and Abcg1 expression might be a significant contributing factor. Combined deficiency of Abca1 and Abcg1 has been unequivocally linked to enhanced atherosclerosis and formation of more complex plaques.14,16 This phenotype can be even recapitulated in the absence of enhanced monocytosis and expansion of hematopoietic stem cells, effects that typically arise when Abca1 and Abcg1 are uniformly deleted in myeloid cells.14,15,25 This is evidenced by increased atherosclerosis in mice with macrophage-specific deficiency of ABCA1 and ABCG1.26 Therefore, the reduced Abca1 and Abcg1 expression in MC1-R–deficient mice might impair the cholesterol handling of lesional macrophages and amplify inflammatory responses, the net outcome of which would be the formation of more complex plaques. Importantly, the current findings highlight that the integrity of MC1-R is an important determinant of plaque vulnerability.
Aside from the plaque phenotype, we observed increased plasma LDL/VLDL cholesterol levels and hepatic cholesterol accumulation in MC1-R–deficient mice, which, in turn, accelerates atherosclerosis. This strengthens our previous finding of reduced plasma LDL/VLDL cholesterol as a therapeutic response to MC1-R activation and further consolidates that MC1-R signaling is involved in the control of circulating cholesterol.12 In the quest of causal factors for the elevated plasma cholesterol, we found that bile acid metabolism was disturbed in Apoe−/− Mc1re/e mice. Because bile acid synthesis accounts for a major fraction of daily cholesterol turnover, it is conceivable that the disturbed bile acid metabolism is linked with hypercholesterolemia. Apoe−/− Mc1re/e mice showed a distinct phenotype with reduced primary and increased secondary bile acid levels. Taking into account that ≈95% of bile acids are reabsorbed in the ileum and that this process is strictly regulated, it is possible that Apoe−/− Mc1re/e mice are recycling more bile acids without significant replenishment by de novo synthesis, which then increases the amount of secondary bile acids. Unfortunately, we were unable to pinpoint the exact molecular-level mechanism for this phenotype, but it might involve changes in the biotransformation and absorption of bile acids in the gut. In parallel with the change in bile acid metabolism, Abcg5 and Abcg8 mRNA levels were upregulated in chow-fed Apoe−/− Mc1re/e mice, reflecting potentially a compensatory response to reduced cholesterol catabolism. The expression of Abcg5 and Abcg8 was then downregulated in HFD-fed Apoe−/− Mc1re/e mice, suggesting that the compensatory mechanism might become exhausted when hypercholesterolemic conditions are prolonged. This, in turn, manifests as increased cholesterol accumulation in the liver and circulation.
Previous studies have demonstrated that human and mouse monocytes express functional MC1-R,9,27,28 but the effects of this receptor on monocyte production and trafficking are largely unknown. We therefore immunophenotyped the Apoe−/− Mc1re/e mouse model to explore whether MC1-R deficiency affects homeostatic monocyte counts within tissues and circulation. We observed that blood Ly6Chigh monocyte count was unchanged in Apoe−/− Mc1re/e mice compared with control group, whereas the splenic pool of this subset was reduced with the concurrent appearance of higher cell number in the aorta. These data imply that the circulating classical monocytes were in equilibrium and that the reservoir of these cells in the spleen was mobilized to fuel the enhancement of arterial monocyte accumulation in MC1-R–deficient mice. These events occurred without evidence of increased mobilization from the bone marrow. After 4-week HFD, monocyte production was augmented in the bone marrow of Apoe−/− Mc1re/e mice. This could be linked to the observed increase in plasma cholesterol levels rather than to the reduction of bone marrow Abcg1 expression because only combined deficiency of ABCA1 and ABCG1 is likely to accelerate myeloid monocyte production.25,26 Strikingly, however, HFD induced blood monocytosis equally in both genotypes, but led to a slight increase in aortic monocyte and macrophage accumulation only in control mice. Accordingly, MC1-R deficiency seems to affect tissue monocyte counts more drastically in chow-fed conditions, when dietary cholesterol is not producing an overwhelming trigger for monocytosis and arterial leukocyte accumulation. Defective MC1-R signaling might therefore accelerate monocyte recruitment specifically during the early phases of atherosclerosis. Monocyte influx and differentiation indeed dictate macrophage accumulation in early atherosclerosis,3,4,20,29 whereas in more advanced lesions, replenishment of macrophages is predominantly dependent on local proliferation.30 The current finding of increased monocyte accumulation in the aorta of MC1-R–deficient mice is well in line with our recent study, where pharmacological activation of MC1-R elicited an opposite effect, that is, reduced monocyte accumulation.12 In addition, chronic MC1-R treatment led to an increase in circulating Ly6Clow count,12 supporting the finding of reduced number of Ly6Clow monocytes in Apoe−/− Mc1re/e mice. Considering that the short-lived Ly6Chigh monocytes are precursors of Ly6Clow monocytes,31 one possible explanation for these findings is that the increased arterial accumulation of Ly6Chigh monocytes is compromising the conversion of these cells into Ly6Clow monocytes and thus reducing their number in the blood. We also found that Ly6Clow monocytes of MC1-R–deficient mice express less CD18, which is part of the LFA-1 integrin complex and contributes to the patrolling behavior of these cells. These notions warrant further research to investigate the significance and mechanisms of this MC1-R–driven modulation of blood Ly6Clow monocyte count.
Monocytes and particularly classical Ly6Chigh monocytes accumulate within atherosclerotic lesions through a process that is regulated at multiple levels, for example, mobilization from sites of production, recruitment, and life span in the lesions. Recruitment and consequent extravasation require coordinated actions of chemokines and their receptors and selectins and adhesion molecules. It was previously postulated that CCR2, CCR5, and CX3CR1 are fundamental mediators of monocyte chemotaxis to inflamed arteries,20,21 but a recent study highlighted the importance of CCR1 and CCR5 over CCR2 and CX3CR1 in monocyte recruitment.32 Although classical monocyte recruitment to atherosclerotic lesions does not require CCR2, it has an important role in monocyte mobilization from the bone marrow. Consequently, we were interested to screen the expression of these receptors in classical monocytes of MC1-R–deficient mice. Surprisingly, we found reduced monocyte Ccr2 expression, although the mice displayed normal circulating monocyte counts, but elevated plasma CCL2 levels and increased Ly6Chigh monocyte accumulation in the aorta. We may only speculate that Ccr2 might be downregulated as a compensatory mechanism in the presence of excess CCL2 or that other recruitment pathways are dominating and damping its expression. In light of this possibility, we analyzed the expression of adhesion molecules that are major determinants of monocyte trafficking and entry to sites of atherosclerosis. Ly6Chigh monocytes of MC1-R–deficient mice showed increased levels of CD62L and PSGL-1, the latter of which is especially crucial in the recruitment of Ly6Chigh monocytes and has a significant impact on atherosclerosis susceptibility.22,33,34 Furthermore, despite that MC1-R deficiency did not affect CD49d expression, we found that the integrin dimer VLA-4, which is composed of CD49d and CD29 and forms a binding partner with VCAM-1, was functionally more active in the Ly6Chigh monocytes of MC1-R–deficient mice as evidenced by increased binding to soluble VCAM-1. The interaction between VLA-4 and VCAM-1 regulates monocyte adhesion to inflamed endothelium,19,35 thus giving another mechanistic explanation for increased arterial monocyte accumulation in MC1-R–deficient mice. Collectively, these data give a new perspective on the role of MC1-R in controlling monocyte recruitment and adhesion molecule expression during atherogenesis.
In conclusion, our data establish the impact of MC1-R on atheroprogression by demonstrating that its dysfunction exacerbates atherosclerosis and plaque vulnerability. The acceleration of atherosclerosis is likely driven by disturbed cholesterol conversion into bile acids and the consequent increase of plasma cholesterol levels. The disturbed cholesterol and bile acid metabolism in MC1-R–deficient mice are the key findings of this study that merit further research to investigate the exact mechanism by which MC1-R regulates cholesterol metabolism.
Acknowledgments
We thank Erica Nyman, Marja-Riitta Kajaala (Histology Unit, Turku Center for Disease Modelling), Sanna Bastman, Satu Mäkelä, and Elina Kahra for their excellent technical assistance. We also thank Helena Gylling and Leena Kaipiainen for technical and scientific help with the gas–liquid chromatographic analyses. Ketlin Adel and the Cell Imaging Core (Turku Centre for Biotechnology, Turku, Finland) are acknowledged for cell sorting.
Sources of Funding
This work was supported by grants from the Academy of Finland (grant 274852 to P. Rinne), the Finnish Foundation for Cardiovascular Research (to P. Rinne and E. Savontaus), the Paavo Nurmi Foundation (to P. Rinne), The Diabetes Research Foundation (to E. Savontaus), the Deutsche Forschungsgemeinschaft (STE-1053/3-1 to S. Steffens), and the Else Kroener Foundation (to S. Steffens).
Disclosures
None.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- ABCA1
- ATP-binding cassette transporter subfamily A member 1
- ABCG1
- ATP-binding cassette transporter subfamily G member 1
- ApoE
- apolipoprotein E
- CCL2
- chemokine (C-C motif) ligand 2
- CCR2
- C-C chemokine receptor type 2
- CCR5
- C-C chemokine receptor type 5
- Col3A1
- collagen type III α 1
- CX3CR1
- C-X3-C motif chemokine receptor 1
- HFD
- high-fat diet
- ICAM-1
- intercellular adhesion molecule 1
- IFN-γ
- interferon-γ
- IL-1β
- interleukin-1β
- LDL
- low-density lipoprotein
- LFA-1
- lymphocyte function–associated antigen 1
- MC1-R
- melanocortin 1 receptor
- PECAM-1
- platelet and endothelial cell adhesion molecule 1
- POMC
- proopiomelanocortin
- PSGL-1
- P-selectin glycoprotein ligand 1
- RCT
- reverse cholesterol transport
- TNF-α
- tumor necrosis factor-α
- VCAM-1
- vascular cell adhesion molecule 1
- VLA-4
- very late antigen 4
- VLDL
- very-low-density lipoprotein
These authors contributed equally to this article.
These authors contributed equally to this article.
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.117.310418/-/DC1.
Highlights.
Lack of MC1-R (melanocortin 1 receptor) signaling accelerates the formation of atherosclerotic plaques in APOE (apolipoprotein E)-deficient mice.
Atherosclerotic plaques in MC1-R–deficient mice showed a more vulnerable phenotype with increased necrotic core size and reduced expression of α-SMA (α-smooth muscle actin).
MC1-R deficiency also impairs bile acid metabolism and thereby aggravates diet-induced hypercholesterolemia and hepatic lipid accumulation.
Ly6Chigh monocytes from MC1-R–deficient mice express higher levels of L-selectin and PSGL-1 (P-selectin glycoprotein ligand 1), which is associated with increased accumulation of these cells in the aorta.
References
- 1.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–519. doi: 10.1038/nri1882. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 2.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–721. doi: 10.1038/nri3520. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swirski FK, Pittet MJ, Kircher MF, Aikawa E, Jaffer FA, Libby P, Weissleder R. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Natl Acad Sci USA. 2006;103:10340–10345. doi: 10.1073/pnas.0604260103. doi: 10.1073/pnas.0604260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 6.Hilgendorf I, Swirski FK, Robbins CS. Monocyte fate in atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35:272–279. doi: 10.1161/ATVBAHA.114.303565. doi: 10.1161/ATVBAHA.114.303565. [DOI] [PubMed] [Google Scholar]
- 7.Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J Lipid Res. 2009;50(suppl):S382–S387. doi: 10.1194/jlr.R800032-JLR200. doi: 10.1194/jlr.R800032-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rinne P, Silvola JM, Hellberg S, Ståhle M, Liljenbäck H, Salomäki H, Koskinen E, Nuutinen S, Saukko P, Knuuti J, Saraste A, Roivainen A, Savontaus E. Pharmacological activation of the melanocortin system limits plaque inflammation and ameliorates vascular dysfunction in atherosclerotic mice. Arterioscler Thromb Vasc Biol. 2014;34:1346–1354. doi: 10.1161/ATVBAHA.113.302963. doi: 10.1161/ATVBAHA.113.302963. [DOI] [PubMed] [Google Scholar]
- 9.Catania A, Gatti S, Colombo G, Lipton JM. Targeting melanocortin receptors as a novel strategy to control inflammation. Pharmacol Rev. 2004;56:1–29. doi: 10.1124/pr.56.1.1. doi: 10.1124/pr.56.1.1. [DOI] [PubMed] [Google Scholar]
- 10.Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257:1248–1251. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 11.Dessinioti C, Antoniou C, Katsambas A, Stratigos AJ. Melanocortin 1 receptor variants: functional role and pigmentary associations. Photochem Photobiol. 2011;87:978–987. doi: 10.1111/j.1751-1097.2011.00970.x. doi: 10.1111/j.1751-1097.2011.00970.x. [DOI] [PubMed] [Google Scholar]
- 12.Rinne P, Rami M, Nuutinen S, Santovito D, van der Vorst EPC, Guillamat-Prats R, Lyytikäinen LP, Raitoharju E, Oksala N, Ring L, Cai M, Hruby VJ, Lehtimäki T, Weber C, Steffens S. Melanocortin 1 receptor signaling regulates cholesterol transport in macrophages. Circulation. 2017;136:83–97. doi: 10.1161/CIRCULATIONAHA.116.025889. doi: 10.1161/CIRCULATIONAHA.116.025889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robbins LS, Nadeau JH, Johnson KR, Kelly MA, Roselli-Rehfuss L, Baack E, Mountjoy KG, Cone RD. Pigmentation phenotypes of variant extension locus alleles result from point mutations that alter MSH receptor function. Cell. 1993;72:827–834. doi: 10.1016/0092-8674(93)90572-8. [DOI] [PubMed] [Google Scholar]
- 14.Westerterp M, Bochem AE, Yvan-Charvet L, Murphy AJ, Wang N, Tall AR. ATP-binding cassette transporters, atherosclerosis, and inflammation. Circ Res. 2014;114:157–170. doi: 10.1161/CIRCRESAHA.114.300738. doi: 10.1161/CIRCRESAHA.114.300738. [DOI] [PubMed] [Google Scholar]
- 15.Soehnlein O, Swirski FK. Hypercholesterolemia links hematopoiesis with atherosclerosis. Trends Endocrinol Metab. 2013;24:129–136. doi: 10.1016/j.tem.2012.10.008. doi: 10.1016/j.tem.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yvan-Charvet L, Ranalletta M, Wang N, Han S, Terasaka N, Li R, Welch C, Tall AR. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest. 2007;117:3900–3908. doi: 10.1172/JCI33372. doi: 10.1172/JCI33372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Out R, Hoekstra M, Habets K, Meurs I, de Waard V, Hildebrand RB, Wang Y, Chimini G, Kuiper J, Van Berkel TJ, Van Eck M. Combined deletion of macrophage ABCA1 and ABCG1 leads to massive lipid accumulation in tissue macrophages and distinct atherosclerosis at relatively low plasma cholesterol levels. Arterioscler Thromb Vasc Biol. 2008;28:258–264. doi: 10.1161/ATVBAHA.107.156935. doi: 10.1161/ATVBAHA.107.156935. [DOI] [PubMed] [Google Scholar]
- 18.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–1262. doi: 10.1172/JCI11871. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194. doi: 10.1172/JCI28549. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Combadière C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, Merval R, Proudfoot A, Tedgui A, Mallat Z. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649–1657. doi: 10.1161/CIRCULATIONAHA.107.745091. doi: 10.1161/CIRCULATIONAHA.107.745091. [DOI] [PubMed] [Google Scholar]
- 22.An G, Wang H, Tang R, Yago T, McDaniel JM, McGee S, Huo Y, Xia L. P-selectin glycoprotein ligand-1 is highly expressed on Ly-6Chi monocytes and a major determinant for Ly-6Chi monocyte recruitment to sites of atherosclerosis in mice. Circulation. 2008;117:3227–3237. doi: 10.1161/CIRCULATIONAHA.108.771048. doi: 10.1161/CIRCULATIONAHA.108.771048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–774. doi: 10.1038/nri3070. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ley K, Miller YI, Hedrick CC. Monocyte and macrophage dynamics during atherogenesis. Arterioscler Thromb Vasc Biol. 2011;31:1506–1516. doi: 10.1161/ATVBAHA.110.221127. doi: 10.1161/ATVBAHA.110.221127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, Tall AR. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328:1689–1693. doi: 10.1126/science.1189731. doi: 10.1126/science.1189731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westerterp M, Murphy AJ, Wang M, et al. Deficiency of ATP-binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ Res. 2013;112:1456–1465. doi: 10.1161/CIRCRESAHA.113.301086. doi: 10.1161/CIRCRESAHA.113.301086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rajora N, Ceriani G, Catania A, Star RA, Murphy MT, Lipton JM. alpha-MSH production, receptors, and influence on neopterin in a human monocyte/macrophage cell line. J Leukoc Biol. 1996;59:248–253. doi: 10.1002/jlb.59.2.248. [DOI] [PubMed] [Google Scholar]
- 28.Bhardwaj R, Becher E, Mahnke K, Hartmeyer M, Schwarz T, Scholzen T, Luger TA. Evidence for the differential expression of the functional alpha-melanocyte-stimulating hormone receptor MC-1 on human monocytes. J Immunol. 1997;158:3378–3384. [PubMed] [Google Scholar]
- 29.Swirski FK, Weissleder R, Pittet MJ. Heterogeneous in vivo behavior of monocyte subsets in atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:1424–1432. doi: 10.1161/ATVBAHA.108.180521. doi: 10.1161/ATVBAHA.108.180521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robbins CS, Hilgendorf I, Weber GF, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19:1166–1172. doi: 10.1038/nm.3258. doi: 10.1038/nm.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soehnlein O, Drechsler M, Döring Y, et al. Distinct functions of chemokine receptor axes in the atherogenic mobilization and recruitment of classical monocytes. EMBO Mol Med. 2013;5:471–481. doi: 10.1002/emmm.201201717. doi: 10.1002/emmm.201201717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo W, Wang H, Ohman MK, Guo C, Shi K, Wang J, Eitzman DT. P-selectin glycoprotein ligand-1 deficiency leads to cytokine resistance and protection against atherosclerosis in apolipoprotein E deficient mice. Atherosclerosis. 2012;220:110–117. doi: 10.1016/j.atherosclerosis.2011.10.012. doi: 10.1016/j.atherosclerosis.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tedder TF, Steeber DA, Pizcueta P. L-selectin-deficient mice have impaired leukocyte recruitment into inflammatory sites. J Exp Med. 1995;181:2259–2264. doi: 10.1084/jem.181.6.2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galkina E, Ley K. Vascular adhesion molecules in atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:2292–2301. doi: 10.1161/ATVBAHA.107.149179. doi: 10.1161/ATVBAHA.107.149179. [DOI] [PubMed] [Google Scholar]