

Abstract
Background
We performed a statistical analysis of a previously published set of gene expression microarray data from six different brain regions in two mouse strains. In the previous analysis, 24 genes showing expression differences between the strains and about 240 genes with regional differences in expression were identified. Like many gene expression studies, that analysis relied primarily on ad hoc 'fold change' and 'absent/present' criteria to select genes. To determine whether statistically motivated methods would give a more sensitive and selective analysis of gene expression patterns in the brain, we decided to use analysis of variance (ANOVA) and feature selection methods designed to select genes showing strain- or region-dependent patterns of expression.
Results
Our analysis revealed many additional genes that might be involved in behavioral differences between the two mouse strains and functional differences between the six brain regions. Using conservative statistical criteria, we identified at least 63 genes showing strain variation and approximately 600 genes showing regional variation. Unlike ad hoc methods, ours have the additional benefit of ranking the genes by statistical score, permitting further analysis to focus on the most significant. Comparison of our results to the previous studies and to published reports on individual genes show that we achieved high sensitivity while preserving selectivity.
Conclusions
Our results indicate that molecular differences between the strains and regions studied are larger than indicated previously. We conclude that for large complex datasets, ANOVA and feature selection, alone or in combination, are more powerful than methods based on fold-change thresholds and other ad hoc selection criteria.
Background
Genome-wide expression data such as that obtained with microarrays presents a significant challenge for analysis. A frequent goal of expression analysis is to identify genes whose expression is altered by an experimental condition. In general, automated methods are needed to accomplish this task owing to the large amount of data involved. A good example of an expression dataset requiring detailed, complex analysis comes from the work of Sandberg et al. [1]. In their study, the expression of 13,067 genes and expressed sequence tags (ESTs) was assayed using oligonucleotide arrays [2] for each of six brain regions in two different inbred strains of mice, with each condition measured twice. This data is a potentially rich source of targets for investigting behavioral and neurophysiological differences between the mouse strains (C57B1/6 and 129SvEv), as well as structural and functional differences among brain regions.
Both regional and strain variations in gene expression are relevant to questions in neuroscience. Inbred mouse strains are used in many studies relating to human neurological and neuropsychiatric disorders such as stroke and alcoholism; however, it has long been recognized that different strains vary significantly in the results obtained in such studies [3]. Expression analysis is one approach to exploring the underlying molecular causes of these differences. Similarly, expression analysis can provide insight into one potential source of functional differences between brain regions. Traditional expression analysis (for example, northern blots and in situ hybridization) can be used to uncover such expression patterns one gene at a time, but the high-throughput nature of array technology allows researchers to take a much wider view.
Given the data of Sandberg et al., and the questions one can address with it, the issue becomes how to proceed with the analysis. Many researchers use an 'ad hoc' approach: for example, the 'fold change' for each gene is considered, where fold change is defined as the ratio of gene expression in a test condition to that in a control condition. Changes in expression above a certain fold-change threshold are deemed to be significant. The problem with such methods is that they typically do not take into account the variability of the measurements being considered. When using the Microarray Analysis Suite (Affymetrix), one also has the option of using the 'absent/present' calls, determined by the software, to make discriminations as to whether a gene is expressed in one set of samples and not others. The trouble with this method is that the absent/present threshold is essentially arbitrary, and there is no easy way to estimate the numbers of false positives and false negatives obtained. Typically in such studies, genes are identified as 'changed' or 'not changed', often without a quantitative estimate of the statistical confidence in that conclusion.
Because of the shortcomings of ad hoc methods, some researchers have tackled the problem of how to apply more robust statistical methods to the problem of identifying changed genes in a dataset [4,5,6]. Most such methods deal with the case of a two-way comparison, for example between a wild-type organism and one that is mutant in a gene of interest. But, many experimenters are studying complex systems and generate large datasets with more than one experimental variable. Such is the case for Sandberg et al. [1], where both strain and brain region were varied in the same experiment. Here, we investigate methods appropriate for analyzing this type of dataset and determine how the methods affect the interpretation of the data of Sandberg et al.
There are several desirable properties of a method for analysis of microarray data. First, it should provide an estimate of the confidence that the gene-expression pattern observed would occur by chance, that is, a p-value. Failing the ability to generate accurate p-values, the method should at least automatically rank the genes in order of interest. Second, the method should be flexible enough to allow for complex experimental designs. Methods that permit us to compare only two conditions are insufficient. Finally, an ideal method is computationally simple and fast. Mathematical simplicity and familiarity are also advantages when the methods must be used and interpreted by biologists with little statistical background.
A standard technique for analyzing such multivariate datasets, which fulfills the above criteria, is analysis of variance (ANOVA) [7]. For the data of Sandberg et al., the appropriate method is a two-way ANOVA because two variables - strain and region - were involved. ANOVA is a well understood and powerful method, but has two drawbacks that can make its application difficult in practice. First, when more than two categories are present for a variable, as in the case of the work of Sandberg et al., where six brain regions were studied, additional work must be done to determine which categories underlie the effects observed. Another drawback of ANOVA is that in its standard form, the number of replicates of each condition should be equal. Adjustments can be made when small differences in the number of replicates occur, but large deviations are difficult to correct for.
We note that some workers have previously applied one-way ANOVA to expression data [8], and that the use of t-tests, which is one-way ANOVA for the limiting case of two replicated conditions, is becoming a more common method for analyzing expression data [6]. Kerr et al. [9] applied ANOVA to two-color cDNA arrays in an experiment involving a single biological variable (tissue type). Their model encompassed all the genes on the array, as well as experimental factors such as dye and array effects. Their complex model differs from the gene-specific application for experiments with more than one biological variable that we discuss here.
An alternative way of approaching the analysis problem is to view it as one of feature selection. In machine learning, feature selection seeks to identify the data points (here, expression profiles) that are most informative when trying to learn a classification problem. In the context of the data of Sandberg et al., we are seeking genes that differentiate between the genotypes and/or brain regions in our samples. While learning to predict this classification is not relevant here, the underlying task is effectively one of feature selection. The features in this situation are the individual samples (arrays). A recent application of feature selection to microarray data was described by Golub et al. [10]. They sought to identify genes that best differentiated between two types of tumor. Because feature selection methods can be quite simple, can be used to differentiate between any number of categories, and permit ranking genes in accordance with how well they differentiate between regions or strains, feature selection fulfills all three criteria for a suitable analysis method. A similar method was used by Chu et al. [11] to perform clustering: they created a set of idealized expression patterns based on the data from hand-selected genes and identified additional genes that were similar to the desired patterns.
Sandberg et al. used criteria based on absent/present calls and fold-change thresholds for their analysis, and applied multiple t-tests to each gene to detect additional regional variations in expression [1,12]. With these methods they identified 24 genes showing strain variation, and about 240 genes showing some type of regional variation. An examination of the data and experimental design suggests that methods such as ANOVA and feature selection could be profitably applied. Here we describe the identification of additional genes showing possible strain- and region-dependent patterns of expression from the data of Sandberg et al. We find that ANOVA and feature selection are complementary, and can be combined to form a powerful method for detecting interesting genes in a complex dataset.
Results
The dataset of Sandberg et al. [1] consists of duplicate analysis of six brain regions (amygdala, cerebellum, cortex, entorhinal cortex, hippocampus and midbrain) in two strains of mice (129SvEv and C57BL/6), for 13,067 genes and ESTs. We performed two-factor ANOVA and feature selection to look for strain- and/or region-specific variation in gene expression in this data. Our feature-selection strategy, which we call 'template matching', is depicted schematically in Figure 1.
Figure 1.
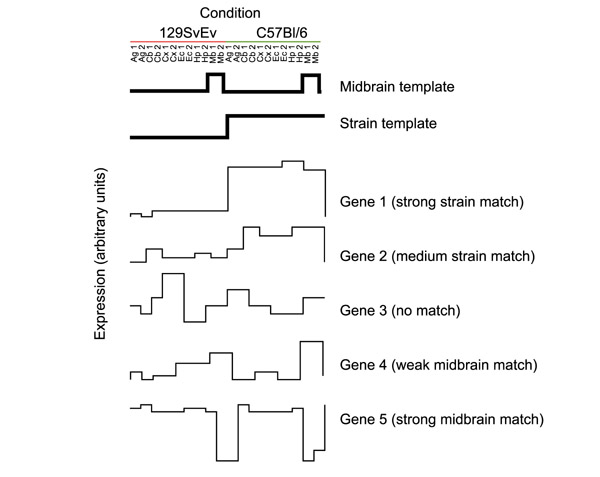
Schematic of the template match method. Hypothetical templates and genes are depicted as graphs of arrays (horizontal axis) vs expression (arbitrary units, vertical axis). The replicates for each condition are listed as 1 and 2 (for example, 129SvEv - Ag1 and 129SvEv - Ag2 are the two amygdala samples from 129SvEv). Two example templates are depicted schematically, and represent the profiles of idealized genes showing the patterns of interest. The top template is an idealized profile designed to select for genes showing midbrain enrichment or depletion, and the lower one is designed to select for genes showing strain differences across all regions. Each gene in the dataset is then ranked by its correlation with the template. In the hypothetical examples shown, gene 1 most closely resembles the strain template, with gene 2 giving the next best match. Gene 4 is a fairly weak match to the midbrain template. Gene 5 is a strong match to the reverse of the midbrain template; the use of the absolute value of the correlation coefficient allows the midbrain template to select both genes 4 and 5. Gene 3 resembled neither template shown here. In the actual implementation, all 13,067 genes are compared to a given template and ranked by the quality of the match. Ag, amygdala; Cb, cerebellum; Cx, cortex; Ec, entorhinal cortex; Hp, hippocampus; Mb, midbrain.
An overview of the results for both the ANOVA and template-matching methods is given in Table 1, which shows the number of genes identified by each method at three different p-value cut-offs. These cut-offs were selected to represent varying levels of false-positive risk (described in the Materials and methods section). The least stringent cut-off (p < 0.001) is almost sure to yield significant numbers of false positives, whereas the most stringent (p < 10-6) will most probably yield very few false positives. The middle threshold of p < 10-5 is a value we use as a compromise for the purpose of comparing our results to those of the previous studies. Table 1 also shows the number of genes identified in the work of Sandberg et al. A more detailed view of part of the data is provided in Figure 2, which enumerates some genes identified by both methods, ranked by template-match p-value. Some (but not all) genes identified by Sandberg et al. [1] and Sandberg [12] are also given high scores (low p-values) by our methods, as indicated in Figure 2. As indicated in Table 1 and Figure 2, even at conservative p-value cut-offs we identified additional genes not noted in [1].
Table 1.
Comparison of numbers of genes identified as showing strain and/or regional variation in gene expression for different methods
| Number of genes selected at each p-value | ||||
| Condition | Method | p < 0.001 | p < 10-5 | p < 10-6 |
| Strain | Sandberg | 24 | 24 | 24 |
| ANOVA | 213 | 65 | 36 | |
| Template | 150 | 56 | 34 | |
| Strain x region | Sandberg | 49 | 49 | 49 |
| ANOVA | 17 | 1 | 0 | |
| All strain effects | Sandberg | 73 | 73 | 73 |
| ANOVA | 230 | 66 | 36 | |
| Template | 150 | 56 | 34 | |
| Amygdala | Sandberg | 1(3) | 1(3) | 1(3) |
| Template | 74 | 2 | 0 | |
| Hippocampus | Sandberg | 2(10) | 2(10) | 2(10) |
| Template | 63 | 16 | 11 | |
| Cortex* | Sandberg | 0 | 0 | 0 |
| Template | 35 | 0 | 0 | |
| Forebrain* | Sandberg | 58 | 58 | 58 |
| Template | 771 | 191 | 112 | |
| Midbrain | Sandberg | 13 | 13 | 13 |
| Template | 181 | 44 | 23 | |
| Cerebellum | Sandberg | 150 | 150 | 150 |
| Template | 890 | 339 | 227 | |
| Entorhinal cortex | Sandberg | 2(10) | 2(10) | 2(10) |
| Template | 37 | 3 | 1 | |
| All tissue effects† | Sandberg | 242 | 242 | 242 |
| ANOVA | 874 | 289 | 168 | |
| Template | 2051 | 595 | 374 | |
The number of genes identified by each method, and by Sandberg et al. [1], for each condition or set of conditions is listed if appropriate. For ANOVA and template match, values for three different p-value thresholds are shown. For the various brain regions, the template-match counts include both matches (enriched genes) and anti-matches (genes expressed at lower levels in the region listed). None of the values given is corrected for genes dominated by negative average difference values. After filtering such genes (see Materials and methods) values would be about 5-10% lower. The values in the 'Sandberg' rows are the same in each column because Sandberg et al. did not generate confidence measures for the genes they selected. The totals for 'Sandberg' include genes listed both in Sandberg et al. [1] and Sandberg [12], and group together genes that were 'restricted', 'enriched', 'absent' and 'decreased'. Numbers in parentheses are the totals obtained if one includes genes mentioned in the text of [1] but not listed by name. *Genes specific to the cortex alone. Because Sandberg et al. do not appear to have specifically sought genes expressed only in the cortex (instead they compared cortex only to cerebellum and midbrain [12]), the number of genes in this category for Sandberg et al. are listed as zero. 'Forebrain' is the combination of cortex, hippocampus, entorhinal cortex and amygdala, which most closely matches the 'Cortex' group chosen in [1,12]. It is included here to allow a fair comparison to their methods. †This category does not include strain x region effects.
Figure 2.

Visualizations and detailed information about some genes identified in this study. For each region or for the strain distinction, up to 40 genes are shown (for space considerations), all of which have template-match p-values < 10-4. The genes are ranked by template-match p-value. Other rankings are possible on the basis of our data. The p-value cut-off (10-4) was chosen so that some genes would be listed for all categories; note that the 'best' genes in some categories are only comparable in specificity to the 'worst' genes in others. For two categories (cerebellum and midbrain), genes showing enrichment and depletion expression in the area of interest are separated. The results have been filtered to remove genes having primarily negative average difference values as described in Materials and methods. Besides the ANOVA and template-match p-values (p-values greater than 10-4 are not displayed), for each gene, the GenBank accession number of the sequence used to derive the probes is shown, along with the UniGene cluster number (if applicable), and annotations drawn primarily from UniGene and FANTOM databases [29]. UniGene cluster identity and annotations were based on the current information rather than the annotations originally provided by Affymetrix and may differ from those described in Sandberg et al. [1]. A bullet appears in the column Sb (Sandberg) if the gene was identified in Sandberg et al. [1] or Sandberg [12]. The need to limit the number of genes shown may give the misleading impression that relatively few new genes were identified, especially for the cerebellum-enriched genes. On the basis of accession number, UniGene ID, and annotation, some genes appear more than once on the same list. This duplication reflects multiple probe sets targeting the same sequence, or multiple probe sets targeting different sequences in the same UniGene cluster. The visualizations were prepared as described in Materials and methods, and we emphasize that the scale shown is normalized and thus arbitrary for each gene. Abbreviations are the same as for Figure 1. The complete lists of genes with annotations and p-values can be obtained from our website [32].
One goal of our study was to compare ANOVA and template matching, and their combination. In many cases the results from the two methods are comparable, and both methods give low p-values for many of the genes listed in Figure 2. An overall comparison of the strain-specific analysis using ANOVA and template matching is shown in Figure 3, where we plotted the p-values calculated for each gene by ANOVA against those calculated for the strain-specific template. Most points lie near the diagonal, showing that in general, the p-values agree well, though ANOVA has a tendency to give more low p-values and there are some significant outliers. In agreement with the trend illustrated in Figure 3, the number of genes that make each cut-off in Table 1 are very similar for strain specificity. The template-matching method, however, tends to give low p-values to many more genes on the basis of region-specificity. For all tissue effects, at a p-value of 10-5, the template method identifies more than twice as many genes as ANOVA, and at less conservative cut-offs the difference is even greater (Table 1). An example of a discrepancy is synuclein (GenBank C79089), which had a p-value of 8.6 × 10-7 based on a template match to the 'midbrain' pattern, but only 0.0011 for a region affect using ANOVA. The relatively poor ANOVA p-value appears to be due to a lower value for one of the four midbrain samples (Figure 2, last gene shown for 'Midbrain enriched'). Thus under some conditions ANOVA may be more conservative than template matching.
Figure 3.
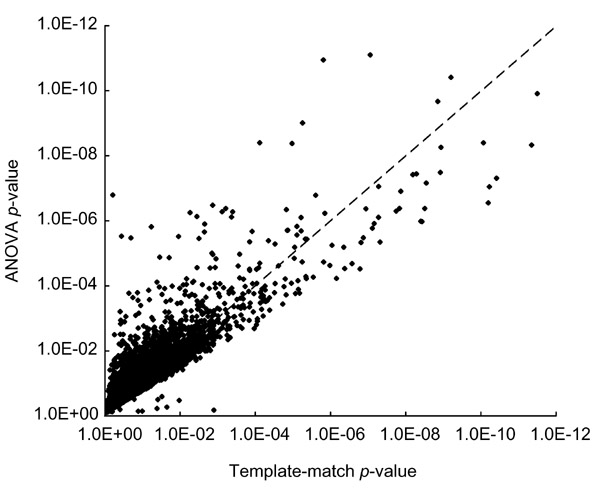
Comparison between template match and ANOVA p-values for strain-specific changes. The p-values for all 13,067 genes are plotted, with template-match p-values on the x-axis and ANOVA p-values on the y-axis. For most genes, the two methods are in good agreement.
The combined data shown in Figure 2 takes advantage of both methods. The genes shown were selected on the basis of both ANOVA and template-match p-values (see Materials and methods). By listing both p-values, we can choose to pay the most attention to genes in which we have good confidence on the basis of two different methods. For example, the genes listed for 'cortex enriched or deficient' in Figure 2 have fairly unremarkable template-match p-values; the p-values from the generally more conservative ANOVA method are substantially worse. This leads us to suggest that few, if any, of these genes are likely to have 'real' expression patterns that are in good agreement with our expectation for a cortex-specific gene. This is borne out by the visualizations shown in Figure 2. Compared to the genes shown for other regions such as cerebellum or hippocampus, the 'cortex enriched or deficient' genes have less striking expression patterns. Thus the numerical ranking appears to be a reasonable guide to making judgments about the significance of the expression patterns.
Strain-specific expression
At a p-value cut-off of 10-5, we found about 65 probe sets that show strain-specific variation in expression across all areas, compared to 24 genes showing overall differences between strains identified by Sandberg et al. [1]. The genes near the top of our ranking include many of those chosen by Sandberg [12], and all 24 genes identified by Sandberg et al. have p-values less than 10-3 by either ANOVA, template match, or both. Two examples of strain-specific genes not identified in the previous work are shown in more detail Figure 4a. The first is Sparc/osteonectin (testican), which was detected at lower levels in the C57B16 strain. The second is phosphatase ACP1/ACP2, with the opposite expression pattern. We rank ACP1/ACP2 and Sparc/osteonectin as having the 9th and 23rd strongest strain differences, respectively (both ACP1/ACP2 and Sparc/osteonectin are also shown in Figure 3). For comparison, Figure 4b shows β-globin, which was identified in [1] and was ranked 67th by template match. Overall, we rank 46 previously unrecognized genes as having stronger strain differences than β-globin.
Figure 4.
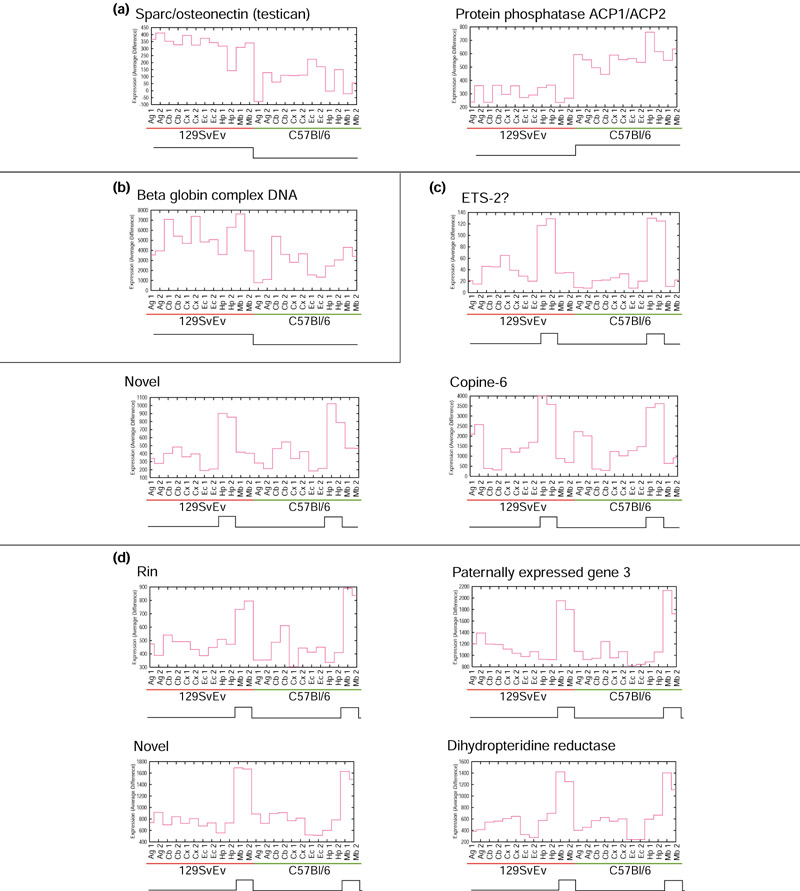
Unadjusted graphs of expression for examples of genes identified in this study. Arrays are plotted along the x-axis, and expression (average difference) is plotted on the y-axis. Below the x-axis of each graph is a schematic of the corresponding template. (a) Two typical examples of high-scoring genes showing strain-specific variation. (b) A gene identified by Sandberg et al. as having strain-specific variation, for comparison with (a). We gave this gene a much lower ranking than the genes shown in (a). (c) Three high-scoring genes showing hippocampus enrichment. (d) Four high-scoring genes showing midbrain enrichment.
Sandberg et al. [1] also identified an additional 49 genes that varied between strains for particular regions. We have not specifically searched for such genes; the ANOVA interaction effects would correspond most closely to such a category, but very few genes were found to have significant interaction effects (Table 1). Preliminary results (see Materials and methods: Availability of data and software) using templates that select specifically for genes expressed in one region in one strain suggest that this strategy will be useful for identifying more complex expression patterns as well as the simple ones we describe in detail here.
Region-specific expression
As with the strain-specific genes, we identify a number of putative region-specific genes in addition to those identified by Sandberg et al. [1]. The two methods lead to an estimate that, at a p-value of 10-5, up to approximately 600 genes show variability of expression with respect to brain region (Table 1), or around 6% of the total, compared to the 242 region-specific genes identified by Sandberg [12] and Sandberg et al. [1]. As for the strain-specific genes, some genes identified in [1] do not receive particularly high scores in our assays (Figure 2, and see Additional data files). For example, both hippocampal genes identified in [1] have p-values greater than 0.001 and do not appear in Figure 3. In general, however, most of the genes selected in [1] appear fairly high on our lists, although additional genes are often given even higher scores by our assays.
In agreement with Sandberg et al. [1], we find that the cerebellum is by far the most distinct of the brain regions studied. The bulk of the genes identified as showing region-specific effects appear to be due to cerebellum enrichment or depletion (339 out of 595 at template-match p < 10-5). The midbrain was the second most distinct single region (Table 1). In contrast, the cortex had the least distinct expression pattern, being very similar to other forebrain regions, as pointed out by Sandberg et al. When considered together, the forebrain (represented by cortex, amygdala, hippocampus and entorhinal cortex) has many genes that distinguish it from the midbrain and cerebellum (Table 1). Taken separately, the cortex, amygdala, hippocampus and entorhinal cortex had relatively few genes with distinct patterns (Table 1, Figure 2). The single amygdala-specific gene (ARP-1) identified by Sandberg et al. appears at the top of our list of amygdala-specific genes, and although we identify a few other high-scoring genes in that category, none approaches the specificity of ARP-1 (Figure 2). Details of a few examples of high-scoring genes we identified for hippocampus and midbrain are shown in Figure 4c and d, respectively.
In many cases, the expression patterns we report seem to be novel, but in others supporting evidence is found in the literature. We did not attempt a comprehensive literature survey for each gene, but support was readily found for the hippocampus enrichment of copine 6 [13], neuropeptide Y receptor Y2 [14], Atonal 2 [15], and kallikrein 8 (neuropsin) [16]. The same can be said for the midbrain enrichment of Rin [17], Peg3 [18], and nexin [19]. Some cerebellar-enriched genes for which we found supporting evidence include calsequestrin [20,21] and aldolase 3 (zebrin) [22].
Discussion
Our most important finding is that by using ANOVA and feature selection at even quite conservative cut-offs, we identify genes with strain- and region-dependent variation in gene expression that were not identified in the original analysis of the data [1]. These newly identified genes expand the range of genes that can be considered as candidates for underlying behavioral differences between the two strains and functional differences among brain regions. We were able to confirm the expression patterns of some of the genes we identified by examining the published literature. Although this examination was not exhaustive, we readily found supporting evidence for the tissue-specific expression patterns of a number of genes which were not identified in the previous surveys of the data. These results also show that the new patterns we identify with high confidence are unlikely to be spurious statistical artifacts, and give an indication that the sensitivity (the ability to avoid missing significant results) of our methods is higher than the ad hoc approach. We also note that our criteria are not necessarily less stringent than those of Sandberg et al. as many genes they selected do not rank particularly high in our results. Furthermore, in some cases, such as for the entorhinal cortex at more conservative p-value cut-offs, we select fewer genes than Sandberg et al. even though at the same cut-off for other regions we select more (Table 1). Thus, our increase in sensitivity does not come at the cost of selectivity (the ability to avoid spurious positive findings) compared to the ad hoc approach. In the remaining discussion, we evaluate our methodology in more detail, and then consider the biological significance of the results.
Evaluation of methodology
The difference in the genes we selected compared to Sandberg et al. [1] seems to depend on two factors: ignoring the absent/present calls, and using statistical methods that provide a score for each gene, rather than simply choosing to assign genes to the group of 'changed' genes. Sandberg et al. appear to have used criteria that were conservative given the measures used (that is, three out of four absent/present calls must be 'present' to consider a gene expressed). But, the fact that the standard ANOVA often does not support the evaluation based on absent/present calls supports the idea that absent/present calls, even if used conservatively, are neither specific nor sensitive enough.
There are two particular drawbacks to using absent/present calls for this type of analysis. Most important, each absent/present call is based on only a single array. If one has only a single array, such a guideline for the ability to detect a given gene may be useful. When multiple measurements are made, however, as in the dataset considered here, there is no set method for evaluating the data. Is a gene 'present' in the experiment when it is 'present' on three of four arrays, or are two out of four arrays sufficient? Instead of such ad hoc criteria, we used statistical comparisons of the samples. This means that low-magnitude signals, which might have been called 'absent', can turn out to be reliable and meaningful when several observations are made. A second problem with the absent/present calls is that they are based on essentially arbitrary thresholds, with no simple estimate of the risk of false positives or false negatives. The use of methods that provide p-values allows a much finer-grained analysis. For these reasons, we feel that absent/present calls should generally not be used to analyze experiments where replicates were performed.
Despite the advantages of ignoring absent/present calls, the 'absolute difference' expression value provided by the Affymetrix software is not ideal. In particular, large negative values occur at a much higher rate than expected (data not shown). These negative values are probably due to problems with mismatch probes, as described in the Materials and methods section. A more thorough analysis would remove, or otherwise correct for, probe pairs containing such problematic probes from the raw data, which we did not have access to here. At least two groups have described alternative methods that start with the 'perfect match/mismatch' pair data [23,24], which may be better at identifying and eliminating such troublesome data points.
The region-specific genes listed in Figure 2 reflect a combination of the two methods we used. This combination takes advantage of desirable features of both methods: ANOVA can detect any effect of region on expression, whereas template matching looks specifically for one pattern at a time. By sorting ANOVA hits into regions based on template-match p-values, we identify some genes that were missed by template matching. In general, this is caused by the ability of ANOVA to detect significant expression patterns that do not fit any particular template very well, but when examined manually reveal interesting patterns related to the template. We note that other methods of post hoc testing ANOVA data exist [7]. A benefit of the template-match method is that it is very simple to apply, and we believe we found some patterns that would be fairly difficult to find using other methods. Thus we consider the template-match method a useful complement to standard post hoc tests.
In the search for region-specific genes, ANOVA tended to be more conservative than template matching. This is probably because ANOVA retains information about the experimental design; for the example of synuclein given in the results, it 'knows' that there is a large disagreement between two matched replicates in the midbrain samples, whereas the template match based on the correlation coefficient considers all variations to be essentially equivalent. The fact that there are only four samples for each tissue, while there are twelve for each strain, means that tests for tissue-specific genes are even more susceptible to the effect of noise in the data. This helps explain why the discrepancies between template match and ANOVA was larger for the region-specific genes than for strain-specific genes.
The methods we describe have some limitations. ANOVA loses power when the number of replicates is not the same for all the conditions, and the experiment must also follow a complete block design, where all possible combinations of factors are tested. Even in cases where these conditions are not adhered to, multiple t-tests (which are generally proscribed in multivariate statistics in favor of ANOVA) would be preferable to resorting to ad hoc methods for assessing statistical significance of the results. The template-match method has the potential drawback that if used alone, one would miss potentially interesting patterns that do not resemble the template(s) being tested. This may be acceptable if there is a clear idea of what is being sought in the experiment, but is possibly detrimental in studies relying on exploration and serendipity to find patterns. The ANOVA/template-match combination ameliorates this problem as one can choose to be guided primarily by the ANOVA p-value (which is not pattern specific) while using the template to reduce the complexity of the analysis by partitioning the data into groups based on interesting patterns.
Biological significance
Our results resemble those of Sandberg et al. [1] in that many of the genes given high scores have previously defined roles in the function of the nervous system. At the same time, many of the high scoring genes have 'housekeeping' roles or more general cellular functions that are not specific to the nervous system. Currently this conclusion is based on a manual examination of the annotations and the literature associated with top-scoring genes. In the future, as annotations improve, we hope to be able to perform such assessments quantitatively and objectively. Clearly, the ranked list of genes we provide can only act as an input to further studies of nervous system function, and does not provide any ready answers to questions about behavioral or functional differences.
As already indicated, another general conclusion from our study is that the variation between regions and strains appears to be greater than originally indicated by the analysis of Sandberg et al. In a commentary on the original study, Geschwind [25] sought explanations for the apparent paucity of region-specific and strain-specific gene expression. Although the sensitivity of the microarray assay itself may be at issue, our results clearly show that the use of different computational analysis methods has a dramatic effect on the interpretation of the data. Geschwind [25] also expressed surprise that the cerebellum, which is histologically more homogeneous than the other brain regions assayed, was discovered to have the largest number of uniquely expressed genes. This interpretation of the data assumes that the other regions such as cortex and hippocampus, being more complex and heterogeneous, should have more differences between each other as well as from the cerebellum. However, because hippocampus, neocortex and entorhinal cortex are all cortical structures with many cell types in common, it is not surprising that they have similar gene expression profiles. In contrast, the cerebellum has a distinct developmental origin, as well as a unique cellular composition, compared to all the other regions assayed. Thus it is not at all surprising that the cerebellum is the most distinct region. In agreement with this line of logic, the mid-brain, which is histologically and developmentally distinct from the cortex and cerebellum, was the next most distinct region. We also note that, contrary to the suggestion of Geschwind, the fact that the cerebellum had the largest number of 'uniquely expressed genes' is not equivalent to stating that the cerebellum has the highest molecular complexity [25]. To assess this type of complexity, the total number of mRNA species expressed in the tissue would have to be measured, but such information is not readily available. If we take the number of 'present' calls provided by the Affymetrix software as a first approximation of a complexity measure, we find no evidence that the cerebellum expresses more genes than other regions assayed by Sandberg et al. (data not shown).
A recent study by Zirlinger et al. [26] also analyzed region-specific gene expression in the amygdala, cerebellum and hippocampus, as well as the olfactory bulb and periaqueductal gray, for a single mouse strain. Our results, and those of Sandberg et al., can be compared to theirs, though there were several important differences in their study that need to be taken into account. First, unlike the study of Sandberg et al., no replicates were performed for each region, although in situ hybridization was used to confirm some of the array results. In addition, the strain Zirlinger et al. used was not one used by Sandberg et al., and there were also differences in the methods for isolating the tissues used by the two groups. Finally, Zirlinger et al. assayed a larger number of genes and ESTs (approximately 34,000) than were in the dataset we studied (approximately 13,000). Some comparisons can be made, however.
In agreement with our results and the results of Sandberg et al., Zirlinger et al. found that the cerebellum was most distinct from the amygdala and hippocampus. Some of the genes they identified are also given high scores by our tests, adding further support to the validity of our results. Of the cerebellar genes identified by Zirlinger et al., 35 of the 70 which are on the Mu11K arrays have ANOVA p-values less than 10-4, while for the template match the number is 39/70. For the amygdala genes, the numbers are 2/10 (ANOVA) and 1/10 (template match). (None of the hippocampal genes they identified appears to be on the Mu11K arrays.) One possible source of discrepancies here is that Zirlinger et al. did not assay neocortex, midbrain or entorhinal cortex. Expression in these regions would decrease their significance in our assays but would not affect their results. The differences in our results suggest that caution is advisable in making broad interpretations based on large-scale gene expression data from a limited sample of tissues and genetic backgrounds, and that confirmation by another method is desirable.
In summary, our results expand the number of candidate genes for investigation of the genetic basis of behavioral differences between the C57BL/6 and 129SvEv mouse strains, as well as the number of genes that might be responsible for structural and functional differences between brain regions in the mouse. More sophisticated methods than ours could be envisioned, but ANOVA and template matching have much to recommend them. They yield results with high sensitivity and selectivity, and are fast, simple and based on commonly understood statistical principles. They should also be readily applicable to other complex expression datasets.
Materials and methods
Data
Expression data was downloaded from the FTP site established by Sandberg et al. [1]. The data had already been normalized and analyzed using the Affymetrix software. Affymetrix arrays use sets of around 20 pairs of oligonucleotide probes to represent a single gene or EST. Each pair consists of a 'perfect-match' primer and a 'mismatch' primer that has a single base change from the perfect match and is meant to control for nonspecific hybridization effects. For each gene (probe set), the Affymetrix software calculates an 'average difference' between the intensities of the perfect-match and mismatch primers, and additionally makes an overall determination as to the absence or presence of a gene (absent/present call) in the sample. For our methods, we used only the average difference measures. We assembled the average difference data for the 24 arrays into a single matrix of 24 columns and 13,067 rows. Updated annotations for the sequences on the arrays were obtained from the UniGene [27] and the FANTOM [28,29] databases.
Feature selection
A simple method of selecting genes that display particular characteristics across m experiments is to compare each profile to a template representing the pattern being sought. We define a template as a binary vector of length m with a value of o corresponding to one expression value, and a value of 1 corresponding to a contrasting expression value. For example, a search for genes showing strain-specific variation in expression could be performed by comparing each profile to the following template, given that the data columns are grouped by strain (12 in 129SvEv, followed by 12 in C57BL/6):
0 0 0 0 0 0 0 0 0 0 0 0 1 1 1 1 1 1 1 1 1 1 1 1
The Pearson correlation coefficient of the profile with the template was used as a simple measure of the agreement of the profile with the pattern. For two profiles X and Y, the correlation coefficient r is calculated using the formula
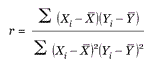
where 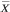 and
and 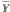 are the means of each profile, and sums are taken for i ranging over all m values in each profile. If in the template a value of o is interpreted as low expression and 1 as high expression, the example pattern would identify samples with lower expression in the 129SvEv than C57BL/6. The correlation is only sensitive to the shape of the profile, not the amplitude, so all profiles having a shape like the template will be given higher scores regardless of the actual expression values. To allow both matches and 'anti-matches', in some cases we used the absolute value of the correlation coefficient, effectively allowing the meanings of ones and zeroes in the template to be reversed for any particular comparison. We note that templates that contain continuous values instead of binary ones could be applied in the same way, allowing one to search for specific patterns of greater complexity, but binary vectors were sufficient for the purposes of this study. High correlation values correspond to a good match. The p-values were calculated using a t test on the correlation coefficient [30]. We prepared templates corresponding to each of the regions assayed by Sandberg et al., as well as using the strain-distinguishing template shown in the example above.
are the means of each profile, and sums are taken for i ranging over all m values in each profile. If in the template a value of o is interpreted as low expression and 1 as high expression, the example pattern would identify samples with lower expression in the 129SvEv than C57BL/6. The correlation is only sensitive to the shape of the profile, not the amplitude, so all profiles having a shape like the template will be given higher scores regardless of the actual expression values. To allow both matches and 'anti-matches', in some cases we used the absolute value of the correlation coefficient, effectively allowing the meanings of ones and zeroes in the template to be reversed for any particular comparison. We note that templates that contain continuous values instead of binary ones could be applied in the same way, allowing one to search for specific patterns of greater complexity, but binary vectors were sufficient for the purposes of this study. High correlation values correspond to a good match. The p-values were calculated using a t test on the correlation coefficient [30]. We prepared templates corresponding to each of the regions assayed by Sandberg et al., as well as using the strain-distinguishing template shown in the example above.
Analysis of variance (ANOVA) and combining ANOVA with feature selection
A standard linear two-way analysis of variance [7,30] was performed independently on the data for each gene. The effects examined for each gene were strain, region, and interactions between strain and region. This is expressed by the following linear equation:
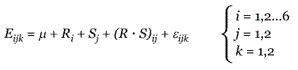
where Eijk is the measured expression level of the gene in replicate k from one of six regions i in one of two strains j, μ is the overall mean expression level of the gene, and R and S are effects due to region and strain, respectively. The R·S term represents interactions between strain and region, which might be detected if a gene is expressed only in specific regions in only one strain, for example. The error term (ε) accounts for random variation from replicate to replicate; that is, any effect not accounted for by the other terms in the model. Thus this model assumes that the measured expression level of a gene can be described as a linear combination of effects due to measurement error, strain, region, and interactions between strain and region. The null hypothesis is that a given factor does not have an effect on expression, meaning that the corresponding term in the equation is zero. The extent to which the fitted parameters R, S and R·S for a gene are non-zero provides the basis of the ANOVA. We generated p-values for each of the three effects for each gene using a standard F-test [7].
As discussed, ANOVA does not allow one to directly distinguish between effects due to particular levels of one parameter. Specifically, ANOVA might indicate that there was a significant effect of region on expression, but does not directly reveal which regions show different expression from any others. Thus, one needs to apply a post hoc test to the ANOVA results to determine which regions, if any, show differences from others. Typically this type of multiple comparison is done using statistical tests such as the Tukey or Scheffé methods [7]. In our study, each region must be compared to each other region, so 15 comparisons must be made in total for each gene. In practice, however, interpretation of these results is more complicated: an additional stage of analysis must be performed to generate, for example, genes expressed at higher levels in the cerebellum than in all other regions, as opposed to any one other region (as is given by Tukey or Scheffé methods). For our study we used a simple strategy suggested by the idea of template matching: we choose genes fitting a pattern in which we are interested from a subset of genes with low ANOVA p-values for region effects. Specifically, we first selected all genes which had region-effect ANOVA p-values < 0.001, and then applied template matching to this set to select genes which fit a particular pattern as described above, at a p-value of 0.0001 or less. These arbitrary, and fairly lax, thresholds were used because they proved to give reasonable numbers of genes in each partition. The resulting groups of genes can then be ranked according to either their ANOVA p-value or by the correlation with the template.
Interpretation of p-values
An important issue in interpreting the p-values generated by tests on single genes is the effect of multiple tests. Specifically, a test performed with a chosen p-value cut-off of 0.01, repeated 13,000 times, will on average generate 130 false positives if each test is independent. Corrections for this multiple testing problem are well known in statistics but are somewhat controversial [31]. Methods used by Tusher et al. [4] and Callow et al. [5] attempt to provide appropriate corrections for microarray experiments, but there is, as yet, no consensus on which methods to apply. For the purposes of presenting our data here we report results at three different thresholds, which would, by the Bonferroni criteria, yield on average about 13, 0.13, or 0.013 experiment-wide false positives under the assumptions of our tests (that is, p-values of 0.001, 10-5 and 10-6, respectively). Because these cut-offs assume that each gene is independent, which is clearly not the case, these false positive estimates are probably conservative with respect to the multiple testing issue. Because we assume here that the standard F and t distributions are adequate for describing the statistics we use, however, using a conservative cut-off provides some additional protection from type I errors due to potential inaccuracy of the uncorrected p-values.
Filtering selected genes
All the analysis described above was performed without adjusting or filtering the raw average difference values. But, a difficulty one commonly encounters with Affymetrix data is that many probe sets yield large negative expression (average difference) measures. Large negative expression values occur when one or more mismatch primers in a probe set have much higher intensities than their corresponding perfect match primers. This may occur because the mismatch primer is binding to some other nucleotide in the sample which does not represent the gene for which a measurement is indented. Because the Affymetrix software determines the presence or absence of expression of a gene in part on the basis of how many probe pairs give high intensities, not just the average across the probe pairs, some genes may be flagged as present despite having negative average differences. Obviously this is not desirable; one would like to correct for problematic mismatch probes before calculating the average difference. A stop-gap measure, which we used here as we did not have access to the original raw data files, is to remove from consideration entire probe sets which have this problem. Specifically, genes with more than 6 values out of 24 that were less than -20 were rejected; in practice this method is quite conservative, removing only the most problematic genes. We note that a few genes (about 20 of approximately 240) selected by Sandberg et al. would be removed from consideration by our filter.
Comparison with the results of Sandberg et al. [1]
A list of the genes identified in the previous work on this dataset were obtained from Tables 1 and 2 of Sandberg et al. [1] and Table 4 of Sandberg [12]. A few groups of genes referred to in these studies are not specifically identified in the publications: the selection of eight hippocampus-enriched, two amygdala-decreased, five entorhinal cortex-enriched and three entorhinal cortex-decreased genes is mentioned, but the genes are not specifically identified in [1]. From our other comparisons, it is fairly likely that some of these genes were given high scores in our analysis, so our comparison to the results of Sandberg et al. for those areas should be regarded as incomplete.
Visualization of the data
In several of our figures, microarray data is displayed as a matrix of rectangles, each corresponding to an individual measurement of one probe set/gene (rows) in one sample (columns). The color, using a 'black-body' scale (from low values represented by black to dark red to orange to yellow to high values represented by white) indicates the relative level of expression of the gene across the different conditions. The values for each gene have been adjusted to a mean of zero and a variance of one to facilitate comparisons between genes, and values > 1 (or < -1) have been adjusted to 1 (or -1), increasing the contrast of the images.
Availability of software
The software used to create the visualizations of the data and to perform template matching and ANOVA is available on request from the authors.
Additional data files
The following text files containing the results of our analyses are available with this article:
Anova results (the first column is the probe name and the final results are listed as "ff" (first factor) "sf" (second factor) and fxsf (interaction); the first factor is strain, the second is region. The numbers in those columns are F values; the other columns are intermediate values used to calculate F). This file may take some time to open.
Template match results (each file contains three columns. The first column is the probe name, the second is the correlation coefficient with the template. The third column is the p-value): cerebellum; hippocampus; midbrain; cortex; fore-brain; entorhinal cortex; amygdala; strain.
All data and results, including detailed browseable tables of the selected genes, and supplementary data and information, are also available from our website [32].
Supplementary Material
Anova results
Template match results: cerebellum
Template match results: hippocampus
Template match results: midbrain
Template match results: cortex
Template match results: fore-brain
Template match results: entorhinal cortex
Template match results: amygdala
Template match results: strain
Acknowledgments
Acknowledgements
We thank Anthony Ferrante for comments on an early version of the manuscript. W.S.N. is funded by an Award in Bioinformatics from the PhRMA Foundation, and by National Science Foundation grants DBI-0078523 and IIS-0093302.
References
- Sandberg R, Yasuda R, Pankratz DG, Carter TA, Del Rio JA, Wodicka L, Mayford M, Lockhart DJ, Barlow C. Regional and strain-specific gene expression mapping in the adult mouse brain. Proc Natl Acad Sci USA. 2000;97:11038–11043. doi: 10.1073/pnas.97.20.11038. ftp://ftp.gnf.org/pub/papers/brainstrain/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipshutz RJ, Fodor SP, Gingeras TR, Lockhart DJ. High density synthetic oligonucleotide arrays. Nat Genet. 1999;21(Suppl):20–24. doi: 10.1038/4447. [DOI] [PubMed] [Google Scholar]
- Crawley JN, Belknap JK, Collins A, Crabbe JC, Frankel W, Henderson N, Hitzemann RJ, Maxson SC, Miner LL, Silva AJ, et al. Behavioral phenotypes of inbred mouse strains: implications and recommendations for molecular studies. Psychopharmacology (Berl) 1997;132:107–124. doi: 10.1007/s002130050327. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callow MJ, Dudoit S, Gong EL, Speed TP, Rubin EM. Microarray expression profiling identifies genes with altered expression in HDL-deficient mice. Genome Res. 2000;10:2022–2029. doi: 10.1101/gr.10.12.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long AD, Mangalam HJ, Chan BY, Tolleri L, Hatfield GW, Baldi P. Improved statistical inference from DNA microarray data using analysis of variance and a Bayesian statistical framework. Analysis of global gene expression profiling in Escherichia coli K12. J Biol Chem. 2001;276:19937–19944. doi: 10.1074/jbc.M010192200. [DOI] [PubMed] [Google Scholar]
- Sahai H, Agell MI. The Analysis of Variance Boston: Birkhauser, 2000.
- Hill AA, Hunter CP, Tsung BT, Tucker-Kellogg G, Brown EL. Genomic analysis of gene expression in C. elegans. Science. 2000;290:809–812. doi: 10.1126/science.290.5492.809. [DOI] [PubMed] [Google Scholar]
- Kerr MK, Martin M, Churchill GA. Analysis of variance for gene expression microarray data. J Comput Biol. 2000;7:819–837. doi: 10.1089/10665270050514954. [DOI] [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown PO, Herskowitz I. The transcriptional program of sporulation in budding yeast. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- Sandberg R. Gene expression profiling of brain regions in inbred mouse strains reveals candidate genes for phenotypic variation Masters Thesis in Biomedicine Stockholm: Karolinska Institute; 2000.
- Nakayama T, Yaoi T, Kuwajima G. Localization and subcellular distribution of N-copine in mouse brain. J Neurochem. 1999;72:373–379. doi: 10.1046/j.1471-4159.1999.0720373.x. [DOI] [PubMed] [Google Scholar]
- Gackenheimer SL, Schober DA, Gehlert DR. Characterization of neuropeptide Y Y1-like and Y2-like receptor subtypes in the mouse brain. Peptides. 2001;22:335–341. doi: 10.1016/s0196-9781(01)00335-7. [DOI] [PubMed] [Google Scholar]
- Schwab MH, Druffel-Augustin S, Gass P, Jung M, Klugmann M, Bartholomae A, Rossner MJ, Nave KA. Neuronal basic helix-loop-helix proteins (NEX, neuroD, NDRF): spatiotemporal expression and targeted disruption of the NEX gene in transgenic mice. J Neurosci. 1998;18:1408–1418. doi: 10.1523/JNEUROSCI.18-04-01408.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZL, Yoshida S, Kato K, Momota Y, Suzuki J, Tanaka T, Ito J, Nishino H, Aimoto S, Kiyama H, et al. Expression and activity-dependent changes of a novel limbic-serine protease gene in the hippocampus. J Neurosci. 1995;15:5088–5097. doi: 10.1523/JNEUROSCI.15-07-05088.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Della NG, Chew CE, Zack DJ. Rin, a neuron-specific and calmodulin-binding small G-protein, and Rit define a novel subfamily of ras proteins. J Neurosci. 1996;16:6784–6794. doi: 10.1523/JNEUROSCI.16-21-06784.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Keverne EB, Aparicio SA, Ishino F, Barton SC, Surani MA. Regulation of maternal behavior and offspring growth by paternally expressed Peg3. Science. 1999;284:330–333. doi: 10.1126/science.284.5412.330. [DOI] [PubMed] [Google Scholar]
- Kury P, Schaeren-Wiemers N, Monard D. Protease nexin-1 is expressed at the mouse met-/mesencephalic junction and FGF signaling regulates its promoter activity in primary met-/mesencephalic cells. Development. 1997;124:1251–1262. doi: 10.1242/dev.124.6.1251. [DOI] [PubMed] [Google Scholar]
- Volpe P, Gorza L, Brini M, Sacchetto R, Ausoni S, Clegg DO. Expression of the calsequestrin gene in chicken cerebellum Purkinje neurons. Biochem J. 1993;294:487–490. doi: 10.1042/bj2940487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei K, Stukenbrok H, Metcalf A, Mignery GA, Sudhof TC, Volpe P, De Camilli P. Ca2+ stores in Purkinje neurons: endoplasmic reticulum subcompartments demonstrated by the heterogeneous distribution of the InsP3 receptor, Ca(2+)-ATPase, and calsequestrin. J Neurosci. 1992;12:489–505. doi: 10.1523/JNEUROSCI.12-02-00489.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes R, Herrup K. Aldolase C/zebrin II and the regionalization of the cerebellum. J Mol Neurosci. 1995;6:147–158. doi: 10.1007/BF02736761. [DOI] [PubMed] [Google Scholar]
- Schadt EE, Li C, Su C, Wong WH. Analyzing high-density oligonucleotide gene expression array data. J Cell Biochem. 2000;80:192–202. [PubMed] [Google Scholar]
- Efron B, Tibshirani R, Goss V, Chu G. Microarrays and their use in a comparative experiment Stanford: Stanford University Department of Statistics. 2000.
- Geschwind DH. Mice, microarrays, and the genetic diversity of the brain. Proc Natl Acad Sci USA. 2000;97:10676–10678. doi: 10.1073/pnas.97.20.10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirlinger M, Kreiman G, Anderson DJ. Amygdala-enriched genes identified by microarray technology are restricted to specific amygdaloid subnuclei. Proc Natl Acad Sci USA. 2001;98:5270–5275. doi: 10.1073/pnas.091094698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCBI-UniGene http://www.ncbi.nlm.nih.gov/UniGene/
- FANTOM: Functional Annotation of Mouse http://www.gsc.riken.go.jp/e/FANTOM/
- Kawai J, Shinagawa A, Shibata K, Yoshino M, Itoh M, Ishii Y, Arakawa T, Hara A, Fukunishi Y, Konno H, et al. Functional annotation of a full-length mouse cDNA collection. Nature. 2001;409:685–690. doi: 10.1038/35055500. [DOI] [PubMed] [Google Scholar]
- Zar JH. Biostatistical Analysis 4th edn Upper Saddle River, NJ: Prentice Hall. 1999.
- Westfall PH, Young SS. Resampling-based Multiple Testing New York: John Wiley & Sons. 1993.
- Mouse brain data http://rbp1sun.cpmc.columbia.edu/pub/mousebrain/web/index.html
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Anova results
Template match results: cerebellum
Template match results: hippocampus
Template match results: midbrain
Template match results: cortex
Template match results: fore-brain
Template match results: entorhinal cortex
Template match results: amygdala
Template match results: strain