

Abstract
Cells constantly adapt their metabolism to meet their energy needs and respond to nutrient availability. Eukaryotes have evolved a very sophisticated system to sense low cellular ATP levels via the serine/threonine kinase AMP-activated protein kinase (AMPK) complex. Under conditions of low energy, AMPK phosphorylates specific enzymes and growth control nodes to increase ATP generation and decrease ATP consumption. In the past decade, the discovery of numerous new AMPK substrates has led to a more complete understanding of the minimal number of steps required to reprogramme cellular metabolism from anabolism to catabolism. This energy switch controls cell growth and several other cellular processes, including lipid and glucose metabolism and autophagy. Recent studies have revealed that one ancestral function of AMPK is to promote mitochondrial health, and multiple newly discovered targets of AMPK are involved in various aspects of mitochondrial homeostasis, including mitophagy This Review discusses how AMPK functions as a central mediator of the cellular response to energetic stress and mitochondrial insults and coordinates multiple features of autophagy and mitochondrial biology.
Cells constantly need to manage their energy consumption depending on the availability of nutrients and on their capacity to produce ATP. ATP is produced by catabolic processes such as the breakdown of glucose or lipids, and the majority of ATP production in cells occurs in mitochondria, the sites of oxidative phosphorylation (OXPHOS). When cells use this stored energy for all manner of cellular processes, ATP is broken down into ADP, which can be further converted to AMP. When cellular ATP levels decrease, it is essential for cells to minimize energy consumption to avoid exhausting what is left of their resources. At the same time, emergency measures have to be taken to restore the cellular energy supply, such as increasing nutrient intake, activating alternative energy-producing pathways or turning over existing macromolecules into nutrients.
Eukaryotes have evolved a very elegant system that allows them to modulate their metabolism based on nutrient availability; the key player in this system is AMP-activated protein kinase (AMPK). In single-cell eukaryotes, such as the yeast Saccharomyces cerevisiae, the AMPK homologue is responsible for activating alternative carbon source utilization pathways when glucose is absent1,2. In higher eukaryotes, AMPK has acquired the capacity to sense the amount of energy available in the cell by directly binding adenine nucleotides3. Upon changes in energy availability, and thus changes in the ATP-to-ADP or ATP-to-AMP ratio, AMPK is activated by an allosteric mechanism that stimulates its kinase activity. Once activated, AMPK redirects metabolism towards increased catabolism and decreased anabolism through the phosphorylation of key proteins in multiple pathways, including mTOR complex 1 (mTORC1)4,5, lipid homeostasis6–9, glycolysis10–13 and mitochondrial homeostasis14–17. In addition to directly regulating key enzymes involved in these pathways, AMPK also rewires cellular metabolism in a prolonged manner by targeting transcriptional regulators17–26. A number of recent phospho-proteomics studies have uncovered even more AMPK substrates27–29, and excellent recent reviews have focused on the regulation of various aspects of metabolism by AMPK30,31.
Here, we summarize the mechanism of action of AMPK and discuss its effects on metabolism, and then focus on recent advances in research on the regulation of autophagy and mitochondrial homeostasis by AMPK. A thorough understanding of AMPK functions and its downstream targets is necessary not only to understand its role as a master regulator of metabolism but also because modulation of AMPK is an attractive avenue for therapeutic intervention in many diseases, including cancer and type 2 diabetes (BOX 1).
Box 1. Targeting AMPK for therapeutic applications.
AMP-activated protein kinase (AMPK) has received a lot of attention as a potential target for treating diseases associated with metabolic perturbation. This includes diabetes, obesity and fatty liver diseases and also cancer, which is often associated with changes in metabolism.
The type 2 diabetes drug metformin had been used for decades when a study showed that its mechanism of action involved the activation of AMPK in hepatocytes223. Mechanistically, metformin induces energy stress through inhibition of complex I of the respiratory chain in mitochondria. This leads to a change in the ATP-to-AMP ratio and canonical AMPK activation. There has been extensive research to evaluate the contribution of AMPK to the effect of metformin on circulating glucose and lipids. AMPK phosphorylation of acetyl-CoA carboxylases (ACCs) has been proposed as a master contributor to the changes in lipid synthesis that are induced by metformin, which in turn modulates insulin sensitivity and glucose uptake in muscle. One key piece of evidence in favour of this hypothesis came from the generation of a mouse knock-in mutant lacking the AMPK site on both ACC1 and ACC2 (REF. 105). This mouse model revealed that these phosphorylation events mediate the insulin-sensitizing effect of metformin, thus establishing AMPK as a relevant target in the action of metformin. Despite controversy224, AMPK is widely viewed as an essential component of the action of metformin at physiological concentrations219,225. Given the role of the AMPK–ACC pathway in regulating fatty acid synthesis, AMPK activation is also an attractive treatment option for conditions associated with increased fatty acid production, such as nonalcoholic fatty liver disease (NAFLD)85. In addition to diabetes, retrospective studies have revealed that patients taking metformin had a decreased occurrence of cancer226. However, whether direct activation of AMPK would be sufficient to replicate the beneficial effect of metformin was not known until recent advances in the generation of potent and specific small-molecule activators of AMPK. Most notably, two recent studies have revealed that direct AMPK activation indeed improves symptoms of type 2 diabetes in multiple animal models. One study revealed that a pan-specific AMPK activator improved diabetes symptoms in several animal models, including rodents and monkeys84. In another study, a direct AMPK activator increased glucose uptake in muscle and reduced blood glucose in type 2 diabetes models83. Interestingly, inactivation of AMPK in the liver had no effect on the efficacy of the drug, whereas muscle-specific deletion of AMPK abolished the effect of the drug, establishing muscle AMPK as a key therapeutic target in type 2 diabetes83.
AMPK activation has been suggested to have tumour-suppressive, anticancer properties in some contexts and may mediate some of the cell-autonomous benefits of mitochondrial inhibitors on tumour cell growth227–234, although AMPK activation may also allow tumour cell survival under the metabolic stress conditions that many tumour cells may face235–238. Additional studies with genetic loss of AMPK and the new bioavailable small-molecule activators of AMPK will shed new light on the cancer settings in which AMPK activation or inhibition may be most desirable.
AMPK structure and activation
AMPK is a heterotrimeric complex composed of a catalytic α-subunit and two regulatory subunits, β and γ (FIG. 1). Vertebrates possess multiple isoforms of each subunit, encoded by different genes. In humans, there are two α-subunits, α1 and α2, encoded by the genes PRKAA1 and PRKAA2 (REF. 32), two β-subunits, β1 and β2, encoded by PRKAB1 and PRKAB2 (REF. 33), and three γ-subunits, γ1, γ2 and γ3, encoded by PRKAG1, PRKAG2 and PRKAG3 (REF. 34). Each AMPK complex is composed of one α-subunit, one β-subunit and one γ-subunit, and all combinations are possible, thus potentially creating 12 distinct AMPK complexes35. One of the remaining questions in the field is whether these different combinations really represent functionally distinct complexes with different substrate specificities or subcellular localization, as discussed below.
Figure 1. AMPK structure and activation.

Domain structure of the AMP-activated protein kinase (AMPK) trimer, showing the α-, β- and γ-subunits with their respective domains. The upstream kinases CAMKK2 and liver kinase B1 (LKB1) are shown above the AMPK complex. LKB1 is a heterotrimer composed of LKB1, STRAD and MO25; CAMKK2 is activated by intracellular calcium. Several factors lead to AMPK activation, such as mitochondrial poisons and oxygen or glucose starvation, as well as exercise. Drugs that activate AMPK include the AMP mimetic AICAR and several small-molecule allosteric activators (listed on the left-hand side). The effect of AMPK activation is to rewire metabolism to decrease anabolic processes (that is, ATP consumption) and increase catabolism (that is, ATP production) to restore a more favourable energy balance. Downstream substrates are grouped by biological functions. ATP-producing processes are activated, while ATP-consuming processes are inhibited. AID, auto-inhibitory domain; CBM, carbohydrate-binding module; CBS, cystathionine-β-synthase.
Subunit composition of AMPK and their functions
The α-subunit contains the kinase domain and a critical residue, Thr172, that is phosphorylated by an upstream kinase. The β-subunit contains a carbohydrate- binding module that allows AMPK to associate with glycogen36. The γ-subunit enables AMPK to respond to changes in the ATP-to-AMP ratio as it contains four tandem cystathionine-β-synthase (CBS) domains that bind adenine nucleotides37. Binding of AMP, and to a lesser extent ADP, to the γ-subunit stimulates AMPK activity38–40 (FIG. 1).
AMP binding to the γ-subunit enhances AMPK activity through three distinct mechanisms. First, AMP has been proposed to stimulate phosphorylation of Thr172 by directly stimulating the activity of the upstream kinase41 or by an allosteric mechanism that would render AMPK a better substrate for the upstream kinase42; both of these models have been challenged by other results showing no effect of AMP on the phosphorylation of Thr172 by the upstream kinase in vitro40,43–45. Second, AMP inhibits the dephosphorylation of Thr172 by protecting it from phosphatases46. Last, AMP causes allosteric activation of AMPK already phosphorylated on Thr172 (REFS 39,44). Even though both ADP and AMP have been shown to bind to the CBS domains and to stimulate AMPK activity, careful evaluation of their respective binding affinity to the γ-subunit, their concentration in cells and how much their concentration varies during energy stress revealed that AMP is likely to be the major contributor to changes in AMPK activity in vivo39. It should be noted that ATP is also able to bind to the CBS domains on the γ-subunit, which allows AMPK to respond to changes in the ATP-to-AMP ratio rather than changes in total nucleotide levels and thus to act as a real sensor of energy levels38.
There are 12 possible combinations of αβγ complexes depending on which subunits constitute the complex. In vivo, some cell types only contain a subset of these combinations, suggesting that some complexes have specific roles. For example, in muscle, only the α1β2γ1, α2β2γ1 and α2β2γ3 complexes are detected. Moreover, following exercise, only the α2β2γ3 complex is activated by Thr172 phosphorylation, even though this complex is not as abundant as the γ1-containing complexes47. This specificity for the γ3-containing complex is in contrast to the effect of the mitochondrial inhibitor PT-1, which specifically activates γ1-containing complexes in muscle48. Even though the consequences of specific complex activation are not known, this observation suggests that specific subunit compositions allow distinct AMPK complexes to respond to different types of stress stimuli. It has been recently shown that the different complex compositions affect activity, activation by nucleotides, sensitivity to phosphatases and allosteric regulation of phosphatase sensitivity by AMP49.
Different subunits have also been reported to have different subcellular localization in cells; for example, the nuclear localization of α2 is increased compared with α1 in muscle during exercise50 and following leptin treatment of myoblasts in culture51. Furthermore, circadian oscillation of α1 nuclear localization has been observed in the mouse liver21, and differential localization of γ-subunits has been reported in muscle fibres52, whereas myristoylation of β-subunits has been reported to trigger association with intercellular membranes45 or mitochondria53 and to result in nuclear exclusion51.
AMPK at the lysosome
It has recently been proposed that AMPK is localized at lysosomes through an inter action with axin, a protein best characterized for its role in WNT pathway regulation54. In this context, axin localizes to lysosomes, where it interacts with liver kinase B1 (LKB1; also known as STK11). In response to energy stress, AMP stimulates interaction with axin, which allows Thr172 phosphorylation by LKB1 and AMPK activation. This is proposed to allow co- regulation of mTOR, a master regulator of cell growth, and AMPK under starvation conditions55. mTOR plays an opposing role to AMPK: it stimulates anabolic pathways under high-nutrient conditions. AMPK and mTOR are both components of ancient conserved pathways that have evolved as a yin-yang-like antagonistic mechanism controlling catabolism and anabolism. A recent study suggests that this regulation of AMPK by axin could play an important role in the activation of AMPK following glucose starvation56. In this study, the authors propose that glucose starvation activates AMPK independently of the canonical AMP-dependent allosteric mechanism. Rather, binding of the glycolytic enzyme aldolase to its substrate fructose-1,6-bisphosphate (FBP) regulates AMPK–axin binding and AMPK activity56. Thus, under conditions of glucose starvation, FBP levels decrease, which is sensed by aldolase and stimulates the formation of the AMPK–axin complex at the lysosome56.
Whether this same axin complex controls AMPK activation in response to distinct metabolic stresses remains to be determined, as does whether specific AMPK substrates in different subcellular locations are controlled by a lysosome-targeted pool of AMPK. However, to date, no substrate is known to be exclusively or even preferentially phosphorylated by a specific AMPK complex, so this remains an open question for future studies.
AMPK activation by upstream kinases
The AMPK complex is activated by phosphorylation on Thr172 in the activation loop of the catalytic (α) subunit by an upstream kinase (FIG. 1).
The discovery that one of the upstream kinases for AMPK is the tumour suppressor LKB1 provided a direct connection between cancer and metabolism42,43,57. Notably, like AMPK, LKB1 functions as a constitutive heterotrimer with the kinase-dead STE20-related kinase STRAD and the STE20 family scaffolding protein MO25 (REFS 58,59) (FIG. 1). Studies of mice in which Lkb1 (also known as Stk11) has been genetically inactivated reveal that LKB1 is responsible for the majority of AMPK activation under energy stress in most mammalian tissues examined, including crucial metabolic tissues such as the liver and muscle60–64. Studies of cells devoid of LKB1 indicate that LKB1 mediates nearly all of the activation of AMPK in response to mitochondrial insults and low-energy conditions65.
However, it should be noted that AMPK can also be directly phosphorylated on Thr172 in response to calcium flux by the calcium-sensitive kinase CAMKK2 (also known as CAMKKβ)66–68, thus linking calcium signalling to the regulation of energy metabolism by AMPK69. Increased intracellular calcium activates CAMKK2 and leads to the activation of AMPK independently of LKB1 and nucleotide levels (FIG. 1). This has been shown to have a role particularly in response to hormones and stresses that acutely control intracellular calcium, including processes such as the regulation of food intake by hypothalamic neurons in mice70,71, activation of antigen and thrombin receptors in immune and endothelial cells, respectively72,73, and stimulation of mitochondrial activity by thyroid hormone74,75. Notably, CAMKK2 has also been shown to activate AMPK following cellular stresses, including amino acid starvation76, hypoxia77,78 and cell detachment from the matrix79. In LKB1-null cells, CAMKK2 is able to maintain some AMPK activity that is still sensitive to changes in the ATP-to-AMP ratio80. This CAMKK2-dependent activation of AMPK explains the observation of residual AMPK activity in LKB1-deficient tumours.
A variety of physiological conditions lead to the activation of AMPK in vivo, including mitochondrial inhibition, nutrient starvation and exercise (FIG. 1). It is likely that these conditions induce AMPK activation through the modulation of the ATP-to-AMP ratio. In addition, several small-molecule activators of AMPK have been developed, which bind to the interface of the α- and β-subunits and activate AMPK kinase activity independently of nucleotide levels (FIG. 1). The first of these drugs was A-769662 (REF. 81). Subsequently, other related compounds with increased potency and specificity have been developed, such as compounds 991, PF-739 and MK-8722 (REFS 82–84).
Regulation of metabolism by AMPK
During energy stress, AMPK directly phosphorylates key factors involved in multiple pathways to restore energy balance. The effect of AMPK on metabolism can be broadly divided into two categories: the inhibition of anabolism to minimize ATP consumption and the stimulation of catabolism to stimulate ATP production. We briefly review the main components of these categories below and then focus on the emerging roles of AMPK in mitochondrial processes and autophagy.
Inhibition of anabolic pathways
AMPK inhibits multiple biosynthetic pathways under conditions of energy shortage (FIG. 2). The first pathway to be identified was the inhibition of lipid and sterol synthesis by AMPK through inhibitory phosphorylation of the acetyl-CoA carboxylases ACC1 and ACC2, which catalyse the first step in de novo lipid synthesis, and inhibitory phosphorylation of HMG-CoA reductase (HMGCR), which catalyses the rate-limiting step in cholesterol synthesis6,7. This function of AMPK has major implications for the use of direct activators in the treatment of diseases associated with excess fatty acid production, such as nonalcoholic fatty liver disease (NAFLD)85, and is consistent with the lipid-lowering effects seen upon liver-specific expression of activated AMPK86. AMPK also prevents the storage of glycogen by inhibitory phosphorylation of the glycogen synthases GYS1 and GYS2 (REF. 87) and inhibits hexosamine synthesis and O-GlcNAc modification of cellular proteins via phosphorylation of GFAT1 (REFS 88,89) (FIG. 2).
Figure 2. AMPK regulates a variety of metabolic processes.

Once activated, the AMP-activated protein kinase (AMPK) complex phosphorylates key targets to rewire metabolism. The direct targets of AMPK are shown in the first concentric circle. The arrow indicates whether the phosphorylation is activating or inhibitory for the function of the target protein. The general process in which each target is involved is indicated in the next circle, and the box colour indicates whether that general process is activated (green) or inhibited (red). For certain targets, an intermediate mediator of the effect is indicated between the two circles. The pathways modulated by AMPK are grouped into four general categories — protein metabolism, lipid metabolism, glucose metabolism, and autophagy and mitochondrial homeostasis — denoting the wide range of processes that are controlled by AMPK. mTOR is modulated by AMPK while also modulating several direct or indirect targets of AMPK. This is illustrated by arrows from mTOR to its targets and serves to emphasize the complex relationship between these two signalling pathways. Transcriptional regulators are denoted by an asterisk. It is important to note that only a subset of AMPK substrates is included in the figure. ChREBP, carbohydrate-responsive element-binding protein; CREB, cAMP response element-binding protein; FOXO, forkhead box protein O; HDAC, histone deacetylase; HMGCR, HMG-CoA reductase; HNF4α, hepatocyte nuclear factor 4α; MFF, mitochondrial fission factor; PGC1α, peroxisome proliferator-activated receptor-γ co-activator 1α; PLD1, phospholipase D1; SREBP1, sterol regulatory element-binding protein 1; TFEB, transcription factor EB.
Under prolonged conditions of reduced energy, AMPK reprogrammes metabolism through transcriptional regulation of biosynthetic pathways. AMPK inhibits gluconeogenesis by phosphorylating CRTC2 (REF. 19) and class IIA histone deacetylases (HDACs)24, which are co-activators of the cAMP response elementbinding protein (CREB) and forkhead box protein O (FOXO) pathways, respectively. AMPK also controls metabolism at the transcriptional level by phosphorylating sterol regulatory element-binding protein 1 (SREBP1)23, carbohydrate-responsive elementbinding protein (ChREBP)90 and hepatocyte nuclear factor 4α (HNF4α)91, which are key transcriptional regulators of lipid and glucose metabolism. AMPK has been reported to phosphor late histone acetyltransferase p300 to regulate nuclear hormone receptor transcriptional activity18.
Cell growth and protein translation are major consumers of ATP in cells. Accordingly, AMPK regulates these processes through the modulation of the master regulator of growth mTOR. AMPK activation leads to the inhibition of mTORC1 by two independent mechanisms: the activation of the negative mTORC1 regulator TSC2 (also known as tuberin)4 and the inhibition of the mTORC1 subunit RAPTOR5. Under nutrient-rich conditions, AMPK is inactive and mTOR is active, whereas under energy-shortage conditions, increased AMPK activity leads to a decrease in mTOR activity. Lower mTOR activity leads to decreased cell growth and to decreased protein synthesis. AMPK also directly regulates protein synthesis by phosphorylating eEF2K, a negative regulator of protein elongation92. eEF2K is also a downstream target of the mTOR pathway93, further reinforcing the many connections and crosstalk between these two pathways controlling cell growth and metabolism and illustrating the ancient yin-yang relationship between these two signalling hubs. In summary, AMPK-dependent inhibition of anabolic glucose, lipid and protein synthesis pathways limits ATP consumption under energy stress (FIG. 2). It should be noted that AMPK has also been reported to regulate multiple additional growth-related pathways, including Hedgehog signalling94, the Hippo pathway95–97, the JAK–signal transducer and activator of transcription (STAT) pathway98 and the p53 tumour suppressor pathway99,100, although many details remain to be fully defined. This Review focuses on metabolic targets of AMPK.
Stimulation of catabolic pathways
In order to replenish ATP stores, AMPK also actively stimulates the breakdown of macromolecules to produce energy. The stimulated pathways include increased glucose utilization, mobilization of lipid stores and turnover of macromolecules by autophagy (FIG. 2). AMPK stimulates glucose utilization by phosphorylating targets involved in the trafficking of glucose transporters to increase glucose uptake into cells. Phosphorylation of TXNIP13 and TBC1D1 (REF. 101) increases the plasma membrane localization of GLUT1 and GLUT4, respectively. Phosphorylation of phospholipase D1 (PLD1) by AMPK has also been shown to indirectly increase glucose uptake102. AMPK also increases flux through the glycolytic pathway by phosphorylating PFKFB3 (6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase 3), which affects the activity of PFK1, a rate-limiting enzyme in glycolysis11 (FIG. 2). In addition to stimulating glucose utilization, AMPK also pushes cells towards using their lipid stores. This is achieved by stimulating lipases such as adipose triglyceride lipase (ATGL; also known as PNPLA2) to release fatty acids from triglyceride stores9. The free fatty acids are then imported into mitochondria for β-oxidation. Import of fatty acids into mitochondria relies on a transport system that requires the acyl transferase CPT1. Interestingly, CPT1 activity seems to be modulated by AMPK activity. Indeed, malonyl-CoA, generated by ACC1 and ACC2, is a potent inhibitor of CPT1 (REFS 103,104). Therefore, by phosphorylating and inhibiting ACC, AMPK decreases the pool of malonyl-CoA, which results in decreased lipid synthesis and increased fatty acid import into mitochondria for β-oxidation. Studies using mouse models with knock-in mutations of Ser79Ala in ACC1 and Ser221Ala in ACC2 revealed that these sites are indeed important for the regulation of lipid homeostasis and the therapeutic effects of metformin downstream of AMPK105 (BOX 1). In addition, both ACC1 and ACC2 contributed to the regulation of lipid homeostasis, showing redundancy in function between ACC1 and ACC2.
Transcriptional effects of AMPK also participte in redirecting metabolism towards increased catabolism. AMPK transcriptionally upregulates genes involved in mitochondrial biogenesis, autophagy and lysosomal degradation (FIG. 2). These processes, as well as the direct connections between AMPK and mitochondria, are discussed in detail in the following sections.
AMPK and mitochondria
A growing body of evidence points to a specific regulation of various aspects of mitochondrial biology and homeostasis by AMPK (FIG. 3). These aspects include control of mitochondrial number through stimulation of mitochondrial biogenesis, regulation of the shape of the mitochondrial network in cells, and mitochondrial quality control through regulation of autophagy and mitophagy (BOX 2).
Figure 3. Regulation of mitochondrial homeostasis by AMPK.

AMP-activated protein kinase (AMPK) directly phosphorylates mitochondrial fission factor (MFF) to regulate mitochondrial fission through dynamin-like protein DRP1 and activates ULK1, the upstream kinase in autophagy and mitophagy. Mitochondrial fission is required for mitophagy and allows damaged mitochondria to be degraded by mitophagy. During energy stress, AMPK also activates peroxisome proliferator-activated receptor-γ (PPARγ) co-activator 1α (PGC1α), which activates mitochondrial biogenesis genes through interaction with PPARγ or oestrogen-related receptors (ERRs). The dashed arrow indicates potentially indirect regulation.
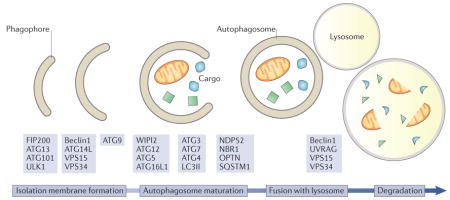
Box 2. Autophagy.
Autophagy is a process by which cells digest their own components using a specialized machinery of adaptors and effectors. It begins with the generation of the autophagosome and recognition of the cargo, followed by the maturation of the autophagosome and fusion with lysosomes (see the figure). The term itself means ‘self-eating’ and was first coined by Belgian scientist and Nobel Prize laureate Christian de Duve. Autophagy serves two main functions: it enables the degradation of cellular structures that are too large for other surveillance pathways, such as the ubiquitin–proteasome system, and it allows cells to survive starvation by recycling building blocks such as amino acids to sustain essential cell functions. Autophagy can be either a bulk recycling of cytosolic components or a targeted removal of macromolecules and even organelles. In particular, removal of mitochondria by autophagy, a process called mitophagy, has been shown to require the canonical autophagy machinery as well as specific markers at the surface of damaged mitochondria that signal their removal. Genes essential for autophagy (ATGs) have been discovered by screening for genes that are required for autophagosome formation in the yeast Saccharomyces cerevisiae during nitrogen starvation 239,240. Yoshinori Ohsumi was awarded the Nobel Prize in physiology or medicine in 2016 for the discovery of the ATG genes. Since the 1990s, the molecular events controlling autophagy execution have been characterized, and the role of ATG genes in controlling various steps of the autophagy pathway has been demonstrated241. The first ATG gene to be cloned, ATG1, encodes a protein kinase required for the initiation of autophagy. Its mammalian homologue, ULK1, plays a similar role. ULK1 forms a complex with ATG13, ATG101 and FIP200. Upon autophagy initiation, ULK1 phosphorylates and activates the class III PI3K complex I, composed of VPS34, VPS15, ATG14L and Beclin1. This complex generates phosphatidylinositol-3-phosphate (PtdIns3P) at the nascent autophagosomal membrane to help shape the phagophore membrane that also contains ATG9, the only transmembrane ATG protein. PtdIns3P recruits ATG16L1 and ATG5-ATG12 to the autophagosome through interaction with the PtdIns3P-binding protein WIPI2. The ATG16L1-ATG5-ATG12 complex then catalyses the covalent attachment of a phosphatidylethanolamine moiety to the carboxy-terminal glycine of LC3, which was previously activated by the action of ATG4, ATG7 and ATG3 (LC3I), generating LC3II156. LC3II is used as a marker of autophagy to visualize autophagosomes and to quantify autophagy in cells242 but it also allows binding of autophagy receptors such as SQSTM1, NDP52, NBR1 and OPTN243. Subsequently, autophagosomes mature and fuse with lysosomes, a process mediated by the class III PI3K complex II composed of VPS34, VPS15, Beclin1 and UVRAG156, to induce degradation of the cargo by lysosomal enzymes.
AMPK promotes mitochondrial biogenesis
Mitochondrial biogenesis occurs in response to increased energy expenditure to produce more ATP. Mitochondrial biogenesis occurs via the growth and division of pre-existing mitochondria. New material is added to the existing mitochondrial network in order to increase mitochondrial mass. Mitochondrial biogenesis requires the upregulation of mitochondrial proteins and an increase in lipid production and transfer to sustain the expansion of the surface area of the inner and outer mitochondrial membranes. The mitochondrial genome encodes some mitochondrial proteins, but the vast majority of mitochondrial proteins are encoded by the nuclear genome. Thus, the signal to stimulate mitochondrial biogenesis needs to be relayed to the nucleus in order for the transcription factors to stimulate the expression of genes encoding mitochondrial proteins, as is the case for retrograde signalling106.
A variety of experimental and physiological conditions result in increased mitochondrial mass, one of the best-studied being exercise. Early observations in the 1950s correlated muscle activity in birds with muscle fibre mitochondrial content and indicated that active muscles contained more mitochondria than less-active muscles107. It is now well established that exercise and muscle activity induce a mitochondrial biogenesis programme aimed at increasing the oxidative capacity of muscles108. Interestingly, exercise is also a potent activator of AMPK, thus raising the possibility that AMPK could participate in this process. This is further supported by the observations that chronic AMPK activation leads to increased mitochondrial biogenesis109 and that the AMPK-activating drug AICAR acts as an exercise mimetic110. Furthermore, over expression of a constitutively active AMPK γ3 subunit in mice induced mitochondrial biogenesis111. Loss-of-function experiments also revealed a role for AMPK in the maintenance of mitochondrial biogenesis. Mice expressing a dominant-negative mutant of AMPK failed to induce mitochondrial biogenesis under energy stress16. Mice lacking the AMPK β1/β2 sub units112 or the upstream kinase LKB1 (REFS 113,114) show decreased mitochondrial content in muscle, and mice with muscle-specific LKB1 knockout fail to increase mitochondrial biogenesis after exercise113. Muscle-specific knockout of AMPK α-subunits also induces defects in mitochondrial biogenesis and function115. In addition, AMPK has also been shown to control mitochondrial content in other tissues, including adipocytes116, macro phages117 and hepatocytes118. Collectively, these studies establish AMPK as a central regulator of mitochondrial biogenesis.
Several downstream effectors of AMPK contribute to the regulation of mitochondrial biogenesis, with no single substrate yet known to mediate the bulk of the effects. Most genes involved in mitochondrial metabolism seem to be under the control of the peroxisome proliferator-activated receptor-γ co-activator 1 (PGC1) family119,120 through interaction with oestrogen-related receptors (ERRs) and to a lesser extent the peroxisome proliferator-activated receptor (PPAR) family of transcription factors121 (FIG. 3). Overexpression of PGC1α in muscle is sufficient to convert type IIb (glycolytic) fibres into mitochondria-rich type II and type I fibres, establishing PGC1α as a master regulator of mitochondrial bio genesis122. PGC1α is regulated by several post-translational mechanisms, such as acetylation123, methylation124 and phosphorylation by various kinases, including AKT125 and p38 MAPK126. Interestingly, it has been reported that the transcriptional upregulation of several genes involved in oxidative metabolism upon AMPK activation required PGC1α17 and that overexpression of the constitutively active AMPK γ3 subunit increased PGC1α expression111. Two sites on PGC1α have been reported to be phosphorylated by AMPK in vitro: Thr177 and Ser538 (REF. 17). Additional studies are needed to define whether they are AMPK target sites in vivo. AMPK may also regulate PGC1α by several indirect mechanisms involving AMPK-dependent modulation of p38 MAPK127, HDAC5 (REFS 24,128) or SIRT1 (REF. 129) (reviewed in REF. 130), so it seems likely that through a combination of mechanisms, AMPK communicates with PGC1α (FIG. 3). In addition, AMPK promotes the activation of transcription factor EB (TFEB; discussed below), and TFEB has been shown to directly bind to and activate the promoter of the gene encoding PGC1α, PPARGC1A, in some contexts131. In addition to PGC1α, AMPK has been shown to increase the expression of the mRNAs encoding PGC1β and ERRα in KRAS-driven colon cancer132, and TFEB and TFE3 have also been connected to PGC1β expression in some contexts133.
The effect of AMPK on mitochondrial biogenesis is not only important for understanding the effects of exercise on muscle mitochondria but it also has therapeutic implications in multiple pathological states, including Duchenne muscular dystrophy134 and mitochondrial myopathy135. AMPK activation also increases exercise capacity in obese mice136, improves muscle regeneration following injury137 and protects muscle from age-related myopathies through the activation of the autophagy pathway, as discussed below138.
Regulation of mitochondrial dynamics by AMPK
Mitochondria are highly mobile organelles that constantly undergo fusion and division events to generate a dynamic and interconnected network139. Different mitochondrial morphologies have been proposed to modulate OXPHOS, mitophagy and apoptosis. In particular, elongated mitochondria have been hypothesized to sustain ATP production during stress140, protect mitochondria from mitophagy141,142 and distribute biomaterials including lipids143 during nutrient starvation. By contrast, in response to other cellular insults, including mitochondrial respiratory chain inhibitors, mitochondria undergo fragmentation, which is thought to facilitate mitophagy of mitochondrial fragments lacking proper membrane potential144, as well as to allow efficient and timely apoptosis145.
It has long been known that environmental conditions, including mitochondrial depolarization and inhibition of mitochondrial ATP synthesis, trigger mitochondrial fragmentation by increasing mitochondrial fission rates and/or decreasing mitochondrial fusion rates146. However, the mechanism by which mitochondrial inhibitors affect the shape of the mitochondrial network was not clearly established, especially for mitochondrial inhibitors that do not affect the mitochondrial membrane potential147. Strikingly, stimuli that induce mitochondrial fragmentation, such as inhibitors of the respiratory chain, are also potent activators of AMPK. AMPK was recently shown to be required for the fragmentation of mitochondria induced by rotenone and antimycin A, which are inhibitors of complex I and complex III, respectively, of the mitochondrial respiratory chain. Surprisingly, AMPK activation through direct small-molecule activators, in the absence of mitochondrial damage, is sufficient to induce mitochondrial fission15. This finding established AMPK as a crucial and direct regulator of mitochondrial dynamics. Proteomics and bioinformatics screens to identify new substrates of AMPK identified two phosphorylation sites, Ser155 and Ser173, on a core component of the mitochondrial fission pathway, mitochondrial fission factor (MFF)15,28. MFF is the primary receptor for the dynamin-like protein DRP1 on the mitochondrial outer membrane, which mediates constriction of mitochondria during fission148–150. Phosphorylation of MFF on Ser155 and Ser172 by AMPK is therefore one potential mechanism that could explain the requirement of AMPK for mitochondrial fragmentation after ETC inhibition and the sufficiency of AMPK activators alone to promote mitochondrial fragmentation. Indeed, activation of AMPK results in increased localization of DRP1 at the mitochondria, an effect that is dependent on the presence of the AMPK phosphorylation sites in MFF15. Thus, AMPK acutely regulates the shape of the mitochondrial network during energy stress by phosphorylating MFF and controlling the localization of DRP1 (REF. 151) (FIG. 3).
Other mitochondrial targets of AMPK
As mentioned above, ACC1 and ACC2 are phosphorylated by AMPK. ACC1 is cytosolic, whereas ACC2 has an additional amino-terminal hydrophobic sequence that specifically targets it to the mitochondrial outer membrane and therefore represents the first identified mitochondrial substrate of AMPK152. Upon activation of AMPK, phosphorylation of Ser79 in ACC1 and Ser221 in ACC2 results in the inactivation of ACC activity. This is thought to not only inhibit the first step in fatty acid synthesis but also stimulate lipid β-oxidation in mitochondria. Specific inhibition of ACC2 at mitochondria is therefore predicted to relieve the inhibition of CPT1 by malonyl-CoA and to result in increased lipid β-oxidation. Studies have revealed that ACC1 and ACC2 both participate in the regulation of lipid homeostasis105. However, the presence of ACC2 at mitochondria suggests that, under certain conditions, localized regulation of ACC function at mitochondria by AMPK could play a role in metabolic adaptation to energy stress. For example, muscles from ACC2 Ser221Ala knock-in mice have decreased basal fatty acid oxidation compared with wild-type muscle and are resistant to the fatty acid oxidation-promoting effect of AICAR153. Interestingly, upon exercise, these differences were lost, suggesting that exercise induces changes in fatty acid oxidation that are independent of the AMPK–ACC–malonyl-CoA axis154.
The intricate relationship between AMPK and mitochondria is probably underestimated. As a master regulator of energy homeostasis, AMPK is very likely to target more aspects and functions of mitochondria. For example, AMPK has recently been shown to affect the movement of mitochondria at the leading edge of the cell during cell migration155, a process probably involving a yet-to-be-identified substrate of AMPK involved in the process of mitochondrial transport. AKAP1, a mitochondria-localized protein known to act as a PKA-anchoring factor, has recently been shown to be phosphorylated by AMPK, providing a potential mechanism for localized PKA activity regulation by AMPK at mitochondria27.
AMPK regulates autophagy and mitophagy
Autophagy is a process by which cellular components such as proteins, macromolecules, organelles and pathogens are recycled by a specialized cellular machinery that mediates the engulfment of the cargo in membranous structures called autophagosomes, their fusion with lysosomes, and subsequent degradation of the cargo in autophagolysosomes (BOX 2 and reviewed in REF. 156). The autophagy machinery involves several multi-protein complexes that control every step, and AMPK regulates various aspects of this machinery.
Autophagy mainly serves two distinct functions: the turnover of old or damaged molecules and the replenishment of nutrient stores during times of starvation. These two functions are similar in their execution but differ in their mechanism of initiation. While the turnover of molecules is thought to be a continuous process in cells, the induction of autophagy during starvation involves the nutrient-sensing pathway mediated by mTORC1. Upon nutrient starvation, mTORC1 is inactivated, which relieves the inhibitory phosphorylation on ULK1, a kinase essential for autophagy induction157,158. mTORC1 also phosphorylates and inhibits other components of the autophagy pathway, such as ATG14L (also known as Barkor)159 and the ULK1-interacting protein ATG13 (REF. 160). These phosphorylation events participate in the inhibition of autophagy under nutrient-rich conditions. Upon activation, ULK1 phosphorylates several proteins involved in the execution of autophagy, including its binding partners FIP200, ATG101 (REF. 161) and ATG13 (REF. 162), the downstream effectors ATG9 (REF. 163), ATG14L159, VPS34 (also known as PI3K type 3), Beclin1 and AMBRA1 (REFS 161,164), and the trafficking protein Sec16A165 (BOX 2, FIG. 4 and reviewed in REF. 166).
Figure 4. Details of the regulation of autophagy by mTOR, AMPK and ULK1.

ULK1-mediated phosphorylation events are shown in green, mTOR-mediated phosphorylation events in purple and AMP-activated protein kinase (AMPK)-mediated phosphorylation events in red. Dashed arrows indicate reported phosphorylation events by AMPK that are insufficiently documented or that involve sites that do not conform to the AMPK motif. Upon activation, AMPK phosphorylates TSC2 and RAPTOR, which leads to the downregulation of mTOR complex 1 (mTORC1) (purple complex, composed of mTOR, RAPTOR, mLST8, DEPTOR and PRAS40 (also known as AKT1S1); not all shown) activity. AMPK phosphorylates ULK1 on at least four serines to promote its activity. ULK1 is part of a complex with ATG101, ATG13 and FIP200. All of these proteins have been shown to be targets of ULK1, while ATG13 has also been reported as a target of both AMPK and mTORC1. PI3K complex I mediates the conversion of phosphatidylinositol (PI) to phosphatidylinositol-3-phosphate (PtdIns3P) at the surface of the forming autophagosome (beige membrane shown engulfing cellular components, including a mitochondrion), a step required for proper recruitment of cargo and adaptor proteins. The PI3K complex I components (light blue) and accessory factors (dark blue) are shown with their reported phosphorylation. ATG9 is an integral membrane protein localized at the autophagosome membrane. The arrows indicate whether the phosphorylation event is activating or inhibitory for the function of the protein.
Regulation of autophagy by AMPK
AMPK has been shown to regulate autophagy both in yeast167 and in mammalian cells168,169. Molecular characterization of this regulation has revealed that AMPK controls autophagy at different steps. First, AMPK directly phosphorylates the mTOR upstream regulator TSC2 on Thr1227 and Ser1345 (REF. 4) and the mTORC1 subunit RAPTOR on Ser722 and Ser792 (REF. 5). Both of these phosphorylation events participate in reducing mTOR activity under conditions of energy stress. As mentioned above, reduced mTOR activity relieves the inhibitory phosphorylation on ULK1 to activate autophagy.
A direct link between AMPK and the core autophagy pathway was established when it was shown that AMPK directly phosphorylates ULK1 on at least four residues: Ser467, Ser555, Thr574 and Ser637 (REFS 14,170) (FIG. 4). Using ULK1-knockout cell lines and reconstitution with non-phosphorylatable mutants, phosphorylation of ULK1 by AMPK was shown to be important for autophagy and cell survival during starvation and metabolic stress. Strikingly, cells expressing ULK1 mutants that cannot be phosphorylated by AMPK also appear to accumulate defective mitochondria, suggesting that the AMPK–ULK1 axis is important not only for general autophagy but also specifically for the selective removal of damaged mitochondria14. Subsequent studies confirmed an essential role for ULK1 in mitophagy under various conditions75,162,171–177. Moreover, efficient removal of damaged mitochondria following AMPK activation has been shown to be important in various contexts, such as in brown adipose tissue116 and during fasting and ageing in muscles138, as well as in macrophages178 and hepatocytes179. In addition to phosphorylating ULK1, AMPK has been reported to phosphorylate residues on other core components of the autophagy pathway, such as Ser761 on ATG9 (REF. 180), Thr133 and Ser135 on VPS34 (REF. 181), Ser91 and Ser94 on Beclin1 (REFS 181,182), Thr50 on the VPS34-associated protein RACK1 (REF. 183) and Thr32 on PAQR3, an ATG14L–VPS34 scaffolding protein184. Some of these phosphorylation events represent suboptimal AMPK sites and have not been independently validated, so many of the molecular details of AMPK and ULK1 control of autophagy remain to be elucidated (FIG. 4).
Another pathway controlling mitophagy is the PINK1–Parkin-mediated removal of depolarized mitochondria185. This pathway involves the stabilization of PINK1 at mitochondria upon loss of the mitochondrial membrane potential186. PINK1 in turn recruits and phosphorylates Parkin, an E3 ubiquitin ligase that marks mitochondria for degradation by the autophagy machinery177. In addition, PINK1 also phosphorylates ubiquitin on mitochondria, a process that amplifies the signal by stimulating Parkin activity187,188. This pathway requires complete mitochondrial depolarization to be activated and is therefore different from the basal surveillance of mitochondria carried out by the AMPK–ULK1 axis. However, it remains to be established whether AMPK and ULK1 play a role in the regulation of mitophagy by the PINK1–Parkin pathway. It should be noted that inducers of PINK1–Parkin-mediated mitophagy, such as the proton ionophore CCCP, are strong inducers of AMPK through the inhibition of ATP synthesis in mitochondria. Therefore, it is conceivable that some targets downstream of AMPK and/or ULK1 could be involved in the PINK1–Parkin pathway.
Mitochondrial localization of AMPK
The presence of at least two well-characterized substrates of AMPK at mitochondria, MFF and ACC2, raises the possibility that a pool of AMPK exists at or close to mitochondria. However, it must be noted that MFF and ACC2 are localized in the mitochondrial outer membrane and are therefore exposed to the cytosol, where they could be phosphorylated by freely floating cytosolic AMPK. Given the central role of mitochondria in energy production, one could hypothesize that a mitochondrial pool of AMPK would be able to sense and respond more rapidly and efficiently to changes in energy status. This hypothesis is supported by some experimental evidence suggesting that a mitochondrial pool of AMPK indeed exists. One such piece of evidence is the observation that myristoylation of the β-subunit of AMPK causes AMPK to localize to mitochondria, where it could regulate the specific removal of damaged mitochondria by mitophagy53. Interestingly, phosphorylation of ULK1 on Ser555 by AMPK has been shown to regulate recruitment of ULK1 to mitochondria during mitophagy, suggesting that localized signalling at mitochondria regulates the removal of damaged mitochondria189.
Small-peptide probes for AMPK activity also suggest the existence of a mitochondrial pool of AMPK that can specifically regulate ATP levels independently of cytosolic AMPK190. However, biochemical fractionations are hard to interpret because of the inherent nature of organelles; they tend to associate in cells and are therefore almost impossible to purify without contamination from other organelles or membrane structure. Imaging techniques allow for a better resolution and identification of subcellular organelles but rely on the specificity of the antibodies used and need to be appropriately controlled. Careful evaluation of the subcellular localization of the seven endogenous AMPK subunits is required to establish the existence and regulation of a potential mitochondrial pool of AMPK.
AMPK acts as a signal integration platform to control mitochondrial health
AMPK regulates at least three essential aspects of mitochondrial homeostasis: biogenesis, fission and mitophagy. During amino acid starvation, autophagy is activated by inhibition of mTORC1. Under these conditions, mitochondria elongate via a process called mitochondrial hyperfusion140 to evade degradation by mitophagy and maximize OXPHOS141,142. This process is beneficial only if the mitochondria are functional and the cell contains the appropriate substrates to support OXPHOS. However, efficient removal of damaged mitochondria and segregation of dysfunctional parts of the mitochondrial network have been reported to rely on mitochondrial fission144,191,192. Therefore, in response to mitochondrial damage, as well as under other low ATP conditions where AMPK is activated, phosphorylation of MFF and ULK1 could facilitate the segregation of damaged mitochondria and their removal by mitophagy, while activation of mitochondrial biogenesis would ensure that mitochondrial mass is restored by the generation of new, healthier mitochondria. The response to starvation therefore depends on whether cells lack amino acids and growth signals, which leads to mTORC1 inactivation but does not activate AMPK, or whether cells lack both nutrients and energy, leading to activation of AMPK, mitochondrial fragmentation and subsequent mitophagy (FIG. 3). It remains to be established whether the strength and/or duration of the signal governs the AMPK response, from a rescue effort to a dismantling and rebuilding mission.
Role of AMPK in autophagy beyond mitochondria
Autophagy is also involved in the removal of intracellular pathogens, a process called xenophagy193. The AMPK pathway has been shown to be activated and to modulate the autophagy response during viral194,195, bacterial178 and prion infections196, thus expanding the repertoire of conditions that involve the regulation of autophagic flux by AMPK. Interestingly, to sustain their replication, several pathogens either induce or repress AMPK activity depending on their specific needs, such as evasion of xenophagy, increased glucose or lipid oxidation, or nutrient availability (reviewed in REF. 197).
Transcriptional regulation of autophagy by AMPK
In addition to regulating general transcriptional regulators such as p300 (REF. 18) and class IIa HDACs24, AMPK activation leads to selective regulation of a subset of genes involved in metabolism, autophagy and lysosomal function. This is achieved through direct and indirect mechanisms that could participate in long-term modulation of autophagy flux by AMPK. First, AMPK directly phosphorylates members of the FOXO family of transcription factors20 that regulate genes involved in autophagy198. AMPK phosphorylation of FOXO3 stimulates its transcriptional activity under conditions of energy stress20. Interestingly, two other members of the FOX transcription factor family, FOXK1 and FOXK2, compete with FOXO3 to repress genes involved in autophagy199. FOXK1 and FOXK2 are phosphorylated by mTOR under conditions of high nutrient availability. Upon phosphorylation by mTOR, FOXK proteins translocate to the nucleus, where they compete with FOXO3 for binding to the same genomic regulatory sites and repress autophagy genes199. As a result, when AMPK is activated, phosphorylation of FOXO3 activates autophagy genes, while repression of mTOR through phosphorylation of TSC2 (REF. 4) and RAPTOR5 induces FOXK exclusion from the nucleus to allow for maximal activation of FOXO3 targets199 (FIG. 5). In addition to FOXK, mTOR also regulates TFEB and TFE3, which are master regulators of lysosomal function200–203. Recently, it was shown that AMPK activity controls lysosomal biogenesis through TFEB during embryonic stem cell differentiation into embryoid bodies26. Under the conditions stated above, when AMPK is active and mTOR is inhibited, TFEB dephosphorylation drives its import into the nucleus, where it turns on a network of genes called the CLEAR network200. Products of genes in the CLEAR network control lysosomal activity, which is required for autophagy.
Figure 5. Modulation of the transcription of autophagy and lysosome genes by AMPK.
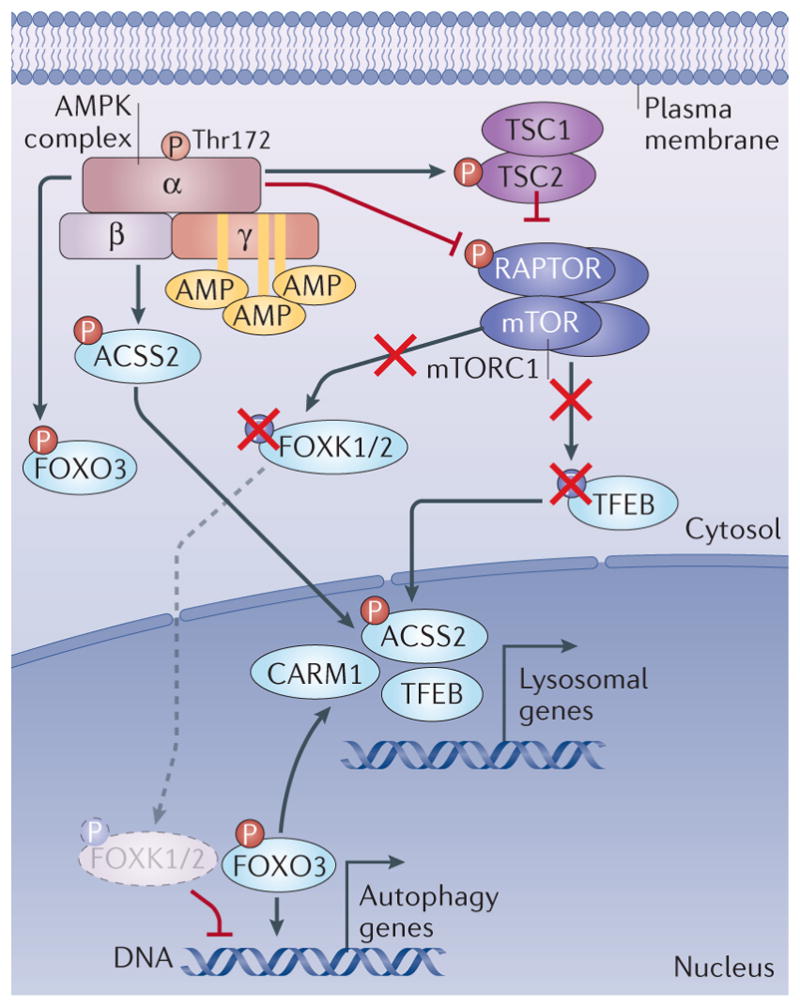
AMP-activated protein kinase (AMPK) regulates the activity of forkhead box protein O3 (FOXO3) through direct phosphorylation. In parallel, AMPK decreases mTOR complex 1 (mTORC1) (purple complex, composed of mTOR, RAPTOR, mLST8, DEPTOR and PRAS40; not all shown) activity through phosphorylation of RAPTOR and the upstream regulator TSC2. This leads to downregulation of mTORC1 activity. This results in dephosphorylation of mTORC1 targets FOXK1 and FOXK2, as well as transcription factor EB (TFEB). As a result, dephosphorylated FOXK1 and FOXK2 can no longer act as transcriptional repressors of FOXO3 targets, allowing higher transcription of autophagy genes downstream of FOXO3 binding. However, dephosphorylation of TFEB allows its nuclear translocation and activation of its target genes, including genes involved in lysosome biogenesis. In addition, an increased CARM1 protein level resulting from FOXO3-dependent gene activation further enhances TFEB-dependent gene expression. Acetyl-CoA carboxylase 2 (ACC2) phosphorylation by AMPK stimulates its nuclear import where it enhances TFEB target gene expression.
In addition to control of TFEB via suppression of mTORC1, two additional mechanisms by which AMPK may activate TFEB-dependent gene expression were reported. AMPK phosphorylation of FOXO3 was shown to repress the expression of the mRNA encoding SKP2, which is part of the SCF E3 ubiquitin ligase complex that degrades the histone demethylase CARM1 (REF. 25). The net effect of AMPK activation was increased levels of CARM1, which acted as a co-activator for TFEB to promote the expression of lysosomal genes (FIG. 5). Very recently, it was reported that AMPK-dependent phosphorylation of the lipogenic enzyme ACSS2 induced its translocation to the nucleus, where it stimulated TFEB-dependent gene expression204. ACSS2 has been hypothesized to control the local synthesis of acetyl- CoA pools in the nucleus to control acetylation of his-tones and other transcriptional regulators205 (FIG. 5). Like CARM1, it is curious that potentially broad effects on histone modification could yield a selective induction of a TFEB-dependent gene expression programme. This is a very interesting developing area at the intersection of AMPK and the control of gene expression under starvation conditions.
Conclusions and perspectives
AMPK has been identified in the past few years to act as a central integrator of mitochondrial homeostasis by controlling various aspects of the mitochondrial life cycle, from biogenesis and dynamics to removal by mitophagy. Given the function of mitochondria as the major source of ATP production in the cell, it makes perfect sense that AMPK, a low ATP sensor that restores ATP homeostasis, would engage downstream effectors to ensure optimal mitochondrial function. Almost every mitochondrial insult or defect activates AMPK, including respiratory chain complexes inhibitors such as the diabetes drug metformin, the Parkinson disease- causing herbicide rotenone, proton ionophores, ATP synthase inhibitors and mitochondrial DNA depletion or mutation. AMPK therefore acts as a guardian of the mitochondria. This is an ancient and conserved role of AMPK, with connections between AMPK and mitochondria occurring in yeast206, Caenorhabditis elegans207–209 and flies210.
Given that across all eukaryotes, AMPK is activated under conditions in which mitochondrial function and mitochondrial production of ATP are compromised, it is surprising that there are not more AMPK substrates dedicated to restoring mitochondrial health, as mitochondrial integrity is a central mediator of cellular energetics. Several questions remain and deserve careful evaluation in the coming years. For example, it remains to be established whether specific AMPK complex compositions preferentially localize at or close to mitochondria and respond to specific stresses involving mitochondria. How such mitochondrial pools of AMPK might interact with or relate to the recently described populations of AMPK and LKB1 at lysosomes56 will be an interesting area for future study. The interplay between AMPK, ULK1 and other kinases activated by mitochondrial and energetic stress (for example, PINK1 and TBK1)177,211–213 and how distinct phosphorylation events dictate mitophagy and fission and/or fusion versus apoptotic cell fate decisions remain poorly defined. Interestingly, recent findings implicate mitochondrial dynamics and mitochondrial health in immune cell function and stem cell pluripotency214–218.
Finally, expansion of the repertoire of known and confirmed substrates of AMPK at mitochondria will help to identify novel aspects of mitochondrial function regulated by AMPK. Given that metformin is a potent AMPK activator, these questions are not only important in understanding fundamental biological processes but also have real-life applications for the more than 150 million people worldwide who take metformin219. Moreover, recent efforts to generate direct activators of AMPK for the treatment of cancer and metabolic diseases (BOX 1) also highlight the importance of understanding how AMPK affects metabolism in general and mitochondrial biology in particular. Regulation of mitochondrial health could represent one of the ways by which metformin and other AMPK activators improve the symptoms of type 2 diabetes105,220, as well as how exercise and calorie restriction improve health and lifespan221,222.
Note added in proof
In an elegant study244 in skeletal muscle of mice bearing a fluorescent reporter gene pMitoTimer that measures oxidative stress and mitophagy in vivo, the authors demonstrated that dominant-negative AMPK suppresses acute-exercise-induced ULK1 phosphorylation, mitophagy and lysosomal biogenesis. Conditional deletion of Ulk1 resulted in loss of acute-exercise-induced mitophagy but had no effect on lysosomal biogenesis, collectively suggesting a broad role for AMPK in restoring metabolic homeostasis after exercise, and a crucial role of ULK1 downstream of AMPK in mitophagy after exercise.
Acknowledgments
S.H. is supported by an Advanced PostDoc.Mobility fellowship of the Swiss National Science Foundation. R.J.S. holds the William R. Brody Chair. The work from the authors’ laboratory described in this Review was supported by grants from the US National Institutes of Health (R01DK080425, R01CA172229, P01CA120964) and The Leona M. and Harry B. Helmsley Charitable Trust (grant #2012-PGMED002).
Glossary
- Allosteric mechanism
Modulation of protein activity by the binding of a molecule to a specific site, often associated with a change in conformation.
- Axin
A protein involved in WNT pathway signalling regulation and in mTOR signalling at the lysosome.
- Acetyl-CoA carboxylases
Enzymes that catalyse the first step in de novo lipid synthesis, the carboxylation of acetyl-CoA to malonyl-CoA.
- Metformin
A widely prescribed type 2 diabetes drug. Mechanistically, metformin inhibits complex I of the respiratory chain and leads to changes in the ATP-to-AMP ratio and activation of AMP-activated protein kinase (AMPK).
- Mitophagy
Specific removal of mitochondria by autophagy.
- Complex I and complex III
Complexes of the respiratory chain in the mitochondrial inner membrane that couple the transfer of electrons to proton pumping. The proton gradient created by the respiratory chain is used to produce ATP, while the electrons are transferred to molecular oxygen.
- Dynamin-like protein DRP1
A protein necessary for mitochondrial fission. DRP1 is recruited to mitochondria at sites of future division and mediates the constriction of mitochondria.
Footnotes
Author contributions
S.H. and R.J.S. researched data for the article, contributed to discussion of the content, wrote the article and reviewed and/or edited the manuscript before submission.
Competing interests statement
The authors declare no competing interests.
References
- 1.Celenza JL, Carlson M. A yeast gene that is essential for release from glucose repression encodes a protein kinase. Science. 1986;233:1175–1180. doi: 10.1126/science.3526554. [DOI] [PubMed] [Google Scholar]
- 2.Gancedo JM. Carbon catabolite repression in yeast. Eur J Biochem. 1992;206:297–313. doi: 10.1111/j.1432-1033.1992.tb16928.x. [DOI] [PubMed] [Google Scholar]
- 3.Crozet P, et al. Mechanisms of regulation of SNF1/AMPK/SnRK1 protein kinases. Front Plant Sci. 2014;5:190. doi: 10.3389/fpls.2014.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 5.Gwinn DM, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987;223:217–222. doi: 10.1016/0014-5793(87)80292-2. [DOI] [PubMed] [Google Scholar]
- 7.Munday MR, Campbell DG, Carling D, Hardie DG. Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. Eur J Biochem. 1988;175:331–338. doi: 10.1111/j.1432-1033.1988.tb14201.x. [DOI] [PubMed] [Google Scholar]
- 8.Watt MJ, et al. Regulation of HSL serine phosphorylation in skeletal muscle and adipose tissue. Am J Physiol Endocrinol Metab. 2006;290:E500–E508. doi: 10.1152/ajpendo.00361.2005. [DOI] [PubMed] [Google Scholar]
- 9.Ahmadian M, et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 2011;13:739–748. doi: 10.1016/j.cmet.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marsin AS, et al. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10:1247–1255. doi: 10.1016/s0960-9822(00)00742-9. [DOI] [PubMed] [Google Scholar]
- 11.Bando H, et al. Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6- bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin Cancer Res. 2005;11:5784–5792. doi: 10.1158/1078-0432.CCR-05-0149. [DOI] [PubMed] [Google Scholar]
- 12.Sakamoto K, Holman GD. Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am J Physiol Endocrinol Metab. 2008;295:E29–E37. doi: 10.1152/ajpendo.90331.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu N, et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell. 2013;49:1167–1175. doi: 10.1016/j.molcel.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egan DF, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toyama EQ, et al. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351:275–281. doi: 10.1126/science.aab4138. This study identifies AMPK as necessary and sufficient to rapidly promote mitochondrial fission in response to ETC inhibitors and identifies the DRP1 receptor MFF as a direct substrate of AMPK involved in this process. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zong H, et al. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci USA. 2002;99:15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci USA. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang W, et al. Regulation of transcription by AMP-activated protein kinase: phosphorylation of p300 blocks its interaction with nuclear receptors. J Biol Chem. 2001;276:38341–38344. doi: 10.1074/jbc.C100316200. [DOI] [PubMed] [Google Scholar]
- 19.Koo SH, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- 20.Greer EL, et al. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007;282:30107–30119. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 21.Lamia KA, et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science. 2009;326:437–440. doi: 10.1126/science.1172156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bungard D, et al. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science. 2010;329:1201–1205. doi: 10.1126/science.1191241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13:376–388. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mihaylova MM, et al. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell. 2011;145:607–621. doi: 10.1016/j.cell.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shin HJR, et al. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature. 2016;534:553–557. doi: 10.1038/nature18014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Young NP, et al. AMPK governs lineage specification through Tfeb-dependent regulation of lysosomes. Genes Dev. 2016;30:535–552. doi: 10.1101/gad.274142.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffman NJ, et al. Global phosphoproteomic analysis of human skeletal muscle reveals a network of exercise-regulated kinases and AMPK substrates. Cell Metab. 2015;22:922–935. doi: 10.1016/j.cmet.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ducommun S, et al. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: identification of mitochondrial fission factor as a new AMPK substrate. Cell Signal. 2015;27:978–988. doi: 10.1016/j.cellsig.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 29.Schaffer BE, et al. Identification of AMPK phosphorylation sites reveals a network of proteins involved in cell invasion and facilitates large-scale substrate prediction. Cell Metab. 2015;22:907–921. doi: 10.1016/j.cmet.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hardie DG, Schaffer BE, Brunet A. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 2016;26:190–201. doi: 10.1016/j.tcb.2015.10.013. This review comprehensively examines all reported AMPK substrates up to late 2016, annotating phosphorylation sites and criteria met to support classification as a substrate. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carling D. AMPK signalling in health and disease. Curr Opin Cell Biol. 2017;45:31–37. doi: 10.1016/j.ceb.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 32.Stapleton D, et al. Mammalian AMP-activated protein kinase subfamily. J Biol Chem. 1996;271:611–614. doi: 10.1074/jbc.271.2.611. [DOI] [PubMed] [Google Scholar]
- 33.Thornton C, Snowden MA, Carling D. Identification of a novel AMP-activated protein kinase beta subunit isoform that is highly expressed in skeletal muscle. J Biol Chem. 1998;273:12443–12450. doi: 10.1074/jbc.273.20.12443. [DOI] [PubMed] [Google Scholar]
- 34.Cheung PC, Salt IP, Davies SP, Hardie DG, Carling D. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J. 2000;346:659–669. [PMC free article] [PubMed] [Google Scholar]
- 35.Ross FA, MacKintosh C, Hardie DG. AMP-activated protein kinase: a cellular energy sensor that comes in 12 flavours. FEBS J. 2016;283:2987–3001. doi: 10.1111/febs.13698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hudson ER, et al. A novel domain in AMP-activated protein kinase causes glycogen storage bodies similar to those seen in hereditary cardiac arrhythmias. Curr Biol. 2003;13:861–866. doi: 10.1016/s0960-9822(03)00249-5. [DOI] [PubMed] [Google Scholar]
- 37.Xiao B, et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature. 2007;449:496–500. doi: 10.1038/nature06161. [DOI] [PubMed] [Google Scholar]
- 38.Hardie DG, Carling D, Gamblin SJ. AMP-activated protein kinase: also regulated by ADP? Trends Biochem Sci. 2011;36:470–477. doi: 10.1016/j.tibs.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 39.Gowans GJ, Hawley SA, Ross FA, Hardie DG. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013;18:556–566. doi: 10.1016/j.cmet.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ross FA, Jensen TE, Hardie DG. Differential regulation by AMP and ADP of AMPK complexes containing different γ subunit isoforms. Biochem J. 2016;473:189–199. doi: 10.1042/BJ20150910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hawley SA, et al. 5′-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J Biol Chem. 1995;270:27186–27191. doi: 10.1074/jbc.270.45.27186. [DOI] [PubMed] [Google Scholar]
- 42.Hawley SA, et al. Complexes between the LKB1 tumor suppressor, STRAD α/β and MO25 α/β are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woods A, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 44.Suter M, et al. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J Biol Chem. 2006;281:32207–32216. doi: 10.1074/jbc.M606357200. [DOI] [PubMed] [Google Scholar]
- 45.Oakhill JS, et al. β-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK) Proc Natl Acad Sci USA. 2010;107:19237–19241. doi: 10.1073/pnas.1009705107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davies SP, Helps NR, Cohen PT, Hardie DG. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2Cα and native bovine protein phosphatase-2AC. FEBS Lett. 1995;377:421–425. doi: 10.1016/0014-5793(95)01368-7. [DOI] [PubMed] [Google Scholar]
- 47.Birk JB, Wojtaszewski JFP. Predominant α2/β2/γ3 AMPK activation during exercise in human skeletal muscle. J Physiol. 2006;577:1021–1032. doi: 10.1113/jphysiol.2006.120972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jensen TE, et al. PT-1 selectively activates AMPK-γ1 complexes in mouse skeletal muscle, but activates all three γ subunit complexes in cultured human cells by inhibiting the respiratory chain. Biochem J. 2015;467:461–472. doi: 10.1042/BJ20141142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rajamohan F, et al. Probing the enzyme kinetics, allosteric modulation and activation of α1- and α2-subunit-containing AMP-activated protein kinase (AMPK) heterotrimeric complexes by pharmacological and physiological activators. Biochem J. 2016;473:581–592. doi: 10.1042/BJ20151051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McGee SL, et al. Exercise increases nuclear AMPKα2 in human skeletal muscle. Diabetes. 2003;52:926–928. doi: 10.2337/diabetes.52.4.926. [DOI] [PubMed] [Google Scholar]
- 51.Suzuki A, et al. Leptin stimulates fatty acid oxidation and peroxisome proliferator-activated receptor alpha gene expression in mouse C2C12 myoblasts by changing the subcellular localization of the α2 form of AMP-activated protein kinase. Mol Cell Biol. 2007;27:4317–4327. doi: 10.1128/MCB.02222-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pinter K, Grignani RT, Watkins H, Redwood C. Localisation of AMPK γ subunits in cardiac and skeletal muscles. J Muscle Res Cell Motil. 2013;34:369–378. doi: 10.1007/s10974-013-9359-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liang J, et al. Myristoylation confers noncanonical AMPK functions in autophagy selectivity and mitochondrial surveillance. Nat Commun. 2015;6:7926. doi: 10.1038/ncomms8926. [DOI] [PubMed] [Google Scholar]
- 54.Zhang YL, et al. AMP as a low-energy charge signal autonomously initiates assembly of AXIN-AMPK- LKB1 complex for AMPK activation. Cell Metab. 2013;18:546–555. doi: 10.1016/j.cmet.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 55.Zhang CS, et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 2014;20:526–540. doi: 10.1016/j.cmet.2014.06.014. [DOI] [PubMed] [Google Scholar]
- 56.Zhang C-S, et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature. 2017;548:112–116. doi: 10.1038/nature23275. This study discovers a provocative new AMP-independent mechanism for glucose sensing by AMPK that involves a super-complex of LKB1, axin, AMPK, the LAMTOR–Ragulator complex and the glycolytic enzyme aldolase on the surface of the lysosome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shaw RJ, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boudeau J, Miranda-Saavedra D, Barton GJ, Alessi DR. Emerging roles of pseudokinases. Trends Cell Biol. 2006;16:443–452. doi: 10.1016/j.tcb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 59.Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- 60.Ikeda Y, et al. Cardiac-specific deletion of LKB1 leads to hypertrophy and dysfunction. J Biol Chem. 2009;284:35839–35849. doi: 10.1074/jbc.M109.057273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jessen N, et al. Ablation of LKB1 in the heart leads to energy deprivation and impaired cardiac function. Biochim Biophys Acta. 2010;1802:593–600. doi: 10.1016/j.bbadis.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shan T, Zhang P, Bi P, Kuang S. Lkb1 deletion promotes ectopic lipid accumulation in muscle progenitor cells and mature muscles. J Cell Physiol. 2015;230:1033–1041. doi: 10.1002/jcp.24831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ollila S, Mäkelä TP. The tumor suppressor kinase LKB1: lessons from mouse models. J Mol Cell Biol. 2011;3:330–340. doi: 10.1093/jmcb/mjr016. [DOI] [PubMed] [Google Scholar]
- 64.Shaw RJ, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hurley RL, et al. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- 67.Hawley SA, et al. Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 68.Woods A, et al. Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 69.Marcelo KL, Means AR, York B. The Ca2+/calmodulin/CaMKK2 axis: nature’s metabolic CaMshaft. Trends Endocrinol Metab. 2016;27:706–718. doi: 10.1016/j.tem.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anderson KA, et al. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 2008;7:377–388. doi: 10.1016/j.cmet.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 71.Yang Y, Atasoy D, Su HH, Sternson SM. Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell. 2011;146:992–1003. doi: 10.1016/j.cell.2011.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tamás P, et al. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J Exp Med. 2006;203:1665–1670. doi: 10.1084/jem.20052469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stahmann N, Woods A, Carling D, Heller R. Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase β. Mol Cell Biol. 2006;26:5933–5945. doi: 10.1128/MCB.00383-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yamauchi M, et al. Thyroid hormone activates adenosine 5′-monophosphate-activated protein kinase via intracellular calcium mobilization and activation of calcium/calmodulin-dependent protein kinase kinase-β. Mol Endocrinol. 2008;22:893–903. doi: 10.1210/me.2007-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sinha RA, et al. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy. 2015;11:1341–1357. doi: 10.1080/15548627.2015.1061849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ghislat G, Patron M, Rizzuto R, Knecht E. Withdrawal of essential amino acids increases autophagy by a pathway involving Ca2+/calmodulin-dependent kinase kinase-β (CaMKK-β) J Biol Chem. 2012;287:38625–38636. doi: 10.1074/jbc.M112.365767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mungai PT, et al. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol Cell Biol. 2011;31:3531–3545. doi: 10.1128/MCB.05124-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sallé-Lefort S, et al. Hypoxia upregulates Malat1 expression through a CaMKK/AMPK/HIF-1α axis. Int J Oncol. 2016;49:1731–1736. doi: 10.3892/ijo.2016.3630. [DOI] [PubMed] [Google Scholar]
- 79.Sundararaman A, Amirtham U, Rangarajan A. Calcium-oxidant signaling network regulates AMP-activated protein kinase (AMPK) activation upon matrix deprivation. J Biol Chem. 2016;291:14410–14429. doi: 10.1074/jbc.M116.731257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fogarty S, et al. Calmodulin-dependent protein kinase kinase-β activates AMPK without forming a stable complex: synergistic effects of Ca2+ and AMP. Biochem J. 2010;426:109–118. doi: 10.1042/BJ20091372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cool B, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 82.Xiao B, et al. Structural basis of AMPK regulation by small molecule activators. Nat Commun. 2013;4:3017. doi: 10.1038/ncomms4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cokorinos EC, et al. Activation of skeletal muscle AMPK promotes glucose disposal and glucose lowering in non-human primates and mice. Cell Metab. 2017;25:1147–1159.e10. doi: 10.1016/j.cmet.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 84.Myers RW, et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science. 2017;357:507–511. doi: 10.1126/science.aah5582. This study, together with reference 83, demonstrates that small-molecule AMPK activators can restore insulin sensitivity and reduce glucose levels in diabetic rodent models and in primate models. The elegant use of liver-specific AMPK double knockout mice and skeletal muscle-specific AMPK double knockout mice demonstrates that only skeletal muscle AMPK is required for the glucose-lowering and insulin-sensitizing effects of these AMPK activators. [DOI] [PubMed] [Google Scholar]
- 85.Smith BK, et al. Treatment of nonalcoholic fatty liver disease: role of AMPK. Am J Physiol Endocrinol Metab. 2016;311:E730–E740. doi: 10.1152/ajpendo.00225.2016. [DOI] [PubMed] [Google Scholar]
- 86.Woods A, et al. Liver-specific activation of AMPK prevents steatosis on a high-fructose diet. Cell Rep. 2017;18:3043–3051. doi: 10.1016/j.celrep.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bultot L, et al. AMP-activated protein kinase phosphorylates and inactivates liver glycogen synthase. Biochem J. 2012;443:193–203. doi: 10.1042/BJ20112026. [DOI] [PubMed] [Google Scholar]
- 88.Eguchi S, et al. AMP-activated protein kinase phosphorylates glutamine: fructose-6-phosphate amidotransferase 1 at Ser243 to modulate its enzymatic activity. Genes Cells. 2009;14:179–189. doi: 10.1111/j.1365-2443.2008.01260.x. [DOI] [PubMed] [Google Scholar]
- 89.Zibrova D, et al. GFAT1 phosphorylation by AMPK promotes VEGF-induced angiogenesis. Biochem J. 2017;474:983–1001. doi: 10.1042/BCJ20160980. [DOI] [PubMed] [Google Scholar]
- 90.Kawaguchi T, Osatomi K, Yamashita H, Kabashima T, Uyeda K. Mechanism for fatty acid ‘sparing’ effect on glucose-induced transcription: regulation of carbohydrate-responsive elementbinding protein by AMP-activated protein kinase. J Biol Chem. 2002;277:3829–3835. doi: 10.1074/jbc.M107895200. [DOI] [PubMed] [Google Scholar]
- 91.Hong YH, Varanasi US, Yang W, Leff T. AMP-activated protein kinase regulates HNF4α transcriptional activity by inhibiting dimer formation and decreasing protein stability. J Biol Chem. 2003;278:27495–27501. doi: 10.1074/jbc.M304112200. [DOI] [PubMed] [Google Scholar]
- 92.Leprivier G, et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell. 2013;153:1064–1079. doi: 10.1016/j.cell.2013.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Faller WJ, et al. mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature. 2015;517:497–500. doi: 10.1038/nature13896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li YH, et al. AMP-activated protein kinase directly phosphorylates and destabilizes Hedgehog pathway transcription factor GLI1 in medulloblastoma. Cell Rep. 2015;12:599–609. doi: 10.1016/j.celrep.2015.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mo JS, et al. Cellular energy stress induces AMPKmediated regulation of YAP and the Hippo pathway. Nat Cell Biol. 2015;17:500–510. doi: 10.1038/ncb3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.DeRan M, et al. Energy stress regulates Hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell Rep. 2014;9:495–503. doi: 10.1016/j.celrep.2014.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang W, et al. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol. 2015;17:490–499. doi: 10.1038/ncb3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rutherford C, et al. Phosphorylation of Janus kinase 1 (JAK1) by AMP-activated protein kinase (AMPK) links energy sensing to anti-inflammatory signaling. Sci Signal. 2016;9:ra109. doi: 10.1126/scisignal.aaf8566. [DOI] [PubMed] [Google Scholar]
- 99.Jones RG, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 100.He G, et al. AMP-activated protein kinase induces p53 by phosphorylating MDMX and inhibiting its activity. Mol Cell Biol. 2014;34:148–157. doi: 10.1128/MCB.00670-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chavez JA, Roach WG, Keller SR, Lane WS, Lienhard GE. Inhibition of GLUT4 translocation by Tbc1d1, a Rab GTPase-activating protein abundant in skeletal muscle, is partially relieved by AMP-activated protein kinase activation. J Biol Chem. 2008;283:9187–9195. doi: 10.1074/jbc.M708934200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kim JH, et al. Phospholipase D1 mediates AMP-activated protein kinase signaling for glucose uptake. PLoS ONE. 2010;5:e9600. doi: 10.1371/journal.pone.0009600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.McGarry JD, Leatherman GF, Foster DW. Carnitine palmitoyltransferase I. The site of inhibition of hepatic fatty acid oxidation by malonyl-CoA. J Biol Chem. 1978;253:4128–4136. [PubMed] [Google Scholar]
- 104.Saggerson D. Malonyl-CoA, a key signaling molecule in mammalian cells. Annu Rev Nutr. 2008;28:253–272. doi: 10.1146/annurev.nutr.28.061807.155434. [DOI] [PubMed] [Google Scholar]
- 105.Fullerton MD, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med. 2013;19:1649–1654. doi: 10.1038/nm.3372. This tour-de-force study using compound knock-in mice demonstrates that AMPK phosphorylation of ACC1 and ACC2 suppresses lipid accumulation in mice under normal dietary conditions and that AMPK-dependent suppression of ACC1 and ACC2 is required for metformin to reduce blood glucose levels. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Quiros PM, Mottis A, Auwerx J. Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol. 2016;17:213–226. doi: 10.1038/nrm.2016.23. [DOI] [PubMed] [Google Scholar]
- 107.Paul MH, Sperling E. Cyclophorase system. XXIII Correlation of cyclophorase activity and mitochondrial density in striated muscle. Proc Soc Exp Biol Med. 1952;79:352–354. doi: 10.3181/00379727-79-19375. [DOI] [PubMed] [Google Scholar]
- 108.Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. 2010;47:69–84. doi: 10.1042/bse0470069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bergeron R, et al. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab. 2001;281:E1340–E1346. doi: 10.1152/ajpendo.2001.281.6.E1340. [DOI] [PubMed] [Google Scholar]
- 110.Narkar VA, et al. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Garcia-Roves PM, Osler ME, Holmström MH, Zierath JR. Gain-of-function R225Q mutation in AMP-activated protein kinase γ3 subunit increases mitochondrial biogenesis in glycolytic skeletal muscle. J Biol Chem. 2008;283:35724–35734. doi: 10.1074/jbc.M805078200. [DOI] [PubMed] [Google Scholar]
- 112.O’Neill HM, et al. AMP-activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci USA. 2011;108:16092–16097. doi: 10.1073/pnas.1105062108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tanner CB, et al. Mitochondrial and performance adaptations to exercise training in mice lacking skeletal muscle LKB1. Am J Physiol Endocrinol Metab. 2013;305:E1018–E1029. doi: 10.1152/ajpendo.00227.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jeppesen J, et al. LKB1 regulates lipid oxidation during exercise independently of AMPK. Diabetes. 2013;62:1490–1499. doi: 10.2337/db12-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lantier L, et al. AMPK controls exercise endurance, mitochondrial oxidative capacity, and skeletal muscle integrity. FASEB J. 2014;28:3211–3224. doi: 10.1096/fj.14-250449. [DOI] [PubMed] [Google Scholar]
- 116.Mottillo EP, et al. Lack of adipocyte AMPK exacerbates insulin resistance and hepatic steatosis through brown and beige adipose tissue function. Cell Metab. 2016;24:118–129. doi: 10.1016/j.cmet.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Galic S, et al. Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J Clin Invest. 2011;121:4903–4915. doi: 10.1172/JCI58577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hasenour CM, et al. 5-Aminoimidazole-4- carboxamide-1-β-D-ribofuranoside (AICAR) effect on glucose production, but not energy metabolism, is independent of hepatic AMPK in vivo. J Biol Chem. 2014;289:5950–5959. doi: 10.1074/jbc.M113.528232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Puigserver P, et al. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 120.Wu Z, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 121.Eichner LJ, Giguère V. Estrogen related receptors (ERRs): a new dawn in transcriptional control of mitochondrial gene networks. Mitochondrion. 2011;11:544–552. doi: 10.1016/j.mito.2011.03.121. [DOI] [PubMed] [Google Scholar]
- 122.Lin J, et al. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 123.Rodgers JT, et al. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 124.Teyssier C, Ma H, Emter R, Kralli A, Stallcup MR. Activation of nuclear receptor coactivator PGC-1α by arginine methylation. Genes Dev. 2005;19:1466–1473. doi: 10.1101/gad.1295005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1α transcription coactivator. Nature. 2007;447:1012–1016. doi: 10.1038/nature05861. [DOI] [PubMed] [Google Scholar]
- 126.Puigserver P, et al. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARγ coactivator-1. Mol Cell. 2001;8:971–982. doi: 10.1016/s1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- 127.Wu Y, et al. Activation of AMPKα2 in adipocytes is essential for nicotine-induced insulin resistance in vivo. Nat Med. 2015;21:373–382. doi: 10.1038/nm.3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Czubryt MP, McAnally J, Fishman GI, Olson EN. Regulation of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) and mitochondrial function by MEF2 and HDAC5. Proc Natl Acad Sci USA. 2003;100:1711–1716. doi: 10.1073/pnas.0337639100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cantó C, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.O’Neill HM, Holloway GP, Steinberg GR. AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: implications for obesity. Mol Cell Endocrinol. 2013;366:135–151. doi: 10.1016/j.mce.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 131.Settembre C, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15:647–658. doi: 10.1038/ncb2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fisher KW, et al. AMPK promotes aberrant PGC1β expression to support human colon tumor cell survival. Mol Cell Biol. 2015;35:3866–3879. doi: 10.1128/MCB.00528-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wada S, et al. The tumor suppressor FLCN mediates an alternate mTOR pathway to regulate browning of adipose tissue. Genes Dev. 2016;30:2551–2564. doi: 10.1101/gad.287953.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ljubicic V, Jasmin BJ. AMP-activated protein kinase at the nexus of therapeutic skeletal muscle plasticity in Duchenne muscular dystrophy. Trends Mol Med. 2013;19:614–624. doi: 10.1016/j.molmed.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 135.Peralta S, et al. Sustained AMPK activation improves muscle function in a mitochondrial myopathy mouse model by promoting muscle fiber regeneration. Hum Mol Genet. 2016;25:3178–3191. doi: 10.1093/hmg/ddw167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Marcinko K, et al. The AMPK activator R419 improves exercise capacity and skeletal muscle insulin sensitivity in obese mice. Mol Metab. 2015;4:643–651. doi: 10.1016/j.molmet.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mounier R, Théret M, Lantier L, Foretz M, Viollet B. Expanding roles for AMPK in skeletal muscle plasticity. Trends Endocrinol Metab. 2015;26:275–286. doi: 10.1016/j.tem.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 138.Bujak AL, et al. AMPK activation of muscle autophagy prevents fasting-induced hypoglycemia and myopathy during aging. Cell Metab. 2015;21:883–890. doi: 10.1016/j.cmet.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. 2016;212:379–387. doi: 10.1083/jcb.201511036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tondera D, et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009;28:1589–1600. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA. 2011;108:10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell. 2015;32:678–692. doi: 10.1016/j.devcel.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shirihai OS, Song M, Dorn GW. How mitochondrial dynamism orchestrates mitophagy. Circ Res. 2015;116:1835–1849. doi: 10.1161/CIRCRESAHA.116.306374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 146.Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27:105–117. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 147.Mishra P, Carelli V, Manfredi G, Chan DC. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014;19:630–641. doi: 10.1016/j.cmet.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Otera H, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Losón OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24:659–667. doi: 10.1091/mbc.E12-10-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wang C, Youle R. Cell biology: form follows function for mitochondria. Nature. 2016;530:288–289. doi: 10.1038/530288a. [DOI] [PubMed] [Google Scholar]
- 152.Abu-Elheiga L, et al. The subcellular localization of acetyl-CoA carboxylase 2. Proc Natl Acad Sci USA. 2000;97:1444–1449. doi: 10.1073/pnas.97.4.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.O’Neill HM, et al. AMPK phosphorylation of ACC2 is required for skeletal muscle fatty acid oxidation and insulin sensitivity in mice. Diabetologia. 2014;57:1693–1702. doi: 10.1007/s00125-014-3273-1. [DOI] [PubMed] [Google Scholar]
- 154.O’Neill HM, et al. Skeletal muscle ACC2 S212 phosphorylation is not required for the control of fatty acid oxidation during exercise. Physiol Rep. 2015;3:e12444. doi: 10.14814/phy2.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cunniff B, McKenzie AJ, Heintz NH, Howe AK. AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol Biol Cell. 2016;27:2662–2674. doi: 10.1091/mbc.E16-05-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bento CF, et al. Mammalian autophagy: how does it work? Annu Rev Biochem. 2016;85:685–713. doi: 10.1146/annurev-biochem-060815-014556. [DOI] [PubMed] [Google Scholar]
- 157.Chan EYW, Kir S, Tooze S. A siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem. 2007;282:25464–25474. doi: 10.1074/jbc.M703663200. [DOI] [PubMed] [Google Scholar]
- 158.Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res. 2014;24:42–57. doi: 10.1038/cr.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Park JM, et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy. 2016;12:547–564. doi: 10.1080/15548627.2016.1140293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Puente C, Hendrickson RC, Jiang X. Nutrient-regulated phosphorylation of ATG13 inhibits starvation-induced autophagy. J Biol Chem. 2016;291:6026–6035. doi: 10.1074/jbc.M115.689646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Egan DF, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 2015;59:285–297. doi: 10.1016/j.molcel.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Joo JH, et al. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol Cell. 2011;43:572–585. doi: 10.1016/j.molcel.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhou C, et al. Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res. 2017;27:184–201. doi: 10.1038/cr.2016.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Russell RC, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741–750. doi: 10.1038/ncb2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Joo JH, et al. The noncanonical role of ULK/ATG1 in ER-to-Golgi trafficking is essential for cellular homeostasis. Mol Cell. 2016;62:491–506. doi: 10.1016/j.molcel.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wang B, Kundu M. Canonical and noncanonical functions of ULK/Atg1. Curr Opin Cell Biol. 2017;45:47–54. doi: 10.1016/j.ceb.2017.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wang Z, Wilson WA, Fujino MA, Roach PJ. Antagonistic controls of autophagy and glycogen accumulation by Snf1p, the yeast homolog of AMP-activated protein kinase, and the cyclin-dependent kinase Pho85p. Mol Cell Biol. 2001;21:5742–5752. doi: 10.1128/MCB.21.17.5742-5752.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Meley D, et al. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. 2006;281:34870–34879. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- 169.Høyer-Hansen M, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 170.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci. 2012;125:1488–1499. doi: 10.1242/jcs.094110. [DOI] [PubMed] [Google Scholar]
- 172.Zhu Y, et al. ULK1 and JNK are involved in mitophagy incurred by LRRK2 G2019S expression. Protein Cell. 2013;4:711–721. doi: 10.1007/s13238-013-3910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Honda S, et al. Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat Commun. 2014;5:4004. doi: 10.1038/ncomms5004. [DOI] [PubMed] [Google Scholar]
- 174.Wu W, et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014;15:566–575. doi: 10.1002/embr.201438501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhu H, et al. PRKAA1/AMPKα1 is required for autophagy-dependent mitochondrial clearance during erythrocyte maturation. Autophagy. 2014;10:1522–1534. doi: 10.4161/auto.29197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Li J, et al. Mitochondrial outer-membrane E3 ligase MUL1 ubiquitinates ULK1 and regulates selenite-induced mitophagy. Autophagy. 2015;11:1216–1229. doi: 10.1080/15548627.2015.1017180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lazarou M, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yang CS, et al. The AMPK-PPARGC1A pathway is required for antimicrobial host defense through activation of autophagy. Autophagy. 2014;10:785–802. doi: 10.4161/auto.28072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Inokuchi-Shimizu S, et al. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J Clin Invest. 2014;124:3566–3578. doi: 10.1172/JCI74068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Weerasekara VK, et al. Metabolic-stress-induced rearrangement of the 14-3-3ζ interactome promotes autophagy via a ULK1- and AMPK-regulated 14-3-3ζ interaction with phosphorylated Atg9. Mol Cell Biol. 2014;34:4379–4388. doi: 10.1128/MCB.00740-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kim J, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152:290–303. doi: 10.1016/j.cell.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhang D, et al. AMPK regulates autophagy by phosphorylating BECN1 at threonine 388. Autophagy. 2016;12:1447–1459. doi: 10.1080/15548627.2016.1185576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhao Y, et al. RACK1 promotes autophagy by enhancing the Atg14L-Beclin 1-Vps34-Vps15 complex formation upon phosphorylation by AMPK. Cell Rep. 2015;13:1407–1417. doi: 10.1016/j.celrep.2015.10.011. [DOI] [PubMed] [Google Scholar]
- 184.Xu DQ, et al. PAQR3 controls autophagy by integrating AMPK signaling to enhance ATG14L-associated PI3K activity. EMBO J. 2016;35:496–514. doi: 10.15252/embj.201592864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Nguyen TN, Padman BS, Lazarou M. Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol. 2016;26:733–744. doi: 10.1016/j.tcb.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 186.Narendra DP, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Koyano F, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- 188.Kane LA, et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205:143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tian W, et al. Phosphorylation of ULK1 by AMPK regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Lett. 2015;589:1847–1854. doi: 10.1016/j.febslet.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 190.Miyamoto T, et al. Compartmentalized AMPK signaling illuminated by genetically encoded molecular sensors and actuators. Cell Rep. 2015;11:657–670. doi: 10.1016/j.celrep.2015.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Twig G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Pryde KR, Smith HL, Chau KY, Schapira AHV. PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J Cell Biol. 2016;2213:163–171. doi: 10.1083/jcb.201509003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Xie N, et al. PRKAA/AMPK restricts HBV replication through promotion of autophagic degradation. Autophagy. 2016;12:1507–1520. doi: 10.1080/15548627.2016.1191857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lv S, Xu QY, Sun EC, Zhang JK, Wu DL. Dissection and integration of the autophagy signaling network initiated by bluetongue virus infection: crucial candidates ERK1/2, Akt and AMPK. Sci Rep. 2016;6:23130. doi: 10.1038/srep23130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Fan XY, et al. Activation of the AMPK-ULK1 pathway plays an important role in autophagy during prion infection. Sci Rep. 2015;5:14728. doi: 10.1038/srep14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Brunton J, Steele S, Ziehr B, Moorman N, Kawula T. Feeding uninvited guests: mTOR and AMPK set the table for intracellular pathogens. PLoS Pathog. 2013;9:e1003552. doi: 10.1371/journal.ppat.1003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zhao J, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6:472–483. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 199.Bowman CJ, Ayer DE, Dynlacht BD. Foxk proteins repress the initiation of starvation-induced atrophy and autophagy programs. Nat Cell Biol. 2014;16:1202–1214. doi: 10.1038/ncb3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Sardiello M, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 201.Settembre C, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Settembre C, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Roczniak-Ferguson A, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5:ra42. doi: 10.1126/scisignal.2002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Li X, et al. Nucleus-translocated ACSS2 promotes gene transcription for lysosomal biogenesis and autophagy. Mol Cell. 2017;66:684–697.e9. doi: 10.1016/j.molcel.2017.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Mews P, et al. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature. 2017;17:1217–1386. doi: 10.1038/nature22405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Friis RMN, et al. Rewiring AMPK and mitochondrial retrograde signaling for metabolic control of aging and histone acetylation in respiratory-defective cells. Cell Rep. 2014;7:565–574. doi: 10.1016/j.celrep.2014.03.029. [DOI] [PubMed] [Google Scholar]
- 207.Apfeld J, O’Connor G, McDonagh T, DiStefano PS, Curtis R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 2004;18:3004–3009. doi: 10.1101/gad.1255404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Curtis R, O’Connor G, DiStefano PS. Aging networks in Caenorhabditis elegans: AMP-activated protein kinase (aak-2) links multiple aging and metabolism pathways. Aging Cell. 2006;5:119–126. doi: 10.1111/j.1474-9726.2006.00205.x. [DOI] [PubMed] [Google Scholar]
- 209.Moreno-Arriola E, El Hafidi M, Ortega-Cuéllar D, Carvajal K. AMP-activated protein kinase regulates oxidative metabolism in Caenorhabditis elegans through the NHR-49 and MDT-15 transcriptional regulators. PLoS ONE. 2016;11:e0148089. doi: 10.1371/journal.pone.0148089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Mandal S, Guptan P, Owusu-Ansah E, Banerjee U. Mitochondrial regulation of cell cycle progression during development as revealed by the tenured mutation in Drosophila. Dev Cell. 2005;9:843–854. doi: 10.1016/j.devcel.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 211.Moore AS, Holzbaur ELF. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci USA. 2016;113:E3349–E3358. doi: 10.1073/pnas.1523810113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Richter B, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci USA. 2016;113:4039–4044. doi: 10.1073/pnas.1523926113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. 2015;60:7–20. doi: 10.1016/j.molcel.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Luchsinger LL, de Almeida MJ, Corrigan DJ, Mumau M, Snoeck HW. Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature. 2016;529:528–531. doi: 10.1038/nature16500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Ho TT, et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature. 2017;543:205–210. doi: 10.1038/nature21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Forni MF, Peloggia J, Trudeau K, Shirihai O, Kowaltowski AJ. Murine mesenchymal stem cell commitment to differentiation is regulated by mitochondrial dynamics. Stem Cells. 2016;34:743–755. doi: 10.1002/stem.2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.West AP, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Buck MD, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. 2016;166:63–76. doi: 10.1016/j.cell.2016.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.An H, He L. Current understanding of metformin effect on the control of hyperglycemia in diabetes. J Endocrinol. 2016;228:R97–R106. doi: 10.1530/JOE-15-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Coughlan KA, Valentine RJ, Ruderman NB, Saha AK. AMPK activation: a therapeutic target for type 2 diabetes? Diabetes Metab Syndr Obes. 2014;7:241–253. doi: 10.2147/DMSO.S43731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Burkewitz K, Weir HJM, Mair WB. AMPK as a pro-longevity target. EXS. 2016;107:227–256. doi: 10.1007/978-3-319-43589-3_10. [DOI] [PubMed] [Google Scholar]
- 222.Burkewitz K, Zhang Y, Mair WB. AMPK at the nexus of energetics and aging. Cell Metab. 2014;20:10–25. doi: 10.1016/j.cmet.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Zhou G, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Foretz M, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Howell JJ, et al. Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab. 2017;25:463– 471. doi: 10.1016/j.cmet.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Quinn BJ, Kitagawa H, Memmott RM, Gills JJ, Dennis PA. Repositioning metformin for cancer prevention and treatment. Trends Endocrinol Metab. 2013;24:469–480. doi: 10.1016/j.tem.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 227.Svensson RU, et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med. 2016;22:1108–1119. doi: 10.1038/nm.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Shackelford DB, et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013;23:143–158. doi: 10.1016/j.ccr.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Vila IK, et al. A UBE2O-AMPKα2 axis that promotes tumor initiation and progression offers opportunities for therapy. Cancer Cell. 2017;31:208–224. doi: 10.1016/j.ccell.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Pineda CT, et al. Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell. 2015;160:715–728. doi: 10.1016/j.cell.2015.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Faubert B, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17:113–124. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Zadra G, et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol Med. 2014;6:519–538. doi: 10.1002/emmm.201302734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Lee KH, et al. Targeting energy metabolic and oncogenic signaling pathways in triple-negative breast cancer by a novel adenosine monophosphate-activated protein kinase (AMPK) activator. J Biol Chem. 2011;286:39247–39258. doi: 10.1074/jbc.M111.264598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Huang X, et al. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. 2008;412:211–221. doi: 10.1042/BJ20080557. [DOI] [PubMed] [Google Scholar]
- 235.Saito Y, Chapple RH, Lin A, Kitano A, Nakada D. AMPK protects leukemia-initiating cells in myeloid leukemias from metabolic stress in the bone marrow. Cell Stem Cell. 2015;17:585–596. doi: 10.1016/j.stem.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Chan LN, et al. Metabolic gatekeeper function of B-lymphoid transcription factors. Nature. 2017;542:479–483. doi: 10.1038/nature21076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Kishton RJ, et al. AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-ALL cell stress and survival. Cell Metab. 2016;23:649–662. doi: 10.1016/j.cmet.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333:169–174. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- 240.Mizushima N, et al. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 241.Ohsumi Y. Historical landmarks of autophagy research. Cell Res. 2014;24:9–23. doi: 10.1038/cr.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Birgisdottir ÅB, Lamark T, Johansen T. The LIR motif — crucial for selective autophagy. J Cell Sci. 2013;126:3237–3247. doi: 10.1242/jcs.126128. [DOI] [PubMed] [Google Scholar]
- 244.Laker RC, et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun. 2017;8:548. doi: 10.1038/s41467-017-00520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]