

Abstract
Little is known about the genetic influence on BMI trajectory throughout adulthood. We created a genetic risk score (GRS) comprising 97 adult BMI-associated variants among 9,971 women and 6,405 men of European ancestry. Serial measures of BMI were assessed from 18 (women) or 21 (men) years to 85 years of age. We also examined BMI change in early (from 18 or 21 to 45 years of age), middle (from 45 to 65 years of age), and late adulthood (from 65 to 80 years of age). GRS was positively associated with BMI across all ages, with stronger associations in women than in men. The associations increased from early to middle adulthood, peaked at 45 years of age in men and at 60 years of age in women (0.91 and 1.35 kg/m2 per 10-allele increment, respectively) and subsequently declined in late adulthood. For women, each 10-allele increment in the GRS was associated with an average BMI gain of 0.54 kg/m2 in early adulthood, whereas no statistically significant association was found for BMI change in middle or late adulthood or for BMI change in any life period in men. Our findings indicate that genetic predisposition exerts a persistent effect on adiposity throughout adult life and increases early adulthood weight gain in women.
Introduction
Overweight and obesity represent a major global health challenge, affecting ∼40% of adults worldwide (1). Although the obesity epidemic is thought to be due mainly to environmental changes that favor a positive energy balance and weight gain, substantial evidence from twin studies supports high heritability and a strong genetic basis for individual predisposition to obesity. Increasing data suggest that the genetic influence on obesity may vary according to age (2). A recent systematic review of twin studies showed that the heritability estimates of BMI increased steadily from childhood until ∼20 years old, at which point it reached its peak (3). However, this age-dependent pattern is identified based on only a few studies that collected BMI data only in early to middle adulthood.
In contrast, several studies have examined the effects of common genetic variants on BMI profiles at different life periods. However, these studies are limited by cross-sectional design (4–7) and inclusion of only a few adult BMI-associated single nucleotide polymorphisms (SNPs) (7–10). For studies using a longitudinal design with repeated measures of BMI, many have focused on childhood and adolescence, with findings indicating an influence of BMI-associated loci on accelerating childhood weight gain (11–17). In contrast, little is known about the genetic influences on BMI trajectory in middle and late adulthood (18–21). Most of the existing studies are limited by the small number of assessed SNPs (7,8,22) and BMI assessments at only two time points (7,22–25). Given that BMI tracking weakens from childhood to late adulthood (26) and most weight gain occurs in middle adulthood, possibly through gene-by-environment interaction (27), a better understanding about the genetics that underlie the trajectory of adiposity throughout the entire adult life will provide critical insights into the origins of the obesity epidemic and help identify the optimal time window for effective interventions to modify genetic risk (28).
Therefore, we performed a longitudinal analysis for the aggregate effect of individual obesity-associated variants on BMI profiles from early to late adult life. We created a genetic risk score (GRS) using the 97 SNPs associated with adult BMI in the recent genome-wide association study (GWAS) meta-analyses (Supplementary Table 1) (29). We associated the GRS with BMI level and BMI change throughout adulthood among participants from two large ongoing cohort studies in the U.S., the Nurses’ Health Study (NHS) and the Health Professionals Follow-Up Study (HPFS).
Research Design and Methods
Study Population
The NHS enrolled 121,700 female registered nurses aged 30–55 years in 1976, and the HPFS enrolled 51,529 male professionals aged 40–75 years in 1986. In both cohorts, follow-up questionnaires were mailed to participants biennially to collect updated information about their physical characteristics, lifestyle, and medical information. Between 1989 and 1990, 32,826 women from the NHS and between 1993 and 1995, 18,225 men from the HPFS returned a blood specimen on ice packs by overnight courier. For the current study, we used data from 9,980 initially healthy women and 6,406 initially healthy men of European ancestry who had been genotyped in previous nested case-control GWASs for various outcomes, including colorectal cancer, gout, kidney stone, type 2 diabetes, glaucoma, coronary heart disease, percent mammographic density (NHS only), endometrial cancer (NHS only), breast cancer (NHS only), and prostate cancer (HPFS only). After excluding 9 women and 1 man who did not report their BMI at any age, a total of 9,971 women and 6,405 men were included in the final analysis. The study protocol was approved by the institutional review boards of Brigham and Women’s Hospital and Harvard T.H. Chan School of Public Health.
BMI Assessment
In both cohorts, participants reported their height at baseline enrollment and reported their body weight in biennial questionnaires. Women in the NHS recalled their weight at 18 years of age in 1980, and men in the HPFS recalled weight at 21 years of age in 1986. Based on these data, we calculated BMI as weight in kilograms divided by the square of height in meters at 18 or 21 years and from 45 to 85 years of age in a 5-year interval. To minimize random variation, we used the average BMI within 2 years for each of the ages since 45 years of age. We also calculated BMI at menopause in women using the weight information reported on the questionnaire when participants had undergone menopause.
Self-reported weight has shown excellent accuracy in pervious validation studies of 140 women and 123 men drawn from the two cohorts (30). The correlation coefficient was 0.97 between self-reported weight and the average of two weight measurements taken by technicians ∼6 months apart. Recalled weight at 18 years of age has also been validated in the NHS II cohort, with a correlation coefficient of 0.87 with weight recorded on college or nursing school records at 18 years of age (31). Although not validated in the HPFS, recalled weight during early adulthood in men has also been shown to be accurate (r = 0.80) in other studies (32).
Genotyping and Computation of GRS
Details about genotyping and imputation of SNPs included in the GRS have been described elsewhere (33). All of the 97 SNPs were genotyped or had a high imputation quality score (r2 ≥ 0.8), as assessed by the MACH software (version 1.0.16; Center for Statistical Genetics, University of Michigan). The GRS was created using an established weighting method. Each of the 97 SNPs was recoded as 0, 1, or 2 according to the number of risk alleles (BMI increasing alleles) and weighted by its relative effect size (β coefficient) derived from the most recent meta-analysis of GWAS (29). The GRS was calculated using the equation: 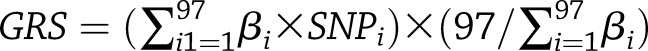 , where βi is the effect size of the SNPi on BMI. Each unit of the GRS represented one risk allele, and the GRS could range from 0 to 194, with a higher score indicating greater genetic predisposition to obesity.
, where βi is the effect size of the SNPi on BMI. Each unit of the GRS represented one risk allele, and the GRS could range from 0 to 194, with a higher score indicating greater genetic predisposition to obesity.
In a sensitivity analysis, we created an alternative GRS based on the SNPs that have been associated with BMI in adolescents and young adults in a previous GWAS (5). Because none of these 8 SNPs was included in our 97-SNP pool, we first identified proxy SNPs using the LDlink online tool (34) and successfully found proxies for 6 of the 8 SNPs with D’ >0.80 (Supplementary Table 2). We then used these six SNPs to create a GRS for early-life BMI weighted by the effect size derived from the GWAS.
Statistical Analysis
To assess potential sex difference, all of the statistical analyses were first performed in each cohort separately, and P for GRS-sex interaction was then calculated in the pooled cohorts by including a product term between GRS and sex in the model. Because not all participants had BMI measurements at each age, to control for the influence of birth cohort when assessing the gene-by-age effects, we adjusted BMI at each age for year of birth using linear regression. To facilitate interpretation, we added to the regression residuals a constant, the predicted mean BMI for the median birth of year in the study population. We used these adjusted BMIs throughout our analysis.
We examined the association of GRS with BMI at each age using linear regression. Longitudinal effects of GRS on BMI were examined by a linear mixed-effects model with age as a covariate. To accommodate any potential nonlinear relationship between age and BMI, we also included a quadratic term of age in the model. A random effect of age was also included to account for repeated measures of BMI at different ages. To evaluate the modifications of genetic effects by age, we calculated the P for GRS-age interaction via a likelihood ratio test by comparing the models with and without the product terms between age and quadratic age and GRS (degree of freedom = 2).
Given the potential influence of menopause on body weight (35), we also examined the associations of GRS with adulthood BMI according to age at menopause in women (<45, 45–49, 50–54, and ≥55 years) and tested the interaction between GRS and age at menopause via the F test.
We calculated BMI change during three life periods: 1) early adulthood (from 18 or 21 to 45 years of age); 2) middle adulthood (from 45 to 65 years of age); and 3) late adulthood (from 65 to 80 years of age). In women, we also studied BMI change from 18 years to menopause and from menopause to 65 years of age. We examined the effects of GRS on BMI change in different life periods using linear regression models. To assess whether the genetic effects on weight change vary by baseline adiposity, we further stratified by BMI at the starting age of each period (<22.5, 22.5–24.9, and ≥25.0 kg/m2). In addition to BMI of 25.0 kg/m2, which is a generally accepted cutoff for overweight, we used 22.5 kg/m2 to identify the lean group. This cutoff, albeit somewhat arbitrary, was chosen to ensure that each BMI stratum had a reasonable sample size for the stratified analysis. We also calculated the P for GRS-baseline BMI interaction using the Wald test for the product term between GRS and the starting BMI for each age period (continuous).
To account for multiple testing, we corrected the statistical significance level to α = 0.05/45 = 0.001 by the Bonferroni correction for the 45 hypothesis tests, including the main associations of GRS with BMI at 10 different ages in each sex and the interactions with sex (10 × 3 = 30), the main associations of GRS with BMI change in three different life periods in each sex and the interactions with sex (3 × 3 = 9), and the interactions between GRS and baseline BMI for BMI change in three different life periods in each sex (3 × 2 = 6). We recognized the use of multiple comparisons and interpreted our data cautiously. SAS 9.4 was used for all analyses (SAS Institute Inc., Cary, NC). All statistical tests were two-sided with the corrected type I error rate of α = 0.001.
Results
Table 1 shows the basic characteristics of study participants. For both women and men, the mean GRS was 88, and the median year of birth was 1931. The mean BMI increased from early adulthood, peaked at 65 years of age, and then started declining thereafter. Similar BMI measurements were observed between our study participants and the overall cohorts (Supplementary Table 3), indicating the representativeness of our study sample.
Table 1.
Descriptive statistics of GRS and BMI at different ages in participants from the NHS (women) and HPFS (men)
| Variable | Women |
Men |
||
|---|---|---|---|---|
| n | Mean (SD) or median (interquartile range) | n | Mean (SD) or median (interquartile range) | |
| GRS | 9,971 | 88 (6) | 6,405 | 88 (6) |
| Year of birth, median (interquartile range) | 9,971 | 1931 (1926–1937) | 6,405 | 1931 (1924–1938) |
| BMI at different ages (years), kg/m2 | ||||
| 18 for women, 21 for men | 9,490 | 21.5 (3.0) | 6,190 | 23.1 (2.8) |
| 45 | 6,249 | 24.5 (4.9) | 1,733 | 25.5 (3.5) |
| 50 | 8,800 | 25.5 (5.0) | 2,595 | 25.6 (3.6) |
| 55 | 9,799 | 26.2 (5.1) | 3,778 | 26.0 (3.7) |
| 60 | 9,699 | 26.8 (5.4) | 4,901 | 26.3 (3.6) |
| 65 | 9,232 | 27.1 (5.4) | 5,789 | 26.5 (3.7) |
| 70 | 7,667 | 26.6 (5.2) | 4,773 | 26.3 (3.7) |
| 75 | 5,506 | 26.1 (5.1) | 4,126 | 26.0 (3.6) |
| 80 | 3,023 | 25.2 (4.8) | 2,417 | 25.4 (3.4) |
| 85 | 891 | 24.3 (4.5) | 1,331 | 24.6 (3.3) |
Figure 1 presents the associations of GRS with BMI at each age (detailed data are reported in Supplementary Table 4). GRS was positively associated with BMI across all ages, and the associations were stronger in women than in men (P for GRS-sex interaction ranged from <0.0001 to 0.15). In women, the mean BMI difference (β) per 10-allele increment in the GRS increased from 0.76 kg/m2 (95% CI 0.66–0.85) at 18 years of age to 1.30 kg/m2 (95% CI, 1.11–1.49) at 45 years of age and remained largely stable until 60 years of age (β = 1.35 kg/m2; 95% CI 1.18–1.52), after which the association decreased and reached the lowest at 85 years of age (β = 0.86 kg/m2; 95% CI 0.38–1.33). When stratified by age at menopause (Supplementary Table 5), we found that the GRS-BMI relationship tended to peak earlier for women who had an early menopause. For example, for women who had menopause at <45 years of age, the strongest association between GRS and BMI occurred at 45 years old (β = 1.39 kg/m2; 95% CI 0.79–1.99), whereas for those who had menopause at >55 years old, the strongest association occurred at 70 years of age (β = 1.19 kg/m2; 95% CI 0.51–1.87).
Figure 1.

Relationship of GRS with BMI at different ages in women and men (detailed data are presented in Supplementary Table 4).
In men, the mean increase in BMI associated with 10-allele increment in the GRS was 0.57 kg/m2 (95% CI 0.46–0.68) at 21 years old, increased to its peak at 45 years of age (β = 0.91 kg/m2; 95% CI 0.65–1.16), and then gradually decreased to its lowest at 85 years of age (β = 0.48 kg/m2; 95% CI 0.20–0.76). Using the linear mixed-effects model, we found a statistically significant interaction between age and GRS in relation to the longitudinal BMI profile (P for GRS-age interaction <0.001 for both men and women).
Table 2 presents the relationship of GRS with BMI change at different periods of adulthood, as assessed by the mean BMI change per 10-allele increment in the GRS (Δ). In women, higher GRS was positively associated with BMI gain from 18 to 45 years and from 18 years of age to menopause, with the Δ of 0.54 kg/m2 (95% CI 0.38–0.71; P < 0.001) and 0.60 kg/m2 (95% CI 0.42–0.77; P < 0.001), respectively. The positive association with BMI gain from 45 years or menopause to 65 years of age was much weaker, with the Δ of 0.17 kg/m2 (95% CI 0.03–0.31; P = 0.02) and 0.07 kg/m2 (95% CI −0.04 to 0.19; P = 0.21), respectively. In contrast, GRS was nonsignificantly associated with more BMI loss in late adulthood, with the Δ of −0.22 kg/m2 (95% CI −0.39 to −0.05; P = 0.01) from 65 to 80 years of age. To examine whether this inverse association was a consequence of an elevated risk of obesity-related morbidity due to weight gain in middle age, we performed a sensitivity analysis by excluding individuals who were diagnosed with cancer, diabetes, or cardiovascular disease (including coronary heart disease, stroke, and coronary artery bypass grafting) before 80 years old. The results remained essentially unchanged (Δ = −0.21 kg/m2; 95% CI, −0.39 to −0.03; P = 0.02).
Table 2.
Relationship of GRS with BMI change at different age periods
| Age period (years) for BMI change | n | Mean BMI change (SD), kg/m2 | Mean BMI change per 10 alleles, Δ (95% CI), kg/m2 | P for main effect of GRS | P for GRS-sex interaction* |
|---|---|---|---|---|---|
| Women | |||||
| From 18 to 45 | 5,956 | 3.0 (4.1) | 0.54 (0.38–0.71) | <0.001 | 0.06 |
| From 45 to 65 | 5,640 | 2.7 (3.4) | 0.17 (0.03–0.31) | 0.02 | 0.06 |
| From 65 to 80 | 2,942 | −0.9 (3.0) | −0.22 (−0.39 to −0.05) | 0.01 | 0.09 |
| From 18 to menopause | 6,705 | 4.2 (4.5) | 0.60 (0.42–0.77) | <0.001 | |
| From menopause to 65 | 6,436 | 1.5 (3.0) | 0.07 (−0.04 to 0.19) | 0.21 | |
| Men | |||||
| From 21 to 45 | 1,699 | 2.5 (2.8) | 0.23 (0.02–0.44) | 0.03 | |
| From 45 to 65 | 1,634 | 1.0 (2.5) | −0.10 (−0.29 to 0.09) | 0.30 | |
| From 65 to 80 | 2,020 | −0.3 (2.1) | −0.01 (−0.16 to 0.14) | 0.90 |
*P value was calculated for the product term between sex and GRS (continuous).
We did not find any statistically significant association between GRS and weight change in men. Despite the difference between sexes, the interaction test for sex did not achieve statistical significance for any of the BMI changes (P for GRS-sex interaction >0.05).
In the sensitivity analysis, we examined the alternative GRS that included six SNPs associated with early-life BMI. We found that this GRS was similarly associated with adulthood BMI and BMI change as the primary GRS (Supplementary Table 6).
Figure 2 shows the stratified associations of GRS with BMI change according to baseline BMI for each life period. GRS conferred a much stronger impact on early adulthood weight gain among men with a BMI of at least 25 kg/m2 at 21 years of age than those with a BMI of <22.5 kg/m2 (Δ = 0.71 vs. 0.14 kg/m2; P for GRS-baseline BMI interaction <0.001). In contrast, an opposite interaction pattern was observed for BMI change during middle adulthood (P for GRS-baseline BMI interaction <0.001 for both sexes). GRS was associated with BMI gain for individuals with a BMI of <22.5 kg/m2 at 45 years old (Δ = 0.22 kg/m2 in women and 0.49 kg/m2 in men), but associated with BMI loss for those who were already overweight at that age (Δ = −0.20 kg/m2 in women and −0.40 kg/m2 in men). No statistically significant interaction was found between GRS and BMI at 65 years of age for BMI change in late adulthood.
Figure 2.

Relationship of GRS with BMI change at different age periods according to categories of BMI (<22.5, 22.5–24.9, and ≥25 kg/m2) at the starting age of each period in women (A–C) and men (D–F). P for interaction was calculated using Wald test for the product term between GRS and the starting BMI for each age period (continuous).
Discussion
To our knowledge, this is the largest longitudinal study to comprehensively examine the effects of genetic obesity variants on adiposity throughout adulthood. We found that the association of GRS with BMI did not peak until middle adulthood (∼45 years in men and 60 years of age in women) and then started declining with aging. The associations appeared to be stronger across all ages in women than in men. We also studied how genetic risk may influence BMI change at different periods of adulthood. Although GRS was positively associated with weight gain in early adulthood in women, no statistically significant association was found in men. These findings provide critical insights into how established genetic risk variants for adult BMI affect changes in weight across adult ages.
Limited data suggest a persistent impact of genetic susceptibility on longitudinal profiles of BMI in adulthood. In the Fels Longitudinal Study, the GRS based on 32 adult BMI-related SNPs showed a positive association with BMI at 19, 23, 30, and 40 years of age (36). In line with our data, they also found that the positive association tended to increase over ages, although formal testing for gene-by-age interactions did not achieve statistical significance. In another study of two cohorts, the 32-SNP–based GRS was associated with higher mean BMI and a steeper increase in BMI until 65 years of age (21). Moreover, in a recent study, we examined the relationship of GRS with the trajectory groups of adiposity that were identified on the basis of body fatness status from 5 up to 65 years of age (37). We found that individuals carrying more genetic obesity variants were more likely to maintain a heavy body shape in early life and gain weight during young and middle adulthood. These findings agree with our current study and collectively support a predictive relationship of GRS with accelerated increases in BMI throughout the midlife period. This may not be surprising because most studies included in the GWAS for derivation of the GRS were conducted in subjects aged 45–65 years. However, our sensitivity analysis using the alternative GRS for early-life BMI showed similar associations as the primary GRS, supporting the robustness of our primary findings of a persistent effect of genetic susceptibility on BMI trajectory across early to middle adulthood.
Some studies have reported contrary findings, including two based on the British National Survey of Health and Development (NSHD), which included ∼2,500 participants with repeated measurements of BMI from 2 to 53 years of age (8,12). The associations with BMI for either major individual genetic variants (8) or composite GRS (12) strengthened during childhood and adolescence, peaked at 20 years of age, and subsequently weakened during middle adulthood. These observations differ from our findings in that we observed that the association of GRS with BMI did not peak until middle adulthood. Such discrepancy may be, at least partly, due to the birth cohort and period effects. Compared with our study participants who were mostly born in the U.S. between 1925 and 1940, the NSHD participants were all born in 1946. Given the recent data suggesting that the association of GRS with adult BMI is modified by the timing of exposure to an obesogenic environment (38), it is possible that later entry into the time period associated with the obesity epidemic in our participants led to an extended penetrance of the genetic influence into middle adulthood, resulting in the achievement of the peak for the GRS-BMI association at an older age than what was observed in the NSHD cohort. Moreover, the NSHD cohort experienced postwar food rationing from birth to age 8 years, which may have contributed to stronger genetic effects on childhood versus later weight gain (12).
Moreover, we noted a sex difference in the GRS-BMI relationship, with a stronger association observed in women than in men across all ages. Although some (39–41) but not all (3) twin studies have reported greater heritability of adult BMI in women than in men, most studies of common genetic variants did not provide sex-specific estimates for the associations with BMI. One possible explanation for our observed sex difference may be related to greater variation in BMI among women than men in all adult ages, particularly in middle adulthood (Table 1), when the GRS-BMI association also showed the largest sex difference. It is also possible that different sets of genes may influence adult BMI in men and women, and the extent of differences in the genes influencing BMI for men and women is more pronounced from early adulthood (20–29 years) to late middle age (60–69 years), as indicated by a recent twin study (42). In addition, considering the potential influence of menopause on body weight and composition, menopause may represent a time point at which the genetic impact on BMI in women may differ. Indeed, we found that GRS was associated with BMI gain from early adulthood to menopause but not after menopause, and the GRS-BMI relationship tended to peak earlier for women who had early menopause, suggesting that the potential influence of genetic susceptibility on BMI weakens after menopause. Given the well-known male–female differences associated with multiple aspects of adiposity and body composition, such as fat distribution, deposition, and accumulation (43), further studies are needed to identify potential biological mechanisms for the sex difference in the genetic origin of obesity.
In addition to BMI, we also examined BMI change at different life periods. Consistent with the well-known role of early-life weight gain in middle adulthood obesity and our observation that the magnitude of the GRS-BMI association increased up to middle life, we found a predominant effect of genetic susceptibility on promoting weight gain from early to middle adulthood. Existing data about the genetic influence on longitudinal change in adiposity measures are very limited. Although twin studies reported a moderate-to-high genetic contribution to rate of change in adulthood BMI (heritability ∼60%) (44,45), a few studies using either FTO variants (7,22) or GRS (25) did not find any association with body weight or BMI change over 5–10 years during adulthood. However, because participants in these studies were already at their middle adulthood (average 45–60 years of age), these null findings are not incompatible with our findings that GRS was only modestly associated with BMI change in middle adulthood (from 45 to 65 years of age) in women.
Interestingly, we observed that the association of GRS with BMI change varied by baseline BMI, with different interaction patterns identified for early versus middle adulthood. Genetically conferred weight gain in early adulthood was more evident among individuals who were heavy at ∼20 years of age, whereas genetically determined weight gain in middle adulthood was more evident among those who were lean at 45 years old. These divergent interactions may indicate distinct pathways for weight gain at different adult ages. Give that the pro-obesity effect of genetic predisposition may not reach its peak until middle adulthood, individuals who are heavy at 20 years of age probably remain at high genetic risk of gaining more weight. In contrast, environmental factors may exert a greater effect on body weight variation in middle adulthood, which can dilute the influence of genetics. Therefore, genetic susceptibility for weight gain may be more evident among individuals who remain lean at 45 years of age through possible interactions with the obesogenic environmental exposures in middle adulthood (20).
Furthermore, in contrast to the strong effect of GRS on weight gain in early adulthood, we found that a higher number of BMI-increasing risk alleles were associated with more weight loss in late adulthood in women, although the statistical test was nonsignificant at the stringent α = 0.001 level. Similar paradoxical observation has been reported in another study (19). These findings suggest that the negative effects of weight gain before middle age may lead to increased age-related weight loss among individuals with high genetic susceptibility for obesity. However, the underlying mechanisms remain unclear. Our sensitivity analysis suggests that higher obesity-related morbidity is unlikely to explain the association of GRS with increasing weight loss in elderly women. Other aging-related factors, such as accelerated loss of muscle mass, should to be investigated in further studies.
The strengths of our study include the large sample size, use of the GRS encompassing all known common genetic variants associated with adult BMI, and longitudinal assessments of BMI for ∼60 years, which allow us to associate GRS with both BMI level and BMI change across the entire adulthood. The major limitation of our study is that not all participants had provided BMI data at each of the studied ages, and therefore, the age-specific analysis may include different subsets of individuals. However, because our study participants were born within a narrow window of time and we further adjusted all BMI measurements for year of birth, the influence of birth cohort or period effects was minimized. Another limitation is that body weight data were self-reported and subject to measurement error. However, the influence of such error is unlikely to be substantial because the self-reported weight data have been shown highly accurate in our previous validation studies.
In conclusion, our study indicates that the GRS comprising 97 BMI-associated SNPs is associated with BMI at all adult ages, with the magnitude of the association being stronger in women than in men and achieving its peak in middle adulthood. Genetic risk alleles may also increase early adulthood weight gain in women. These findings extend our knowledge about the genetic contributions to adulthood adiposity. Further studies are needed to elucidate the mechanisms for the age-dependent genetic effects on BMI and potential sex differences, as well as to understand how environmental factors may modify the genetic effects on BMI trajectory throughout adulthood to better control the obesity epidemic.
Supplementary Material
Article Information
Acknowledgments. The authors thank the participants and staff of the Nurses' Health Study and the Health Professionals Follow-Up Study for valuable contributions as well as the following state cancer registries for help: AL, AR, AZ, CA, CO, CT, DE, FL, GA, IA, ID, IL, IN, KY, LA, ME, MD, MA, MI, NC, ND, NE, NH, NJ, NY, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, and WY.
Funding. This work was supported by the 2017 AACR-AstraZeneca Fellowship in Immuno-oncology Research (grant 17-40-12-SONG) and the National Institutes of Health (K24-DK-098311, UM1-CA-186107, R01-CA-49449, UM1-CA-167552, and U54-CA-155626). A.T.C. is a Stuart and Suzanne Steele MGH Research Scholar.
The funders had no role in design and conduct of the study, collection, management, analysis, and interpretation of the data, preparation, review, or approval of the manuscript, and the decision to submit the manuscript for publication.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. M.S. researched data and wrote the manuscript. Y.Z. researched data and reviewed and edited the manuscript. L.Q. researched data and reviewed and edited the manuscript. F.B.H. reviewed and edited the manuscript. A.T.C. reviewed and edited the manuscript. E.L.G. reviewed and edited the manuscript and supervised the study. M.S. is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db17-1156/-/DC1.
References
- 1.Ng M, Fleming T, Robinson M, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014;384:766–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silventoinen K, Kaprio J. Genetics of tracking of body mass index from birth to late middle age: evidence from twin and family studies. Obes Facts 2009;2:196–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Min J, Chiu DT, Wang Y. Variation in the heritability of body mass index based on diverse twin studies: a systematic review. Obes Rev 2013;14:871–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graff M, Gordon-Larsen P, Lim U, et al. The influence of obesity-related single nucleotide polymorphisms on BMI across the life course: the PAGE study. Diabetes 2013;62:1763–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graff M, Ngwa JS, Workalemahu T, et al.; GIANT Consortium . Genome-wide analysis of BMI in adolescents and young adults reveals additional insight into the effects of genetic loci over the life course. Hum Mol Genet 2013;22:3597–3607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winkler TW, Justice AE, Graff M, et al.; CHARGE Consortium; DIAGRAM Consortium; GLGC Consortium; Global-BPGen Consortium; ICBP Consortium; MAGIC Consortium . The influence of age and sex on genetic associations with adult body size and shape: a large-scale genome-wide interaction study. PLoS Genet 2015;11:e1005378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hertel JK, Johansson S, Sonestedt E, et al. FTO, type 2 diabetes, and weight gain throughout adult life: a meta-analysis of 41,504 subjects from the Scandinavian HUNT, MDC, and MPP studies. Diabetes 2011;60:1637–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hardy R, Wills AK, Wong A, et al. Life course variations in the associations between FTO and MC4R gene variants and body size. Hum Mol Genet 2010;19:545–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaakinen M, Läärä E, Pouta A, et al. Life-course analysis of a fat mass and obesity-associated (FTO) gene variant and body mass index in the Northern Finland Birth Cohort 1966 using structural equation modeling. Am J Epidemiol 2010;172:653–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ong KK, Elks CE, Wills AK, et al. Associations between the pubertal timing-related variant in LIN28B and BMI vary across the life course. J Clin Endocrinol Metab 2011;96:E125–E129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belsky DW, Moffitt TE, Houts R, et al. Polygenic risk, rapid childhood growth, and the development of obesity: evidence from a 4-decade longitudinal study. Arch Pediatr Adolesc Med 2012;166:515–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elks CE, Loos RJ, Hardy R, et al. Adult obesity susceptibility variants are associated with greater childhood weight gain and a faster tempo of growth: the 1946 British Birth Cohort Study. Am J Clin Nutr 2012;95:1150–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elks CE, Loos RJ, Sharp SJ, et al. Genetic markers of adult obesity risk are associated with greater early infancy weight gain and growth. PLoS Med 2010;7:e1000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steinsbekk S, Belsky D, Guzey IC, Wardle J, Wichstrøm L. Polygenic risk, appetite traits, and weight gain in middle childhood: a longitudinal study. JAMA Pediatr 2016;170:e154472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warrington NM, Howe LD, Paternoster L, et al. A genome-wide association study of body mass index across early life and childhood. Int J Epidemiol 2015;44:700–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warrington NM, Howe LD, Wu YY, et al. Association of a body mass index genetic risk score with growth throughout childhood and adolescence. PLoS One 2013;8:e79547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warrington NM, Wu YY, Pennell CE, et al. Modelling BMI trajectories in children for genetic association studies. PLoS One 2013;8:e53897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hohenadel MG, Baier LJ, Piaggi P, et al. The impact of genetic variants on BMI increase during childhood versus adulthood. Int J Obes 2016;40:1301–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rukh G, Ahmad S, Ericson U, et al. Inverse relationship between a genetic risk score of 31 BMI loci and weight change before and after reaching middle age. Int J Obes 2016;40:252–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang T, Huang T, Heianza Y, et al. Genetic susceptibility, change in physical activity, and long-term weight gain. Diabetes 2017;66:2704–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dahl AK, Reynolds CA, Fall T, Magnusson PK, Pedersen NL. Multifactorial analysis of changes in body mass index across the adult life course: a study with 65 years of follow-up. Int J Obes 2014;38:1133–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vimaleswaran KS, Ängquist L, Hansen RD, et al. Association between FTO variant and change in body weight and its interaction with dietary factors: the DiOGenes study. Obesity (Silver Spring) 2012;20:1669–1674 [DOI] [PubMed] [Google Scholar]
- 23.Ahmad S, Poveda A, Shungin D, et al. Established BMI-associated genetic variants and their prospective associations with BMI and other cardiometabolic traits: the GLACIER Study. Int J Obes 2016;40:1346–1352 [DOI] [PubMed] [Google Scholar]
- 24.Ishola AF, Gerstein HC, Engert JC, et al. Longitudinal relationships between glycemic status and body mass index in a multiethnic study: evidence from observational and genetic epidemiology. Sci Rep 2016;6:30744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sandholt CH, Allin KH, Toft U, et al. The effect of GWAS identified BMI loci on changes in body weight among middle-aged Danes during a five-year period. Obesity (Silver Spring) 2014;22:901–908 [DOI] [PubMed] [Google Scholar]
- 26.Aarestrup J, Bjerregaard LG, Gamborg M, et al. Tracking of body mass index from 7 to 69 years of age. Int J Obes 2016;40:1376–1383 [DOI] [PubMed] [Google Scholar]
- 27.Xiang L, Wu H, Pan A, et al. FTO genotype and weight loss in diet and lifestyle interventions: a systematic review and meta-analysis. Am J Clin Nutr 2016;103:1162–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franks PW, McCarthy MI. Exposing the exposures responsible for type 2 diabetes and obesity. Science 2016;354:69–73 [DOI] [PubMed] [Google Scholar]
- 29.Locke AE, Kahali B, Berndt SI, et al.; LifeLines Cohort Study; ADIPOGen Consortium; AGEN-BMI Working Group; CARDIOGRAMplusC4D Consortium; CKDGen Consortium; GLGC; ICBP; MAGIC Investigators; MuTHER Consortium; MIGen Consortium; PAGE Consortium; ReproGen Consortium; GENIE Consortium; International Endogene Consortium . Genetic studies of body mass index yield new insights for obesity biology. Nature 2015;518:197–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rimm EB, Stampfer MJ, Colditz GA, Chute CG, Litin LB, Willett WC. Validity of self-reported waist and hip circumferences in men and women. Epidemiology 1990;1:466–473 [DOI] [PubMed] [Google Scholar]
- 31.Troy LM, Hunter DJ, Manson JE, Colditz GA, Stampfer MJ, Willett WC. The validity of recalled weight among younger women. Int J Obes Relat Metab Disord 1995;19:570–572 [PubMed] [Google Scholar]
- 32.Rhoads GG, Kagan A. The relation of coronary disease, stroke, and mortality to weight in youth and in middle age. Lancet 1983;1:492–495 [DOI] [PubMed] [Google Scholar]
- 33.Hunter DJ, Kraft P, Jacobs KB, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet 2007;39:870–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 2015;31:3555–3557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis SR, Castelo-Branco C, Chedraui P, et al.; Writing Group of the International Menopause Society for World Menopause Day 2012 . Understanding weight gain at menopause. Climacteric 2012;15:419–429 [DOI] [PubMed] [Google Scholar]
- 36.Choh AC, Lee M, Kent JW, et al. Gene-by-age effects on BMI from birth to adulthood: the Fels Longitudinal Study. Obesity (Silver Spring) 2014;22:875–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song M, Zheng Y, Qi L, Hu FB, Chan AT, Giovannucci EL. Associations between genetic variants associated with body mass index and trajectories of body fatness across the life course: a longitudinal analysis. Int J Epidemiol. 2 December 2017. [Epub ahead of print]. DOI: 10.1093/ije/dyx255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walter S, Mejía-Guevara I, Estrada K, Liu SY, Glymour MM. Association of a genetic risk score with body mass index across different birth cohorts. JAMA 2016;316:63–69 [DOI] [PubMed] [Google Scholar]
- 39.Hur YM. Sex difference in heritability of BMI in South Korean adolescent twins. Obesity (Silver Spring) 2007;15:2908–2911 [DOI] [PubMed] [Google Scholar]
- 40.Herskind AM, McGue M, Sørensen TI, Harvald B. Sex and age specific assessment of genetic and environmental influences on body mass index in twins. Int J Obes Relat Metab Disord 1996;20:106–113 [PubMed] [Google Scholar]
- 41.Harris JR, Tambs K, Magnus P. Sex-specific effects for body mass index in the new Norwegian twin panel. Genet Epidemiol 1995;12:251–265 [DOI] [PubMed] [Google Scholar]
- 42.Silventoinen K, Jelenkovic A, Sund R, et al. Differences in genetic and environmental variation in adult BMI by sex, age, time period, and region: an individual-based pooled analysis of 40 twin cohorts. Am J Clin Nutr 2017;106:457–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schousboe K, Willemsen G, Kyvik KO, et al. Sex differences in heritability of BMI: a comparative study of results from twin studies in eight countries. Twin Res 2003;6:409–421 [DOI] [PubMed] [Google Scholar]
- 44.Hjelmborg Jv, Fagnani C, Silventoinen K, et al. Genetic influences on growth traits of BMI: a longitudinal study of adult twins. Obesity (Silver Spring) 2008;16:847–852 [DOI] [PubMed] [Google Scholar]
- 45.Ortega-Alonso A, Sipilä S, Kujala UM, Kaprio J, Rantanen T. Genetic influences on change in BMI from middle to old age: a 29-year follow-up study of twin sisters. Behav Genet 2009;39:154–164 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.